

Abstract
Transmission electron microscopy reveals ultrastructural details of cells, and it is a valuable method for studying cell organelles. That is why we used this method for detailed morphological description of different adult tissue-derived stem cells, focusing on the morphological signs of their functions (proteosynthetic activity, exchange with external environment, etc.) and their comparison. Preparing a specimen from the cell culture suitable for transmission electron microscopy is, however, much more challenging than routine tissue processing for normal histological examination. There are several issues that need to be solved while working with cell pellets instead of solid tissue. Here we describe a simple protocol for the isolation and culture of mesenchymal stem cells from different adult tissues, with applications to stem cell biology and regenerative medicine. Since we are working with population of cells that was obtained after many days of passaging, very efficient and gentle procedures are highly necessary. We demonstrated that our semi-conservative approach regarding to histological techniques and processing of cells for transmission electron microscopy is a well reproducible procedure which results in quality pictures and images of cell populations with minimum distortions and artifacts. We also commented about riskiest steps and histochemical issues (e.g., precise pH, temperature) while preparing the specimen. We bring full and detailed procedures of fixation, post-fixation, infiltration, embedding, polymerization and contrasting of cell obtained from in vitro cell and tissue cultures, with modifications according to our experience. All this steps are essential for us to know more about adult stem cells derived from different sources or about other random cell populations. The knowledge about detailed ultra-structure of adult stem cells cultured in vitro are also essential for their using in regenerative medicine and tissue engineering.
Keywords: Adult stem cells, in vitro cultivation, transmission electron microscopy, histological techniques, cell suspension
Introduction
The challenges of sample preparation can limit a researcher’s selection of transmission electron microcopy (TEM) for analysis of rare cell populations after in vitro cultivation. There are few efficient methods that have been used for processing of different cell populations in the suspension for TEM; here, we want to share our experience with cell populations of human mesenchymal stem cells derived from different sources.
There are several peculiarities and issues to be solved while working with cell pellets instead of solid tissues. Adding the fact that here we are working with a relatively rare populations of cells that were obtained after many days of passaging. Therefore, very efficient and gentle procedures are highly necessary while working with these.1,2 If we wanted to get the most from our limited sources of cells, we were forced to make some modifications of the routine process of tissue preparation for TEM analysis. Each step of the sample preparation can have great impact on the quality of the resulting sample.1 Because of this challenging procedure, ultrastructural details and organelle content of the analyzed cell population remained elusive in the past. We selected semi-conservative approach consisting in usage of centrifugal force and aspiration after each step of specimen preparation with slightly modified concentrations of some solutes and time periods. Even though, at the beginning of our work, we discussed other approaches such as pre-embedding the suspensions or pellets in bovine serum albumin (BSA) and bis-acrylamide (BA), cross-linked and polymerized for localization of cells for TEM processing,3 or creation of cytospin preparations (low-speed centrifuge can deposit a uniform monolayer of cells onto an area of a glass slide while maintaining cell morphology), followed by TEM analysis. Mainstream research of adult stem cells (especially of mesenchymal origin) focuses mainly on histochemical and immunohistochemical approach,4,5 sometimes incorporates cellular imaging technologies (like micro-CT or MRI)6-9 or flow cytometry analysis10,11 and analyzes these cells using weapons from repertoire of these disciplines.
Classical TEM analysis can provide us with a lot of details and knowledge about cell’s ultrastructure and therefore to define many of the morphological and functional parameters of various types of adult mesenchymal stem cells.12 We were encouraged to draft presented work, because, in recent literature, there is only a small number of papers regarding electron microscope analysis of adult tissue-derived stem cells13 and further less information about technical issues of their analysis. In the methodological blocks of these texts, histological procedures prior to electron microscopy analysis are not mentioned or mentioned only briefly.10 Hence, we decided to bring forward our experience and problems we had to deal with during the whole process.
In our manuscript, we described our own experiences with ultrastructural analysis of adult mesenchymal stem cells after in vitro cultivation.
