

Abstract
Acute myeloid leukemia (AML) is the most common acute leukemia in adults, which is heterogeneous in terms of morphological, cytogenetic and clinical features. Cytogenetic abnormalities, including karyotype aberrations, gene mutations and gene expression abnormalities are the most important diagnostic tools in diagnosis, classification and prognosis in acute myeloid leukemias. Based on World Health Organization (WHO) classification, acute myeloid leukemias can be divided to four groups. Due to the heterogeneous nature of AML and since most therapeutic protocols in AML are based on genetic alterations, gathering further information in the field of rare disorders as well as common cytogenetic abnormalities would be helpful in determining the prognosis and treatment in this group of diseases. Recently, the role of microRNAs (miRNAs) in both normal hematopoiesis and myeloid leukemic cell differentiation in myeloid lineage has been specified. miRNAs can be used instead of genes for AML diagnosis and classification in the future, and can also play a decisive role in the evaluation of relapse as well as response to treatment in the patients. Therefore, their use in clinical trials can affect treatment protocols and play a role in therapeutic strategies for these patients. In this review, we have examined rare cytogenetic abnormalities in different groups of acute myeloid leukemias according to WHO classification, and the role of miRNA expression in classification, diagnosis and response to treatment of these disorders has also been dealt with.
Key words: rare abnormalities, microRNAs, therapy, acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is a type of myeloid lineage malignancy characterized by proliferation of blast cells in bone marrow or peripheral blood.1 AML is the most common acute leukemia in adults, and its incidence is increased with increase in age (mostly over 60). It is heterogeneous in terms of morphology, immunophenotype as well as cytogenetic, molecular genetics and clinical features.1,2 Acute leukemias are classified in four groups based on World Health Organization (WHO) classification: i) AML with recurrent genetic abnormalities (AML-RGA); ii) AML with myelodysplasia-related changes (AML-MRC); iii) therapy-related acute myeloid leukemia (t-AML); iv) AML with normal or non-specific karyotype (AML-NOS).2
Cytogenetic abnormalities, including karyotype aberrations, gene mutations and disruption of gene expression are important tools to detect, classify and determine the prognosis of acute leukemia. These abnormalities form the basis of targeted and appropriate treatment of leukemia to prevent additional treatment in case of cytogenetic abnormalities with good prognosis.2,3 Overall, cytogenetic abnormalities are grouped in three categories: i) favorable; ii) intermediate; iii) adverse. Some of these abnormalities are common and some are rare.2,3
To date, several rare cytogenetic abnormalities have been identified in various AML varieties. Myeloid leukemias result from a number of genomic alterations in target genes. These somatic mutations are often cytogenetically detectable, and take the form of balanced or unbalanced chromosomal aberrations. Several myeloid hematologic malignancies are associated with balanced chromosomal aberrations, resulting in development of chimeric genes with a high genetic, diagnostic and prognostic value.4 In fact, over 200 different genes are known to have been developed by translocations in myeloid leukemias, including promyelocytic leukemia gene (PML), Fms-related tyrosine kinase 3 (FLT3) and homeobox protein-A (HOXA), rearrangement of which is responsible for various disorders in AML via development of fusion proteins. In fact, these genes encode heterogeneous disorders in different subtypes of myeloid leukemia.4 Given that the majority of treatment protocols in AML are based on certain genetic alterations, obtaining more information on rare diseases in this group of diseases would be useful in treatment of patients.5 Only a few cases of some of these disorders have been reported to date, and only one case has been reported from others in different AML subgroups (Figure 1).
Figure 1.
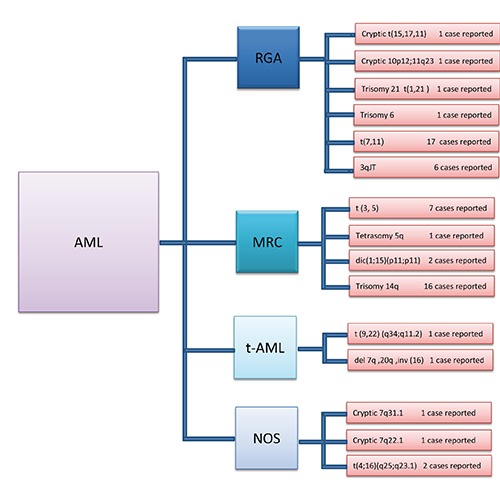
The number of cases reported in rare cytogenetic abnormalities of 4 different groups in acute myeloid leukemia (AML). The most interesting rare cytogenetic abnormalities in each group of AML have different distribution in the world. In some of abnormalities, several cases but in others just one case has been reported. RGA, recurrent genetic abnormalities; MRC, myelodysplasia-related changes; t-AML, therapy-related AML; NOS, non-specific karyotype; JT, jumping translocation; dic, dicentric chromosome.
MicroRNAs (miRNAs) constitute a group of noncoding, small and single stranded 18-25 nucleotide long RNAs regulating gene expression in two ways: i) mRNA targeting and its degradation; and ii) affecting post-translation mechanisms of proteins.6,7
Expression of miRNAs is altered in many human cancers, including acute myeloid leukemia, indicating their role in pathogenesis of this type of leukemia. Research on the role of miRNAs in AML has been increasing, suggesting the application of miRNA expression in classification of myeloid leukemias as well as in detection of rare diseases to determine the appropriate therapy for these patients (Table 1).8-44
Table 1.
Rare cytogenetic abnormalities in 4 different acute myeloid leukemia groups with miRNA involved.
