

Abstract
Renin progenitors appear early and are found in multiple tissues throughout the embryo. Besides their well known role in blood pressure and fluid homeostasis, renin progenitors participate in tissue morphogenesis, repair, and regeneration, and may integrate immune and endocrine responses. In the bone marrow, renin cells offer clues to understand normal and neoplastic hematopoiesis.
In the traditional, endocrine view of the renin angiotensin system (RAS), renin is synthesized and released by a few cells located at the tip of afferent arterioles at the entrance to the glomeruli, the juxtaglomerular (JG) cells (FIGURE 1). Upon release from JG cells, renin reaches the circulation and triggers an enzymatic cascade that culminates in the generation of angiotensin II (Ang II), a powerful vasoconstrictor and regulator of sodium reabsorption, responsible for most of the known actions of the RAS, including control of renal hemodynamics, blood pressure, and fluid-electrolyte balance. Tight regulation of renin release is therefore crucial for the precise operation of the RAS and maintenance of homeostasis. In addition to this classical view, it is becoming increasingly evident that other cells outside the kidney express renin and that the function of renin cells may not be limited to the regulation of blood pressure and fluid-electrolyte homeostasis. This brief review will explore the origin, regulation, and fate of renin cells in the kidney and advance the hypotheses that renin cells may have additional homeostatic roles in renal and extra-renal tissues.
FIGURE. 1.

Renin expression in the developing kidney and in response to homeostatic stress
During early development, renin cells are found along the length of the afferent arterioles and interlobular arteries, inside the glomeruli and in the kidney interstitium (top), whereas in adult life they are restricted to the juxtaglomerular (JG) area (bottom). Throughout development, renin cells differentiate into vascular smooth muscle cells (VSMC), mesangial cells, and pericytes, and those cells retain the ability to reexpress renin in response to homeostatic stress resembling the fetal pattern. Right: representative immunostaining for renin (in brown) in the embryonic kidney at 15.5 days of gestation (top) and in the adult kidney (bottom). CEI, converting enzyme inhibitors; Neo UUO, neonatal unilateral ureteral obstruction; RAS, renin angiotensin system.
Development of Renin Cells
In the mammalian embryo, renin cells emerge before organogenesis has been initiated. As development progresses, the cells appear in multiple organs and tissues, such as adrenal glands, testis, sympathetic ganglia, cartilage, stomach, and thymus (10, 44). Their appearance in the metanephros, the definitive kidney of mammals, occurs later than in most other embryonic tissues. For instance, renin cells in the fetal adrenal are numerous and precede their appearance in the kidney (20).
The progenitors for renin cells in the kidney have recently been identified (45) (FIGURE 2). Early in the development of the metanephric kidney (E12), three major structures can be recognized: 1) the ureteric bud that gives rise to the collecting duct and the ureter; 2) the condensing cap mesenchyme, an inner layer immediately adjacent to the ureteric bud that gives rise to most of the epithelial nephron including glomerular epithelium, proximal tubules, loops of Henle, and distal and connecting tubules; and 3) the loose mesenchyme, an outer layer that harbors Foxd1+ stromal cells and precursors for endothelial cells. The Foxd1+ stromal cells give rise not only to renin precursor cells (RPCs) but also to all the mural cells of the kidney vasculature, including smooth muscle cells and perivascular fibroblasts. Foxd1 cells are also progenitors for interstitial pericytes and glomerular mesangial cells (FIGURE 2). RPCs themselves, in turn, differentiate into smooth muscle cells, glomerular mesangial cells, and interstitial pericytes, as previously described (44). Thereafter, RPCs participate in the assembling and branching of the kidney vasculature, where they are distributed extensively along large intrarenal arteries, in newly generated arteriolar branches, and inside the glomeruli in what later will become the glomerular mesangium (41, 44). Thus, throughout fetal and early postnatal life, there is a continuous and stereotypical changing of renin distribution with the appearance of every new arteriolar branch. During this process and until the arterioles mature, renin distribution is extensive throughout the renal vasculature (FIGURE 1). When arteriolar development is completed, renin cells are confined to their “classical” JG localization, as usually seen in the adult unstressed animal. This pattern of development is reminiscent of the changes in renin distribution throughout phylogeny from the time of the first emergence of these cells in bony fish (31, 42), suggesting that the ontogeny of renin replicates its phylogenetic history.
FIGURE. 2.