Materials and Methods
Isolation and cultivation
We observed several different types of adult tissue-derived stem cells. We passaged primarily isolated cells three times, maintaining their biological features; thus, we did not use stabilized cell lines. Their isolation and cultivation was described in our previous works. We were working with muscle-tissue derived stem cells,12 dental pulp-derived stem cells,14 adipose tissue-derived stem cells15 and human mesenchymal stem cells derived from bone marrow.16 In case of solid tissues we used combination of mechanical disaggregation and enzymatic digestion. In case of suspension tissues we used centrifugation in density gradient for separation of stem cells. Obtained cells were rinsed in culture medium and seeded into plastics Petri dishes. Cells were cultured in several modifications of minimum essential medium (MEM), including alpha-MEM, D-MEM and D-MEM/F12 (Sigma-Aldrich, St. Louis, MO, USA) always supplemented with 10% fetal bovine serum (PAA Laboratories GmbH, Pasching, Austria) and antibiotics (100 U/mL penicillin, 100 mg/mL streptomycin, 180 g/mL gentamycin) (PAA Laboratories GmbH). Cells were sub-cultured when reached 80% confluency.
Fixation
The advantage of working with cell suspension lies in the fact, that there is no need for excision and related problems such as impaired tissue integrity and its topographical connections, and damage from faulty removal. Another advantage is that we can perform fixation of cells immediately and completely, discarding a major problem in tissue fixation, the auto-degradation and even disintegration processes due to incomplete and/or imperfect fixation process and consequent asphyxial changes.
We worked always with at least 500 000 cells which after centrifugation created pellet, macroscopically visible at the bottom of the test-tube. There is not a well-defined and broadly accepted speed to centrifuge cells into a pellet for TEM processing, however, high centrifugation speeds may damage cells,17 so, after the enzymatic digestion, cells were centrifuged at 1200 RPM for 10 min. It is important to maintain a pellet throughout the fixation and subsequent procedures to have enough cells for proper embedding. Repeated centrifugation can be carried out if the pellet is disrupted.1 Cell pellet was fixated at the room temperature for 3 h in 2.5% glutaraldehyde solution in phosphate buffer (pH 7.2).18 The crucial part was maintaining a steady pH by adding sodium hydrogen phosphate with water or potassium dihydrogen phosphate, when needed. Then, cells with fixative were rinsed with the cooled phosphate buffer with 10% saccharose, 3 times for 30 min and left sealed for one night in the refrigerator.
During fixation, the aldehydes rapidly react, in particular with amino groups of amino acids (mainly lysine), and other compounds to form the so-called Schiff’s reagents. Aldehyde moiety remains bound to the molecules which are fixated and becomes part of the fixed structure. Therefore, aldehydes belong to the group of additive fixing agents. There was no need for using methods of fractional vacuum distillation for removing undesirable degradation products and impurities in solutions of glutaraldehyde as glutaraldehyde with extremely high purity was used. We never used old fixative, because of its quick deterioration. Of course, with selected chemical fixation, we did not avoid the problem of reduced dispersity of the liquid colloidal system in which, the dispersed particles clump together into larger structures what results in their coagulation.
These problems should be taken into the consideration during further processing of the preparation and also in TEM analysis. Also, impaired aldehyde fixation can lead to production of myelin figures.19 Nevertheless, we consider general conformational changes as a small (in morphological image one needs to count with detectable graininess, swelling or, on the contrary, with shrinkage of structures) and therefore, glutaraldehyde fulfills its role in cell fixation. As an alternative to a chosen fixation, glutaraldehyde cacodylate buffered solution20 or glutaraldehyde with acrolein can be used.21 All methods of increasing the level of free oxygen (adding of hydrogen peroxide, aeration) in the solution increase the level of fixation as well.19
Postfixation
For postfixation we used Millonig buffer containing 1% osmium tetroxide (OsO4) (pH 7.2) for 2 h in refrigerator, to provide an enhanced contrast for TEM analysis. After simple fixation with glutaraldehyde, more than 95% of neutral lipid content within the tissue or cell is lost due to the unsatisfactory reaction of glutaraldehyde with lipids and proteoglycans.19 Although this is just a minor problem when working with adult tissue-derived stem cells, we prevented this situation performing postfixation with OsO4, when loss of lipids was markedly reduced.