| WHO category | Rare karyotype abnormality | Genes involved | Prognosis | Signaling pathway/mutation target | miRNA up regulation | miRNA down regulation | References |
|---|---|---|---|---|---|---|---|
| AML-RGA | Cryptic t(15,17,11) | PML/RARA | Favorable | PML-RARA, BAK1 | miR-378; miR-30a, b, c; let-7a-3; miR-337-3p; miR-625; miR-218; miR-27b; miR-625; miR-125B | 8-12 | |
| t(12;15;17)(q24;q24;q11) | PML/RARA | Favorable | PML-RARA, BAK1 | miR-378; miR-30a, b, c;let-7a-3; miR-337-3p; miR-625; miR-218; miR-27b; miR-625; miR-125b | 9-13 | ||
| Cryptic 10p12;11q23 | MLL/MLLPTD | Adverse | PBX3 | miR-30a, b, c;let-7a-3;miR-196b; miR-17h, g | miR-210; miR-23a; miR-181b;miR-495 | 10,11,14 | |
| t(8;21)(q22;q22) | AML1/ETO | Favorable | RUNX1/RUNX1T1 | miR-30a, b, c;miR-378; let-7a-3; miR-223 | 9-11,15 | ||
| t(8;21)(q13;q22) | Favorable | 16 | |||||
| t(1;8;21)(p35;q22;q22) | AML1/ETO | Favorable | RUNX1/RUNX1T1 | miR-30a, b, c; miR-378; let-7a-3 | 9-11,17 | ||
| Trisomy 6 | Favorable | 18 | |||||
| t(7,11) | NUP98/HOXA | Adverse | 19 | ||||
| 3qJT | RPN1/EV11 | Adverse | NOTCH1, BCL2 | miR-449a | 10,20 | ||
| Trisomy 21 t(1,21) | RBM15/EVI1 | Adverse | 21 | ||||
| t(16;21)(q24;q22) | AML1/MTG16 | Favorable | 22 | ||||
| t(8;17)(q23;q24.2), t(9;22)(p24.1;q12.2) | Unknown/BCR/ABL | Adverse | 23 | ||||
| t(7;13)(p11;q34) | ABCA13/ITGBL1 | Adverse | 24 | ||||
| t(4;12)(q12;p13) | ETV6/CHIC2 | Adverse | 25 | ||||
| (1;3)(p36;q21) | Adverse | 26 | |||||
| *i(17q) without t(15;17) | Adverse | 27 | |||||
| t(7;21)(p22;q22) | RUNX1/USP42 | Favorable | 28 | ||||
| inv(11)(p15q22) | NUP98/DDX10 | Favorable | 28 | ||||
| t(11;17)(q23;p12) | MLL/TOP3A | Adverse | PBX3 | miR-30a, b, c; let-7a-3; miR-196b; miR-17h, g | miR-210;miR-23a; miR-181b; miR-495 | 9-11,29 | |
| t(9;11)(p22;q23) | MLI/AF9 | Favorable | PBX3 | miR-30a, b, c; let-7a-3; miR-196b; miR-17h, g | miR-210; miR-23a; miR-181b; miR-495 | 9-11,30 | |
| t(8;16)(p11;p13) | MYST3/CREBBP | Adverse | CREBBP | miR-218; miR-128; miR-27b; miR-15a; miR-195 | 9-11,31 | ||
| Trisomy 10 | Favorable | 32 | |||||
| Trisomy 11 | FLT3/NRAS | Favorable | PIM1/NRAS | let-7a-3; miR-378; miR-337-3p, miR-625; miR-155; miR-181a, b, d; miR-193a-3p; miR-411; let-7a, b, d, f, g, l | miR-34b | 9-12,33 | |
| t(15;17) and t(8;21) | PML-RARA/ | Favorable | RUNX1/RUNX1T1, BAK1, PML-RARA | miR-30a, b, c; miR-378; let-7a-3; miR-337-3p, | miR-16 | 9-12,33 | |
| AML-MRC | t (3, 5) | NPM1/MLF1 | Favorable | E2F3/KLF4, RB1CC1/MN1, CLCN3, CRKL/IRF2, KIT/SERPINB9 | miR-378; let-7a-3-M; miR10a; miR15a; miR-20a; miR-29a | miR-34a | 9-12,34 |
| Tetrasomy 5q | Favorable | 35 | |||||
| dic(1;15)(p11;p11) | CEP1/CEP15 | Adverse | 36 | ||||
| Trisomy 14q | FLT3-ITD/KRAS | Adverse | PIMI/KRAS | miR-378; let-7a-3; miR-155; miR-181a, b, d; miR-193a-3p; miR-411 | miR-16 | 9-12,37 | |
| t-AML | t (9,22) (q34;q11.2) (mixed phenotype) | Favorable | 38 | ||||
| de1 7q(q22q34), 20q (q11.2q13.1), inv(16))(p13.1;q22) inv(12)(p13;q13) | Adverse | miR-30a, b, c | 39,13 | ||||
| ETV6(TEL) | Adverse | 40 | |||||
| t(11;18)(q13;q21.1) | MLLPTD | Adverse | PBX3 | miR-378; let-7a-3 | miR-210; miR-23a; miR-181b; miR-495 | 9-11,40 | |
| t(1;16)(p31;q24) | CBFA2T3/RUNX1 | Adverse | 41 | ||||
| AML-NOS | Cryptic 7q31.1 | DOCK4/FLT3-ITD | Favorable | PIM1 | miR-378; let-7a-3 | miR-16 | 9-11,42 |
| Cryptic 7q22.1 | CUX1 | Favorable | 42 | ||||
| NUP98/NSD1 | Adverse | 43 | |||||
| FAM65A/CTCF | Adverse | ||||||
| KIAA1267/ARL17 | Adverse | ||||||
| BC041636/GGCT | Adverse | ||||||
| CRNKL1/NAA20 | Adverse | 44 | |||||
| R3HDM2/INHBC | Adverse | ||||||
| ABHD16A/LY6G5C | Adverse | ||||||
| HSPA14/SUV39H2 | Adverse | ||||||
| t(4;16)(q25;q23.1) | EGF/ELOVL6 | Adverse |
PML/RARA, promyelocytic leukemia gene/retinoic acid receptor α; BAK1, BCL2-antagonist/killer 1; MLL/MLLTPD, mixed-lineage leukemia/partial tandem duplication; PBX3, pre-B-cell leukemia homeobox 3; AML1/ETO, acute myeloid leukemia 1 protein/eight-twenty one oncoprotein; RUNX1/RUNX1T1, Runt-related transcription factor 1; NUP98//HOXA, nucleoporin 98 gene/homeobox protein-A; RPN1/EVI1, ribophorin I/ecotropic viral integration-1; NOTCH1, Notch (Drosophila) homolog 1; BCL2, B-cell lymphoma 2; RBM15, RNA binding motif protein 15; MTG16, myeloid translocation gene 16; BCR/ABL, breakpoint cluster region/"Abelson; ABCA13/ITGBL1, ATP-binding cassette, sub-family A (ABC1)/integrin, β-like 1 (with EGF-like repeat domains); ETV6/CHIC2, Ets variant 6/cysteine-rich hydrophobic domain 2; USP42, ubiquitin specific peptidase 42; DDX10, DEAD (Asp-Glu-Ala-Asp) box polypeptide 10; TOP3A, topoisomerase (DNA) III α; AF9, ALLl-fused gene from chromosome 9; MYST3/CREBBP, MYST histone acetyltransferase (monocytic leukemia) 3/CREB binding protein; FLT3/NRAS, Fms-related tyrosine kinase 3/neuroblastoma RAS viral (V-Ras) oncogene homolog; PIM1, Pim-1 oncogene; MLF1, myeloid leukemia factor 1; E2F3, E2F transcription factor 3; KLF4, Kruppel-like factor 4; RB1CC1, RB1-inducible coiled-coil 1; MN1, meningioma (disrupted in balanced translocation) 1; CLCN3, chloride channel, voltage-sensitive 3; CRKL,V-Crk avian sarcoma virus CT10 oncogene homolog-like; IRF2, interferon regulatory factor 2; SERPINB9, serpin peptidase inhibitor, clade B (Ovalbumin), member 9; CEP1/CEP15, C. elegans p53-like protein 1,15; KRAS, Kirsten rat sarcomaviral oncogene homolog; CBFA2T3, core-binding factor, runt domain, α subunit 2; DOCK4, dedicator of cytokinesis 4; cux-1, cut-like homeobox 1; NSD1, nuclear receptor binding set domain protein 1; FAM65A, family with sequence similarity 65, member A; CTCF, CCCTC-binding factor (zinc finger protein); KIAA1267, KAT8 regulatory NSL complex subunit 1; ARL17, ADP-ribosylation factor-like 17B; GGCT, γ-glutamylcyclotransferase; CRNKL1, Crooked neck pre-MRNA splicing factor 1; NAA20N(α)-acetyltransferase 20, NatB catalytic subunit; R3HDM2, R3H domain containing 2; INHBC, inhibin, β C; ABHD16A, abhydrolase domain containing 16A; LY6G5C, lymphocyte antigen 6 complex, locus G5C; HSPA14, heat shock 70kDa protein 14; SUV39H2, suppressor of variegation 3-9 homolog 2 (Drosophila); EGF, epidermal growth factor; ELOVL6, ELOVL fatty acid elongase 6; AML-RGA, acute myeloid leukemia with recurrent genetic abnormalities; AML-MRC, acute myeloid leukemia with myelodysplasia-related changes; t-AML, therapy-related acute myeloid leukemia; AML-NOS, acute myeloid leukemia with normal karyotype or nonspecific karyotype abnormalities.