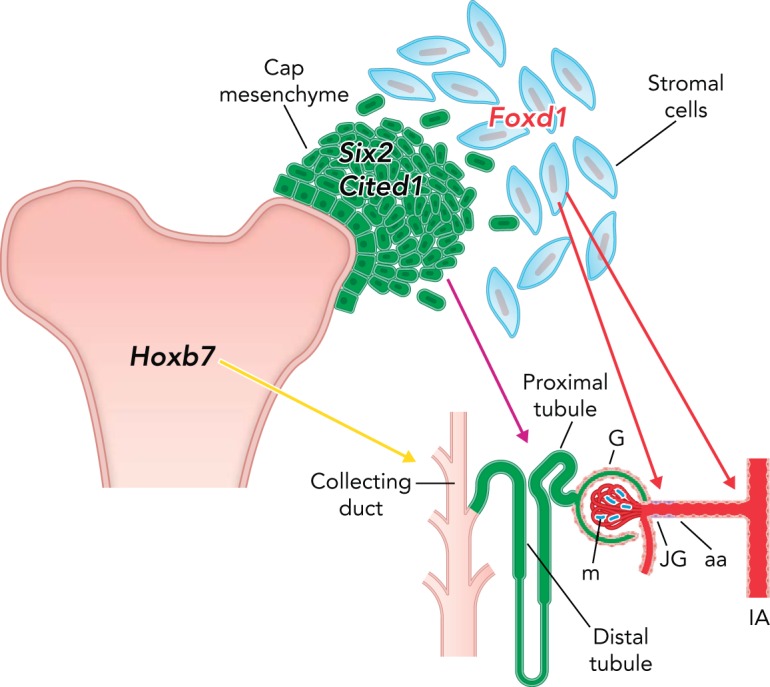
Metanephric kidney progenitors and their derivatives
The ureteric bud, which expresses the transcription factor Hoxb7, gives rise to the ureter and collecting duct; the cap mesenchyme, which expresses the transcription factors Six2 and Cited1, gives rise to all the epithelial nephrons from podocytes and Bowman's capsule to the distal tubule; the loose stromal cells, which express the transcription factor Foxd1, are the progenitors for renin cells, vascular smooth muscle cells, mesangial cells, and interstitial pericytes. G, glomerulus; m, mesangium; JG, juxtaglomerular cells; aa, afferent arteriole; IA, interlobular artery.
The Juxtaglomerular Cell
Surprisingly, although we know a fair amount about the aspartyl protease renin, progress regarding the actual cells that manufacture it has been slow. The reasons are multiple: 1) renin cells in the adult unstressed mammal constitute a small fraction (0.01%) of the total kidney cell mass, 2) the cells have been very difficult to isolate to purity, and 3) when placed in culture, renin cells lose the ability to express renin. To solve these problems, the generation of a variety of reporter mice and cell models was necessary to interrogate with precision the molecular and signaling events that control the identity and fate of these cells (5, 19, 35, 37, 44, 51).
The JG cell is perhaps the best known and most specialized of the repertoire of cells that at some point in life manufacture renin. As their name indicates, JG cells are localized at the entrance to the glomeruli within the wall of the afferent arterioles. They are plump compared with the flat smooth muscle cells situated along and upstream the afferent renal arterioles. JG cells also have a characteristic epithelioid appearance. Because they have granules that contain renin and myofibrils, they are often described as “granular myoepitheliod” cells.
Recently, the transcriptome of the JG cell was described and compared with the transcriptomes of practically every cell in the kidney (5). As expected, JG cells express abundant renin transcripts and a complex array of smooth muscle genes such as smooth muscle myosin heavy chain, calponin, smooth muscle alpha actin, and several transcriptional (SRF, Mef2c, KLFs) and posttranscriptional (miR 145, miR 330, miR 125b-5p) regulators of the smooth muscle program (5). It is interesting to ascertain from a physiological standpoint why these cells need to maintain a dual endocrine-contractile phenotype. We speculated that to maintain homeostasis JG cells should be able to contract and release renin on demand. We observed that JG cells maintain expression of smooth muscle and renin even under very challenging physiological situations. When renin expression is chronically stimulated, smooth muscle cells along afferent vessels re-acquire the renin phenotype. As a consequence, renin cells progressively cover the renal arterioles, a phenomenon that is conserved in mammals including humans (16, 49). Even then, renin cells maintain expression, albeit diminished, of smooth muscle genes preventing the vasculature from behaving as a passive circulation. In a passive circulation, every change in blood pressure would result in a parallel, linear change in renal blood flow and glomerular filtration rate. In this situation, even small increases in blood pressure could result in proportional changes in GFR and diuresis with the resultant volume depletion and/or dehydration. From an evolutionary standpoint, the situation would be disadvantageous and eventually incompatible with individual survival. Usually, if the challenge is transient, even under extreme physiological situations, the pre-glomerular arterioles maintain the ability to contract and autoregulate renal blood flow and maintain GFR constant. However, in pathological situations such as the endocrine kidney generated by stenosis of the renal artery, autoregulation is defective.
Renal Arterioles, Glomeruli, and Interstitium Possess Cells Capable of Synthesizing Renin When Homeostasis is Threatened
Afferent arterioles, and interlobular and arcuate arteries are invested with smooth muscle cells that are interconnected with each other and with surrounding interstitial pericytes. These cells retain the capacity to reexpress renin in response to a variety of physiological demands.
