

Abstract
Background
In cardiorenal syndrome type 2 (CRS2), the role of systemic congestion in heart failure (HF) is still obscure. We studied a model of CRS2 [monocrotaline (MCT)-treated rats] secondary to pulmonary hypertension and right ventricular (RV) failure in order to evaluate the contribution of prevalent congestion to the development of kidney injury.
Methods
Ten animals were treated with MCT for 4 weeks until they developed HF. Eleven animals were taken as controls. Signs of hypertrophy and dilatation of the right ventricle demonstrated the occurrence of HF. Brain natriuretic peptide (BNP), serum creatinine (sCreatinine), both kidney and heart neutrophil gelatinase-associated lipocalin (NGAL), matrix metallopeptidase 9 (MMP9), serum cytokines as well as kidney and heart cell death, as assessed by TUNEL, were studied.
Results
Rats with HF showed higher BNP levels [chronic HF (CHF) 4.8 ± 0.5 ng/ml; controls 1.5 ± 0.2 ng/ml; p < 0.0001], marked RV hypertrophy and dilatation (RV mass/RV volume: CHF 1.46 ± 0.31, controls 2.41 ± 0.81; p < 0.01) as well as pleural and peritoneal effusions. A significant increase in proinflammatory cytokines and sCreatinine was observed (CHF 3.06 ± 1.3 pg/ml vs. controls 0.54 ± 0.23 pg/ml; p = 0.04). Serum (CHF 562.7 ± 93.34 ng/ml vs. controls 245.3 ± 58.19 ng/ml; p = 0.02) as well as renal and heart tissue NGAL levels [CHF 70,680 ± 4,337 arbitrary units (AU) vs. controls 32,120 ± 4,961 AU; p = 0.001] rose significantly, and they were found to be complexed with MMP9 in CHF rats. A higher number of kidney TUNEL-positive tubular cells was also detected (CHF 114.01 ± 45.93 vs. controls 16.36 ± 11.60 cells/mm2; p = 0.0004).
Conclusion
In this model of CHF with prevalent congestion, kidney injury is characterized by tubular damage and systemic inflammation. The upregulated NGAL complexed with MMP9 perpetuates the vicious circle of kidney/heart damage by enhancing the enzymatic activity of MMP9 with extracellular matrix degradation, worsening heart remodeling.
Key Words: Cardiorenal syndromes, Neutrophil gelatinase-associated lipocalin, Matrix metallopeptidase 9, Heart failure, Cytokines, Kidney injury
Introduction
Cardiorenal syndrome (CRS) is defined as a complex pathophysiological disorder of the heart and kidneys in which acute or chronic dysfunction of one organ may induce acute or chronic dysfunction of the other organ [1]. CRS type 2 (CRS2) is characterized by chronic abnormalities of cardiac function leading to kidney injury or dysfunction [2,3]. It is difficult to extract specific mechanisms for CRS2 based on human studies, which are mainly observational. Therefore, animal studies may provide important insights into the pathogenesis of CRS2 by mimicking human phenotypes. The predominant mechanisms proposed include neurohormonal activation, hemodynamic factors such as renal hypoperfusion and venous congestion as well as inflammation and oxidative stress [2,3,4,5].
A decreased glomerular filtration rate may be the result of reduced cardiac output in systolic heart failure (HF), but in congestive and diastolic HF, this may not be the case. A high venous pressure is an alternative and important cause of worsening kidney function; in this case, tubular damage plays a major role [6] and simultaneously constitutes a stimulus for peripheral synthesis and the release of inflammatory mediators [7]. Moreover, inflammation could be an additional nonhemodynamic mechanism for the progression of chronic kidney disease [5,8] triggered by local kidney factors or by increased gut absorption of endotoxin [9,10]. In fact, patients with severe HF feature high levels of tumor necrosis factor (TNF)-α and interleukins (IL) [11].