*Isochromosome.
In this article, we examine rare cytogenetic abnormalities and assess their prognosis and response to treatment in different acute myeloid leukemia subtypes according to WHO classification as well as the role and expression of miRNA types in classification, diagnosis and response to treatment in this type of leukemia.
Rare cytogenetic abnormalities in acute myeloid leukemia with recurrent genetic aberrations
This group of leukemias includes seven neoplasms with common cytogenetic abnormalities with at least 20% blasts in peripheral blood or bone marrow (BM). If the blast level is lower than 20%, detection of molecular abnormalities would be helpful in their diagnosis. Rare cytogenetic abnormalities in this group will be discussed below.1,2,5
Acute promyelocytic leukemia with cryptic PML/retinoic acid receptor (PML/RARA) rearrangement in chromosomes 11,15 and 17 is a rare cytogenetic disorder. The primary mechanism of this disorder is addition of PML gene on chromosome 15 to RARA gene locus on chromosome 17.
The resulting PML/RARA in chromosome 17 causes cryptic rearrangement involving t(15,17,11) by translocation and movement on chromosome 11.8 Molecular techniques like real-time polymerase chain reaction (RT-PCR), FISH, and DNA sequencing are useful for understanding a range of cryptic translocations. PML/RARA has been found to be responsible for this malignancy using the above-mentioned techniques. PML and RARA genes generate a protein and affect DNA, causing transcription inhibition and lack of normal differentiation of myeloid cells. In general, this gene rearrangement has a good prognosis and responds well to chemotherapy with ATRA, idarubicin and cytarabine.8,45
Cryptic insertion from chromosome 10 to chromosome 11 is another rare disorder in this group, which leads to rearrangement on 11q23 and 10p12 chromosomes and development of mixed-lineage leukemia (MLL)/MLLTP gene, which is a complex karyotype disorder. Evaluation of PCR products in this disorder has indicated the presence of a similar breakpoint between MLL/MLLTP, which is caused by alternative splicing of the MLL gene.14
MLL fusion gene is seen in 95% of 11q23 rearrangements in AML. MLL fusion proteins function as a general transcription factor capable of activating various promoters in cellular genes such as HOX.46,47
MLL fusion gene affects HOX gene and deregulate it, inducing leukemogenesis. HOX expression is high in stem cells, and the expression of it should be decreased during maturation. 47
HOX gene is controlled by MLL and its fusion proteins, and is in fact a downstream target for HOX gene. HOX proteins are the major hematopoietic oncoproteins overexpressed in a wide range of leukemias, and mediate their function via activating promoters of such genes as C-myeloblastosis (C-Myb).47,48 In fact, C-Myb is a downstream target for HOX gene. HOX gene causes C-Myb overexpression, and uses it as access point to affect the malignant cells.
Continuous HOX and HOX gene mutation inhibits the differentiation and proliferation of pre-leukemic precursors. This disorder occurs in children, and has a poor prognosis and response to treatment.14,48
Acute megakaryoblastic leukemia with trisomy 21 is a rare disorder in children, which creates a complex karyotype leading to acquired chromosome t(1,21)/RBM15/EVI1. Chromosome abnormalities involving RNA binding motif protein 15 (RBM15) are associated with acute megakaryoblastic leukemia.21 This means that RBM15 acts as an oncogene, and the fusion protein produced from this gene disrupts Notch and cytokine signaling in megakaryoblast lineages.
Normal expression of RBM15 results in myeloid differentiation, and its overexpression inhibits the differentiation of hematopoietic cells. This leads to increased Notch signaling.21,49 Notch ligands and receptors facilitate the proliferation and survival of bone marrow stem cells. RBM15 overexpression enhances notch signaling and thereby stimulates the transcription activity of hes family bHLH transcription factor 1 (HES1) promoters, suppressing the activity of globin transcription factor 1 (GATA-1), which is essential for megakaryocyte lineage differentiation.49 Acute megakaryoblastic leukemia and acute lymphoid leukemia are two common subtypes of acute leukemia in children associated with increased copies of chromosome 21. However, unlike trisomy 21 in acute megakaryoblastic leukemia, this trisomy is associated with a good prognosis in acute lymphoblastic leukemia (ALL).21,49
Trisomy 6 is a rare disorder in acute magakaryoblastic leukemia in children. The first mechanism involved in its leukemogenesis is gene dosage effect, leading to increased gene expression and copies. The second mechanism is the gene mutations involved in leukemogenesis in trisomy chromosome.18,50 For example, mutation in core binding factor genes is associated with acquired chromosome 21.
Core-binding factor (CBF) complex is a transcription factor, which plays an important role in normal hematopoiesis by expression regulation of several genes, including granulocyte-colony stimulating factor receptor and interleukin-3.51
Involvement of MLL gene in trisomy 11 has also been reported to play a role in pathogenesis of these trisomies. Gene mutations involved in trisomy 6 are not yet well understood, but their recognition will contribute to elucidate the role of these genes in pathogenesis of this type of leukemia. In general, this type of leukemia has a poor prognosis and response to treatment.18,51
t(7,11)/NUP98//HOXA is a rare cytogenetic abnormality in acute myeloid leukemias. The protein product of the involved genes causes degradation of nuclear pore complexes and deregulation of HOXA transcription factor.52 Overexpression and deregulation of the function of HOX gene leads to altered transcription of genes in hematopoietic precursors, inhibition of normal cell differentiation and leukemogenesis.
HOX overexpression is accelerated by simultaneous expression and interaction with Meis homeobox 1 (MEIC1). MEIC1 is involved in normal hematopoiesis, and plays a role in AML development via interaction with HOX overexpression. Moreover, generation of NUP98-HOXA9 fusion gene as an initial event in development of leukemia causes sustained tumorigenesis by activating HOX, ectopic virus integration site 1 (EVI1) and FLT3, leading to development of AML.19
This type of leukemia is often observed in young females, and is associated with poor prognosis. No specific chemotherapy drug has been recognized for treatment of this disorder. Treatment with high dose daunorubicin has been associated with relapse in most patients (80%). Relapse has been observed in 3% of patients undergoing stem cell transplantation. Research on the role of transplantation in treatment and survival of the patients is underway.19,53
3qJT with trisomy 8 is a rare type of jumping translocations (JTs). This disorder is caused by duplication of 3q21, production of gene segments containing RPN1 (ribophorin I) gene in long arm of chromosome 3 as donor chromosome and its transfer to the short arm of recipient chromosome 8. Donor breakpoint 3qJT sites are close to RPN1 gene, and the recipient breakpoint is located in telomere sequence.53 The telomere is short in the cells afflicted with 3qJT. Association of this disorder with trisomy 8 causes further reduction of the telomere length. Shortening the telomeric sequences is a multistep process that leads to genomic instability and amplification of 3q segment, which causes the stability of telomere ends after attachment to it. JTs are clonal abnormalities in which telomere repeats are stabilized and not shortened after fusion, while telomere ends in other chromosomes remain short during cell division. In general, this disorder has a poor prognosis and response to treatment (Table 1).20,54 Overall, depending on chromosome abnormalities and the involvement of respective genes, different pathways will be followed in the cell for development of leukemia, some of which will have a good prognosis and some poor prognosis. For example, involvement of PML/RARA genes inhibits transcription in normal cells and converts them to leukemic cells with a favorable prognosis and response to treatment. RBM15 gene causes some changes in the signaling pathways, and is associated with poor prognosis and response to treatment. Involvement of MLL and NUP98 genes also leads to deregulation of the function of HOX family members, changing transcription of the cell and its conversion to leukemic cell with unfavorable prognosis and response to treatment.
