

Abstract
Nitration of both protein-bound and free tyrosine by reactive nitrogen species results in the formation of nitrotyrosine (NT). We previously reported that free NT impairs microtubule polymerization and uncouples endothelial nitric oxide synthase (eNOS) function in pulmonary artery endothelial cells (PAEC). Because microtubules modulate mitochondrial function, we hypothesized that increased NT levels during inflammation and oxidative stress will lead to mitochondrial dysfunction in PAEC. PAEC isolated from fetal lambs were exposed to varying concentrations of free NT. At low concentrations (1–10 μM), NT increased nitration of mitochondrial electron transport chain (ETC) protein subunit complexes I–V and state III oxygen consumption. Higher concentrations of NT (50 μM) caused decreased microtubule acetylation, impaired eNOS interactions with mitochondria, and decreased ETC protein levels. We also observed increases in heat shock protein-90 nitration, mitochondrial superoxide formation, and fragmentation of mitochondria in PAEC. Our data suggest that free NT accumulation may impair microtubule polymerization and exacerbate reactive oxygen species-induced cell damage by causing mitochondrial dysfunction.
Keywords: microtubules, nitric oxide, mitochondria, bioenergetics
mitochondria function as power plants for mammalian cells by generating ATP through oxidative phosphorylation. Superoxide anion (O2−) is formed as a byproduct during oxidative phosphorylation, from mitochondrial complexes I and III electron leakage (3). Increased mitochondrial O2− generation is a major contributor to oxidative stress in cells, and the adaptive mechanisms that decrease mitochondrial O2− levels, including manganese superoxide dismutase (MnSOD), are critical to cell survival. Recent studies provided evidence that nitric oxide (NO) may affect mitochondrial O2− formation by modulating oxidative phosphorylation (40) and mitochondrial biogenesis (37). This interaction is believed to play a critical role in vasomotor control.
Gao et al. demonstrated that disrupting endothelial nitric oxide synthase (eNOS) association with mitochondrial outer membrane leads to an increase in mitochondrial oxygen consumption in endothelial cells (17). NO regulates mitochondrial function by providing a tonic inhibitory control on the oxidative phosphorylation (5, 7, 11). This modulation of oxygen consumption prevents excess O2− formation, thus decreasing reactive oxygen species (ROS)-induced cell damage (3, 18).
Intimate association between mitochondria and microtubules has been described for more than 30 years (21). Microtubules interact with voltage-dependent anion channel (porin) of mitochondrial outer membrane (10) to regulate mitochondrial function (41). Agents that disturb microtubule polymerization can affect mitochondrial biogenesis (24) and mitochondrial ROS formation (51). It is likely that microtubule-active agents can disturb eNOS-mitochondrial interaction.
We previously reported that free nitrotyrosine (NT) incorporates into microtubules posttranslationally, disturbs microtubule polymerization, and uncouples eNOS function in pulmonary artery endothelial cells (PAEC) obtained from fetal lambs (48). Free NT levels rapidly build up with infection but are cleared by the kidneys. However, the longer than expected half-life in plasma suggests that most of the free NT in plasma is probably coming from degradation of nitrated proteins (15, 23). It is reasonable to assume that local concentration of free NT during infection is higher than plasma obtained distantly.
Plasma free NT levels are normally undetectable in healthy volunteers (16), but levels above 20 μM have been reported in patients with chronic renal failure and higher than 100 μM in cases with superimposed sepsis (26). Because free NT can affect polymerization of microtubules in endothelial cells (48), it is possible that increased plasma free NT levels during infections can disturb mitochondrial function. This is clinically relevant since nosocomial infection is one of the known factors that impairs lung growth, especially in premature infants, while an adequate endothelial cell function is considered to be vital to lung growth. We hypothesize that 1) elevated free NT levels decrease eNOS-mitochondria association; 2) decreased eNOS-mitochondria association results in an increase in mitochondrial oxygen consumption; and 3) the increase in oxygen consumption is associated with increased O2− formation with mitochondrial oxidative damage.
MATERIALS AND METHODS
Sheep is the most commonly used animal model to study lung disorders in human neonates, and our group has worked with this model for several years (26). Studies were performed in PAEC isolated from fetal lambs (n = 6) between passages 3 and 6 to maintain phenotype as suggested in the literature. PAEC from adult sheep and aortic endothelial cells (AEC) from fetal lambs were also used in some experiments. The use of animals was approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. PAEC were isolated from late premature fetal lambs as we previously described (26). Endothelial cell identity was confirmed by factor VIII staining and acetylated-low density lipoprotein uptake for each experiment (26).
Goat anti-hexokinase-II (Hek2), rabbit anti-eNOS, anti-heat shock protein-90 (hsp90), and anti-mitofusin-1 and -2 (MFN-1/-2) antibodies were from Santa Cruz Biotechnology (Dallas, TX). Rabbit anti-porin antibody and mouse anti-mitochondrial complex protein cocktail were from Abcam (Cambridge, MA). Rabbit anti-MnSOD antibody was from Assay Designs (Ann Arbor, MI). Rabbit anti-serine-637-phospho-dynamin-related protein 1 (DRP1) and DRP1 antibodies were from GeneTex (Irvine, CA). The mitochondria isolation kit was from ThermoFisher Scientific (Pittsburg, PA). The 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetra-ethyl-benzimid-azolyl-carbocyanine iodide (JC-1) mitochondrial membrane potential kit was from Cayman Chemical (Ann Arbor, MI). Hanks' balanced salt solution (HBSS), endothelial-serum-free medium (SFM), MitoSOX, and MitoTracker Orange were from Invitrogen (Carlsbad, CA). All other chemicals, mouse anti-α-tubulin (B5-1-2 and D-M-1-A), anti-acetylated-α-tubulin (6-11B-1), and rabbit anti-NT (N0409) antibodies were from Sigma-Aldrich (St. Louis, MO).