The aim of our study was to evaluate the contribution of prevalent congestion to the development of kidney injury in a model of CRS2 secondary to pulmonary hypertension and right ventricular (RV) failure. Specifically, the aim of the study was (1) to evaluate the occurrence and mechanism of kidney damage and (2) to analyze the role of neutrophil gelatinase-associated lipocalin (NGAL), a product of tubular damage currently used as an early biomarker of kidney dysfunction, in worsening heart remodeling, contributing to a perpetuation of the vicious circle of heart/kidney damage.
Materials and Methods
Animals and Development of Pulmonary Hypertension and RV Hypertrophy and Failure
RVHF was induced in 10 male Sprague-Dawley rats (90-100 g) by intraperitoneal injection of 30 mg/kg monocrotaline (MCT) according to Dalla Libera et al. [12] and Vescovo et al. [13] [chronic HF (CHF) group]. MCT is a well-established model of RVHF which mimics the congestive HF syndrome in humans [12,13]. Eleven rats treated with saline served as a control group.
Twenty-eight days after MCT injection, all animals were euthanized; their hearts and kidneys were excised, and parts were frozen in liquid nitrogen or fixed in paraformaldehyde and then embedded in paraffin. Blood was also drawn. The Biological Ethics Committee of the University of Padua approved the experiments. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published in 1996 by the US National Institutes of Health.
Assessment of RV Hypertrophy and Failure
To ensure that the MCT rats developed RVHF beyond the well-known postmortem signs of congestion, such as pericardial, pleural and peritoneal effusions, the following measurements were taken: left ventricular mass/RV mass index (LVM/RVM; an index of hypertrophy) and RVM/RV volume index (RVM/RVVol; an index of dilatation).
The two indices were calculated by computerized planimetry (Image PRO-Plus 4.0; Media Cybernetics, Silver Spring, Md., USA) on formalin-fixed transverse sections of the heart taken from the middle portion of the interventricular septum [14,15].
Histological Study
At the end of the experiments, the hearts and kidneys were removed from the animals, fixed in neutral buffered formalin and then processed for histology. H&E, Masson trichrome (for fibrosis) and periodic acid-Schiff stains were done to define the heart and kidney damage.
Assessment of Cell Death
In situ nick-end labeling (TUNEL) of fragmented DNA was performed on cryosections of the heart and kidney using an in situ cell death detection kit (POD; Boehringer Mannheim). Labeled nuclei were identified from the negative nuclei counterstained with TO-PRO-3 and counted after being photographed. The total number of positive nuclei was determined by counting (magnification ×250) all the labeled nuclei present in the entire specimen [14,15]. The number of positive nuclei was then expressed as the number of TUNEL-positive nuclei per square millimeter [14,15,16].
Brain Natriuretic Peptide Assessment
Brain natriuretic peptide (BNP) was measured on sera with an enzyme-linked immunoassay (ELISA) kit (BNP-45 cat. No. EK-011-17; Phoenix Pharmaceuticals Inc., Burlingame, Calif., USA) following the manufacturer's instructions. The antibody was specific for rat BNP [14,15].
Creatinine Assessment
Creatinine was measured on sera with an ELISA (Rat Creatinine ELISA kit; Cusabio Biotech Co., Tema Ricerca, Castenaso, Italy) following the manufacturer's instructions. The antibody was specific for rat creatinine. Values are expressed in picograms per milliliter.
NGAL Assessment on Sera
NGAL was measured on sera with an ELISA (Rat NGAL ELISA kit; Bioporto Diagnostics, Bio Exe Research Technology, Verona, Italy) following the manufacturer's instructions. The antibody was specific for rat NGAL.
Semiquantitative NGAL mRNA Assessment on Kidney Tissues
The total RNA from rat kidneys was extracted by the acid guanidinium thiocyanate method and the phenol-chloroform extraction technique. One microgram of RNA was reverse transcribed with MuLV reverse transcriptase (Life Technologies, Monza, Italy) in the presence of random hexamers according to the manufacturer's instructions. The cDNA was amplified for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to evaluate the quality of the extraction.