Millonig buffer consists of 2.26% sodium phosphate dibasic solution (2.26 g NaH2PO4 + 100 mL of double-distilled water); 2.52% sodium hydroxide solution (2.52 g NaOH + 100 mL of double-distilled water); 5.4% glucose solution (5.4 g C6H12O6 + 100 mL of double-distilled water). The procedure is as follow: 41.5 mL of 2.26% sodium phosphate dibasic solution is mixed together with 8.5 mL of 2.52% sodium hydroxide solution, the result needs to have a pH of 7.3 (adding drop by drop sodium hydroxide or sodium phosphate dibasic solution when needed). Then, to 45 mL of obtained solution, 5 mL of 5.4% glucose solution is added. The resulting solution, 50 mL of Millonig solution with pH 7.3 is stored in a dark sealed bottle in the refrigerator, up to 3 days. Fixation with glutaraldehyde together with postfixation using osmium tetroxide may lead to formation of various artifacts, distortions, displacement of chemical components and anomalous deposits (in the form of electron-dense granules and structures, mainly alongside membranes). Such artifacts, however, can be prevented to some extent by using an extremely pure glutaraldehyde and by using the same buffer during the whole procedure.22 After postfixation, we were able to distinguish cells as a blackish mass, thus orientation in the specimen was adequate.
Fixatives are designed to react with biological tissues and extreme caution should be taken when handling them. Osmium tetroxide can fix the cornea of the eye.
Dehydration
The water which is in the sample would rapidly evaporate in a high vacuum condition in the microscope, and therefore resulting in the collapse of the sample’s structure. Consequently, the water in the tissue needs to be temporarily replaced with some chemical agent that finally recedes to embedding medium. This chemical is in most cases ethanol. Although ethanol is an efficient organic solvent; cells are not impaired thanks to prior stabilization with fixation agent. Dehydration must be perfect; however, a good compromise between a well-processed specimen and conformational changes of proteins must be made. For dehydration, we used procedure in Table 1. The better the fixation, the more macromolecules were stabilized and transferred to form of insoluble complexes, and the less they leached during dehydration. After postfixation with OsO4 and dehydration, when cells were not thoroughly washed, ethanol and residues from fixation solution precipitated and penetrated inside the cells. As dehydration can contribute to protein denaturation and lipid solubilization, exposure should be as brief as possible.
Table 1.
Dehydration procedure.
| Chemical | Time | Note |
|---|---|---|
| 50% alcohol | 5 minutes | Sealed, cooled (4°c) |
| 70% alcohol | 10 minutes | Sealed, cooled (4°c) |
| 80% alcohol | 10 minutes | Sealed, cooled (4°c) |
| 96% alcohol | 15 minutes | Sealed, cooled (4°c) |
| 100% alcohol | 20 minutes | Sealed, room temperature (20°c) |
| 100% alcohol | 20 minutes | Sealed, room temperature (20°c) |
| 100% alcohol | 20 minutes | Sealed, room temperature (20°c) |
| Propylene oxide | 15 minutes | Sealed, room temperature (20°c) |
| Propylene oxide | 20 minutes | Sealed, room temperature (20°c) |
| Propylene oxide + Durcupan I | Overnight | Opened, room temperature (20°C) |
After each step, a precise centrifugation at 1000 RPM for 2 min and a gentle aspiration of fluid is required. When working with 100% ethanol, a dry pipette and a quick processing is recommended to prevent contamination with air humidity. After dehydration with ethanol, cells were then transferred into embedding material through propylene oxide. Propylene oxide is the alkylene oxide (epoxide), and is well miscible with both, ethanol and epoxy resins. Its residues are polymerized together with an embedding material, and therefore propylene oxide is regarded as a perfect transition medium between the dehydration and embedding process.
