

Abstract
Approximately 350 million people worldwide are chronically infected by hepatitis B virus (HBV). HBV causes severe liver diseases including cirrhosis and hepatocellular carcinoma (HCC). In about 25% of affected patients, HBV infection proceeds to HCC. Therefore, the mechanisms by which HBV affects the host cell to promote viral replication and its pathogenesis have been the subject of intensive research efforts. Emerging evidence indicates that both autophagy and microRNAs (miRNAs) are involved in HBV replication and HBV-related hepatocarcinogenesis. In this review, we summarize how HBV induces autophagy, the role of autophagy in HBV infection, and HBV-related tumorigenesis. We further discuss the emerging roles of miRNAs in HBV infection and how HBV affects miRNAs biogenesis. The accumulating knowledge pertaining to autophagy and miRNAs in HBV replication and its pathogenesis may lead to the development of novel strategies against HBV infection and HBV-related HCC tumorigenesis.
Keywords: Hepatitis B virus, Autophagy, MicroRNA, Hepatocellular carcinoma, Viral replication
Core tip: A number of reviews have described autophagy and microRNAs (miRNAs) in the hepatitis B virus (HBV)-related tumorigenesis. However, few reviews have provided insights into the relationships among autophagy, miRNAs, HBV biogenesis and hepatocarcinogenesis. In this review, we describe the emerging role of autophagy and miRNAs in HBV replication and pathogenesis. Recent studies are reviewed in the following sections: (1) the mechanism by which HBV induces autophagy; (2) the effect of HBV-induced autophagy on HBV replication; (3) the relationship between autophagy and HBV in HBV-related hepatocarcinogenesis; (4) the mechanism by which miRNAs affects HBV replication; and (5) the regulation of miRNAs biogenesis by HBV.
INTRODUCTION
Proper autophagic responses protect the cell from stresses and maintain cellular homeostasis. Autophagy progression is regulated by a series of autophagy-related genes (Atg). Atg5, Atg7 and Beclin 1 are essential genes in the autophagic process and silencing any of these genes blocks autophagy[1]. A novel function of autophagy known as secretory autophagy (also called autosecretion or type III secretion) has been the subject of increasing research interest[2]. In contrast to traditional degradative autophagy, the recruited cargo is carried by the double-membrane autophagosome and exported to the extracellular environment (exocytosis). Traditionally, the cargo in the cell is delivered through the endoplasmic reticulum-Golgi apparatus-plasma membrane pathway[3]. However, specific cargo proteins in the cytosol are transported to the extracellular environment without going through the endoplasmic reticulum-Golgi apparatus-plasma membrane pathway. This unconventional secretory autophagy has been shown to be involved in vesicle trafficking and cytokines secretion (IL-1β, IL-6, IL-18 and TNF-α) for innate and adaptive immune responses[4].
Cancer progression triggers diverse metabolic stresses which lead to increased autophagic activity. Aberrant autophagy may cause cell death which is known as type II programmed cell death. Recent evidence indicates that autophagy suppresses tumorigenesis to preserve cellular fitness and genome integrity[5,6]. Therefore, manipulation of autophagic activity may have potential in the development of an alternative therapeutic strategy against cancer development or drug resistance in cancer cells[7-9]. Deficient autophagic responses cause diverse pathologic conditions of the liver, including liver dysfunction and tumorigenesis[9,10]. Autophagic machinery is also important for innate and adaptive immunity. In innate immunity, diverse pathogens including bacteria and viruses are selectively engulfed by the autophagosome followed by fusion with the lysosome to form the autolysosome and autolysosome-mediated clearance[11]. In adaptive immunity, autophagic machinery produces antigenic peptides, which are loaded onto the major histocompatibility complex (MHC) class II molecules and presented to CD4+ T cells[12]. Pathogen infection of the host cells causes cellular stress. To diminish the deleterious impact of stress, the autophagic degradation system is induced to recruit the damaging molecules including proteins, organelles, pathogens, and microRNAs, which are subsequently subjected to autophagic degradation. Meanwhile, pathogens utilize various strategies to escape, suppress or hijack the autophagic degradation pathway. They may also interfere with autophagy-related immune defenses[13,14].
MicroRNAs (miRNAs) are small non-coding RNAs which are initially transcribed as long primary miRNAs which then undergo sequential processing to form precursor miRNAs (pre-miRNA) by RNase III endonucleases. Pre-miRNAs are then transported into the cytoplasm where they are transformed by Dicer processing to become mature miRNAs[15]. MiRNAs suppress their target-gene expression either by transcriptional degradation or by translational inhibition, depending on sequence homology between the miRNA and the target gene. MiRNAs are involved in diverse diseases including viral infections and cancers[16].
Hepatitis B virus (HBV) is characterized by partly double-stranded relaxed circular DNA (rcDNA) and belongs to the hepadnaviridae family. The HBV virion consists of an outer envelope and the inner core proteins, 3.2 kb of rcDNA genome and DNA polymerase[17]. During HBV infection of the hepatocytes, the uncoated HBV rcDNA is transported to the nucleus. In the presence of viral DNA polymerase, the rcDNA is transformed into covalently closed circular DNA (cccDNA), which serves as the template for transcription of viral mRNAs in the presence of host RNA polymerase. The HBV genome contains four overlapping open reading frames (ORFs) comprising the S, C, X and P regions. The S region encodes three envelope proteins (S, M, and L) for viral envelopment and the C region encodes the core protein for the viral capsid. The X region encodes the X protein for viral replication. The P region encodes the proteins for viral RNA reverse transcription and DNA replication[18]. The progeny nucleocapsid harboring the rcDNA then proceeds to viral envelopment and mature virus release. In this review, we conduct an in-depth exploration of the role of autophagy and miRNAs in HBV infection and pathogenesis.
AUTOPHAGY AND HBV
HBV infection can induce autophagy and different genotypes of HBV have shown different increments of autophagic activities[19]. The following sections summarize the underlying mechanisms of HBV-induced autophagy and the roles of autophagy in HBV infection, replication, and HBV-related hepatocellular carcinogenesis.
HBV induces autophagy
HBV induces autophagy mainly through the HBx protein or small surface protein (SHB)-dependent mechanism.