Under normal circumstances, secretion of renin by JG cells suffices to respond to minute-to-minute variations in blood pressure and maintain the renin levels required to support hemodynamic and fluid-electrolyte balance. However, when an adult mammal is exposed to manipulations that threaten homeostasis such as dehydration, hemorrhage, sodium depletion, or pharmacological inhibition of the RAS, circulating renin increases primarily due to an increase in the number of renin cells along the pre-glomerular arterioles (13-15). The increased circulating renin eventually reestablishes blood pressure and body fluid homeostasis. However, if the disequilibrium persists and the need for renin continues, as in mice deficient in angiotensinogen (23) or treated chronically with hypotensive agents, additional smooth muscle-like cells (interstitial pericytes and glomerular mesangial cells) undergo re-transformation and are thus further “recruited” to synthesize renin in a pattern resembling that of the embryo (3, 54, 55). Although this phenomenon has been denominated as “recruitment” (13) and sometimes JG cell “hyperplasia,” it does not involve migration or replication of cells but a re-acquisition of the renin phenotype by interstitial, glomerular, or arteriolar cells that were capable of making renin during embryonic life (FIGURE 1). When this transformation occurs, the cells seem to de-differentiate: they change morphology, become epithelioid, make granules that contain renin, and express a set of genes characteristic of the renin phenotype, including Akr1b7 (aldo-keto-reductase 1B7), an enzyme recently identified as characteristic of the renin endocrine phenotype (5, 27). The cells also express miR-330, a micro-RNA that is only expressed in JG cells when they are subjected to stimuli that demand high production of renin (29). These findings indicate that adult kidney cells retain the plasticity to reenact the renin cell phenotype and suggest that developmental choices made in early life may affect physiological responses in adult life. It is plausible that the ability of renin cell descendants to retain the memory of the renin phenotype is dependent on strong epigenetic marks that are not erased during differentiation and are instead retained throughout the life of cells, even after they have differentiated into other cell types. In support of this hypothesis, the ability to reenact the renin phenotype does not only occur in vivo but is also maintained in long-term culture: we generated a cell model whereby renin cell descendants isolated from the kidney arterioles were labeled genetically with cyan fluorescent protein (CFP). These cells retain CFP expression even when they are transformed into other cell types. They also contain a reporter yellow fluorescent protein (YFP) driven by a transgenic renin promoter in a manner such that when transcription of renin is active, the cells also express YFP. When these CFP+ cells are exposed to cAMP analogs and/or to histone deacetylase (HDAC) inhibitors, a fraction of the CFP+ cells becomes YFP positive again and expresses endogenous renin mRNA, indicating that cAMP activated the renin promoter (37). These changes were facilitated by increased acetylation of histone 4 around the cAMP-responsive element (CRE) of the renin gene opening the chromatin for renin transcription (34). The experiments suggest that the ability of these cells to regain the renin phenotype is inherent to cells derived from the renin lineage. Because the culture contains a pure population of CFP+ clonal cells derived from the renin lineage, the recruitment of renin expression cannot be explained by migration of a few foreign lineage-unrelated cells as it recently has been suggested (56). The results suggest that renin cell descendants retain a poised chromatin landscape favorable for renin expression in response to external stimuli. Therein is likely to reside the mechanism for these cells to switch back and forth between the smooth muscle and the renin phenotype. It has recently been postulated that a small fraction of renal mesenchymal CD44+ cells may be activated during sodium depletion and presumably migrate to the JGA, where they insert themselves as renin-producing cells (56). The contribution of these cells to the recruitment process seems minor compared with the already available pool of cells capable of reexpressing renin on demand. Furthermore, given that JG cells do express CD44 as evidenced by single cell transcriptome analysis (17, 28), it is not clear whether those CD44 cells descend from renin precursors and are simply reactivating the developmental program described above. Further studies are required to better identify those cells and determine the biological importance of this process if at all different from the classical reactivation of renin expression in renin cell descendants.
The cAMP Pathway
cAMP regulates renin synthesis and release. It mediates the actions of numerous physiological signals (i.e., β-adrenoreceptor activation, adenosine, prostaglandins, low calcium) that regulate renin secretion (38). cAMP also positively controls renin gene transcription and augments renin mRNA levels. Isolated renal arterioles and single arteriolar cells from newborn rats release renin and increase renin mRNA levels in response to adenylate cyclase stimulation. The increase in renin release is due to an increased number of renin-secreting and renin-expressing cells (recruited cells) without changes in the amount of renin secreted by individual cells (12). Because the cells are immobilized atop the floor of a Cunningham chamber, renin expression and release occur without participation of a functional macula densa or cell migration. As described above, using cells harboring yellow fluorescent protein (YFP) driven by the renin promoter, we demonstrated an increase in the number of renin-expressing cells in response to manipulations that increase cAMP levels. These results in vitro corroborate numerous reports in whole animals (13, 14, 54, 55) indicating that the control of hormone availability is achieved by regulating the number of cells that express renin.
The renin gene contains cAMP-responsive elements (CRE) that are critical for basal and cAMP-stimulated expression of the renin gene (24, 33, 52). Creb and its associated coactivators CBP/p300 associate with the CRE site and integrate multiple signals that control renin synthesis and release. CBP and p300 are histone acetyl transferases (HATs) that deposit activating acetyl marks in histones, resulting in local relaxation of chromatin, nucleosomal displacement, and access of transcription factors such as Creb 1. In fact, conditional deletion of both HATs in renin cells resulted in a marked reduction in the number of renin+ JG cells, diminished renin expression, and abnormal arteriolar development, indicating that CBP and p300 are necessary for the maintenance of renin cell identity and nephrovascular integrity. Furthermore, mice with deletion of both HATs could not recruit renin-expressing cells or increase circulating renin in response to a challenge to homeostasis.
Attesting to the importance of cAMP signaling in renin regulation, deletion of Gsα, a more proximal component of the cAMP pathway in the renin cells, causes a reduction in renin cells in early life, accompanied by arteriolar branching defects and renal failure (8, 9). These results emphasize the crucial role of the cAMP pathway in regulating renin cell fate and plasticity.
The Notch Pathway: Conveying Instructions Along the Renal Arterioles
Proper functioning and maintenance of the renin cell phenotype requires cell-to-cell communication. The Notch pathway is an ancestral cell-to-cell communication system involved in cell fate decisions during development and in response to physiological challenges in the mature animal (1, 4, 26). Given that Notch receptors, their ligands, and their final transcriptional effector RBP-J are all expressed in renin cells, we hypothesized that Notch/RBP-J may be involved in the acquisition and/or maintenance of the renin phenotype. To test this hypothesis, we deleted RBP-J in renin cells in vivo (6). Mice with the conditional deletion of RBP-J had a severe reduction in the number of renin cells, low circulating renin, and decreased blood pressure. Furthermore, these mice were unable to elicit a recruitment of renin cells in response to a homeostatic challenge, indicating that RBP-J also is necessary to maintain the memory of the renin phenotype.