Rare cytogenetic abnormalities in acute myeloid leukemia with myelodysplasia-related changes
Acute myeloid leukemia with myelodysplastic changes is a feature of this group of leukemias. Cases with a history of myelodysplastic syndromes, cytogenetic abnormalities associated with myelodysplasia, more than 20% myeloblasts in BM and less than 50% dysplasia in two or three cell lines are included in this group of leukemias.2,5
AML with NPM1/MLF1/t (3, 5) is a rare balanced cytogenetic abnormality. Breakpoints are located at 3q25.1 and 5q34. NPM1/MLF1 mutation functions with NPM1 mutation similar to AML, and this can indicate signaling pathways in both leukemias.34 HOX gene is largely associated with the involvement of nucleophosmin (NPM1) gene in AML subtypes. This gene increases the expression of HOXA and HOXB family genes and their protein binding partners like MEIS1 and PBX3, which would lead to lack of normal differentiation in progenitor cells, cell proliferation and leukemia. This disorder has a good prognosis and response to treatment.34,55
In a number of tumors (e.g., colorectal cancer), it has been shown that the ploidy leads to chromosomal abnormality, instability and karyotype changes. Such abnormalities have also been observed in AML patients. Tetraploidy of chromosome 5 is a rare cytogenetic disorder and an important stage in AML-MRC pathogenesis. This tetraploidy causes chromosome instability due to loss of telomere in 5q followed by unbalanced chromosomal translocation, which ultimately leads to a complex karyotype.35,56 Deletion in 5q causes chromosomal translocations known as derivative chromosome involving der (5) t(5; 14). Der (2) t(1; 2) and der (3) t(3; 13) are other karyotype changes observed in 5q deletion due to tetraploidy. This rare disorder has a good prognosis and response to treatment.35
Dicentric chromosome (dic) is connection of two abnormal chromosome pieces, each containing one centromere with satellite DNA. CEP1/CEP15/dic (1; 15) (p11; p11) is a rare form of these disorders, which leads to trisomy in the long arm of chromosome 1. The genes involved in dic (1; 15) are unknown. However, chromosomal translocations of breakpoints in this disorder occur in heterochromatin.36 Heterochromatin rearrangements (relative trisomies and monosomies) cause imbalance by gene silencing in chromosomal breakpoints, leading to displacement of heterochromatin to an ectopic location. They also provoke functional changes in genes and chromatin coupled proteins. In general, this disorder has a poor prognosis and response to treatment with induction chemotherapies.36,37
Trisomy in chromosome 14q is a rare acrocentric chromosomal abnormality. It occurs due to 14q rearrangement of two homologous long arms, and is quite heterogeneous. This trisomy is generated in the onset of tumorigenesis process, and plays an essential role in initiation of the neoplastic process and development of malignancy.37,58 However, increased expression and dosage effect of oncogenes like V-Akt murine thymoma viral oncogene homolog 1 (AKT1), FBJ murine osteosarcoma viral oncogene homolog (FOS) and Jagged 2 (JAG2) cause proliferation of malignant cells in this disorder and continuation of the tumorigenesis process.59,60 Mutation in proto-oncogenes like FLT3-internal tandem duplication (FLT3-ITD) and Kirsten rat sarcoma viral oncogene homolog (KRAS) affects the proliferation and differentiation of myeloid cells, and their interaction with loss-of-function mutations of HOX family members causes lack of hematopoietic transcription factors for normal differentiation of the cells, resulting in increase in malignant cells. These mutations are observed together with myelodysplastic disorders in this abnormality. This disorder has a poor prognosis and response to treatment (Table 1).37,53,61
In general, chromosomal abnormalities in this group, which lead to complex karyotype, have a better prognosis and response to treatment in case of disruption of telomere but have a poor prognosis and response to treatment if the heterochromatin region is disrupted. Ploidy abnormalities that lead to involvement of NPM1, FLT3-ITD and KRAS are at odds with the HOX family genes, and lead to failure of normal cell differentiation and leukemia.
Rare cytogenetic abnormalities in acute myeloid leukemia with therapy-related acute myeloid leukemia
This type of myeloid leukemia is divided into two distinct subgroups: i) those with a history of exposure to radiation or alkaline reagents such as melphalan and cyclophosphamide and associated with a latent phase of 3-10 years; ii) those receiving topoisomerase II inhibitors like etoposide and anthracyclines with a shorter latent period of 1-3 years.1,2,5 t-AML with mixed phenotype is a rare phenomenon, and only one case of it has been reported so far. This disorder appears with (q34; q11.2) t (9,22) translocation after chemotherapy with topoisomerase II inhibitors and radiotherapy of the uterus cancer after a latent phase of 7 years. In bone marrow examination, there is a complex karyotype with/47 idem, + der (22) t(9; 22) t(9; 22) (q34; q11.2), dup (1) (q21q32). Immunophenotype of the patient indicates the involvement of B/myeloid lineage.38 Based on WHO classification, this type of chromosomal abnormality is grouped as t-AML secondary to treatment with a mixed phenotype; however, the biological characteristics of the disease are similar to mixed phenotype. Despite mixed phenotype and t(9, 22) translocation, the disease has a good prognosis and response to therapy during treatment protocol with AML, including induction chemotherapy combined with Imatinib.38,62,63
Del 7q (q22q34) del 20q (q11.2q13.1) with inv (16) (p13.1; q22) is an extremely rare karyotype complex. It is grouped as t-AML caused by topoisomerase II inhibitors in treatment of breast cancer after a latent phase of 3 years, and only one case of it has been reported so far.39 Complex karyotype in this type of t-AML has been reported to be associated with deletion of chromosome 5, but deletion in chromosomes 7 and 20 is observed in this rare disorder. Chromosomal breakage due to topoisomerase II inhibitors causes leukemogenic effects, and accounts for chromosomal translocation and leukemia in this disorder.39,64,65 In general, t-AML has a poor response to treatment and a short improvement course. The mentioned abnormality has a bad prognosis, and does not respond to induction chemotherapies (Table 1).39 Overall, the inhibitory effect of topoisomerase II inhibitors causes chromosomal breakage, complex karyotype and leukemia in this group of disorders. Prognosis and response to treatment is poor in this rare group of disorders, but when these disorders are associated with another disorder with mixed phenotype, the prognosis is good, and response to therapy is more favorable due to the use of AML treatment protocol.