HBx protein induces autophagy: The HBx protein has been demonstrated to be the major molecule involved in inducing autophagy during HBV infection[20-23]. Beclin 1 is an autophagy-related protein (the mammalian orthologue of yeast Atg6), which forms a complex with PI3KC3 to initiate autophagic progression. Beclin 1 is responsible for localization of the autophagic complex proteins (PI3KC3 and UVRAG) to the pre-autophagosomal structure[24]. AMP-activated protein kinase and mTORC1 signaling molecules, the sensors of nutrient and energy, regulate the Beclin 1-PI3KC3 complex to produce phosphatidylinositol 3-phosphate (PI3P), a signaling lipid for the recruitment of autophagy effectors[25]. HBx directly increases Beclin 1 expression through the activation of the -277/+179 region of Beclin 1 promoter, which further enhances starvation-induced autophagy (Figure 1)[20]. HBx can also increase the enzymatic activity of PI3KC3, which mediates PI3P formation and enhances autophagosome formation (Figure 1)[21]. Autophagy can be induced by death-associated protein kinase (DAPK) through phosphorylation of Beclin 1 and protein kinase D, which activates PI3KC3[26]. DAPK is a calcium/calmodulin serine/threonine kinase and is associated with different cell death pathways, including autophagic cell death[27]. DAPK phosphorylates Beclin 1 on the BH3 domain to promote its dissociation from Bcl-XL, which triggers autophagic progression[28]. Zhang et al[23] demonstrated that HBx activates DAPK through dephosphorylation of DAPK, and induces autophagy in a Beclin 1-dependent manner (Figure 1). In summary, the HBx protein induces autophagy at the initiation stage of autophagic progression.
Figure 1.
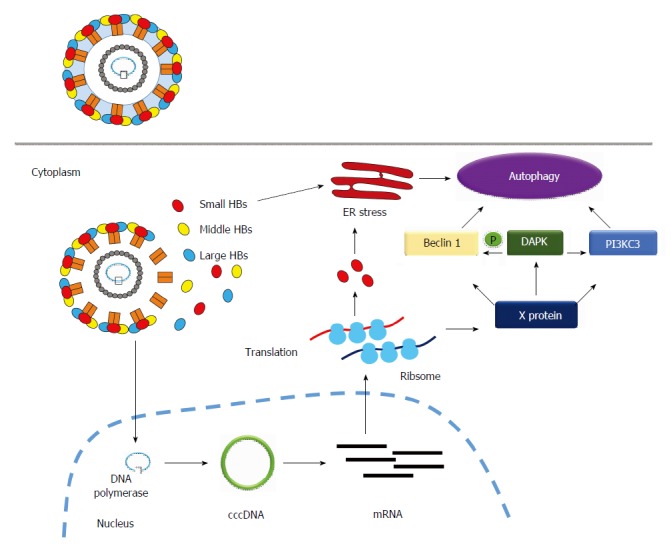
How hepatitis B virus infection induces autophagy. Hepatitis B virus (HBV) induces autophagy mainly through HBx protein or small surface protein (Small HBs)-related mechanism. The former (HBx) induces autophagy by the following routes: (1): Increases mRNA expression of Beclin 1 through transcriptional regulation; (2): Activates enzymatic activity of PI3KC3 to enhance autophagosome formation; (3): Activates DAPK to trigger autophagy in a Beclin 1-PI3KC3-dependent manner. The latter (SHBs) triggers ER stress to induce autophagy. DAPK: Death-associated protein kinase.
HBV small surface protein (SHB) induces autophagy: The HBV envelope proteins also plays a role in HBV-induced autophagy. There are three envelope proteins, including large, medium, and small surface proteins (SHBs), in HBV. Li et al[29] found that deletion of the HBV envelope proteins abrogate HBV-induced autophagosome formation. They further demonstrated that the intracellular SHBs, but not extracellular SHBs, trigger endoplasmic reticulum (ER) stress and unfolding protein responses including ATF-6, PERK and IRE1 signaling pathways to induce autophagy (Figure 1). SHBs do not affect the expression level of Beclin 1, and therefore involves a different mechanism to that of HBx-induced autophagy. The phenomena described above indicate that HBV may use various structure proteins to induce autophagy through different mechanisms.
The effect of HBV-induced autophagy on HBV replication
HBV infection is thought to follow a particular sequence involving virus entry followed by cccDNA synthesis, mRNA transcription, viral protein synthesis, encapsidation, viral DNA replication (reverse transcription), envelopment and release of the mature viruses[30]. HBV-induced autophagy positively or negatively regulates virus replication at different stages of HBV infection.
Promotion of HBV replication - DNA replication: To clarify the effect of HBV-induced autophagy on HBV replication, Sir et al[21] suppressed autophagy using an inhibitor (3-MA) or by knocking out autophagy-related genes using siRNA (si-Atg7 or si-Vps34) during virus infection, and revealed that inhibition of autophagy only slightly decreased viral mRNA synthesis and HBV RNA packaging. HBV DNA replication was significantly suppressed, suggesting that autophagy mainly enhances HBV replication. They further revealed colocalization of the HBV core protein and autophagosome during HBV infection, suggesting that the autophagosome may function as the docking site for viral replication in a similar manner to that observed in poliovirus and dengue virus infection[31,32]. Tain et al[33] utilized the HBV transgenic mice harboring liver-specific knockout of Atg5 gene (Atg5-/-) to demonstrate that autophagy enhances HBV replication in vivo. In summary, these data indicate that autophagy promotes HBV replication through induction of viral DNA replication (Figure 2). Whether unconventional secretory autophagy plays a role in HBV replication and release warrants further investigation.
Figure 2.
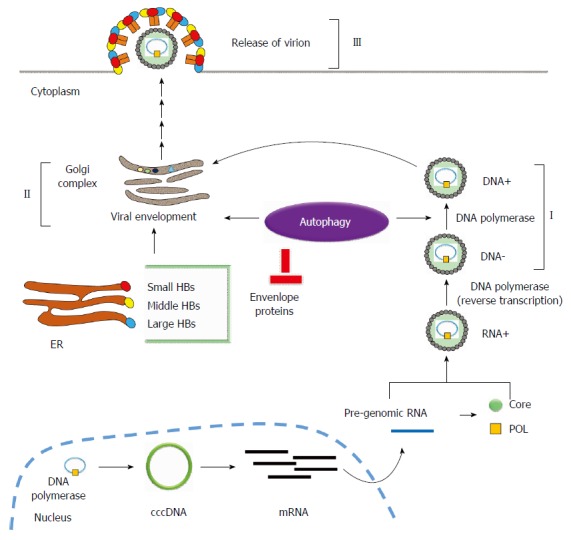
The relationship between hepatitis B virus-induced autophagy and hepatitis B virus replication. Hepatitis B virus (HBV)-induced autophagy regulates virus replication and maturation at different stages of HBV replication. (I): Autophagy enhances HBV replication at the DNA replication stage; (II): HBV-induced autophagy is required for viral envelopment. However, envelope proteins (HBs) could be eliminated via the autophagic degradation pathway (III).