To determine whether RBP-J acted directly on the renin gene, we generated mice bearing either a bacterial artificial chromosome (BAC) reporting enhanced green fluorescent protein (eGFP) under the control of the wild-type renin promoter (WT-BAC-GFP) or a mutant (Mut-BAC-GFP) in which the four core nucleotides in the RBP-J consensus sequence critical for RBP-J binding were mutated. Mutant BAC mice had markedly decreased GFP mRNA and were unable to respond properly to a sodium depletion and captopril challenge, whereas WT-BAC-GFP mice were able to recruit GFP+ cells upstream of the afferent arterioles. Thus RBP-J regulates the renin promoter directly and is also involved in the ability of smooth muscle cells along the arteriole to reacquire the renin phenotype.
RBP-J governs other genes involved in the renin phenotype. Aldo-keto reductase (AKR1b7) is an enzyme coexpressed at high levels in renin cells during normal development and in response to homeostatic challenges (7, 27). Recently, we showed that both genes, renin and Akr1b7, are regulated by cAMP in vitro and in vivo (27). Given its coregulation with renin and the fact that AKR1b7 possesses binding sites for RBP-J, we examined whether Akr1b7 expression pattern is altered in mice with RBP-J deletion. Mice with deletion of RBP-J have significantly fewer AKR1b7 cells. This was accompanied by a frank decrease in the number of renin granules, indicating that RBP-J controls several components of the endocrine renin phenotype (7).
As described above, JG cells retain the ability to contract by expressing a variety of smooth muscle genes. In addition to endocrine genes, RBP-J controls the expression of smooth muscle genes and their master regulators, indicating that RBP-J/Notch controls a genetic program that maintains the endocrine-contractile phenotype of the renin cell (FIGURE 3).
FIGURE. 3.

RBP-J is a master regulator that maintains the identity of the JG cell
RBP-J regulates a network of genes that confers the endocrine-contractile phenotype of the JG cell. Red bars located in the promoters of target genes represent RBP-J binding sites. Endocrine genes, such as renin and Akr1b7, and renin granules are noted on the left. Genes required to maintain the contractile phenotype are indicated on the right. RBP-J regulates smooth muscle genes directly through their promoter regions and indirectly by upregulating the expression of miR-145-5p and SRF. miR-145-5p positively regulates SRF, and they act together to activate the expression of smooth muscle genes. CaM, calmodulin; Cn, calcineurin; P, phosphate; Smtn, smoothelin. Figure is adapted from Ref. 7 and used with permission from the Journal of the American Society of Nephrology.
Renin cell fate tracing studies showed that the marked decrease in the number of renin+/AKR1b7+ cells in RBP-J cKO mice was not due to a decreased endowment of renin precursors or increased cell death. In fact, the distribution of labeled cells was identical in control and mutant kidneys, suggesting that the decrease in renin cells was due to a switch of phenotype of former renin-expressing cells (which are present in the same locations in the kidney vasculature) to another cell type. So what was then the identity of those cells that expressed neither renin nor smooth muscle proteins? Those dually negative cells did not express endothelial, epithelial, or stem cell markers (7). However, arteriolar cells expressed genes characteristic of hematopoietic cells, indicating that, in renin cells, RBP-J normally suppresses the ectopic expression of genes from other lineages (7). These findings are of interest because JG cells normally express transcription factors crucial for the commitment and differentiation of hematopoietic cells, including Early B cell Factor 1 (EBF1), Ikaros, HIF2α, E2A, and RBP-J itself (2, 5). The mechanisms involved in RBP-J suppression of genes from “other” unwanted lineages remain to be studied. FIGURE 3 shows that RBP-J controls the identity of the renin cell by 1) activating genes involved in the dual endocrine-contractile phenotype of the renin cell and 2) preventing the undesirable, ectopic expression of genes from other lineages, which could have severe consequences for the control of homeostasis. The effects of RBP-J deletion are more severe if the mutation occurs at an early stage of differentiation: deletion of RBP-J in the renal stromal Foxd1+ cells, the earliest progenitors for all the mural cells of the kidney vasculature, results in a decreased endowment of renin cells, mesangial cells, and smooth muscle cells, leading to altered vascular formation and glomerular aneurysms. Deletion of RBP-J in cells downstream in the Foxd1 lineage hierarchy (deletion of RBP-J in the renin lineage cells) does not result in overt morphological changes but leads to a remarkable change in the identity of the renin cell. Thus deletion of RBP-J in the early progenitor leads to a decrease of descendants, whereas deletion in the more differentiated renin cell progenitors results in a significant change in cell identity. These results suggest that the effects of RBP-J/Notch depend on the degree of differentiation and maturation of the target cells. A summary of RBP-J actions at different stages in the differentiation of the renin cell is depicted in FIGURE 4.
FIGURE. 4.