Infiltration, embedding, polymerization
For infiltration, we used the procedure in Table 2. Both Durcupan I and Durcupan II are a water-soluble embedding agents, made from Durcupan ACM (Fluka Chemie AG, Buchs, Switzerland), according manufacturer directions. Once more, after each step we centrifuged the suspension at 1000 RPM for 2 min; however, even other speed and rotation time can be applied with satisfactory results. Polymerization took place at 60°C and lasted 3 days. For making a Durcupan I (used in dehydration procedure), components A (100 mL or 104.8 g), B (100 mL or 107.4 g) and D (1.5 mL) should be mixed together. The solution (yellowish viscous liquid) should be mixed well, sealed and stored in the refrigerator. It is essential to prevent its contact with humidity. To get Durcupan II, 0.45 mL to 0.48 mL of component C (accelerator) should be added to 21 g of Durcupan I. The solution should first be mixed well and then sealed. For embedding is better and more convenient to prepare always a new Durcupan II solution. According to our experience, component C in Durcupan II can be used at larger quantities (up to 0.50 mL), to ensure proper solidification of the embedding agent, but taking into account that in the other hand, the excess of component C causes the specimen to become granular. The resulting solution is an orange-pink medium.
Table 2.
Infiltration and embedding procedure.
| Chemical | Time | Note |
|---|---|---|
| Infiltration | ||
| Durcupan I | 120 minutes | Sealed, 50°C |
| Durcupan II | 180 minutes | Sealed, 40°C |
| Durcupan II | 120 minutes | Sealed, 40°C |
| Durcupan II | Overnight | |
| Embedding | ||
| Fresh Durcupan II | ||
Contrasting
Semi-thin sections were cut and stained with toluidine blue. Next steps did not differ from a routine preparation; specimen was cut with ultramicrotome and floating slices on the water or on other appropriate inert medium was collected on TEM grids. The recommended section thickness for embedded cells is between 50-100 nm (sections with a silver or light gold color).1 Since the contrast in the electron microscope depends primarily on the differences in the electron density of the organic molecules in the cell, the efficiency of a stain is determined by the atomic weight of the stain attached to the biological structures. Hence, for contrasting, we picked alcoholic uranyl acetate solution (we tried both methanolic and 98% ethanolic uranyl acetate solution with satisfactory results and comparable picture quality). However, highest contrast was obtained when double contrasting, used in sequence with uranyl acetate and the so-called Reynolds solution was performed.23 The combination of these two contrasting agents gave us nearly uniform contrast of all the structures of the cell. Uranyl acetate delivered good contrasting results of membranes, nucleic acids and conglomerates of nucleic acids together with protein complexes (ribosomes). In our laboratory, Reynolds solution is used for contrasting, with good results. It is made from trisodium citrate, lead (II) nitrate and water that react together, resulting in the formation of lead citrate, which is used routinely for contrasting worldwide. During contrasting with lead, fine precipitates of insoluble PbCO3 can occur. These are difficult to remove from the section and they have an affinity for the cytoplasmic structures, wherein creating artifacts indistinguishable from other cytoplasmic granules.24,19
Prior to the mixing of chemicals together, it is important to prevent the precipitation of lead carbonate in the water and the consecutive contamination of the specimen (which appears as black grains in the electron microscope); this is done by avoiding any carbonate in the solution. In order to degas the water, we put the water under vacuum in an Erlenmeyer flask overnight. The formula for preparing 50 mL of Reynolds solution is as follow: 1.33 g Pb(NO3)2 + 1.76 g Na3C6H5O7.2H2O + 30 mL double-distilled degassed water. Then, the resulting suspension was shaken for 15 min vigorously and intermittently (1 min for further 30 min). Afterwards, the solution was left undisturbed at room temperature for 30 min. The result should appear cloudy. Next step consisted in adding of 8 mL 1N sodium hydroxide and enough water to obtain 50 mL of solution with subsequent stirring. Reynolds solution must be kept in the refrigerator in a dark bottle, up to 6 weeks if sealed tightly (however, we used always fresh one). When there is a precipitation or the solution is not clear, discarding this chemical and making a new one is the only right solution. The pH should be 12.0±0.1. An exact pH proved to be an extremely important condition.