Promotion of HBV replication: viral envelopment: During HBV infection, viral DNA is synthesized through reverse transcription within the nucleocapsid, which then progresses to envelopment in the Golgi apparatus and becomes the mature virion followed by exocytosis to release the mature viruses from the cell. HBV nucleocapsid-associated DNA (I), intracellular enveloped DNA (II) and extracellular enveloped DNA (III) represent the three steps of HBV viral nucleocapsid, envelopment, and secretion. Li et al[29] showed that suppression of HBV-induced autophagy slightly reduced nucleocapsid-associated DNA (II) but significantly decreased intracellular enveloped DNA (II) and extracellular enveloped DNA (III), indicating that HBV-induced autophagy plays a major role during viral envelopment (Figure 2). Finally, colocalization of HBV envelope proteins with the autophagosome supports the notion that autophagosome may serve as the docking site for HBV envelopment.
Suppression of HBV replication - Degradation of envelope proteins: ER plays multiple functions including protein folding and transporting cargo to the Golgi apparatus. Misfolded proteins may accumulate, causing ER stress and unfolded protein responses (UPR)[34]. HBV infection induces ER stress and activation the IRE1-XBP1 signaling pathway of UPR. Accordingly, HBV envelope proteins are translocated into ER for viral envelopment[35,36]. To relieve HBV-induced UPR, EDEM (ER degradation-enhancing, mannosidase-like) proteins recognize and transport the misfolded glycoproteins to the ER-related signaling pathways for degradation[37]. Lazar et al[38] reported that EDEM1 is upregulated to interact with HBV viral envelope proteins for degradation during HBV infection. Furthermore, the degradation of HBV envelope proteins was reversed by an autophagy inhibitor but not by a proteasomal inhibitor, implying that interacted envelope proteins are diminished by the autophagic degradation pathway. Furthermore, EDEM1-mediated degradation of HBV envelope proteins significantly suppressed the secretion of the enveloped and subviral HBV particles (Figure 2).
The relationship between autophagy and HBV in HBV-related tumorigenesis
Although many studies have reported that HBV induces autophagy, which promotes viral replication during HBV infection both in vitro and in vivo, the role of autophagy in HBV-associated tumorigenesis remains unclear. Many reports showed that autophagy plays a suppressive role in HCC tumorigenesis. The mice with mosaic deletion of Atg5 or liver-specific Atg7-/- gene developed multiple liver tumors. In addition, autophagic gene Beclin 1 expression was decreased in HCC tumors compared with adjacent non-tumor tissues[39,40]. However, Tian et al[41] claimed that autophagy shifts from a suppressive role to a promoter role in HCC development at the late stages by inhibiting the expression of various tumor suppressor genes including p53 and p21. In the following section, the current findings with respect to the mechanism by which autophagy influences HBV-related tumorigenesis and the implications for potential therapies are described.
The role of autophagy in HBV-related tumorigenesis: Both the HBV transgenic mouse model and human HCC specimens were analyzed to clarify the role of autophagy in HBV-related tumorigenesis. In contrast to autophagy induction by HBV infection, autophagy is suppressed in the liver tumors of a transgenic mouse model and HBV-related HCC. In the late stages of liver tumor development in the HBx transgenic mice model, autophagic activity is decreased following liver tumor formation. Both mRNA and protein levels of autophagy-related genes are reduced in the tumor tissues[42]. Qu et al[43] demonstrated that heterozygous deletion of Beclin 1 in the HBV transgenic mice increased spontaneous malignancies and accelerated HBV-induced HCC. Furthermore, in a clinical investigation, Kotsafti et al[44] showed that the mRNA of Beclin 1 was significantly lower in HCC tissues than in chronic hepatitis tissues. Consistent with these reports, we demonstrated that the protein levels of Atg5 and Beclin 1 were significantly lower in tumor tissues compared with the adjacent non-tumor tissues of HBV-associated HCC[43]. These results imply that autophagy plays a tumor suppressive role in hepatocarcinogenesis and is inhibited in HBV-associated tumorigenesis. However, the mechanism by which high autophagy during HBV infection shifts to low autophagy in HBV-related tumorigenesis remains unclear.
Autophagy-related potential therapy for HBV-related HCC: Although the role of autophagy in HBV-related tumorigenesis remains uncertain, several research groups have claimed that autophagy exerts a suppressive effect in HBV-related HCC tumorigenesis. Therefore, manipulating autophagic activity may have potential in the development of alternative therapies in the treatment of HCC. Different therapeutic strategies involving the regulation of autophagic activity in HCC treatment are described as: (1) Autophagy induces cell death; One method of suppressing HCC tumorigenesis is to induce hepatoma cell death. In contrast to type I programmed cell death (apoptosis), the checkpoints of type II programmed cell death autophagy are determined using several metabolic stresses[45]. Su et al[46] showed that soybean fermentation products containing live bacteria (SCB) were used to suppress liver tumor formation via induction of apoptosis and autophagy without significantly changing the mean body and liver weight in a syngeneic mouse model. Inhibition of autophagy by the inducer 3-MA suppresses SCB-induced apoptosis, indicating that SCB induced-autophagy promotes apoptotic cell death. Furthermore, Zhang et al[47] designed an inhibitor (NTI-007) to treat HBV-infected cells, which targets the multiple transmembrane transporter as well as the functional receptor for HBV and leads to autophagic cell death of the HBV-infected cells. Taken together, the above findings indicate that some natural and synthetic compounds have chemotherapeutic potential against HBV infection through the induction of autophagic cell death; (2) Autophagy enhances antitumor immune responses: Another therapeutic approach in the treatment of HBV-related HCC involves stimulating antitumor immune responses[48,49]. HBx protein is an oncoprotein that has been detected in 80% of HBV-related HCC and is therefore a potential target for immunotherapy[50,51]. Yan et al[52] reported that significant antitumor immune responses could be induced by irradiation treatment of mice inoculated with the HBx gene expressing tumor cells. Irradiation of the tumor cells harboring the HBx gene induced the cytolytic T lymphocyte response which recognizes and lyses the HBx-expressing hepatoma cells. Accordingly, autophagosomes and autolysosomes were detected in the irradiated tumor cells expressing the HBx gene. These findings suggest that this autophagy-enhanced CD8+ and CD4+ T lymphocyte antitumor response could lead to the development of a promising therapy against HBx-related HCC; and (3) Autophagy degrades oncogenic miRNA: Selective autophagy has been reported to recognize and clean the specific cytosolic targets through autolysosome degradation[53]. Autophagy may suppress tumor formation of various cancers through selective degradation of the oncogenic molecules including proteins, microRNAs, and damaged organelles[54]. Lan et al[42] recently demonstrated that low autophagic activity together with high expression of miR-224 were detected both in HBx transgenic mice and HBV-related HCC patients. Autophagy selectively recruited the oncogenic miR-224, promoting tumor formation and cellular migration activity by silencing its target gene smad4. Furthermore, boosting autophagic activity by amiodarone, an autophagy inducer, suppressed oncogenic miR-224 expression and significantly reduced liver tumor formation in a rat orthotopic liver tumor model. These results demonstrated that besides proteins and organelles, microRNAs could also be degraded by autophagic degradation machinery to suppress HBV-related tumorigenesis.