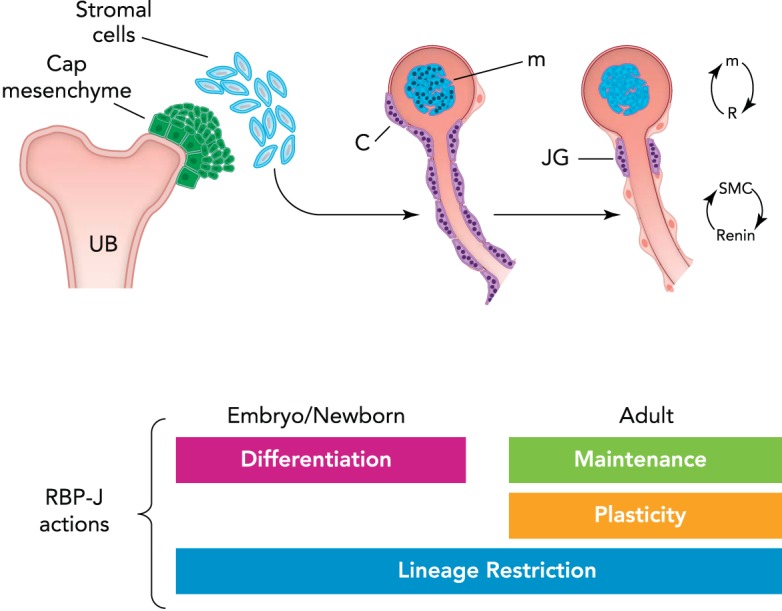
Conceptual summary of RBP-J actions in the kidney vasculature
During early vascular development, RPB-J regulates differentiation and morphogenesis of the kidney vasculature by controlling the differentiation of mesangial cells and the mural cells (arteriole to the left). Later in life, the role of RBP-J/Notch may be to maintain the differentiated state and the plasticity of smooth muscle cells (SMCs) (right arteriole) to regain the renin phenotype. Throughout the continuum of life, RBP-J prevents the expression of unwanted/undesirable genes by lineage restriction. UB, ureteric bud; c, capsule; m, mesangium; JG, juxtaglomerular cell. Cells with dots (granules) indicate renin expression.
Given that both the Notch/RBP-J and the cAMP/Creb/CBP/p300 pathways control the identity and recruitment of renin cells, it would be of considerable interest to understand how these two systems interact to maintain the renin phenotype.
Identification of Renin Progenitors in Hematopoietic Tissues
While investigating the effects of RBP-J deletion on the identity of renin cells, we found that the bone marrow harbors a unique set of renin progenitors of relevance to our understanding of normal and neoplastic hematopoiesis (2). As mice with RBP-J deletion in renin cells aged beyond 6–8 mo, they developed overt signs of a highly penetrant form of pre-B-cell leukemia (2). We performed an extensive series of experiments to characterize the disease and to identify the cell of origin for this striking disease. We found that cells from the bone marrow and spleen normally contain a subset of pre-B lymphocytes that express renin, and refer to these cells as Brenin cells. Brenin cells display remarkable developmental, biochemical, and transcriptional similarities with the more distant kidney JG cell (2). Similar to JG cells, Brenin cells in the bone marrow and spleen decrease in number as development proceeds. Interestingly, at all ages examined, Brenin cells are 10 times more abundant than JG cells. Furthermore, the cells are responsive to physiological and pharmacological cues: they increase in number when angiotensin generation is decreased. Brenin cells and JG cells also have in common the expression of transcription factors, notably EBF1, a well known regulator of lymphocyte development, Notch ligands, and receptors, and RBP-J among others (2). This suggests a conservation of the regulatory machinery and a possible lineage relationship between hematopoietic and kidney renin cells. Both cell types require RBP-J to differentiate properly. Deficiency of RBP-J in kidney results in a significant decrease in the number of renin cells. Brenin cells lacking RBP-J do not differentiate normally; they are held as precursors, lose control of their cell cycle, proliferate, and become neoplastic. In both cases, RBP-J controls cell fate.
The function of renin cells in hematopoietic tissues remains to be determined. In analogy with the kidney, it is possible that Brenin cells control bone marrow structural morphogenesis and/or hematopoietic development. From an integrative physiological standpoint, it is tempting to speculate that hematopoietic renin cells are part of a systemic defense response coordinated not only to sustain blood pressure and fluid/electrolyte homeostasis but also to provide a rapid response to infections and exposure to foreign antigens. If this assumption proves correct, it will widen the defense functions of renin cells linking the endocrine and immune systems, two major ancestral mechanisms to maintain homeostasis.
Renin Expression and Renin Cells are Necessary for Nephrovascular Development and Regeneration
Total body deletion of the renin gene leads to severe kidney developmental defects. The kidneys have hypoplastic papillae, dilated renal pelvis, hydronephrosis, interstitial fibrosis, focal glomerulosclerosis, perivascular mononuclear infiltration, and concentric arteriolar thickening (48). Given the complexity of this phenotype and the fact that renin had also been observed in tubules (40, 43, 50), it was not possible until recently to discriminate whether the phenotype was due to deficiency of renin in the vascular vs. the tubular compartment. Deletion of renin in each compartment clearly indicated that the entirety of the phenotype could be explained solely by the lack of renin in the vasculature (46). In fact the phenotype of mice without vascular renin was identical to the one observed in the total renin KO mice. Those experiments also demonstrated that the major source of circulating renin is the kidney vasculature (46). It should be noted that a similar phenotype was also described in mice deficient in angiotensinogen (21, 22, 30), angiotensin converting enzyme (11, 18, 25), or angiotensin receptors1a/1b (32, 53). Curiously, mice in which renin cells were ablated with diphtheria toxin did not show the concentric hypertrophy of arterioles (36), suggesting that renin-producing cells per se (as opposed to renin) may in part contribute to the vessel thickening, possibly by synthesizing some factor(s) other than renin that stimulates concentric vessel proliferation. This hypothesis is being tested in our laboratory.