Preparation of NT solution.
NT stock solution (10 mM) was freshly prepared in sterile 0.1 N NaOH and filtered through a 0.22-μm syringe filter. The pH of control media was adjusted with 0.1 N NaOH as previously described (48).
Cell cultures.
Endothelial cells were cultured in DMEM with 20% FCS, l-glutamine, and 1% antibiotic/antimycotic in a humidified incubator at 37°C in 21% O2 with 5% CO2.
Mitochondria isolation.
Mitochondria were isolated from PAEC by the differential centrifugation method and a Dounce homogenizer according to the manufacturer's instructions. PAEC were pelleted by centrifuging at ∼850 g for 2 min, supernatant was removed, and mitochondria were isolated from the pellet using reagent from the kit.
Fluorescent staining.
Endothelial cells were plated on four-well chamber slides until ∼60% confluence, and then free NT (0, 1, 5, 10, or 50 μM) was added to selected wells for overnight incubation. ATP (10−5 M) was added for 10 min to activate the cells. Following ATP treatment, culture medium was removed, and cells were washed with HBSS. MitoTracker Orange (200 nM) was then applied for 30 min. Cells were again washed in HBSS and fixed in 4% formalin for 5 min at room temperature. Mitochondrial morphology was imaged by a technician blinded to the treatment using a fluorescent microscope (×60 objective oil lens). Individual mitochondria (particle) were subjected to analysis by ImageJ after convolution, despeckle, and threshold at 3% to acquire value for circularity (4π·area−1·perimeter−2) and length of major and minor axes under the function of “analyze particles” after setting measurements at “shape descriptors.” From these values, form factor (FF: circularity−1) and aspect ratio (AR: major axis/minor axis) were calculated. High AR values represent elongated mitochondria (less fragmented), and high FF values indicate increased mitochondrial complexity (4). Low AR value (fragmented mitochondria) can lead to apoptosis (54).
Immunoblotting and immunoprecipitation.
Endothelial cells were cultured with/without the presence of free NT overnight before immunoblotting or immunoprecipitation. PAEC whole cell lysates were separated and blotted using mouse anti-α-tubulin (D-M-1-A, 1:1,000) and mouse anti-acetylated-α-tubulin (1:1,000) to study the stability of polymerized microtubules. PAEC cell lysates were immunoprecipitated using rabbit anti-hsp90 antibody and then blotted with either mouse anti-hsp90 antibody (1:500) or rabbit anti-NT antibody (1:1,000) to study the extent of hsp90 nitration. Cell lysates obtained after incubation with different levels of NT (0, 1, 5, and 10 μM) for 48 h were immunoprecipitated with 3 μg mouse anti-mitochondrial complex protein cocktail, and the membranes were blotted with either anti-3-NT antibody or anti-mitochondrial complex protein cocktail (1:2,000) to study the extent of complex protein nitration.
To study the interaction between eNOS and microtubules, endothelial cells were washed with microtubule-stabilizing buffer (0.1 M PIPES, pH 7.0, 1 mM EGTA, 1 mM MgSO4, and 2 M glycerol) and then lysed in microtubule lysis buffer (25 mM Tris·HCl, pH 7.4, 0.4 M NaCl, and 0.5% sodium dodecyl sulfate) (52) before immunoprecipitation with anti-α-tubulin antibody (B5-1-2). After separation by electrophoresis, proteins were blotted with rabbit anti-eNOS (1:1,000), mouse anti-hsp90, or mouse anti-α-tubulin (D-M-1-A, 1:1,000) antibodies. Whole cell lysates were blotted for DRP1 (1:500), serine-637 (sheep serine-630), phospho-DRP1 (1:500), and MFN-1/2 (1:500) to study the effect of NT on mitochondrial fission and fusion. Whole cell lysates were blotted for Hek2 (1:1,000) and mitochondrial electron transport chain (ETC) complex antibody cocktail to study protein levels of enzymes involved in bioenergetics.
Mitochondria isolated from PAEC were lysed in radioimmunoprecipitation assay buffer (48) by vigorous vortexing and centrifuging at 13,000 revolutions/min. eNOS-associated proteins were immunoprecipitated using rabbit anti-eNOS antibody and protein A-Sepharose beads. The immunoprecipitated proteins were separated by 10% SDS-PAGE. Rabbit anti-eNOS antibody (1:2,000) and anti-porin antibody (1:500) were used as primary antibodies for immunoblots. Lysates were also immunoprecipitated with rabbit anti-MnSOD antibody and blotted with both rabbit anti-MnSOD (1:1,000) and anti-NT (1:1,000) antibodies to study MnSOD nitration. ImageJ was used to analyze the band densities for all immunoblots, and immunoprecipitated protein (eNOS or MnSOD) was used as loading control.
Mitochondrial DNA copy number.