Grids were placed face-down on the drops of uranyl acetate alcohol solution, covered with the foil and contrasted for 15 min, then removed with forceps and rinsed with the 50% alcohol 5 times. With the filter paper, the excess of alcohol was sucked off. Grids were stowed in the petri dish to prevent the precipitation of the lead citrate by exposure to CO2. Then the grids were placed on the drops of lead citrate for incubation, and left there for 5 min. Each grid was then removed and rinsed using double-distilled water. With the filter paper, the excess of water was sucked off as well. After wicking and drying, the grids could be analyzed. When doing this, it is essential to illuminate samples with the electron beam well spread to avoid damage to the delicate thin sections.1
Results
Generally, all analyzed cells exhibited characteristics typical for mesenchymal stem cells. TEM analysis of first passages often revealed the ultrastructure common for all the cells with high proteosynthetic level. Cells had polymorphic nuclei with deep notches, containing mainly euchromatin and conspicuous nucleoli (Figure 1 A-F), none of the artifacts were observed. This morphology is characteristic for cells with active transcription of genetic information into mRNA. In the cytoplasm, mainly alongside the nucleus, well-developed dilated cisternae of rough endoplasmic reticulum, formed by broad vesicles (Figure 1 C,F), branched protrusions of the plasmatic membranes which resemble filopodia (Figure 1 B,E) and served for attaching to the ground, fatty vacuoles and intracytoplasmic lamellar bodies were present.
Figure 1.
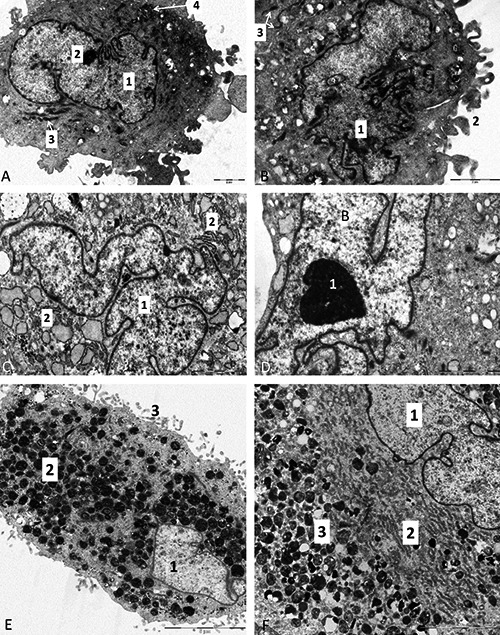
A) Electron micrograph of a dental pulp-derived stem cell from in vitro cell culture; 1, cell nucleus with predominance of euchromatin; 2, nucleolus; 3, mitochondria; 4, aggregation of glycogen granules. B) Dental pulp-derived stem cell; 1, irregular shaped nucleus with deep notches and predominance of euchromatin; 2, plasma membrane create irregular finger-like projections called fillopodia; 3, mitochondria. C) Electron micrograph of skeletal muscle-derived stem cell from in vitro cell culture; 1, pale, euchromatic nucleus; 2, dilated cisternae of rough endoplasmic reticulum. D) Detail on irregular shaped nucleus of skeletal muscle-derived stem cell, 1, large nucleolus. E) Electron micrograph of white adipose tissue-derived stem cell from in vitro cell culture; 1, pale, euchromatic nucleus; 2, residual bodies (cell organelles after degradation); 3, filopodia. F) Detail of white adipose tissue-derived stem cell from in vitro cell culture; 1, pale euchromatic nucleus; 2, cytoplasm is filled with rough endoplasmic reticulum; 3, residual bodies.
In the cytoplasm, in close proximity to the nucleus, well developed rough endoplasmic reticulum and cisterns of Golgi apparatus were found in most cases. When analyzing adipose tissue - derived stem cells, within their cytoplasm, large number of small lipid droplets and between them, sparsely placed, small and inconspicuous, electron-dense, lamellar bodies, which resembled myelin figures were observed. The cells from the later passages were already completely attached to the bottoms, what was visible as a numerous branched projections of their cytoplasm, which resembled filopodia. Cells’ nuclei were, even in these passages, pale with predominance of euchromatin and contained 1 to 2 well-developed nucleoli. This implied a still high transcriptional activity inside of nuclei, in another words, it presented a proof of proteosynthesis. Primary isolated muscle-derived stem cells had a fibroblast-like shape. During subsequent passages they maintained this morphology. In certain parts of the cytoplasm there were aggregates of granules of glycogen. Human dental pulp-derived stem cells (Figure 1A,B) possess abundant cisterns of rough endoplasmic reticulum and numerous coated matrix vesicles as well as granules of glycogen present in their cytoplasm. Nearby the nucleus, small, elongated mitochondria were placed. Most of these cells created and secreted vesicles; in plasmalemma bounded amorphous electron-lucid granules and also few glycogen granules.