The quantity of RNA was evaluated by spectrophotometer (NanoVue; GE Healthcare Life Sciences, EuroClone S.p.A., Pero, Italy). PCR amplification products were exported on agarose gel electrophoresis and stained with ethidium bromide, and gel pictures were taken with Alliance 2.7 (UVITEC Ltd. Cambridge, Eppendorf, Milan, Italy). Quantitative evaluation was done by Alliance 2.7 1D image software, and the results are expressed as arbitrary units (AU). The following oligonucleotide primer pairs were used for NGAL: forward 5′-ACATTCGTTGCAAGCTCCAG-3′ and reverse 5′-TCCGTACAGGGTGACTTTGA-3′. The primer pairs were designed using Primer 3 (free access) based on published sequences (http://www.ncbi.nlm.nih.gov ).
Immunoblotting of NGAL and Matrix Metallopeptidase 9 Proteins on Heart and Kidney Tissues
Heart and kidney samples were homogenized and solubilized in sodium dodecyl sulfate (SDS) buffer [9,15]. Protein quantification was performed using the Qubit® Protein Assay Kit (Life Technologies) according to the manufacturer's instructions. Protein samples were mixed with a nonreducing and a reducing buffer and incubated for 5 min at either room temperature (reducing and nondenaturing conditions) or 95°C (reducing and denaturing conditions).
All samples were subsequently separated on 10% gel in SDS-PAGE and transferred onto a nitrocellulose membrane (Amersham, EuroClone). The membrane was blocked for 1 h with 5% nonfat milk in TBS containing 0.5% (v/v) Triton X-100 (Sigma-Aldrich) and incubated overnight with polyclonal goat antibodies against matrix metallopeptidase 9 (MMP9) or NGAL (1:500; Abcam, Prodotti Gianni, Milan, Italy). Blots were developed using the SuperSignal West Femto ECL substrate (Pierce, EuroClone).
Densitometric software (Alliance 2.7 1D fully automated software) determined the percent distribution of the NGAL and MMP9 antibodies after image acquisition by Alliance 2.7 (UVITEC, Eppendorf, Milan, Italy).
Bio-Plex Multiplex Cytokine Assay
Serum samples were analyzed with a suspension array system (Bio-Plex; Bio-Rad, Hercules, Calif., USA) for the quantification of 9 cytokines [IL-1α, −1β, −2, −4, −6 and −10, interferon-γ (IFN-γ) and TNF-α] following the manufacturer's instructions as previously described [14,15,17].
Statistical Analysis
Means ± SD of the observed data were calculated. Student's t test was used for unpaired data. p < 0.05 was considered statistically significant. Analysis of variance was also used [14,15].
Results
Occurrence of HF
At 28 days, postmortem examination of the CHF rats demonstrated pericardial, pleural and peritoneal effusions characteristic of CHF. LVM/RVM, which is an index of RV hypertrophy, was significantly decreased in CHF animals (1.58 ± 0.16) compared to controls (2.93 ± 0.35; p < 0.001), indicating that the RV developed mass increased (table 1). RVM/RVVol, an index of RV dilatation, showed significantly lower values in CHF rats than in controls (1.46 ± 0.31 vs. 2.41 ± 0.81; p = 0.01), indicating that for a similar RV mass, CHF rats show greater RV dilatation (table 1).
Table 1.
Occurrence of HF and cardiac remodeling
| LVM/RVM | RVM/RVVol | BNP, ng/ml | |
|---|---|---|---|
| Controls (n = 11) | 2.93±0.35** | 2.41±0.81* | 1.5±0.2*** |
| CHF group (n = 10) | 1.85±0.16** | 1.46±0.31* | 4.8±0.5*** |
Values are expressed as means ± SD.
p = 0.01
p < 0.001
p < 0.0001.