Primers for sheep NADH dehydrogenase 1 (ND1; ENSOARG00000000006.1; forward: GGAGGACTGAACCAAACCCA; reverse: GCCTGAGGATAGGGGAATGC) and -2 (ND2; ENSOARG00000000010; forward: TACTTCAACCCATCGCCGAC; reverse: ACGGCTAGGCTTGATATGGC) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; ENSOARG00000004849; forward: CGTGAAGGGCTGTTTACCGA; reverse: ACGTGTCCGTTGTGGATCTG) DNA were designed according to sheep sequence. The polymerase chain reaction cycle started at 95°C for 3 min then 40 cycles of 95°C for 10 s followed by 60°C for 30 s, then 95°C for 1 min, and ended at 55°C for 1 min. The number of the threshold cycle (Ct) for each target mitochondrial DNA (mtDNA) was normalized to the corresponding Ct of GAPDH nuclear DNA to obtain the ΔCt, and then 2−ΔΔCt was calculated against the corresponding control PAEC to obtain the relative mtDNA copy number.
Oxygen consumption.
PAEC treated overnight with/without NT were trypsinized, washed with HBSS, and then suspended in endothelial-SFM. Cells (2×106 in 200 μl) were used for studies with Oxygraph (Hansatech Instruments, Norfolk, UK) containing 174 μl of mitochondrial respiration medium (0.5 mM EGTA, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM K-HEPES, 1 g/l fatty acid-free BSA, and 200 mM sucrose, pH 7.1) at 37°C. The cells were permeabilized with 2 μl digitonin (10 mg/ml) for 2 min. Succinate (5 mM), 4 μl, was injected to stimulate state II respiration followed by 20 μl ADP (50 mM) to stimulate state III respiration. Oxygen consumption (nmol·s−1·2 × 10−6 cells) was continuously recorded, and slopes for state III respiration were calculated for comparisons.
Mitochondrial ROS formation.
Mitochondrial ROS formation was studied using MitoSOX Red (43). PAEC were incubated with/without 50 μM NT overnight at ∼60% confluence. MitoSOX Red (5 μM) was added for 15 min at 37°C, with/without 10−5 M ATP, and then cells were washed two times with HBSS followed by 4% formalin fixation. Polyethylene glycol-conjugated Cu,Zn-SOD (PEG-SOD, 300 U/ml) was used to confirm the signal was mainly due to O2−. Fluorescence was imaged with excitation and emission at 510 and 590 nm, respectively. Fluorescence was quantified using MetaView software and expressed as relative light units.
Mitochondrial membrane potential by flow cytometry.
JC-1 selectively enters healthy mitochondria, with high membrane potential (ΔΨm), to form orange-red fluorescing aggregates but exists as green fluorescing monomers in cells with unhealthy mitochondria (44). The JC-1 orange-red/green ratio was thus used in this study to evaluate ΔΨm. PAEC with/without 50 μM NT overnight treatment were stimulated with ATP (10−5 M) for 10 min and then stained with JC-1 for 15 min before trypsin treatment. The cells were washed with assay buffer two times and analyzed on a FACS Calibur (Becton-Dickinson, San Jose, CA) flow cytometer using CellQuest Pro software. A total of 30,000 cells were measured for each sample.
MnSOD activity.
We measured MnSOD activity by the colorimetric method using a Superoxide Dismutase Assay Kit (Cayman Chemical), based on the principle of inhibition of O2−-mediated tetrazolium conversion into formazan by SOD, according to the manufacturer's instruction. PAEC with/without 50 μM NT overnight treatment were scraped into 20 mM HEPES buffer (pH 7.2, containing 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose). The cell suspensions were centrifuged at 1,500 g for 5 min at 4°C. MnSOD specific activity was measured by absorbance at 450 nm after the addition of KCN, which inhibits Cu,Zn-SOD and was normalized to the protein content.
Statistical analysis.
Data were analyzed by MedCalc Statistical Software version 15.2.1 (MedCalc Software, Ostend, Belgium; http://www.medcalc.org) and GraphPad Prism 6.01 and expressed as means ± SE. One-way ANOVA with Student-Newman-Keul's post hoc test was used, after Levene's test for equality of error variances, for comparison of more than two groups. Student's t-test, or Mann-Whitney U-test, was used for comparing two groups wherever appropriate. A P value <0.05 was considered significant.
RESULTS
Free NT destabilizes microtubules and increases the binding of hsp90 and eNOS to microtubules.
Acetylation of α-tubulin stabilizes polymerized microtubules and promotes intracellular protein transport (29, 42). Free NT did not change the α-tubulin protein level but decreased α-tubulin acetylation in PAEC (0.45 ± 0.04 vs. 0.16 ± 0.01, n = 3, P < 0.05, Fig. 1A). This is consistent with our previous report that free NT depolymerizes microtubules (48). Free NT also increased hsp90 nitration (Fig. 1B), which may contribute to decreased association between eNOS and hsp90 (48). The increased association between eNOS and α-tubulin, and between hsp90 and α-tubulin (Fig. 1C), together with the decreased association between eNOS and hsp90 we reported previously (48) suggested that free NT prevents normal chaperone interaction between hsp90 and eNOS.
Fig. 1.

Free nitrotyrosine (NT) decreases microtubule acetylation but increases endothelial nitric oxide synthase (eNOS)-tubulin and heat shock protein 90 (hsp90)-tubulin associations and hsp90 nitration. Immunoblot of pulmonary artery endothelial cell (PAEC) lysates showed free NT (50 μM) decreased acetylation of α-tubulin (A). Immunoprecipitation using hsp90 antibody shows that free NT increases hsp90 nitration (B). Free NT increases both eNOS-tubulin (□) and hsp90-tubulin (■) associations (C). *P < 0.05 between groups; †P < 0.01 between groups for eNOS-tubulin association; and ‡P < 0.05 between groups for hsp90-tubulin association.
Free NT decreases eNOS interaction with mitochondria.