Discussion
Because transmission electron microscopy provides ultrastructural details of cells, it is a valuable and useful method for studying cell organelles.24 That is why we have used it for a detailed description of adult stem cells as an example of rare cell populations from human body. Preparation of samples from cell culture suitable for the analysis with transmission electron microscope is, however, much more difficult and challenging than routine histological processing of a solid tissue.25
Generally, we were able to describe morphology of long-term cultivated stem cells without distractions; this is where we see the biggest impact of our work, since the potential clinical significance of mesenchymal stem cells is huge. For example, beside their multilineage differentiation potential and the ability to modulate oxidative stress, and secrete various cytokines and growth factors that can have immunomodulatory, angiogenic, anti-inflammatory and anti-apoptotic effects,26 nowadays grows evidence that human mesenchymal stem cells have an intrinsic property for homing towards tumor sites and can be used as tumor-tropic vectors for tumor therapy. Treatment with adipose tissue-derived stem cells conditioned media showed significant upregulation of apoptotic genes and significant downregulation of antiapoptotic genes such as survivin and XIAP in various tumors.27 The antiapoptotic protein survivin, a promising diagnostic biomarker and prognostic parameter, is widely expressed in most human cancers.28
This work can help others authors overcome some problems and obstacles when dealing with processing of cell populations for morphologic analysis. We obtained high quality, reproducible and comparable images. After repeating our approach several times, with stem cell populations from different sources, always with freshly made solutions, and getting the same results, we conducted that one can concentrate on specific problems and features of the cell population, and not distortions and artifacts, when using our protocol regarding processing of different human mesenchymal stem cells after in vitro expansion. Thus, we were able to perform ultrastructural morphological analysis of different adult tissue-derived stem cells and deliver it to the reader.
Funding Statement
Funding: this study was supported by the grant of the Slovak Research and Development Agency No. APVV-0434-12.
References
- 1.Schrand AM, Schlager JJ, Dai L, Hussain SM. Preparation of cells for assessing ultrastructural localization of nanoparticles with transmission electron microscopy. Nat Protoc 2010;5:744-57. [DOI] [PubMed] [Google Scholar]
- 2.Li X, Wu D, Shen J, Zhou M, Lu Y. Rapamycin induces autophagy in the melanoma cell line M14 via regulation of the expression levels of Bcl-2 and Bax. Oncol Letts 2013;5:167-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taupin P. Processing scarce biological samples for light and transmission electron microscopy. Eur J Histochem 2008;52: 135-9. [DOI] [PubMed] [Google Scholar]
- 4.Kisiel AH, McDuffee LA, Masaoud E, Bailey TR, Esparza Gonzalez BP, Nino-Fong R. Isolation, characterization, and in vitro proliferation of canine mesenchymal stem cells derived from bone marrow, adipose tissue, muscle, and periosteum. Am J Vet Res 2012;73:1305-17. [DOI] [PubMed] [Google Scholar]
- 5.König A, Shcherbata HR. Visualization of adult stem cells within their niches using the Drosophila germline as a model system. Methods Mol Biol 2013;1035:25-33. [DOI] [PubMed] [Google Scholar]
- 6.Darkazalli A, Levenson CW. Tracking stem cell migration and survival in brain injury: current approaches and future prospects. Histol Histopathol 2012;27:1255-61. [DOI] [PubMed] [Google Scholar]