Taken together, increased autophagic activity suppresses HBV-related HCC tumorigenesis through the induction of cell death, enhancement of the immune response, or by the degradation of oncogenic factors.
THE EFFECT OF MIRNAS ON HBV
MiRNAs regulate gene expression through post-transcriptional modification by targeting 3’ UTR of mRNA and silencing its translation[55]. During viral infection, the miRNAs either from infected viruses or the host cells could affect the target gene expression of viruses or host cells[56,57]. Here, we explored the mechanisms by which miRNAs regulate HBV replication as well as the underlying mechanism whereby miRNAs biogenesis is regulated by HBV.
MiRNAs affect HBV replication
HBV gene expression and replication can be regulated by host or viral miRNAs. These miRNAs may suppress or promote HBV infection at each stage of viral replication. We investigated the process by which miRNAs regulates HBV replication through direct targeting of mRNA in virus genes or by targeting host genes which are required for virus replication.
MiRNAs directly target HBV genes: Wu et al[58] used clinical HBV patient specimens and identified four human host miRNAs let-7, miR-345, miR-433 and miR-511 using target gene prediction software, which targeted the highly conserved HBV genes including the genes of polymerase, S and preC in different clades of HBV, indicating that these host miRNAs suppress HBV replication. Moreover, Kohno et al[59] used SCID mice harboring humanized hepatocyte cells to identify miRNAs upregulated by HBV infection in the liver tissue. MiR-1231 was the most upregulated miRNA which suppressed HBV replication through the inhibition of HBV core protein expression. In addition, miR-125a-5p, miR-199a-3p and miR-210 have also been reported to be able to suppress HBV replication by targeting and reducing S gene expression[60,61]. In addition, one predicted HBV miRNA located in the precore region (CAUGUCCUACUGUUCAAGCCUC) may target three HBV genes (large S, polymerase and X genes) and serves as the feedback regulation of HBV replication to maintain the latent status[62]. Taken together, miRNAs may suppress HBV replication through the suppression of diverse viral gene expression.
MiRNAs regulate HBV replication by targeting host genes: It has been shown that HBV-synthesized miRNAs may target its own genes to affect its replication, but host miRNAs could also affect virus replication by targeting host genes. Guo et al[63] reported that miR-372/373 was upregulated in HepG2.2.15 cells, stably transfected with a complete HBV genome, and miR-372/373 enhanced the expression levels of HBV core-associated DNA and HBx protein. They found that miR-372/373 silenced the host transcription factor nuclear factor I/B (NFIB), which is a repressor bound to the enhancer 1 region of HBV genome including the S gene promoter, suggesting that HBV enhances its replication through the upregulation of miR-372/373 to suppress NFIB expression. In addition, Jin et al[64] showed that miR-501 is overexpressed in HBV-infected cell lines and in HCC specimens, thereby promoting HBV replication by suppressing expression of HBXIP, which is a host protein interacting with HBx protein at its transactivation domain to repress its function. These findings imply that HBV enhances its replication by regulating host miRNAs expression to suppress anti-HBV proteins from the host cell.
One research group identified potential miRNAs which regulate HBV replication by screening a library of miRNAs. Zhang et al[65] reported that overexpression of miR-1 in hepatoma cells increased HBV replication by silencing histone deacetylase 4 (HDAC4) and increased the expression of farnesoid X receptor (FXRA). FXRA, a transcriptional factor, interacts with RXRA (FXRA/RXRA) and then binds to the HBV core promoter to enhance its activity. HDACs regulate acetylation of H3/H4 histones bound to the cccDNA of HBV and this epigenetic modification regulates the transcription and replication of HBV. In contrast, Hu et al[66] reported that miR-141 identified by screening 64 miRNAs suppressed HBV replication by silencing host transcription factor PPARA, which is an essential factor for HBV pregenomic RNA synthesis and viral replication. These findings summarize the miRNAs which affect virus replication through the suppression of host genes.
MiR-15b promotes HBV replication by silencing hepatocyte nuclear factor 1 alpha, a repressor of the HBV enhancer I region. HBV replication upregulates HBx protein expression, which then suppresses miR-15b expression[67]. The relationship between HBV and microRNA suggests a feedback regulation between HBV replication and host miRNAs expression. MicroRNA may play dual roles in HBV replication. MiR-122 is highly expressed in the liver of HCC patients and is positively associated with HBV infection. Qiu et al[68] showed that miR-122 promotes HBV replication by silencing heme oxygenase-1 which interacts and reduces the stability of the HBV core protein. The above findings are in contrast to the results of a study by Ji et al[69] that showed miR-122 suppresses HBV replication.
HBV regulates the biogenesis of miRNAs
HBV may affect microRNA biogenesis to alter the expression of miRNAs. MiRNAs are initially transcribed by RNA polymerase II and III to form primary miRNA (pri-miRNA, 300-1000 nucleotides) in the nucleus, which is subsequently processed by the microprocessor consisting of RNase, Drosha, and DGCR8 (DiGeorge syndrome critical region gene 8) resulting in a shorter precursor miRNA (pre-miRNA, 70-90 nucleotides). Pre-miRNA is then exported by the nuclear export receptor exportin-5 and RAN-GTP to the cytoplasm, where it is processed by Dicer to generate the mature miRNA. The mature miRNA is incorporated into the RNA-induced silencing complex (RISC) and then regulates gene silencing. HBV affects the biogenesis of miRNAs at various stages through different mechanisms.