BNP confirmed the morphologic data, showing high levels in CHF rats compared to controls and thereby confirming the presence of RVHF in this group (4.8 ± 0.5 vs. 1.5 ± 0.2 ng/ml; p < 0.0001; table 1; fig. 1a).
Fig. 1.

Kinetic and final values for BNP, sNGAL and sCreatinine. a Kinetic curve of BNP levels on rat sera during the 4 weeks of the experiments. At 3 weeks, CHF rats showed significantly higher BNP levels than controls. At 4 weeks, CHF rats showed high levels of BNP (4.8 ± 0.5 ng/ml), confirming the presence of RVHF in CHF rats compared to controls (1.5 ± 0.2 ng/ml; p < 0.0001). b Kinetic curve of NGAL on rat sera. Similarly to BNP levels, sNGAL levels were significantly elevated in MCT-treated rats. Note that 1 week after MCT injection, sNGAL levels reached up to 268.5 ± 75.9 ng/ml. The histogram shows the NGAL values on rat sera at 4 weeks for CHF rats (536.9 ± 53.7 ng/ml) and controls (245.3 ± 58.19 ng/ml; p = 0.02). c Kinetic curve of creatinine on rat sera. One week after MCT injection, sCreatinine was 0.7 ± 0.1 pg/ml and reached up to 3.06 ± 1.3 pg/ml in the fourth week. The histogram shows the sCreatinine values after 4 weeks of MCT treatment (3.06 ± 1.3 pg/ml) compared to controls (0.54 ± 0.23 pg/ml; p = 0.04).
Serum Cytokine Levels
CHF rats showed increased serum levels for all the cytokines measured. This was particularly true for TNF-α (CHF 1,325.37 ± 559.95 pg/ml vs. controls 84.46 ± 64.50 pg/ml), IL-1α (CHF 183.71 ± 51.61 pg/ml vs. controls 78.31 ± 26.07 pg/ml), IL-4 (CHF 731.67 ± 154.83 pg/ml vs. controls 316.62 ± 12.55 pg/ml), IL-6 (CHF 5,314.93 ± 1,632.98 pg/ml vs. controls 1,754.01 ± 188.85 pg/ml) and IL-10 (CHF 2,613.10 ± 934.04 pg/ml vs. controls 847.70 ± 152.19 pg/ml), which showed significantly higher levels (p < 0.05; fig. 2).
Fig. 2.

Serum cytokine levels. Inflammatory/immune mediators IL-1α, −1β, −2, −4, −6 and −10, IFN-γ and TNF-α were determined with a Bio-Plex suspension assay on rat sera. Values are the means of the experiments run in duplicate ± SE. CHF rats show increased levels of all cytokines compared to controls, which very likely is a result of the general inflammation triggered by HF. * p < 0.05.
Serum Creatinine Levels
CHF rats showed a significant rise in serum creatinine (sCreatinine; see fig. 1c for the time course of sCreatinine levels). One week after MCT injection, the sCreatinine level was 0.7 ± 0.1 pg/ml, and it reached up to 3.06 ± 1.3 pg/ml in the fourth week, in contrast to the controls, which showed a value of 0.54 ± 0.23 pg/ml (p = 0.04; fig. 1c) in the fourth week.
Serum NGAL Levels
Similarly, CHF rats showed a significant rise in serum NGAL (sNGAL; see fig. 1b for the time course of sNGAL values). One week after MCT injection, the sNGAL level was 268.5 ± 75.9 ng/ml, and it reached 536.9 ± 53.7 ng/ml in the fourth week of treatment. The sNGAL level in CHF rats was 562.7 ± 93.34 ng/ml in the fourth week, which was significantly higher than that in controls (245.3 ± 58.19 ng/ml; p = 0.02; fig. 1b).