- 7.Vande Velde G, Raman Rangarajan J, Vreys R, Guglielmetti C, Dresselaers T, Verhoye M, et al. Quantitative evaluation of MRI-based tracking of ferritin-labeled endogenous neural stem cell progeny in rodent brain. Neuroimage 2012;62:367-80. [DOI] [PubMed] [Google Scholar]
- 8.Tang KX, Yan JH, Shen YF, Li BY, Chen YM, Liu DY, et al. Tracing type 1 diabetic Tibet miniature pig’s bone marrow mesenchymal stem cells in vitro by magnetic resonance imaging (1). J Diabetes 2014;6:123-31. [DOI] [PubMed] [Google Scholar]
- 9.Zhao YH, Yang Q, Xia Q, Peng J, Lu SB, Guo QY, et al. In vitro cartilage production using an extracellular matrix-derived scaffold and bone marrow-derived mesenchymal stem cells. Chin Med J (Engl) 2013;126:3130-7. [PubMed] [Google Scholar]
- 10.Ferro F, Spelat R, Falini G, Gallelli A, D’Aurizio F, Puppato E, et al. Adipose tissue-derived stem cell in vitro differentiation in a three-dimensional dental bud structure. Am J Pathol 2011;178:2299-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radtke CL, Nino-Fong R, Rodriguez-Lecompte JC, Esparza Gonzalez BP, Stryhn H, McDuffee LA. Osteogenic potential of sorted equine mesenchymal stem cell subpopulations. Can J Vet Res 2015;79:101-8. [PMC free article] [PubMed] [Google Scholar]
- 12.Danisovic L, Varga I, Polák S, Bajciková B, Adamkov M, Vojtassák J. Biological and morphological characterization of in vitro expanded human muscle-derived stem cells. Tsitologiia 2011;53:482-7. [PubMed] [Google Scholar]
- 13.Zhang Z, Li F, Tian H, Guan K, Zhao G, Shan J, et al. Differentiation of adipose-derived stem cells toward nucleus pulposus-like cells induced by hypoxia and a three-dimensional chitosan-alginate gel scaffold in vitro. Chin Med J (Engl) 2014;127:314-21. [PubMed] [Google Scholar]
- 14.Varga I, Hollý D, Vojtaššák J, Böhmer D, Polák Š, Danišovič L. Morphological characterization of in vitro expanded human dental pulp-derived stem cells. Biologia 2011;66:706-11. [Google Scholar]
- 15.Danišovič Ľ, Kuniaková M, Varchulová-Nováková Z, Boháč M, Varga I, Rusnáková J, et al. Comprehensive characterization of human adipose tissue-derived stem cells expanded in vitro. Biologia 2013;68:747-53. [Google Scholar]
- 16.Danišovič L, Varga I, Polák Š, Uličná M, Hlavačková L, Böhmer D, et al. Comparison of in vitro chondrogenic potential of human mesenchymal stem cells derived from bone marrow and adipose tissue. Gen Physiol Biophys 2009;28:56-62. [PubMed] [Google Scholar]
- 17.De Brauwer EI, Jacobs JA, Nieman F, Bruggeman CA, Wagenaar SS, Drent M. Cytocentrifugation conditions affecting the differential cell count in bronchoalveolar lavage fluid. Anal Quant Cytol Histol 2000;22:416-22. [PubMed] [Google Scholar]
- 18.Gurgen SG, Erdogan D, Take-Kaplanoglu G. The effect of histamine on kidney by fasting in rats. Bratisl Lek Listy 2013;114:251-7. [DOI] [PubMed] [Google Scholar]
- 19.Mráz P, Polónyi J. Metódy elektrónovej mikroskopie živočíšnych tkanív [Methods of electron microscopy of animal tissues] [Book in Slovak]. Vydavatel’stvo Slovenskej Akadémie Vied, Bratislava: 1988. [Google Scholar]
- 20.Glauert AM. Practical methods in electron microscopy, vol. 3 Elsevier, Amsterdam: 1974. [Google Scholar]
- 21.Mersey B, McCully ME. Monitoring the course of fixation of plant cells. J. Micr. 1978;114:49-76. [Google Scholar]
- 22.Kuthy E, Csapó Z. Peculiar artefacts after fixation with glutaraldehyde and osmium tetroxide. J Microsc 1976;107:177-82. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 1963;17: 208-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham L, Orenstein JM. Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research. Nat Protoc 2007;2:2439-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar S, Ciraolo G, Hinge A, Filippi MD. An efficient and reproducible process for transmission electron microscopy (TEM) of rare cell populations. J Immunol Methods 2014;404:87-90. [DOI] [PMC free article] [PubMed] [Google Scholar]