HBV affects miRNA transcription and stability: MiR-122 is a liver-specific miRNA which maintains liver function. Dysregulated miR-122 affects virus replication of diverse hepatitis viruses[70]. In HCV infection, miR-122 promotes its replication by interacting with the 5’-non-coding region (5’-NCR) of HCV[71]. In contrast, miR-122 inhibits HBV replication and is significantly downregulated during HBV infection[72]. HBV suppresses miR-122 using various mechanisms. Song et al[73] reported that HBV suppresses miR-122 expression via the HBx protein, which interacts with peroxisome proliferator activated receptor-gamma (PPARγ) and suppresses the transactivation function of PPARγ/RXRα complex on the promoter of miR-122 (Figure 3A). In addition, HBV downregulates miR-122 expression by influencing miRNA stabilization. Peng et al[74,75] demonstrated that HBx suppresses transcription of Gld2 protein (germline development 2) which specifically increases miR-122 stabilization by catalyzing 3’ monoadenylation and reduces miR-122 maturation (Figure 3B). Li et al[76] showed that HBV suppresses miR-122 expression and causes the upregulation of the miR-122 target gene PTTG (pituitary tumor-transforming gene 1 binding factor), which leads to the promotion of HCC tumorigenesis. They further identified the miR-122 complementary targeting site on the four mRNAs of HBV, including pre-C/C, pre-S, S and X gene. Therefore, HBV mRNA functions as a sponge to bind and reduce miR-122 expression (Figure 3C). Taken together, the above findings provide mechanistic insights into the differential regulation of miRNAs by HBV.
Figure 3.
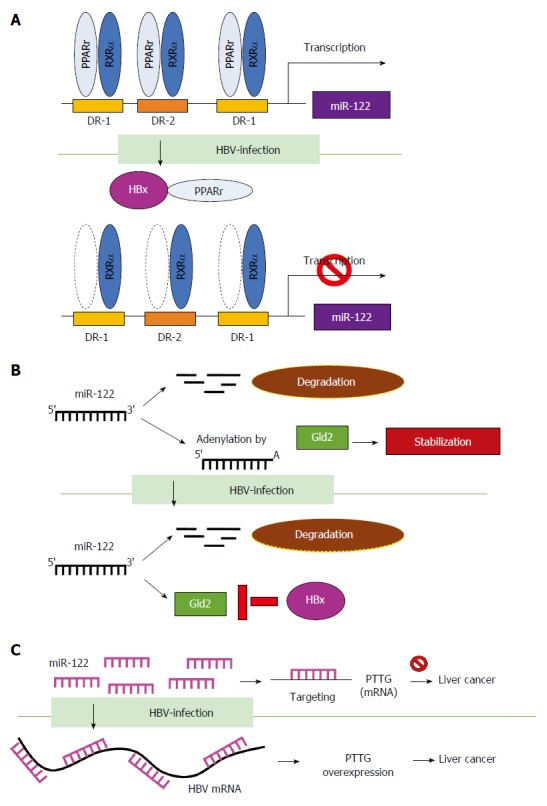
Hepatitis B virus affects microRNA transcription and stability. Hepatitis B virus (HBV) suppresses miR-122 through different mechanisms. A: HBV suppresses miR-122 promoter activity and expression through transcriptional regulation by directly interacting with peroxisome proliferator activated receptor-gamma (PPARγ); B: HBx suppresses transcription of Gld2 to decrease miR-122 stability; and C: HBV functions as a sponge to bind miR-122 and reduces its expression. RXRa: Retinoid X Receptor, Alpha; Gld2: Germline development 2.
HBV affects RNase expression: HBV infection alters miRNA expression profiles in infected cell lines and clinical HCC patients[69,77]. HBV could also regulate miRNA biogenesis through miRNA-related RNases. Drosha and Dicer are two miRNA processing components required for miRNA maturation. Recent evidence showed that HBV dysregulates miRNA biogenesis by affecting these two proteins. Ren et al[78] reported that expression of both Drosha mRNA and protein was suppressed in cells expressing the HBV genome. They revealed that HBx protein downregulates Drosha expression through the reduction of its promoter activity (Figure 4). Furthermore, HBV upregulates the expression of the repressor YY1 to suppress Drosha cofactor DGCR8 expression and subsequently represses DGCR8 promoter activity (Figure 4)[79]. Lund et al[80] identified three single nucleotide polymorphisms (SNPs) including DICER rs1057035 and RAN rs3803012 in HBV-related clinical HCC specimens, and these SNPs were found to be associated with the risk of HBV-related HCC occurrence. They found that the SNPs located on the 3’UTR of DICER and RAN genes affect the binding affinity of miR-574-3p and miR-199a-3p, respectively. Dysregulated DICER and RAN affect miRNA biogenesis in HBV-related diseases (Figure 4)[81]. Taken together, these findings show that HBV suppresses miRNA biogenesis through the repression of the promoter activity of mature miRNA-related genes.
Figure 4.
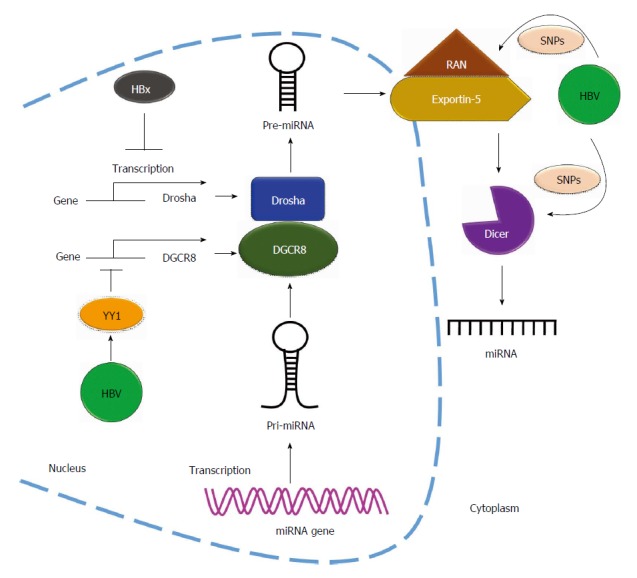
Hepatitis B virus regulates microRNAs biogenesis. Hepatitis B virus (HBV) regulates the biogenesis of miRNA by affecting the miRNA-related Rnases. HBx protein downregulates Drosha expression through reduction of the transcriptional activity of Drosha promoter and upregulates the expression of transcription factor YY1 to suppress DGCR8 expression by repressing DGCR8 promoter activity. Single nucleotide polymorphisms (SNPs) located on 3’UTR of Dicer and RAN genes affect the biogenesis of miRNA in HBV-related diseases. DGCR8: DiGeorge syndrome critical region gene 8.