Rare cytogenetic abnormalities in acute myeloid leukemia with normal or nonspecific karyotype
In this group of leukemias, the features related to other groups (including dysplasia) are absent. Cytogenetic features are either nonspecific, which are not included in other AML subtypes or the patients have a normal karyotype, in which other cytogenetic features like gene expression would be helpful in their diagnosis.2,5
7q31.1 is a cryptic 7q del associated with relative monoallelic deletion of dedicator of cytokinesis 4 (DOCK4) gene, and is an acute leukemia with normal karyotype. DOCK4 is a tumor suppressor gene, which is involved in the activity of GTPases for signal transduction pathways such as Rho GTPase Rac1, which are essential for normal cell proliferation, adhesion, migration and phagocytosis.42,66,67 DOCK4 is also associated with other signaling pathways like β-catenin and GSK-3, and plays an important role in the regulation of cell function in hematopoiesis through molecular interactions with these two pathways. Deletion of this gene is involved in tumorigenesis process in this disorder, and involvement of FLT3-ITD gene contributes to the process of tumorigenesis.42.68 This type of leukemia shows intermediate cytogenetic abnormality, so that it has a poor the prognosis, and relapse occurs after the initial treatment. However, in later stages of treatment, it responds well to treatment, and improvement of the patient is observed.42 7q22.1 abnormality and cut-like homeobox 1 (CUX1) gene mutation is another rare disorder, which shows similar characteristics and treatment response to the previous disorder. The protein product of CUX1 gene is a member of DNA-binding protein family, which regulates gene expression and differentiation, and plays an important role in normal cell cycle.42,69 Mutations in this gene cause down-regulation of phosphoinositide-3-kinase interacting protein (PI3K-IP1) inhibitor, which is an inhibitor of phosphoinositide-3PI3K kinase. This activates the PI3K signaling pathway and increases the proliferation and expansion of tumor cells. PI3K consists of a family of enzymes involved in cell growth, proliferation and differentiation, and increased expression or mutation of it would result in development of tumor. This disorder has an intermediate prognosis and response to therapy similar to the previous one.69,70 Identification of genetic mutations involved in leukemias with normal karyotype contributes to their diagnosis. Recent studies on fusion transcriptions in normal karyotype AML has shown that some fusion genes located on the same chromosome in the human genome are not associated with karyotype status of the AML, and can be useful diagnostic markers in leukemias with normal karyotype.44,71 Comparing these genes with other subtypes of AML shows that they are only observed in normal karyotype AML. These genes include seven rare fusion genes as follows: FAM65A-CTCF, KIAA1267-ARL17, BC041636-GGCT, CRNKL1-NAA20, INHBC-R3HDM2, ABHD16A-LY6G5C, and HSPA14-SUV39H2. Some of these genes like CTCF, INHBC and HSPA14 are involved in other malignancies such as breast and prostate cancers as well as Wilm’s tumor. These mutations have a poor prognosis and response to treatment.44,72,73 NUP98/NSD1 is another rare mutation, which is more common in children. NUP98/NSD1 is a class II oncogene. Involvement of FLT3-ITD gene with this mutation raises a new relationship between class I and II oncogenes. The resulting NUP98-homeobox fusion can bind to DNA and recruit CREBBP/p300, causing histone acetylation and transcription activation of target genes. Nucleoporin 98kDa (NUP98) gene is associated with increased expression of HOXA and HOXB family members, inhibiting normal differentiation of cells, increasing their proliferation and causing leukemia.43,74,75 FLT3-ITD gene is a proto-oncogene, mutations of which cause expansion of malignant cells. Due to the involvement of class II oncogenes in pathogenesis of this disorder, it has a poor overall prognosis, is associated with relapse and is refractory to induction chemotherapy (Table 1).43,76 In general, involvement of DOCK4 and CUX1 genes leads to changes in cell signaling pathways and increased proliferation of tumor cells, which have an intermediate prognosis and response to therapy. As stated about the involvement of AML-RGA gene group, involvement of NUP98 gene is associated with the HOX family of genes, and causes a poor prognosis and response to treatment in this group. As a result, identification of genetic mutations involved in this group is of particular importance in its diagnosis and treatment because of the normal karyotype.
The role and alteration of microRNAs in rare cytogenetic abnormalities in acute myeloid leukemia
Several mechanisms have been identified about mutations, chromosomal translocations and epigenetic changes (including DNA methylation) as the first epigenetic event in AML. Recently, other epigenetic mechanisms such as miRNA have been detected in leukemias, and their role in normal physiology and pathogenesis has been specified by increasing knowledge in this field.10,77,78 miRNAs play important biological roles in cell growth, differentiation, response to stress and apoptosis. Expression evaluation of miRNA is a vastly growing research field, since miRNAs play vital roles in normal hematopoiesis, regulation and differentiation of the myeloid lineage and their ectopic expression is associated with hematologic malignancies and leukemogenesis.13,79 The role of miRNA in AML was for the first time determined by the studies conducted to differentiate between ALL and AML, in which the important role of miRNA in differentiation of AML from ALL was shown.80 To date, few studies have been performed on the role of miRNA in AML, and the mechanisms involved in the regulatory role of miRNA are not yet fully identified due to the high level of heterogeneity in patients with different subtypes of AML. Nevertheless, despite these factors, the role of miRNAs in pathogenicity and myeloid malignancies has been demonstrated.80 Research has indicated the role of a number of miRNAs in chromosomal translocations t(8, 21), t(15, 17), inv (16) and in common molecular abnormalities such as NPM1, CCAAT enhancer binding factor (CEBPA) and FLT3-ITD in AML.81 In addition, several miRNA tumor suppressors, including miR-155, miR-21 and let-7, have been recently identified in various subgroups of AML, and this suggests that miRNA are involved in the pathogenesis of AML, and can contribute to diagnosis of this disease.82 Recent studies have shown that the expression of miR-10a, miR-10b and miR-196a, which are located in internal regions of HOX gene cluster, is associated with the expression of HOXA and HOXB in AML, indicating the ectopic function of these miRNAs in the regulation of proliferation and differentiation in leukemia.82 Correlation between gene expression and miRNA expression has been detected in subtypes with poor prognosis in patients with normal karyotype through the investigation of genes involved in the disease, indicating the malignant phenotype in them by increased or decreased expression of miRNA. A few studies indicate the role of miRNA in response to treatment in myeloid leukemias.83 miR-223 has an important role in myeloid differentiation and granulopoiesis. It has been reported that some fusion proteins in leukemias can inhibit the expression of miR-223. Silencing of miR-223 expression is associated with differentiation arrest of myeloid precursors and leukemogenesis.81 However, disrupted function of miR-223 alleles has indicated its role in granulocytopoiesis in myeloid lineage. Therefore, in general, we can say that miRNAs play an important role independent from gene expression in myeloid cell differentiation of AML subtypes.81 The role of let-7c in granulocyte differentiation in several AML cell lines has been detected. Decreased let-7 expression has been shown in a variety of solid tumors, which indicates its role as a tumor suppressor.12,13 Definition of AML subtypes by a finite number of known miRNAs indicates their strong expression potential in various AML subtypes. However, the number of known human miRNA in leukemia is increasing; therefore, detection of new miRNAs with specific expression in AML subtypes will enable their classification by miRNA.82 miRNAs involved in rare cytogenetic abnormalities and those effective in leukemogenesis, the involved genes as well as the signaling pathways in each of these disorders in four AML groups according to WHO have been summarized in Table 1. In general, miRNAs can play different roles in AML classification, prognosis and response to treatment due to several functions in myeloid lineage.