Renin cells may have additional roles in tissue repair. Very recently, two groups of investigators showed that renin cells from the JG area participate in the replenishment of podocytes and mesangial cells in two different models of podocyte and mesangial cell injury (39, 47).
In an experimental model that closely resembles focal segmental glomerular sclerosis, Pippin and colleagues followed the fate of cells from the renin lineage and showed that, after podocyte depletion induced with a cytotoxic anti-podocyte antibody, cells of the renin lineage repopulated a significant fraction of parietal epithelial cells of the Bowman's capsule and podocytes (39). Thus, under pathological situations, JG cells are able to regenerate injured glomerular cells. Podocytes are said not to regenerate from remaining podocytes. In addition, they are not known to descend from renin cells. The intimate mechanisms as to how podocytes are regenerated from renin cells remains to be studied, but it is possible that, under the stress of injury, direct conversion or transdifferentiation of renin cells to podocytes may occur. In addition, parietal epithelial cells, a fraction of which may descend from renin cells (44), may also give rise to podocytes. In this case, a reenactment of an embryological program, as in the case of the mesangial cells discussed below, may also take place.
Given that, during fetal and early postnatal nephrogenesis, renin precursors give rise to mesangial cells (44), Starke and colleagues recently tested whether renin cells in the adult animal could regenerate the glomerular mesangium in an experimental model of mesangiolysis that resembles mesangial proliferative glomerulonephritis (47). Using mice harboring inducible reporters to track the fate of renin cell precursors, they found that over two-thirds of the regenerating glomeruli were populated by cells derived from the renin lineage. Mesangial cells presumably arose from nearby renin containing JG cells. Upon differentiation, as it occurs during development, the newly differentiated cells acquired mesangial markers such as α8-integrin and platelet-derived growth factor receptor beta, and stopped making renin. It would be interesting to explore whether regeneration of mesangial cells in this instance results from the reenactment of the yet to be discovered molecular program used during differentiation of mesangial cells from renin precursors during normal kidney development.
Altogether, these experiments indicate that renin cells constitute a progenitor niche with the ability to regenerate injured cells from the glomerulus.
These interesting findings open new and exciting questions. The molecular programs underlying this regenerative process remain to be elucidated. Similarly, the signals and processes used by seemingly migrating JG cells to arrive at the site of injury to replace injured cells remain to be identified. It also would be important to determine whether the replenishment of cells is either a transient or a permanent phenomenon, a reaction to injury or a reparative process with long-term consequences accompanied by short or, even better, long-term functional changes such as improvement in proteinuria and/or other markers of renal function.
Overall, the studies briefly discussed above illustrate the versatile nature of renin cells and open new, fertile ground not only to understanding the biology of renin precursors per se but also to comprehending tissue repair and regeneration.
Footnotes
The authors are supported by National Institutes of Health Grants DK-096373 and HL-096735 (to R.A.G.), and DK-091330 and DK-096373 (to M.L.S.S.L.).
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: R.A.G. and M.L.S.S.-L. conception and design of research; R.A.G. and M.L.S.S.-L. prepared figures; R.A.G. and M.L.S.S.-L. drafted manuscript; R.A.G. and M.L.S.S.-L. edited and revised manuscript; R.A.G. and M.L.S.S.-L. approved final version of manuscript.
References
- 1.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science 268: 225–232, 1995. [DOI] [PubMed] [Google Scholar]
- 2.Belyea BC, Xu F, Pentz ES, Medrano S, Li M, Hu Y, Turner S, Legallo R, Jones CA, Tario JD, Liang P, Gross KW, Sequeira-Lopez ML, Gomez RA. Identification of renin progenitors in the mouse bone marrow that give rise to B-cell leukaemia. Nat Commun 5: 3273, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg AC, Chernavvsky-Sequeira C, Lindsey J, Gomez RA, Sequeira-Lopez MLS. Pericytes synthesize renin. World J Nephrol 2: 11–16, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7: 678–689, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Brunskill EW, Sequeira-Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, Potter SS, Gomez RA. Genes that confer the identity of the renin cell. J Am Soc Nephrol 22: 2213–2225, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, Sequeira-Lopez ML, Gomez RA. Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics 43: 1021–1028, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castellanos-Rivera RM, Pentz ES, Lin E, Gross KW, Medrano S, Yu J, Sequeira-Lopez ML, Gomez RA. Recombination signal binding protein for Ig-kappaJ region regulates juxtaglomerular cell phenotype by activating the myo-endocrine program and suppressing ectopic gene expression. J Am Soc Nephrol 26: 67–80, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Faulhaber-Walter R, Wen Y, Huang Y, Mizel D, Chen M, Sequeira Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J. Renal failure in mice with Gsalpha deletion in juxtaglomerular cells. Am J Nephrol 32: 83–94, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, Chen M, Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsα in juxtaglomerular cells. Am J Physiol Renal Physiol 292: F27–F37, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Deschepper CF, Mellon SH, Cumin F, Baxter JD, Ganong WF. Analysis by immunocytochemistry and in situ hybridization of renin and its mRNA in kidney, testis, adrenal, and pituitary of the rat. Proc Natl Acad Sci USA 83: 7552–7556, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esther CR Jr, Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab Invest 74: 953–965, 1996. [PubMed] [Google Scholar]