CONCLUSIONS AND FUTURE DIRECTIONS
In summary, we provide a detailed description of our current understanding of the relationships among HBV, autophagy, and miRNAs. However, the following questions remain unresolved: (1) Based on manipulation of autophagic activity, therapeutic strategies against the diseases of HBV-acute infection and HBV-related HCC tumorigenesis should be different. Because autophagy plays a promoting role in HBV replication but a suppressive role in HBV-associated tumorigenesis, the timing of HBV infection as well as the tumorigenesis status of the HCC patient should be carefully evaluated before considering an autophagy-related therapy. Further studies on combination therapy using autophagy inducers and anti-viral drugs for HBV-related diseases should be considered; (2) The role of secretory autophagy in HBV infection and its related pathogenesis could be further explored. Secretory autophagy has been reported to regulate cytokines secretion including IL-1β, IL-18 and HMGB1 (high-mobility group box 1 protein). Interestingly, IL-1β and HMGB1 were significantly upregulated in the serum of patients with hepatitis B virus-related acute-on-chronic liver failure[82,83], and the expression of IL-18 in peripheral blood mononuclear cells from patients with acute HBV infection was higher compared with the expression in patients with chronic HBV infection[84]. These reports imply that HBV may affect release of these cytokines through HBV-induced secretory autophagy. Furthermore, the HBV gene in liver-specific Atg5 knockout transgenic mice showed significantly lower expression of HBsAg and HBeAg in the serum compared with the wild-type mice[33]. It is possible that HBV-induced secretory autophagy contributes to release of viral antigens. Further studies are needed to clarify the role of secretory autophagy in HBV infection and pathogenesis; (3) The role of HBV-affected miRNA biogenesis in HBV replication remains undetermined. Current evidence indicates that HBV suppresses miRNAs biogenesis by affecting RNases and cofactors of miRNA biogenesis, but whether this is beneficial or harmful for HBV replication remains unclear; and (4) The effect of autophagy and/or miRNAs on HBV recurrence and latency has not been clearly elucidated. During HBV infection, virus infection is defined as acute in the initial period of several months and is defined as chronic when the infection period has lasted for many years. HBV is actively replicated during the acute infection phase followed by recovery in the presence of the host immune responses. However, during the chronic infection, the immune-tolerant phase is characterized by a high level of HBV DNA. When host immunity shifts to the immune-active status, HBV accordingly becomes less replicative. However, HBV may be reactivated to a high replication status in certain HBV carriers. These phenomena imply that HBV continuously undergoes reactivation and latency. Further exploration is needed to clarify the mechanisms by which autophagy and/or miRNAs affect HBV recurrence and latency in HBV-infected patients.
Footnotes
Supported by Ministry of Science and Technology (NSC 101-2320-B-006-025-MY3).
Conflict-of-interest statement: The authors declare no potential conflicts of interest and no financial support.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 29, 2015
First decision: August 31, 2015
Article in press: October 26, 2015
P- Reviewer: Kuramitsu Y, Osna NA S- Editor: Yu J L- Editor: A E- Editor: Liu XM
References
- 1.Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, Xiang Y, Cuervo AM, Czaja MJ. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem. 2008;283:4766–4777. doi: 10.1074/jbc.M706666200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. doi: 10.1016/j.ceb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhattacharya A, Prakash YS, Eissa NT. Secretory function of autophagy in innate immune cells. Cell Microbiol. 2014;16:1637–1645. doi: 10.1111/cmi.12365. [DOI] [PubMed] [Google Scholar]
- 4.Jiang S, Dupont N, Castillo EF, Deretic V. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun. 2013;5:471–479. doi: 10.1159/000346707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakai Y, Oku M, van der Klei IJ, Kiel JA. Pexophagy: autophagic degradation of peroxisomes. Biochim Biophys Acta. 2006;1763:1767–1775. doi: 10.1016/j.bbamcr.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 6.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14:376–391. doi: 10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- 8.Yousefi S, Simon HU. Autophagy in cancer and chemotherapy. Results Probl Cell Differ. 2009;49:183–190. doi: 10.1007/400_2008_25. [DOI] [PubMed] [Google Scholar]
- 9.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin S, White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy. 2008;4:563–566. [PMC free article] [PubMed] [Google Scholar]
- 11.Deretic V, Kimura T, Timmins G, Moseley P, Chauhan S, Mandell M. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75–84. doi: 10.1172/JCI73945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mostowy S, Cossart P. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 2012;22:283–291. doi: 10.1016/j.tcb.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Lee YR, Hu HY, Kuo SH, Lei HY, Lin YS, Yeh TM, Liu CC, Liu HS. Dengue virus infection induces autophagy: an in vivo study. J Biomed Sci. 2013;20:65. doi: 10.1186/1423-0127-20-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 16.Couzin J. MicroRNAs make big impression in disease after disease. Science. 2008;319:1782–1784. doi: 10.1126/science.319.5871.1782. [DOI] [PubMed] [Google Scholar]
- 17.Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, Schluep T. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antiviral Res. 2015;121:47–58. doi: 10.1016/j.antiviral.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Shi Y, Yang H. [Infection with hepatitis B virus enhances basal autophagy] Weishengwu Xuebao. 2010;50:1651–1656. [PubMed] [Google Scholar]
- 20.Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, Li F, Wang Y, Tiollais P, Li T, et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology. 2009;49:60–71. doi: 10.1002/hep.22581. [DOI] [PubMed] [Google Scholar]