Discussion
AML is a heterogeneous disease caused by clonal proliferation of malignant myeloid precursors and disrupted differentiation in these cells. Although the majority of patients with AML recover, most of them demonstrate a poor prognosis, undergo relapse and finally succumb to the disease. Significant changes in gene transcription leading to relapse after chemotherapy are still unknown. Some reports indicate that AML can be classified in different groups based on chromosomal abnormalities, acquired somatic mutations, gene expression, miRNA and methylation status.83 Several cytogenetic abnormalities, such as chromosomal aberrations, gene expression and somatic mutations can contribute to determination of AML subtypes, rare disorders as well as favorable, intermediate and adverse phenotypes, and can be effective in their response to treatment. Upon AML diagnosis, induction chemotherapy of the patients using anthracyclines (daunorubicin, idarubicin) and cytarabine is started until complete remission (CR).84 Response to treatment depends on the cytogenetic characteristics and patient’s age. Therefore, leukemias with monosomal karyotype and cytogenetic abnormalities associated with poor prognosis as well as older patients show a poor response to treatment. Therefore, by targeting the specific genotypic characteristics of AML patients, they can be treated using specific drugs.84 There are some intractable cases that depend on the interaction of leukemic cells and the surrounding microenvironment in the bone marrow. These cells home to microenvironment niche, and are protected against cytotoxic effects of chemotherapy. Interaction between stromal cells and leukemia progenitor cells through adhesion molecules is the reason for this resistance to treatment, which can affect the pathobiology and response to treatment.83,84 Studies on cell lines have shown that increased expression of miRNAs inhibits cell differentiation, and decreased expression of them causes differentiation in these cell lines. Many miRNAs controlling the differentiation of myeloid lineage are not known to date. miRNAs contribute to our understanding of myeloid malignancies.12,13 In most cases of AML, CR is associated with increased expression of miR-15a/16, and it has been observed that this miRNA is decreased during disease relapse. Evidence also suggests a link between miRNA and patient’s prognosis. For example, increased miR-199a and miR-191 in individuals with trisomy of chromosome 8 is associated with a poor prognosis. Increased expression of miR-155 has also been associated with a poor prognosis, but miR-181a is associated with a better prognosis in AML, and indicates the potential to achieve effective treatment for these patients. Reduced expression of miR-181a, b in cytogenetically normal patients has been associated with myeloid leukemia with a severe malignant phenotype.81,83 miR-378 and miR-let7a-3 have recently been shown to be present in most subtypes of AML, and their increased expression is associated with a poor prognosis. Studies have shown that CR is not significantly different in patients with or without these miRNAs. However, the relapse rate is higher in patients with increased expression of this miRNA, which indicates the importance of detection of this miRNA in AML subgroups to contribute to more effective treatment in these patients.10,11
Due to the heterogeneous nature of AML, frequent mutations in its subgroups and development of rare karyotypes in addition to common karyotypes in this leukemia, determination of patients’ prognosis based on cytogenetic features like gene expression and mutations can be helpful. These features can also be good therapeutic targets for the patients. miRNAs have been recently recognized as a new mechanism in normal myeloid differentiation as well as leukemic cell differentiation. miRNAs can be superior to gene expression in diagnosis, classification, prognosis and as biomarkers in AML due to relationship with signaling pathways when the cytogenetic features of the disease are not well known. In addition, using miRNA-based therapeutic approaches, treatment strategies for AML can be designed by determining disease prognosis by miRNA. Moreover, response to treatment and the likelihood of relapse in patients can be predicted. Therefore, miRNAs present a new method of anticancer therapy, which can help obviate the challenges in treatment of AML patients.
References
- 1.Chin YM, Puteri JN, Azli I, et al. Cytogenetic profile of de novo acute myeloid leukemia patients in Malaysia. Int J Biomed Sci 2013;9:26-32. [PMC free article] [PubMed] [Google Scholar]
- 2.Hasserjian RP. Acute myeloid leukemia: advances in diagnosis and classification. Int J Lab Hematol 2013;35:358-66. [DOI] [PubMed] [Google Scholar]
- 3.Brown CMS, Larsen SR, Iland HJ, et al. Leukaemias into the 21st century: part: the acute leukaemias. Intern Med J 2012:1179-86. [DOI] [PubMed] [Google Scholar]
- 4.Panagopoulos I, Gorunova L, Brandal P, et al. Myeloid leukemia with t(7;21)(p22;q22) and 5q deletion. Oncol Rep 2013;30:1549-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin CC, Medeiros LJ, Bueso-Ramos CE. Recent advances in the diagnosis and classification of myeloid neoplasms - comments on the 2008 WHO classification. Int J Lab Hematol 2010;32:461-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iffat K, Anindo M, Yaqinuddin A, et al. Insights into the potential use of microRNAs as biomarker in cancer. Int J Surg 2012;10:443-9. [DOI] [PubMed] [Google Scholar]
- 7.Zhu CL, Wang YG, Kuai WX, et al. Prognostic value of miR-29a expression in pediatric acute myeloid leukemia. Clin Biochem 2013;46:49-53. [DOI] [PubMed] [Google Scholar]
- 8.Sadrzadeh H, Kerr DA, Dal Cin P, et al. A unique PML-RAR rearrangement involving chromosomes 11, 15, and 17 in a patient with acute promyelocyticleukemia. Exp Hematol 2013;41:769-71. [DOI] [PubMed] [Google Scholar]
- 9.Qian J, Lin J, Qian W, et al. Overexpression of miR-378 is frequent and may affect treatment outcomes in patients with acute myeloid leukemia. Leukemia Res 2013;37:765-8. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Lin J, Yang J, et al. Overexpressed let-7a-3 is associated with poor outcome in acute myeloid leukemia. Leukemia Res 2013;37:1642-47. [DOI] [PubMed] [Google Scholar]
- 11.Xiong Q, Yang Y, Wang H, et al. Characterization of miRNomes in acute and chronic myeloid leukemia cell lines. Genom Prot Bioinf 2014;12:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon JEA, Wong JJL, Rasko JE, et al. MicroRNAs in myeloid malignancies. Br J Haematol 2013;162:162-76. [DOI] [PubMed] [Google Scholar]
- 13.Bennour A, Tabka I, Youssef YB, et al. A PML/RARA chimeric gene on chromosome 12 in a patient with acute promyelocytic leukemia (M4) associated with a new variant translocation: t(12;15;17)(q24;q24;q11). Med Oncol 2013;30:1-5. [DOI] [PubMed] [Google Scholar]