The interaction between porin and eNOS is important for regulation of eNOS activity (46) and is especially important for vascular endothelial cell function. Immunoprecipitation of mitochondrial lysates showed decreased association between porin and eNOS in the presence of free NT after ATP stimulation (47.7 ± 14.1%, n = 3, P < 0.05, Fig. 2). According to these observations, we speculate that free NT decreases eNOS docking to the mitochondrial outer membrane during NOS stimulation, secondary to impaired targeting by hsp90 and/or by the microtubules.
Fig. 2.
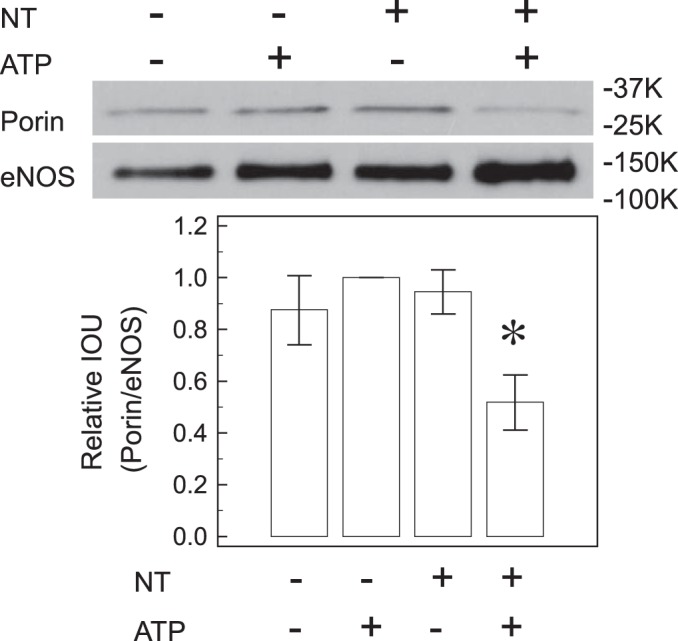
Free NT decreases porin-eNOS association. Immunoprecipitation of eNOS followed by immunoblotting with porin antibody showed that free NT decreases porin-eNOS association after ATP stimulation, indicating that free NT decreases eNOS interaction with mitochondrial outer membrane after activation. The porin-to-eNOS ratio for ATP-treated control was set as “1” for each experiment. *P < 0.05 from the other 3 groups.
Free NT increases mitochondrial oxygen consumption.
The state III oxygen consumption of PAEC increased in the presence of free NT (Fig. 3A). When the NT level reached 10 μM, the state III oxygen consumption by PAEC doubled (0.16 ± 0.03 cells vs. 0.08 ± 0.01 nmol·s−1·2 × 10−6 cells, P < 0.05, n = 5), indicating higher oxygen requirement for ATP synthesis.
Fig. 3.

Free NT alters mitochondrial function. A: free NT increases state III oxygen consumption in PAEC. Oxygen consumption was studied after stimulation with 5 mM succinate. State III oxygen consumption increased after both 1 and 10 μM free NT (inset). The slope of oxygen consumption was doubled by 10 μM free NT. B: mitochondrial electron transport chain complex proteins I–V were immunoprecipitated with the mixture of 5 antibodies, followed by immunoblotting for NT. At low levels (1, 5, and 10 μM) of free NT, there was no significant change in the level of mitochondrial complex proteins (data not shown). However, nitration of all 5 complex proteins increased significantly (B). The largest increase in nitration was seen in complex 4. C: higher concentration of free NT (50 μm) decreases the levels of mitochondrial electron transport chain proteins in whole cell lysates determined by Western blot, but not hexokinase II. D: high concentration of free NT (50 μM) similarly decreases the levels of electron transport chain proteins in fetal aortic endothelial cells (AEC) and adult PAEC whole cell lysates. *P < 0.05 compared with control.
Under a low level of free NT (1–10 μM) there was no significant difference in mitochondrial ETC proteins and Hek2 in whole cell lysates (data not shown), but nitration of all five ETC proteins increased significantly by immunoprecipitation using mitochondrial ETC antibody cocktail to pull down proteins and then blotted with nitrotyrosine antibody (Fig. 3B). When the NT level reached 50 μM, the levels of all five complex proteins of the mitochondrial ETC were decreased in PAEC whole cell lysates, as determined by Western blots; however, there was no change in the level of Hek2, a key regulatory enzyme in the glycolytic pathway (Fig. 3C). Similar changes were also observed in fetal aortic endothelial cells and adult PAEC (Fig. 3D) whole cell lysates except ETC levels started to decrease at 10 μM NT in adult PAEC. These data indicate that different types of endothelial cells may have different susceptibility to free NT.
Free NT increases mitochondrial ROS formation.
We previously showed that free NT increased ROS formation in PAEC probably through eNOS uncoupling (48). In this study, we observed that free NT significantly increased MitoSOX fluorescence in PAEC (n = 7, P < 0.05 by 2-way ANOVA, Fig. 4), indicating increased mitochondrial ROS formation. PEG-SOD significantly decreased the ROS signal, suggesting O2− is the major mitochondrial ROS that generated the MitoSOX fluorescence.
Fig. 4.

Free NT increases mitochondrial superoxide formation. MitoSOX fluorescence was low in control PAEC both without (A) and with (B) ATP stimulation. The presence of free NT increased MitoSOX fluorescence both without (C) or with (D) ATP stimulation. MitoSOX fluorescence in PAEC increased significantly with free NT, and polyethylene glycol-conjugated Cu,Zn-SOD (PEG-SOD) quenched the signals (E). *P < 0.05 compared with control without ATP stimulation (white bar); #P < 0.05 compared with control with ATP stimulation (black bar); **P < 0.05 for PEG-SOD treatment (gray bar) compared with control and free NT-treated PAEC with and without ATP stimulation.