- 21.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci USA. 2010;107:4383–4388. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia Y, Zeng D, Yu F, He J, Zhou Z, Tu W, Deng H, Tian DA, Liu M. Role of autophagy in monokine induced by interferon γ (Mig) production during adenovirus-hepatitis B virus infection. Hepatogastroenterology. 2012;59:1245–1250. doi: 10.5754/hge12089. [DOI] [PubMed] [Google Scholar]
- 23.Zhang HT, Chen GG, Hu BG, Zhang ZY, Yun JP, He ML, Lai PB. Hepatitis B virus x protein induces autophagy via activating death-associated protein kinase. J Viral Hepat. 2014;21:642–649. doi: 10.1111/jvh.12191. [DOI] [PubMed] [Google Scholar]
- 24.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirth M, Joachim J, Tooze SA. Autophagosome formation--the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol. 2013;23:301–309. doi: 10.1016/j.semcancer.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Levin-Salomon V, Bialik S, Kimchi A. DAP-kinase and autophagy. Apoptosis. 2014;19:346–356. doi: 10.1007/s10495-013-0918-3. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Chen GG, Zhang Z, Chun S, Leung BC, Lai PB. Induction of autophagy in hepatocellular carcinoma cells by SB203580 requires activation of AMPK and DAPK but not p38 MAPK. Apoptosis. 2012;17:325–334. doi: 10.1007/s10495-011-0685-y. [DOI] [PubMed] [Google Scholar]
- 28.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol. 2011;85:6319–6333. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 31.Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374:240–248. doi: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol. 2007;81:12543–12553. doi: 10.1128/JVI.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol. 2011;85:13453–13456. doi: 10.1128/JVI.06064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiramatsu N, Chiang WC, Kurt TD, Sigurdson CJ, Lin JH. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am J Pathol. 2015;185:1800–1808. doi: 10.1016/j.ajpath.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W, Cao Y, Wang T, Xiang G, Lu J, Zhang J, Hou P. The N-Glycosylation Modification of LHBs (Large Surface Proteins of HBV) Effects on Endoplasmic Reticulum Stress, Cell Proliferation and its Secretion. Hepat Mon. 2013;13:e12280. doi: 10.5812/hepatmon.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Gao B, Ye L, Han X, Wang W, Kong L, Fang X, Zeng Y, Zheng H, Li S, et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007;124:44–49. doi: 10.1016/j.virusres.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 37.Hirao K, Natsuka Y, Tamura T, Wada I, Morito D, Natsuka S, Romero P, Sleno B, Tremblay LO, Herscovics A, et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J Biol Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- 38.Lazar C, Macovei A, Petrescu S, Branza-Nichita N. Activation of ERAD pathway by human hepatitis B virus modulates viral and subviral particle production. PLoS One. 2012;7:e34169. doi: 10.1371/journal.pone.0034169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, Shi GM, Wang XY, Ke AW, Wu B, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68:9167–9175. doi: 10.1158/0008-5472.CAN-08-1573. [DOI] [PubMed] [Google Scholar]
- 40.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, Ou JH. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015;22:1025–1034. doi: 10.1038/cdd.2014.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014;59:505–517. doi: 10.1002/hep.26659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kotsafti A, Farinati F, Cardin R, Cillo U, Nitti D, Bortolami M. Autophagy and apoptosis-related genes in chronic liver disease and hepatocellular carcinoma. BMC Gastroenterol. 2012;12:118. doi: 10.1186/1471-230X-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256. doi: 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su CL, Chen FN, Won SJ. Involvement of apoptosis and autophagy in reducing mouse hepatoma ML-1 cell growth in inbred BALB/c mice by bacterial fermented soybean products. Food Chem Toxicol. 2011;49:17–24. doi: 10.1016/j.fct.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Fu LL, Tian M, Liu HQ, Li JJ, Li Y, He J, Huang J, Ouyang L, Gao HY, et al. Design and synthesis of a novel candidate compound NTI-007 targeting sodium taurocholate cotransporting polypeptide [NTCP]-APOA1-HBx-Beclin1-mediated autophagic pathway in HBV therapy. Bioorg Med Chem. 2015;23:976–984. doi: 10.1016/j.bmc.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Cheng P, Wen Y, Chen P, Yang L, Zhao X, Lv H, Quan Q, Wu Y, Yang H, et al. T lymphocyte responses against hepatitis B virus-related hepatocellular carcinoma induced by adenovirus vaccine encoding HBx. Int J Mol Med. 2010;26:869–876. doi: 10.3892/ijmm_00000536. [DOI] [PubMed] [Google Scholar]
- 49.Wang YJ, Hou Y, Huang H, Liu GR, White AP, Liu SL. Two oral HBx vaccines delivered by live attenuated Salmonella: both eliciting effective anti-tumor immunity. Cancer Lett. 2008;263:67–76. doi: 10.1016/j.canlet.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 50.Wang C, Yang W, Yan HX, Luo T, Zhang J, Tang L, Wu FQ, Zhang HL, Yu LX, Zheng LY, et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology. 2012;55:108–120. doi: 10.1002/hep.24675. [DOI] [PubMed] [Google Scholar]
- 51.Zhang XD, Wang Y, Ye LH. Hepatitis B virus X protein accelerates the development of hepatoma. Cancer Biol Med. 2014;11:182–190. doi: 10.7497/j.issn.2095-3941.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan Y, Liu N, Lu L, Zang CM, Shao B, Li Y, Wen Y, Wei Y, Cheng P. Autophagy enhances antitumor immune responses induced by irradiated hepatocellular carcinoma cells engineered to express hepatitis B virus X protein. Oncol Rep. 2013;30:993–999. doi: 10.3892/or.2013.2531. [DOI] [PubMed] [Google Scholar]
- 53.Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 54.Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy. 2013;9:1720–1736. doi: 10.4161/auto.26550. [DOI] [PubMed] [Google Scholar]
- 55.Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Staedel C, Darfeuille F. MicroRNAs and bacterial infection. Cell Microbiol. 2013;15:1496–1507. doi: 10.1111/cmi.12159. [DOI] [PubMed] [Google Scholar]
- 57.Guo YE, Steitz JA. Virus meets host microRNA: the destroyer, the booster, the hijacker. Mol Cell Biol. 2014;34:3780–3787. doi: 10.1128/MCB.00871-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu FL, Jin WB, Li JH, Guo AG. Targets for human encoded microRNAs in HBV genes. Virus Genes. 2011;42:157–161. doi: 10.1007/s11262-010-0555-7. [DOI] [PubMed] [Google Scholar]