- 14.De Figueiredo AF, Vieira TP, Liehr T, et al. A rare cryptic and complex rearrangement leading to MLL-MLLT10 gene fusion masked by del(10)(p12) in a child with acute monoblastic leukemia (AML-M5). Leuk Res 2012;36:e74-7. [DOI] [PubMed] [Google Scholar]
- 15.Kumari P, LingappaKavitha B, Reddy CO, et al. A rare cytogenetic presentation of acute myeloid leukemia (AML-M2). Acta Medica Iranica 2012;12:827-30. [PubMed] [Google Scholar]
- 16.Gmidene A, Frikha R, Sennana H, et al. T(1;21;8)(p34;q22;q22): a novel variant of t(8;21) in acute myeloblastic leukemia with maturation. Med Oncol 2011;28:510-2. [DOI] [PubMed] [Google Scholar]
- 17.Park IJ, Park JE, Kim HJ, et al. Acute myeloid leukemia with t(16;21)(q24;q22) and eosinophilia: case report and review of the literature. Cancer Genet Cytogenet 2010;196:105-8. [DOI] [PubMed] [Google Scholar]
- 18.McCullough SJ, Neat MJ, Power M, O’Marcaigh A. Trisomy 6 in a child with acute megakaryoblastic leukemia (AML-M7). Cancer Genet Cytogenet 2004;154:190-92. [DOI] [PubMed] [Google Scholar]
- 19.Takeda A, Goolsby C, Yaseen NR, et al. Cells differentiation of primary human CD34 + hematopoietic NUP98-HOXA9 induces long-term proliferation and blocks. Cancer Res 2006;66:6628-36. [DOI] [PubMed] [Google Scholar]
- 20.Zou YS, Ouahchi K, Lu Y, et al. Jumping translocations of 3q21 in an acute monocytic leukemia (M5) patient reveal mechanisms of multistage telomere shortening in pathogenesis of AML. Leukemia Res 2012;36:31-3. [DOI] [PubMed] [Google Scholar]
- 21.Pauliena S, Busson-Le Coniat M, Berge R. Acute megakaryocytic leukaemia with acquired polysomy 21 and translocation t(1;21). Ann Genet 2000;43:99-104. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad F, Dalvi R, Das BR, Mandava S. Novel t(8;17)(q23;q24.2) and t(9;22)(p24.1;q12.2) in acute megakaryoblastic leukemia AML-M7 subtype inan adult patient. Cancer Genet Cytogenet 2009;193:112-5. [DOI] [PubMed] [Google Scholar]
- 23.Gerbino E, Tapinassi C, Malazzi O, et al. A novel t(7;13)(p12;q33 approximately q34) in AML-M2. Cancer Genet Cytogenet 2009;195:198-200. [DOI] [PubMed] [Google Scholar]
- 24.Manabe M, Nakamura K, Inaba A, et al. A rare t(4;12)(q12;p13) in an adolescent patient with acute myeloid leukemia. Cancer Genet Cytogenet 2010;200:70-2. [DOI] [PubMed] [Google Scholar]
- 25.Lim G, Kim MJ, Oh SH, et al. Acute myeloid leukemia associated with t(1;3)(p36;q21) and extreme thrombocytosis: a clinical study with literature review. Cancer Genet Cytogenet 2010;203:187-92. [DOI] [PubMed] [Google Scholar]
- 26.Duan Y, Nie J, Zhang Z, et al. A rare case with typical acute promyelocytic leukemia morphology associated with isolated isochromosome 17q without RAR rearrangement. Hematol Oncol Stem Cell Ther 2013;6:42-5. [DOI] [PubMed] [Google Scholar]
- 27.Jeandidier E, Gervais C, Radford-Weiss I, et al. A cytogenetic A A cytogenetic study of 397 consecutive acute myeloid leukemia cases identified three with a t(7;21) associated with 5q abnormalities and exhibiting similar clinical and biological features, suggesting a new, rare acute myeloid leukemia entity. Cancer Genet 2012;205:365-72. [DOI] [PubMed] [Google Scholar]
- 28.Gorello P, Nofrini V, Brandimarte L, et al. Inv(11)(p15q22)/NUP98-DDX10 fusion and isoforms in a new case of de novo acute myeloid leukemia. Cancer Genet 2013;206:92-6. [DOI] [PubMed] [Google Scholar]
- 29.Kuipers JE, Coenen EA, Balgobind BV, et al. High IGSF4 expression in pediatric M5 acute myeloid leukemia with t(9;11)(p22;q23). Blood 2011;117:928-35. [DOI] [PubMed] [Google Scholar]
- 30.Coenen EA, Zwaan CM, Reinhardt D, et al. Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity:a collaborative study by the International-Berlin-Frankfurt-Münster AML-study group. Blood 2013;122:2704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan Ji, McDonoughM C, Kulharya A, et al. Isolated trisomy 10 in an infant with acute myeloidleukemia: a case report and review of literature. Int J Clin Exp Pathol 2010;3:718-22. [PMC free article] [PubMed] [Google Scholar]
- 32.Faisal M. Alseraye ZZ, Carlos BRet al. Trisomy 11 as an isolated abnormality in acute myeloideukemia is associated with unfavorable prognosis butnot with an NPM1 or KIT mutation. Int J Clin Exp Pathol 2011;4:371-7. [PMC free article] [PubMed] [Google Scholar]
- 33.Uz B, Eliacik E, Isik A, et al. Co-expression of t(15;17) and t(8;21) in a case of acute promyelocytic leukemia: review of the literature. Turk J Haematol 2013;30:400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dumezy F, Renneville A, Mayeur-Rousse C, et al. Acute myeloid leukemia with translocation t(3;5): new molecular insights. Haematologica 2013;98:52-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarosova M, Nedomova R, Hubacek J, et al. Rare tetraploidy with large 5q deletion in acute myeloid leukemia with myelodysplasia-related changes (AML-MRC). Leukemia Res 2012;36:68-70. [DOI] [PubMed] [Google Scholar]
- 36.Sahin DG, Durak B, Gunduz E, et al. A new case of dic(1;15)(p11;p11) in AML M1: apropos of a case and a review of the literature. Case Rep Genet Rep 2013;1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.some abnormality is associated with older age, a heterogenous group of myeloid neoplasms with dysplasia, and a wide spectrum of disease progression. J Biomed Biotechnol 2010;1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho JH, Hur M, Moon HW, et al. Therapy-related acute leukemia with mixed phenotype and t(9;22)(q32;q11.2): a case report and review of the literature. Hum Pathol 2012;43:605-9. [DOI] [PubMed] [Google Scholar]
- 39.Yonal I, Hindilerden F, Ozcan E, et al. The co-presence of deletion 7q, 20q and inversion 16 in therapy-related acute myeloid leukemia developed secondary to treatment of breast cancer with cyclophosphamide, doxorubicin, and radiotherapy: a case report. J Med Case Rep 2012;6:1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trikalinos NA, Chen Q, Ning Y, et al. Unbalanced 11;18 translocation in an acute erythroid leukemia after radioactive iodine therapy. Cancer Genet 2013;206:252-5. [DOI] [PubMed] [Google Scholar]
- 41.Micci F, Thorsen J, Haugom L, et al. Translocation t(1;16)(p31;q24) rearranging CBFA2T3 is specific for acute erythroid leukemia. Leukemia 2011;25:1510-2. [DOI] [PubMed] [Google Scholar]
- 42.Kjeldsen E, Veigaard C. DOCK4 deletion at 7q31.1 in a de novo acute myeloid leukemia with a normal karyotype. Cell Oncol 2013;36:395-03. [DOI] [PubMed] [Google Scholar]
- 43.Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011;118:3645-56. [DOI] [PubMed] [Google Scholar]
- 44.Wen H, Li Y, Malek SN, et al. New fusion transcripts identified in normal karyotype acute myeloid leukemia. PloS One 2012;7:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raelson JV, Nervi C, Rosenauer A, et al. The PML/RARA oncoprotein is a direct molecular target of retinoic acid in acute promyelocytic leukemia cells. Blood 1996;80:2826-32. [PubMed] [Google Scholar]