- 12.Everett AD, Carey RM, Chevalier RL, Peach MJ, Gomez RA. Renin release and gene expression in intact rat kidney microvessels and single cells. J Clin Invest 86: 169–175, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez RA, Chevalier RL, Everett AD, Elwood JP, Peach MJ, Lynch KR, Carey RM. Recruitment of renin gene-expressing cells in adult rat kidneys. Am J Physiol Renal Fluid Electrolyte Physiol 259: F660–F665, 1990. [DOI] [PubMed] [Google Scholar]
- 14.Gomez RA, Lynch KR, Chevalier RL, Everett AD, Johns DW, Wilfong N, Peach MJ, Carey RM. Renin and angiotensinogen gene expression and intrarenal renin distribution during ACE inhibition. Am J Physiol Renal Fluid Electrolyte Physiol 254: F900–F906, 1988. [DOI] [PubMed] [Google Scholar]
- 15.Gomez RA, Norwood VF. Developmental consequences of the renin-angiotensin system. Am J Kidney Dis 26: 409–431, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Gribouval O, Gonzales M, Neuhaus T, Aziza J, Bieth E, Laurent N, Bouton JM, Feuillet F, Makni S, Ben AH, Laube G, Delezoide AL, Bouvier R, Dijoud F, Ollagnon-Roman E, Roume J, Joubert M, Antignac C, Gubler MC. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 37: 964–968, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Harding SD, Armit C, Armstrong J, Brennan J, Cheng Y, Haggarty B, Houghton D, Lloyd-MacGilp S, Pi X, Roochun Y, Sharghi M, Tindal C, McMahon AP, Gottesman B, Little MH, Georgas K, Aronow BJ, Potter SS, Brunskill EW, Southard-Smith EM, Mendelsohn C, Baldock RA, Davies JA, Davidson D. The GUDMAP database: an online resource for genitourinary research. Development 138: 2845–2853, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilgers KF, Reddi V, Krege JH, Smithies O, Gomez RA. Aberrant renal vascular morphology and renin expression in mutant mice lacking angiotensin-converting enzyme. Hypertension 29: 216–221, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Jones CA, Hurley MI, Black TA, Kane CM, Pan L, Pruitt SC, Gross KW. Expression of a renin/GFP transgene in mouse embryonic, extra-embryonic, and adult tissues. Physiol Genomics 4: 75–81, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Jones CA, Sigmund CD, McGowan RA, Kane-Haas CM, Gross KW. Expression of murine renin genes during fetal development. Mol Endocrinol 4: 375–383, 1990. [DOI] [PubMed] [Google Scholar]
- 21.Kihara M, Umemura S, Sumida Y, Yokoyama N, Yabana M, Nyui N, Tamura K, Murakami K, Fukamizu A, Ishii M. Genetic deficiency of angiotensinogen produces an impaired urine concentrating ability in mice. Kidney Int 53: 548–555, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, Jennette JC, Coffman TM, Maeda N, Smithies O. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci USA 92: 2735–2739, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HS, Maeda N, Oh GT, Fernandez LG, Gomez RA, Smithies O. Homeostasis in mice with genetically decreased angiotensinogen is primarily by an increased number of renin-producing cells. J Biol Chem 274: 14210–14217, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Klar J, Sandner P, Muller MW, Kurtz A. Cyclic AMP stimulates renin gene transcription in juxtaglomerular cells. Pflügers Arch 444: 335–344, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Krege JH, John SW, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O'Brien DA, Smithies O. Male-female differences in fertility and blood pressure in ACE-deficient mice. Nature 375: 146–148, 1995. [DOI] [PubMed] [Google Scholar]
- 26.Lai EC. Notch signaling: control of cell communication and cell fate. Development 131: 965–973, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Lin EE, Pentz ES, Sequeira-Lopez ML, Gomez RA. Aldo-keto reductase 1b7, a novel marker for renin cells, is regulated by cyclic AMP signaling. Am J Physiol Regul Integr Comp Physiol 309: R576–R584, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, Zhang P. GUDMAP: the genitourinary developmental molecular anatomy project. J Am Soc Nephrol 19: 667–671, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Medrano S, Monteagudo MC, Sequeira-Lopez MLS, Pentz ES, Gomez RA. Two microRNAs -miR-330 and miR-125b-5p- mark the juxtaglomerular cell and balance its smooth muscle phenotype. Am J Physiol Renal Physiol 302: F29–F37, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niimura F, Labosky PA, Kakuchi J, Okubo S, Yoshida H, Oikawa T, Ichiki T, Naftilan AJ, Fogo A, Inagami T. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest 96: 2947–2954, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimura H, Ogawa M, Sawyer WH. Renin-angiotensin system in primitive bony fishes and a holocephalian. Am J Physiol 224: 950–956, 1973. [DOI] [PubMed] [Google Scholar]