Free NT decreases ΔΨm.
Free NT decreased the JC-1 orange-red/green ratio in PAEC (n = 5, P < 0.05, Fig. 5), indicating a decrease in ΔΨm, which could lead to increased mitochondrial damage (44).
Fig. 5.
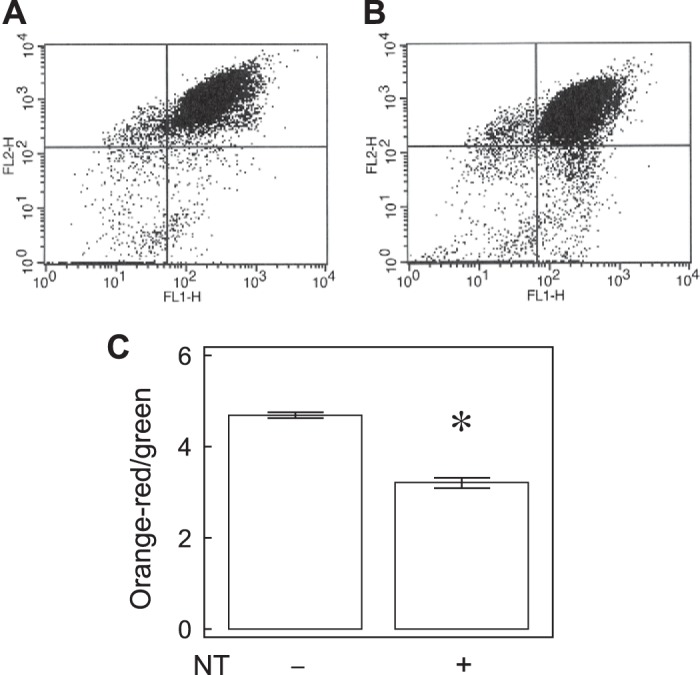
Free NT decreases mitochondrial membrane potential. Most of the cells (95.5 ± 0.2%) were healthy and occupy the top right quadrant, whereas the unhealthy or dying cells occupy the bottom right quadrant (A). NT decreased the percentages (94.1 ± 0.4%) in the top right quadrant (B). NT significantly decreased the excitation (Ex) 560/emission (Em) 595-to-Ex485/Em535 ratios (C, n = 5, P < 0.01). *P < 0.01 compared with control.
Mitochondria are more fragmented in the presence of free NT.
The MitoTracker Orange fluorescence was mainly distributed in the perinuclear zone after ATP stimulation. Free NT-treated PAEC had more fragmented mitochondria with lower AR when the level reached 1–5 μM and were less complex with lower FF when the level reached 10 μM, suggesting mitochondrial dysfunction or damage in the presence of free NT (Fig. 6A) (4, 25) and further supporting the JC-1 observations. Similar changes were also seen in fetal aortic endothelial cells (Fig. 6B) and adult PAEC (Fig. 6C). Phosphorylation of DRP-1 at serine-630 decreased (0.03 ± 0.01 vs. 0.32 ± 0.03, n = 3, P < 0.05, Fig. 6D) while total DRP-1 levels increased (2.05 ± 0.03 vs. 1.45 ± 0.04, n = 3, P < 0.05) in the presence of 50 μM free NT. Free NT also decreased the levels of MFN-1 (0.04 ± 0.00 vs. 0.13 ± 0.02, n = 3, P < 0.05) and MFN-2 (0.03 ± 0.00 vs. 0.15 ± 0.03, n = 3, P < 0.05), which are consistent with the mitochondrial morphological changes. There was no change in DRP-1 levels, but similar changes in phospho-DRP-1, MFN-1, and MFN-2 were observed in adult PAEC and fetal aortic endothelial cells by 50 μM free NT.
Fig. 6.

Free NT affects mitochondrial morphology. Mitochondrial morphology was analyzed after MitoTracker Orange fluorescent staining and processed by Image J. Mitochondria became more fragmented, but less complex, in the presence of free NT, and the severity escalated as free NT level increased (A). Similar changes were also observed in fetal AEC (B) and adult PAEC (C) with difference in susceptibility. The level of dynamin-related protein-1 (DRP1) increased, but the level of phosphorylated DRP1 decreased by 50 μM free NT, indicating less mitochondrial fusion. Free NT also decreased the levels of mitofusin-1 and -2, which are required to facilitate mitochondrial fusion (D). AR, aspect ratio; FF, form factor.
Mitochondrial DNA copy number increases after NT treatment.
Relative mtDNA copy numbers increased by 2.87 ± 0.38-fold for ND1 (n = 4, P < 0.05) and 3.13 ± 0.65-fold for ND2 (n = 4, P < 0.05) in the presence of 50 μM NT, indicating a response to mitochondrial damage.
No change in MnSOD activity and nitration after NT treatment.
We did not see changes in either nitration of MnSOD protein (1.02 ± 0.51 vs. 1.48 ± 0.28, n = 3, P = 0.45) or MnSOD activity (1.22 ± 0.15 vs. 1.19 ± 0.17 U/mg protein, n = 4, P = 0.87) after free NT treatment. These results suggest that the increase in MitoSOX fluorescence with exposure to free NT is not due to changes in MnSOD activity.