- 46.Balgobind BV, Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 2009;114:2489-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slany RK. The molecular biology of mixed lineage leukemia. Haematologica 2009;94:984-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hess J, Bittner B. C-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood 2006;108:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xianyong M, Matthew JR, Lin W, et al. Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol Cell Biol 2007;27:3056-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Virtaneva K, Wright FA, Tanner SM, et al. Expression profiling reveals fundamental biological differences in acute myeloid leukaemia with isolated trisomy 8 and normal cytogenetics. Proc Natl Acad Sci U S A 2001;98:1124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snaddon J, Neat M, Smith ML, et al. Mutations in the runt homology domain of CBF 2 in myeloid malignancies with acquired trisomy 21. Cancer Genet Cytogenet 2002;136:151-2. [DOI] [PubMed] [Google Scholar]
- 52.Wei S, Wang S, Qiu S, et al. Clinical and laboratory studies of 17 patients with acute myeloid leukemia harboring t(7;11)(p15;p15) translocation. Leukemia Res 2013;37:1010-5. [DOI] [PubMed] [Google Scholar]
- 53.Eguchi-Ishimae M, Eguchi M, Ohyashiki K, et al. Enhanced expression of the EVI1gene in NUP98/HOXA-expressing leukemia cells. Int J Hematol 2009;89:253-6. [DOI] [PubMed] [Google Scholar]
- 54.Capper R, Britt-Compton B, Tankimanova M, et al. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev 2007;21:2495-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verhaak RGW, Goudswaard GS, Putten WV, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established geneexpression signatures and their favorable prognostic significance. Blood 2005;106:3747-54. [DOI] [PubMed] [Google Scholar]
- 56.Lengauer C, Kinzler KW, Vogelstein B, et al. Genetic instability in colorectal cancers. Nature 1997;386:623-7. [DOI] [PubMed] [Google Scholar]
- 57.Pidoux AL, Allshire RC. The role of heterochromatin in centromere function. Philos Trans R Soc B 2005;360:569-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heim S, Mitelman F. Numerical chromosome aberrations in human neoplasia. Cancer Genet Cytogenet 1986;22:99-108. [DOI] [PubMed] [Google Scholar]
- 59.Toze CL, Barnett MJ, Naiman SC, Horsman DE. Trisomy 14 is a non-random karyotypic abnormality associated with myeloid malignancies. Br J Haematol 1997;98:177-85. [DOI] [PubMed] [Google Scholar]
- 60.Gal H, Amariglio N, Trakhtenbrot L, et al. Gene expression profiles of AML derived stemcells; similarity to hematopoietic stem cells. Leukemia 2006;20:2147-54. [DOI] [PubMed] [Google Scholar]
- 61.Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet 2002;3:179-98. [DOI] [PubMed] [Google Scholar]
- 62.Killick S, Matutes E, Powles RL, et al. Outcome of biphenotypic acute leukemia. Haematologica 1999;84:699-706. [PubMed] [Google Scholar]
- 63.Kim M, Lim J, Kim Y, et al. A case of therapy-related acutemyeloid leukemia associated with inv(16), with subsequent development of t(9;22). Leukemia 2006;20:746-8. [DOI] [PubMed] [Google Scholar]
- 64.Voso MT, D’Alò F, Greco M, et al. Epigenetic changes in therapy related MDS/AML. Chem Biol Interact 2010;184:46-9. [DOI] [PubMed] [Google Scholar]
- 65.Qian Z, Joslin JM, Tennant TR, et al. Cytogenetic and genetic pathways in therapy-related acutemyeloid leukemia. Chem Biol Interact 2010;184:50-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hiramoto K, Negishi M, Katoh H. Dock4 is regulated by RhoG and promotes Rac-dependent cell migration. Exp Cell Res 2006;312:4205-16. [DOI] [PubMed] [Google Scholar]
- 67.Mulloy JC, Cancelas JA, Filippi MD, et al. Rho GTPases in hematopoiesis and hemopathies. Blood 2010;115:936-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Upadhyay G, Goessling W, North TE, et al. Molecular association between -catenin degradation complex and Rac guanine exchange factor DOCK4 is essential for Wnt/ -catenin signaling. Oncogene 2008;27:5845-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong CC, Martincorena I, Rust AG, et al. Inactivating CUX1 mutations promote tumorigenesis. Nature Genet 2014;46:33-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He X, Zhu Z, Johnson C, et al. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res 2008;68:5591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prakash T, Sharma VK, Adati N, et al. Expression of conjoined genes: another mechanism for gene regulation in eukaryotes. PLoS One 2010;5:e13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kannan K, Wang L, Wang J, et al. Recurrent chimeric RNAs enriched in human prostate cancer identified by deepsequencing. Proc Natl Acad Sci U S A 2011;108:9172-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kasper LH, Brindle PK, Schnabel CA, et al. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol 1999;19:764-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Argiropoulos B, Humphries RK. Hox genes in hematopoiesisand leukemogenesis. Oncogene 2007;26:66-76. [DOI] [PubMed] [Google Scholar]
- 76.Chou WC, Chen CY, Hou HA, et al. Acute myeloid leukemia bearing t(7;11)(p15;p15) is a distinct cytogenetic entity with poor outcome and a distinct mutation profile: comparative analysis of 493 adult patients. Leukemia 2009;23:1303-10. [DOI] [PubMed] [Google Scholar]
- 77.Garcia-Orti L, Cristobal I, Cirauqui C, et al. Integration of SNP and mRNA arrays with microRNA profiling reveals that MiR-370 is upregulated and targets NF1 in acute myeloid leukemia. PloS One 2012;7:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yuan Y, Kasar S, Underbayev C, et al. MicroRNAs in acute myeloid leukemia and other blood disorders. Leuk Res Treat 2012;1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Babashah S, Sadeghizadeh M, Tavirani MR, et al. Aberrant microRNA expression and its implications in the pathogenesis of leukemias. Cell Oncol 2012;35:317-34. [DOI] [PubMed] [Google Scholar]
- 80.Jongen-Lavrencic M, Sun SM, Dijkstra MK, et al. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 2008;111:5078-85. [DOI] [PubMed] [Google Scholar]
- 81.Marcucci G, Radmacher M D, Maharry K, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1919-28. [DOI] [PubMed] [Google Scholar]
- 82.acute myeloid leukemia: evidence of biological heterogeneity. Cancer 2013;119:3076-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Löwenberg B. Acute myeloid leukemia: the challenge of capturing disease variety. Hematology (Am Soc Hematol Educ Program) 2008:1-1052. [DOI] [PubMed] [Google Scholar]
- 84.Rad SMAH, Shanaki Bavarsad M, Arefian E, et al. The role of microRNAs in stemness of cancer stem cells. Oncol 2013;32:1642-47. [DOI] [PMC free article] [PubMed] [Google Scholar]