Tissue NGAL Levels
In the kidneys of CHF rats, the mRNA expression of NGAL was significantly higher than that found in controls (70,680 ± 4,337 vs. 32,120 ± 4,961 AU; p = 0.001; fig. 3).
Fig. 3.

Tissue NGAL expression. a The histogram shows the degree of mRNA expression of NGAL in kidney tissue. Note that in CHF rats, the NGAL level was significantly higher than that found in controls (p = 0.001). b Representative images of PCR mRNA amplification for the NGAL gene and GAPDH gene controls.
Kidney Histology
On histology, no sign of glomerular damage was detectable. Instead, tubular dilatation was evident (fig. 4e–h). Neither signs of fibrosis (fig. 4g) nor of inflammatory infiltrate were observed in the kidneys of CHF rats (fig. 4).
Fig. 4.

Kidney histology. a, b, e, f H&E staining showing the absence of glomerular, interstitial and vascular lesions in controls and CHF rats. Only a tubular dilatation due to congestion is present in CHF rats (red arrows). Original magnification: ×20 (a, e), ×40 (b, f). c, g Masson trichrome staining showing tubular dilatation in CHF rats compared to controls, but no presence of fibrosis in the kidney. Original magnification: ×40. d, h Periodic acid-Schiff (PAS) staining highlighting tubular epithelial denudation, an early histological marker of tubular damage, in CHF rats compared to controls (red square). Original magnification: ×40. Color refers to the online version only.
Cell Death
Cell death was significantly increased in both the heart and the kidney of CHF rats. The number of TUNEL-positive kidney cells was found to be significantly higher in CHF rats than in controls (114.01 ± 45.93 vs. 16.36 ± 11.60 cells/mm2; p = 0.0004; fig. 5a). Cell death occurred almost exclusively in tubular cells, and glomerular cells were in fact spared (fig. 5b). Also in the heart, the number of TUNEL-positive cells was significantly higher in CHF rats than in controls (52.04 ± 26.64 vs. 7.52 ± 4.75 cells/mm2; p = 0.05; fig. 5a).
Fig. 5.

Cell death assay. a The histogram shows the degree of apoptosis [TUNEL-positive (T+) cells] quantified by TUNEL assay in the kidney and heart. In CHF rats, cell death was significantly increased both in the kidney and heart. b Representative images of controls (left) and CHF rats (right) made with the TUNEL technique. White arrows indicate TUNEL-positive cells identified by brown nuclei. Note the lower degree of TUNEL-positive cells in controls for both kidney and heart samples. Color refers to the online version only.
Protein Expression of NGAL and MMP9 in Hearts and Kidneys
Western blotting of heart and kidney samples performed both in reducing and denaturing conditions revealed the presence of 25- and 50-kDa bands, which corresponds to monomers and dimers of NGAL (fig. 6a). Control rats showed a predominance of the 50-kDa form, while the CHF group showed both the 25- and the 50-kDa form. The optical density of heart NGAL was two-fold higher in CHF rats (6,056,000 ± 877,500 AU) than in controls (3,508,000 ± 193,800 AU; p = 0.01; fig. 6d).
Fig. 6.

Protein expression of NGAL and MMP9 on heart and kidney tissue. a Representative Western blots for NGAL detection in kidneys and hearts performed in reducing and denaturing conditions. The Western blots revealed the presence of 25- and 50-kDa bands, which correspond to monomers and dimers of NGAL protein. Also note the presence, in CHF rats, of the 130-kDa band, which represents complexed NGAL/MMP9 (red square). b Representative Western blots of the same sample for MMP9 detection in kidneys and hearts performed in reducing and denaturing conditions. The Western blots revealed the presence of a 50- to 55-kDa band, which corresponds to the MMP9 monomer. In CHF rats, we also found bands at 115, 130 and 220 kDa (red square) in the heart, representing the complexed forms of NGAL/MMP9. This kind of pattern is more pronounced in heart tissue. c Representative Western blots of heart and kidney samples performed both in nonreducing and in nondenaturing conditions (that preserved the native complex form), confirming the presence of complexed NGAL/MMP9 in both kidney and heart samples of CHF rats compared to controls. d Semiquantitative optical density of NGAL in the heart, where NGAL protein expression was twice as high in CHF rats compared to controls (p = 0.01). e Semiquantitative optical density of NGAL in the kidney. Color refers to the online version only.