DISCUSSION
Nitrotyrosine was generally regarded as a signature for nitrosative stress until Kooy and Lewis (27) reported that free NT can also impair the vascular response to adrenoceptor agonists. Mihm et al. later reported that free NT causes vascular endothelial dysfunction and DNA damage (34). It is cytotoxic to mouse endothelial cells with LC50 ∼98 μM (35). Our group has worked on neonatal lung disorders using sheep as the animal model for years. We recently reported that free NT uncouples eNOS and impairs in vitro angiogenesis of PAEC at levels as low as 1 μM (48). In that report we also provided evidence that NT disturbs polymerization of microtubules with increased apoptosis.
Acetylated α-tubulin is abundant in polymerized tubulins (2, 5, 38, 49). This acetylation not only plays an important role in intracellular protein (42) and organelle transport (20, 39) but also contributes to eNOS serine-1177 phosphorylation (47) and hsp90-Akt/PKB interaction (5), all of which are important to angiogenesis by enhancing NO production. Judging from our previous report that free NT uncouples eNOS activity, it is plausible to assume free NT accumulation during inflammation could also impair mitochondrial function. Free NT was first shown to inhibit mitochondrial function in neurons at levels as high as 125 μM (31). In this study similar findings were observed in endothelial cells, with high levels of free NT (10–50 μM) leading to fragmentation of mitochondria.
A wide range of free NT has been used (1 nM∼250 μM) to study NT effects on cells or organs (27, 31, 34, 35). In this study, we showed that free NT decreased α-tubulin acetylation and impaired mitochondrial function in endothelial cells at levels as low as 1 μM, which is a level commonly seen during severe infection (26). The different susceptibilities observed in our studies compared with another report (31) may be secondary to either different phenotypes or different developmental stage. This may be even more important in young infants since impaired mitochondrial function, angiogenesis, and eNOS activity can lead to poor lung growth (1) while nosocomial infection is an important contributing factor to bronchopulmonary dysplasia in prematurely born infants. We also observed that the effect of free NT is not limited to fetal PAEC but also impair mitochondrial function in AEC from fetal sheep and PAEC from adult sheep.
Acetylated α-tubulin is an inherent component of mitochondrial membrane that interacts with porin (10). Porin is a global regulator of mitochondrial function (8). The interaction between microtubules and porin helps to maintain ΔΨm and regulates mitochondrial metabolism (14). We observed decreased α-tubulin acetylation and porin-eNOS association, and increased mitochondrial O2− formation after NT treatment, suggesting that free NT can impair endothelial function by disturbing reversible polymerization of microtubules and mitochondrial function.
In the present study, we observed that increased free NT levels impair eNOS-mitochondrial interaction. These changes were accompanied by disturbed mitochondrial function (19) as shown by decreased ΔΨm and increased mitochondrial fragmentation. Decreased ΔΨm and ATP synthesis usually precede apoptosis and necrosis (30). Even at low levels of free NT (1–10 μM), ETC proteins were nitrated, a process known to decrease ATP formation (33). At a high level of NT (50 μM), we observed both an increase in nitration and a decrease in the levels of all five ETC complex proteins. The increase in DRP1 with decreases in phospho-DRP1 and MFN protein levels suggests impaired mitochondrial fusion and fission at high free NT levels. These findings indicated that high levels of free NT impair mitochondrial morphology and function in endothelial cells.
NO regulates mitochondrial oxygen consumption in a concentration-dependent manner (5, 11). Our data suggest that NT decreases eNOS-mitochondria association, which may decrease NO availability for mitochondria. This hypothesis is supported by the increase in state III oxygen consumption observed in the presence of NT in our studies. The decreased NO availability may also explain the increase in mitochondrial ROS production, collapse of ΔΨm (9), and mitochondrial fragmentation. Previous studies suggested that eNOS-mitochondria association provides a milieu of appropriate NO concentration that regulates oxygen consumption and ROS production in endothelial cells (17).
hsp90 is an important regulator of NO production and angiogenesis. Prolonged NT exposure increases hsp90 nitration in PAEC and further aggravates eNOS uncoupling (53, 54). Similar to the report by Su (45) that microtubule-active agents modified NO production, we previously also reported that NT decreases NO production (48). However, contrary to Su's report, we observed increased eNOS-α-tubulin association in the PAEC. The significance of this increased association with respect to eNOS uncoupling and decreased eNOS docking to mitochondria in the presence of free NT requires further investigation.
Eiserich reported that NT incorporation into microtubules decreases the binding of dynein to microtubules (13). Acetylation of α-tubulin is known to be associated with polymerization of microtubules (36). Dompierre reported that increased α-tubulin acetylation promotes the association of microtubules with dynein and kinesin (12). Dynein and kinesin are microtubule-associated motors that transport cargo along microtubules (22). We observed that free NT decreases α-tubulin acetylation, suggesting that NT may contribute to decreased dynein binding to microtubules (13).
Contrary to the report by Giustiniani that microtubule acetylation favors hsp90 recruitment to microtubules (20) we found an increased hsp90-microtubule association after NT exposure. The reason for these different observations in the two studies requires further investigation. One possible reason for the difference may be the use of trichostatin A to increase microtubule acetylation in Giustiniani's report, which also is known to affect other cell functions (28). Another possible explanation is that we used PAEC, whereas Guistiniani used nonendothelial cell lines, which may respond in different ways. We also observed that NT increases eNOS-microtubule association. The increased eNOS-microtubule binding by NT may decrease eNOS docking to mitochondria by decreasing its translocation.
There are two possible explanations for the increased mitochondrial ROS formation observed in our study, decreased MnSOD activity or increased electron leak from oxidative phosphorylation. We did not see changes in either MnSOD nitration (32) or MnSOD activity by NT. Our findings suggest that the increased mitochondrial ROS formation is primarily due to impaired regulation of oxidative phosphorylation.