- 32.Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA 95: 15496–15501, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan L, Black TA, Shi Q, Jones CA, Petrovic N, Loudon J, Kane C, Sigmund CD, Gross KW. Critical roles of a cyclic AMP responsive element and an E-box in regulation of mouse renin gene expression. J Biol Chem 276: 45530–45538, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Pentz ES, Cordaillat M, Carretero OA, Tucker AE, Sequeira Lopez ML, Gomez RA. Histone acetyl transferases CBP and p300 are necessary for maintenance of renin cell identity and transformation of smooth muscle cells to the renin phenotype. Am J Physiol Heart Circ Physiol 302: H2545–H2552, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pentz ES, Lopez ML, Kim HS, Carretero O, Smithies O, Gomez RA. Ren1d and Ren2 cooperate to preserve homeostasis: evidence from mice expressing GFP in place of Ren1d. Physiol Genomics 6: 45–55, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Pentz ES, Moyano MA, Thornhill BA, Sequeira Lopez ML, Gomez RA. Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am J Physiol Regul Integr Comp Physiol 286: R474–R483, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Pentz ES, Sequeira Lopez ML, Cordaillat M, Gomez RA. Identity of the renin cell is mediated by cAMP and chromatin remodeling: an in vitro model for studying cell recruitment and plasticity. Am J Physiol Heart Circ Physiol 294: H699–H707, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Persson PB, Skalweit A, Mrowka R, Thiele BJ. Control of renin synthesis. Am J Physiol Regul Integr Comp Physiol 285: R491–R497, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Pippin JW, Sparks MA, Glenn ST, Buitrago S, Coffman TM, Duffield JS, Gross KW, Shankland SJ. Cells of renin lineage are progenitors of podocytes and parietal epithelial cells in experimental glomerular disease. Am J Pathol 183: 542–557, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prieto-Carrasquero MC, Botros FT, Pagan J, Kobori H, Seth DM, Casarini DE, Navar LG. Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip goldblatt hypertensive rats. Hypertension 51: 1590–1596, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddi V, Zaglul A, Pentz ES, Gomez RA. Renin-expressing cells are associated with branching of the developing kidney vasculature. J Am Soc Nephrol 9: 63–71, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Rider SA, Mullins LJ, Verdon RF, MacRae CA, Mullins JJ. Renin expression in developing zebrafish is associated with angiogenesis and requires the Notch pathway and endothelium. Am J Physiol Renal Physiol 309: F531–F539, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, Terreros DA, Lalouel JM. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension 34: 1265–1274, 1999. [DOI] [PubMed] [Google Scholar]
- 44.Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 6: 719–728, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Sequeira-Lopez ML, Lin EE, Li M, Hu Y, Sigmund CD, Gomez RA. The earliest metanephric arteriolar progenitors and their role in kidney vascular development. Am J Physiol Regul Integr Comp Physiol 308: R138–R149, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sequeira-Lopez ML, Nagalakshmi VK, Li M, Sigmund CD, Gomez RA. Vascular versus tubular renin: role in kidney development. Am J Physiol Regul Integr Comp Physiol 309: R650–R657, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starke C, Betz H, Hickmann L, Lachmann P, Neubauer B, Kopp JB, Sequeira-Lopez ML, Gomez RA, Hohenstein B, Todorov VT, Hugo CP. Renin lineage cells repopulate the glomerular mesangium after injury. J Am Soc Nephrol 26: 48–54, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi N, Lopez ML, Cowhig JE Jr, Taylor MA, Hatada T, Riggs E, Lee G, Gomez RA, Kim HS, Smithies O. Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J Am Soc Nephrol 16: 125–132, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Taugner R, Hackenthal E. The Juxtaglomerular Apparatus: Structure and Function. Heidelberg, Germany: Springer Verlag, 1989. [Google Scholar]
- 50.Taugner R, Hackenthal E, Inagami T, Nobiling R, Poulsen K. Vascular and tubular renin in the kidneys of mice. Histochemistry 75: 473–484, 1982. [DOI] [PubMed] [Google Scholar]
- 51.Thompson HA, Burson JM, Lang JA, Gross KW, Sigmund CD. Tissue-specific expression of novel messenger ribonucleic acids cloned from a renin-expressing kidney tumor cell line (As4.1). Endocrinology 136: 3037–3045, 1995. [DOI] [PubMed] [Google Scholar]
- 52.Todorov VT, Volkl S, Friedrich J, Kunz-Schughart LA, Hehlgans T, Vermeulen L, Haegeman G, Schmitz ML, Kurtz A. Role of CREB1 and NFκB-p65 in the down-regulation of renin gene expression by tumor necrosis factor α. J Biol Chem 280: 24356–24362, 2005. [DOI] [PubMed] [Google Scholar]
- 53.Tsuchida S, Matsusaka T, Chen X, Okubo S, Niimura F, Nishimura H, Fogo A, Utsunomiya H, Inagami T, Ichikawa I. Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J Clin Invest 101: 755–760, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tufro-McReddie A, Arrizurieta EE, Brocca S, Gomez RA. Dietary protein modulates intrarenal distribution of renin and its mRNA during development. Am J Physiol Renal Fluid Electrolyte Physiol 263: F427–F435, 1992. [DOI] [PubMed] [Google Scholar]
- 55.Tufro-McReddie A, Chevalier RL, Everett AD, Gomez RA. Decreased perfusion pressure modulates renin and ANG II type 1 receptor gene expression in the rat kidney. Am J Physiol Regul Integr Comp Physiol 264: R696–R702, 1993. [DOI] [PubMed] [Google Scholar]
- 56.Yang Y, Gomez JA, Herrera M, Perez-Marco R, Repenning P, Zhang Z, Payne A, Pratt RE, Koller B, Beierwaltes WH, Coffman T, Mirotsou M, Dzau VJ. Salt restriction leads to activation of adult renal mesenchymal stromal cell-like cells via prostaglandin E2 and E-prostanoid receptor 4. Hypertension 65: 1047–1054, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]