The same NGAL samples blotted against anti-MMP9 revealed the presence of a 50- to 55-kDa band, which corresponds to the MMP9 monomer. In CHF rats, we also found bands at 115, 130 and 220 kDa, representing the complexed forms of NGAL/MMP9 (fig. 6b). This kind of pattern was more evident in the heart samples.
Western blots of heart and kidney samples, performed both in nonreducing and in nondenaturing conditions, confirmed the presence of the complexed NGAL/MMP9 in these two kinds of samples of CHF groups as compared to controls (fig. 6c).
Discussion
In this well-characterized model of RV failure and systemic congestion with preserved left ventricular ejection fraction, kidney injury occurred as demonstrated by increased levels of sCreatinine and NGAL. MCT-treated rats developed RVHF and congestion, as demonstrated by the clinical signs (presence of ascites, pleural and pericardial effusions), by the elevated BNP levels and by the morphologic changes of the right ventricle with hypertrophy and dilatation.
In animals with a normal left ventricle at histology and morphometric analysis, no glomerular, vascular and interstitial damage was present (fig. 4). Therefore, we can reasonably exclude a toxic effect of MCT in that it produces kidney damage only at high doses administered for prolonged periods of time [18,19].
A systemic inflammatory response, which is also a consequence of HF, occurred as demonstrated by the elevated levels of circulating proinflammatory cytokines. The inflammatory response was likely to be responsible for the high levels of cell death found in the kidney and the heart.
Cell death in the kidney was almost exclusively confined to tubular cells, and this accounts for tissue and plasma NGAL release. It suggests that a congestion-induced increase in kidney intravenous pressure is responsible for the tubular damage and NGAL release [16,20,21].
However, NGAL release may also be the result of kidney inflammation [3,4,22]. Although serum cytokine levels were high, inflammatory cell infiltrates and fibrosis were not seen in the kidney. In contrast, we found a high number of TUNEL-positive cells both in the kidney and in the heart [4,13,16,23,24]. The source of the inflammation is certainly multifactorial, and either the heart or the kidney could be its active source [21,24]. It is possible that kidney hypoperfusion in systolic HF and congestion in diastolic and congestive HF may form two different pathways by producing an inflammatory milieu, tubular damage and NGAL release. It has been shown that congestion by itself may trigger inflammation, as observed by Colombo et al. [7]; they demonstrated in humans that forearm vein ligation induces the release of proinflammatory cytokines from the endothelium.
In our animals, increased intrarenal venous pressure triggered tubular damage. The released NGAL induced a further burst of inflammatory cytokines that, by endocrine or paracrine mechanisms, produced organ damage to the heart [12,13,16] and kidney, as well as to other organs such as skeletal muscles and the lung, as previously observed by our group [14,15]. Venous hypertension and congestion may also activate the systemic and intrarenal renin-angiotensin-aldosterone system [25]. Neuroendocrine activation, with elevated levels of angiotensin II, catecholamine and BNP, also contributes to organ damage and a further deterioration of heart and kidney function [12,14].
In our model of CRS2, we confirmed the role of NGAL as an early biomarker of kidney injury, in that its appearance in the blood far precedes the rise in sCreatinine. This paper also supports the concept of a perpetuation of heart and kidney damage in CRS2. In fact, we demonstrated that NGAL released by tubuli is complexed with MMP9 protein in both heart and kidney CHF rats. NGAL (a specific MMP9 binder) binds MMP9, protecting it from degradation with enhancement of its enzymatic activity, leading to increased collagen breakdown with consequent negative heart remodeling [26,27,28,29] (fig. 7).