- 59.Kohno T, Tsuge M, Murakami E, Hiraga N, Abe H, Miki D, Imamura M, Ochi H, Hayes CN, Chayama K. Human microRNA hsa-miR-1231 suppresses hepatitis B virus replication by targeting core mRNA. J Viral Hepat. 2014;21:e89–e97. doi: 10.1111/jvh.12240. [DOI] [PubMed] [Google Scholar]
- 60.Zhang GL, Li YX, Zheng SQ, Liu M, Li X, Tang H. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res. 2010;88:169–175. doi: 10.1016/j.antiviral.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Potenza N, Papa U, Mosca N, Zerbini F, Nobile V, Russo A. Human microRNA hsa-miR-125a-5p interferes with expression of hepatitis B virus surface antigen. Nucleic Acids Res. 2011;39:5157–5163. doi: 10.1093/nar/gkr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin WB, Wu FL, Kong D, Guo AG. HBV-encoded microRNA candidate and its target. Comput Biol Chem. 2007;31:124–126. doi: 10.1016/j.compbiolchem.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 63.Guo H, Liu H, Mitchelson K, Rao H, Luo M, Xie L, Sun Y, Zhang L, Lu Y, Liu R, et al. MicroRNAs-372/373 promote the expression of hepatitis B virus through the targeting of nuclear factor I/B. Hepatology. 2011;54:808–819. doi: 10.1002/hep.24441. [DOI] [PubMed] [Google Scholar]
- 64.Jin J, Tang S, Xia L, Du R, Xie H, Song J, Fan R, Bi Q, Chen Z, Yang G, et al. MicroRNA-501 promotes HBV replication by targeting HBXIP. Biochem Biophys Res Commun. 2013;430:1228–1233. doi: 10.1016/j.bbrc.2012.12.071. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Zhang E, Ma Z, Pei R, Jiang M, Schlaak JF, Roggendorf M, Lu M. Modulation of hepatitis B virus replication and hepatocyte differentiation by MicroRNA-1. Hepatology. 2011;53:1476–1485. doi: 10.1002/hep.24195. [DOI] [PubMed] [Google Scholar]
- 66.Hu W, Wang X, Ding X, Li Y, Zhang X, Xie P, Yang J, Wang S. MicroRNA-141 represses HBV replication by targeting PPARA. PLoS One. 2012;7:e34165. doi: 10.1371/journal.pone.0034165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dai X, Zhang W, Zhang H, Sun S, Yu H, Guo Y, Kou Z, Zhao G, Du L, Jiang S, et al. Modulation of HBV replication by microRNA-15b through targeting hepatocyte nuclear factor 1α. Nucleic Acids Res. 2014;42:6578–6590. doi: 10.1093/nar/gku260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y, Chen L, Chen Y, Duan Z, Meng S. miR-122-induced down-regulation of HO-1 negatively affects miR-122-mediated suppression of HBV. Biochem Biophys Res Commun. 2010;398:771–777. doi: 10.1016/j.bbrc.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 69.Ji F, Yang B, Peng X, Ding H, You H, Tien P. Circulating microRNAs in hepatitis B virus-infected patients. J Viral Hepat. 2011;18:e242–e251. doi: 10.1111/j.1365-2893.2011.01443.x. [DOI] [PubMed] [Google Scholar]
- 70.Hu J, Xu Y, Hao J, Wang S, Li C, Meng S. MiR-122 in hepatic function and liver diseases. Protein Cell. 2012;3:364–371. doi: 10.1007/s13238-012-2036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang J, Guo JT, Jiang D, Guo H, Taylor JM, Block TM. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J Virol. 2008;82:8215–8223. doi: 10.1128/JVI.02575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang S, Qiu L, Yan X, Jin W, Wang Y, Chen L, Wu E, Ye X, Gao GF, Wang F, et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology. 2012;55:730–741. doi: 10.1002/hep.24809. [DOI] [PubMed] [Google Scholar]
- 73.Song K, Han C, Zhang J, Lu D, Dash S, Feitelson M, Lim K, Wu T. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology. 2013;58:1681–1692. doi: 10.1002/hep.26514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.D’Ambrogio A, Gu W, Udagawa T, Mello CC, Richter JD. Specific miRNA stabilization by Gld2-catalyzed monoadenylation. Cell Rep. 2012;2:1537–1545. doi: 10.1016/j.celrep.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peng F, Xiao X, Jiang Y, Luo K, Tian Y, Peng M, Zhang M, Xu Y, Gong G. HBx down-regulated Gld2 plays a critical role in HBV-related dysregulation of miR-122. PLoS One. 2014;9:e92998. doi: 10.1371/journal.pone.0092998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li C, Wang Y, Wang S, Wu B, Hao J, Fan H, Ju Y, Ding Y, Chen L, Chu X, et al. Hepatitis B virus mRNA-mediated miR-122 inhibition upregulates PTTG1-binding protein, which promotes hepatocellular carcinoma tumor growth and cell invasion. J Virol. 2013;87:2193–2205. doi: 10.1128/JVI.02831-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, Zhao JJ, Wang CM, Li MY, Han P, Wang L, Cheng YQ, Zoulim F, Ma X, Xu DP. Altered expression profiles of microRNAs in a stable hepatitis B virus-expressing cell line. Chin Med J (Engl) 2009;122:10–14. doi: 10.3901/jme.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 78.Ren M, Qin D, Li K, Qu J, Wang L, Wang Z, Huang A, Tang H. Correlation between hepatitis B virus protein and microRNA processor Drosha in cells expressing HBV. Antiviral Res. 2012;94:225–231. doi: 10.1016/j.antiviral.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 79.Shan X, Ren M, Chen K, Huang A, Tang H. Regulation of the microRNA processor DGCR8 by hepatitis B virus proteins via the transcription factor YY1. Arch Virol. 2015;160:795–803. doi: 10.1007/s00705-014-2286-x. [DOI] [PubMed] [Google Scholar]
- 80.Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 81.Liu L, An J, Liu J, Wen J, Zhai X, Liu Y, Pan S, Jiang J, Wen Y, Liu Z, et al. Potentially functional genetic variants in microRNA processing genes and risk of HBV-related hepatocellular carcinoma. Mol Carcinog. 2013;52 Suppl 1:E148–E154. doi: 10.1002/mc.22062. [DOI] [PubMed] [Google Scholar]
- 82.Wang K, Wu ZB, Ye YN, Liu J, Zhang GL, Su YJ, He HL, Zheng YB, Gao ZL. Plasma Interleukin-10: A Likely Predictive Marker for Hepatitis B Virus-Related Acute-on-Chronic Liver Failure. Hepat Mon. 2014;14:e19370. doi: 10.5812/hepatmon.19370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Duan XZ, Hu JH, Li C, Liu FF, Liu XY, Tong JJ, Xin SJ. Relation between serum levels of high mobility group box 1 and hepatitis B virus-related acute-on-chronic liver failure. Zhonghua Ganzangbing Zazhi. 2013;21:434–437. doi: 10.3760/cma.j.issn.1007-3418.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 84.Wu DL, Xu GH, Lu SM, Ma BL, Miao NZ, Liu XB, Cheng YP, Feng JH, Liu ZG, Feng-Ding WQ, et al. Correlation of AIM2 expression in peripheral blood mononuclear cells from humans with acute and chronic hepatitis B. Hum Immunol. 2013;74:514–521. doi: 10.1016/j.humimm.2013.01.022. [DOI] [PubMed] [Google Scholar]