In summary, our data show that prolonged free NT exposure impairs microtubule function and eNOS docking to mitochondria in PAEC, resulting in increased mitochondrial ROS production and mitochondrial dysfunction. Low levels of NT (1–10 μM) impair oxidative phosphorylation, whereas high levels (50 μM) lead to impaired mitochondrial biogenesis. Our observations provide another NO-mediated mechanism by which severe infections can lead to impaired endothelial cell function. This mechanism may contribute to the impaired lung growth observed with nosocomial infections in premature infants (1, 50). However, there are some limitations that should be considered in the interpretation of our results. Our study was conducted in vitro using isolated endothelial cells, and the responses may be different phenotypically from what will happen in vivo in the vascular endothelium.
GRANTS
The study was supported by CRI Start-up funds from the Children's Hospital of Wisconsin; a 2013 CTSI Pilot Award (UL1TR000055, Advancing a Healthier Wisconsin); NIH Grants R03-HD-073274 (R.-J. Teng), R03-HD-65841, and R01-HL-057268; and a Muma Endowed Chair in Neonatology and Children's Research Institute (to G. G. Konduri).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: R.-J.T., T.-J.W., and G.G.K. conception and design of research; R.-J.T., T.-J.W., and A.J.A. performed experiments; R.-J.T. and T.-J.W. analyzed data; R.-J.T., T.-J.W., A.J.A., and G.G.K. interpreted results of experiments; R.-J.T. prepared figures; R.-J.T., T.-J.W., and A.J.A. drafted manuscript; R.-J.T., T.-J.W., A.J.A., and G.G.K. edited and revised manuscript; R.-J.T., T.-J.W., A.J.A., and G.G.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Ivane Bakhutashvili for performing the Oxygraph study.
REFERENCES
- 1.Abman SH. Bronchopulmonary dysplasia: “a vascular hypothesis.” Am J Respir Crit Care Med 164: 1755–1756, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Black MM, Keyser P. Acetylation of alpha-tubulin in cultured neurons and the induction of alpha-tubulin acetylation in PC12 cells by treatment with nerve growth factor. J Neurosci 7: 1833–1842, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755–767, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol 300: C447–C455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta 1504: 46–57, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta 1658: 44–49, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Brunori M, Giuffrè A, Sarti P, Stubauer G, Wilson MT. Nitric oxide and cellular respiration. Cell Mol Life Sci 56: 549–557, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cambray-Deakin MA, Robson SJ, Burgoyne RD. Colocalisation of acetylated microtubules, glial filaments, and mitochondria in astrocytes in vitro. Cytoskeleton 10: 438–449, 1988. [DOI] [PubMed] [Google Scholar]
- 9.Caro AA, Cederbaum AI. Ca2+-dependent and independent mitochondrial damage in HepG2 cells that overexpress CYP2E1. Arch Biochem Biophys 408: 162–170, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Carré M, André N, Carles G, Borghi H, Brichese L, Briand C, Braguer D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem 277: 33664–33669, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Clementi E, Brown GC, Foxwell N, Moncada S. On the mechanism by which vascular endothelial cells regulate their oxygen consumption. Proc Natl Acad Sci 96: 1559–1562, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci 21: 3571–3583, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eiserich JP, Estévez AG, Bamberg TV, Ye YZ, Chumley PH, Beckman JS, Freeman BA. Microtubule dysfunction by posttranslational nitrotyrosination of α-tubulin: A nitric oxide-dependent mechanism of cellular injury. Proc Natl Acad Sci USA 96: 6365–6370, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J Cell Biol 190: 363–375, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frost MT, Barry Halliwell B, Moore KP. Analysis of free and protein-bound nitrotyrosine in human plasma by a gas chromatography/mass spectrometry method that avoids nitration artifacts. Biochem J 345: 453–458, 2000. [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuyama N, Takebayashi Y, Hida M, Ischda H, Ichimori K, Nakazawa H. Clinical evidence of peroxynitrite formation in chronic renal failure patients with septic shock. Free Radic Biol Med 22: 771–774, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Gao S, Chen J, Brodsky SV, Huang H, Adler S, Lee JH, Dhadwal N, Cohen-Gould L, Gross SS, Goligorsky MS. Docking of endothelial nitric oxide synthase (eNOS) to the mitochondrial outer membrane: a pentabasic amino acid sequence in the autoinhibitory domain of eNOS targets a proteinase K-cleavable peptide on the cytoplasmic face of mitochondria. J Biol Chem 279: 15968–15974, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Giulivi C, Kato K, Cooper CE. Nitric oxide regulation of mitochondrial oxygen consumption I: cellular physiology. Am J Physiol Cell Physiol 291: C1225–C1231, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Giustiniani J, Couloubaly S, Baillet A, Pourci ML, Cantaloube I, Fourniat C, Paul JL, Poüs C. Basal endothelial nitric oxide synthase (eNOS) phosphorylation on ser1177 occurs in a stable microtubule- and tubulin acetylation-dependent manner. Exp Cell Res 315: 3509–3520, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Giustiniani J, Daire V, Cantaloube I, Durand G, Poüs C, Perdiz D, Baillet A. Tubulin acetylation favors Hsp90 recruitment to microtubules and stimulates the signaling function of the Hsp90 clients Akt/PKB and p53. Cell Signal 21: 529–539, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Heggeness MH, Simon M, Sogner SJ. Association of mitochondria with microtubules in cultured cells. Proc Natl Acad Sci USA 75: 3863–3866, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279: 519–526, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Kamisaki Y, Wada K, Ataka M, Yamada Y, Nakamoto K, Ashida K, Kishimoto Y. Lipopolysaccharide-induced increase in plasma nitrotyrosine concentrations in rats. BBA-Mol Basis Dis 1362: 24–28, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Karbowski M, Spodnik JH, Teranishi M, Wozniak M, Nishizawa Y, Usukura J, Wakabayashi T. Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J Cell Sci 114: 281–291, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Diff 10: 870–880, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Konduri GG, Bakhutashvili I, Eis A, Pritchard K Jr. Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 292: H1812–H1820, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Kooy NW, Lewis SJ. Nitrotyrosine attenuates the hemodynamic effects of adrenoceptor agonists in vivo: relevance to the pathophysiology of peroxynitrite. Eur J Pharmacol 310: 155–161, 1996. [DOI] [PubMed] [Google Scholar]
- 28.Kovacs JJ, Murphy PJM, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt TP, Yao WB. HDAC6 regulates hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18: 601–607, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Le Dizet M, Piperno G. Cytoplasmic microtubules containing acetylated alpha-tubulin in Chlamydomonas reinhardtii: spatial arrangement and properties. J Cell Biol 103: 13–22, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator - thinking outside the box. Biochim Biophys Acta 1762: 181–190, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Ma TC, Mihm MJ, Bauer JA, Hoyt KR. Bioenergetic and oxidative effects of free 3-nitrotyrosine in culture: selective vulnerability of dopaminergic neurons and increased sensitivity of non-dopaminergic neurons to dopamine oxidation. J Neurochem 103: 131–144, 2007. [DOI] [PubMed] [Google Scholar]
- 32.MacMillan-Crow L, Cruthirds DL, Ahki KM, Sanders PW, Thompson JA. Mitochondrial tyrosine nitration precedes chronic allograft nephropathy. Free Radic Biol Med 31: 1603–1608, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Maldonado EN, Patnaik J, Mullins MR, Lematers JJ. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res 70: 10192–10201, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mihm MJ, Jing L, Bauer JA. Nitrotyrosine causes selective vascular endothelial dysfunction and DNA damage. J Cardiovasc Pharmacol 36: 182–187, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Mihm MJ, Wattanapitayakul SK, Piao SF, Hoyt DG, Bauer JA. Effects of angiotensin II on vascular endothelial cells: formation of receptor-mediated reactive nitrogen species. Biochem Pharmacol 65: 1189–1197, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Nam HJ, Kang JK, Kim SK, Ahn KJ, Seok H, Park SJ, Chang JS, Potholakis C, Lamont JT, Kim H. Clostridium difficile toxin A decreases acetylation of tubulin, leading to microtubule depolymerization through activation of histone deacetylase 6, and this mediates acute inflammation. J Biol Chem 285: 32888–32896, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA 101: 16507–1651, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orrenius S, Ankarcrona M, Nicotera P. Mechanisms of calcium-related cell death. Adv Neurol 71: 137–151, 1966. [PubMed] [Google Scholar]
- 39.Piperno G, LeDizet M, Chang X. Microtubules containing acetylated α-tubulin in mammalian cells in culture. J Cell Biol 104: 289–302, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poderoso JJ. The formation of peroxynitrite in the applied physiology of mitochondrial nitric oxide. Arch Biochem Biophys 484: 214–220, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Rappaport L, Oliviero P, Samuel JL. Cytoskeleton and mitochondrial morphology and function. Mol Cell Biochem 184: 101–105, 1998. [PubMed] [Google Scholar]
- 42.Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig Verhey KJ J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16: 2166–2172, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protocols 3: 941–947, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 411: 77–82, 1997. [DOI] [PubMed] [Google Scholar]
- 45.Su Y, Zharikov SI, Block ER. Microtubule-active agents modify nitric oxide production in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 282: L1183–L1189, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Sun J, Liao JK. Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc Natl Acad Sci USA 99: 13108–13113, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takemura R, Okabe S, Umeyama T, Kanai Y, Cowan NJ, Hirokawa N. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J Cell Sci 103: 953–964, 1992. [DOI] [PubMed] [Google Scholar]
- 48.Teng RJ, Wu TJ, Bisig CG, Eis A, Pritchard KA, Konduri GG. Nitrotyrosine impairs angiogenesis and uncouples eNOS activity of pulmonary artery endothelial cells isolated from developing sheep lungs. Pediatr Res 69: 112–117, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17: 3–8, 1997. [DOI] [PubMed] [Google Scholar]
- 50.Van Marter LJ, Dammann O, Allred EN, Leviton A, Pagano M, Moore M, Martin C. Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr 140: 171–176, 2002. [DOI] [PubMed] [Google Scholar]
- 51.Varbiro G, Veres B, Gallyas F Jr, Sumegi B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med 31: 548–558, 2001. [DOI] [PubMed] [Google Scholar]
- 52.Vaughan EE, Geiger RC, Miller AM, Loh-Marley PL, Suzuki T, Miyata N, Dean DA. Microtubule acetylation through HDAC6 inhibition results in increased transfection efficiency. Mol Ther 16: 1841–1827, 2008. [DOI] [PubMed] [Google Scholar]
- 53.Ye Y, Quijano C, Robinson KM, Ricart KC, Strayer AL, Sahawneh MA. Prevention of peroxynitrite-induced apoptosis of motor neurons and PC12 cells by tyrosine-containing peptides. J Biol Chem 282: 6324–6337, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 79: 341–351, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]