Fig. 7.
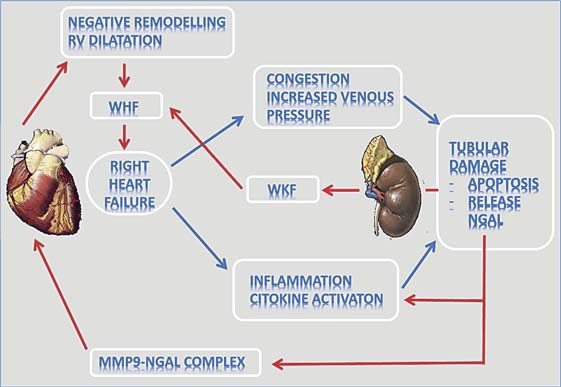
Vicious circle of congestion and heart/kidney damage in CRS2. The image represents the interrelationship between the heart and kidney leading to a vicious circle amplifying the damage. RVHF increases the venous pressure, with consequent tubular damage, inflammation, cytokine activation, kidney NGAL release and NGAL-MMP9 upregulation in the kidney. The upregulated NGAL-MMP9 complex perpetuates the vicious circle of kidney/heart damage by enhancing extracellular matrix degradation and worsening heart remodeling. WHF = Worsening HF; WKF = worsening kidney function.
Conclusions
To the best of our knowledge, this is one of the few studies exploring the pathophysiology of CRS2 in animals, shedding light on the mechanism involved in this syndrome. This paper shows the role of congestion in the development of tubular injury with release of NGAL. On the basis of our findings, we put forward the hypothesis that congestion may lead to development of tubular injury with release of NGAL.
NGAL released from renal tubular cells can be seen not only as an early marker of kidney damage but also as an effector. In fact, NGAL also produces further inflammation and heart damage through modulation of the degradation activity of MMP9 (fig. 7). The discovery of this mechanism can open new avenues for the treatment of HF with drugs such as neprilysin, an endopeptidase inhibitor that interferes also with the cleavage of zinc-dependent metalloprotease, which in clinical trials has been shown to improve mortality and preserve heart and kidney function [30].
Disclosure Statement
The authors declare to have no conflicts of interest.
Acknowledgements
The authors thank Elisabetta Baliello, Alessandra Dubrovich and Giulia Fregonese for their skillful technical assistance. This work was supported by research grants from the Veneto Region (RF-VEN303/09) and from the University of Padua (CPDR099073, CPDA108809, 60A07-5074 and 60A07-8587, as well as GRIC13IH1H).
References
- 1.Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, Bellomo R, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, Hillege H, House AA, Katz N, Maisel A, Mankad S, Zanco P, Mebazaa A, Palazzuoli A, Ronco F, Shaw A, Sheinfeld G, Soni S, Vescovo G, Zamperetti N, Ponikowski P, Acute Dialysis Quality Initiative (ADQI) Consensus Group Cardio-renal syndromes: report from the consensus conference of the Acute Dialysis Quality Initiative. Eur Heart J. 2010;31:703–711. doi: 10.1093/eurheartj/ehp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hebert K, Dias A, Delgado MC, Franco E, Tamariz L, Steen D, Trahan P, Major B, Arcement LM. Epidemiology and survival of the five stages of chronic kidney disease in a systolic heart failure population. Eur J Heart Fail. 2010;12:861–865. doi: 10.1093/eurjhf/hfq077. [DOI] [PubMed] [Google Scholar]
- 3.Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13:422–430. doi: 10.1016/j.cardfail.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Cruz DN, Schmidt-Ott KM, Vescovo G, House AA, Kellum JA, Ronco C, McCullough PA. Pathophysiology of cardiorenal syndrome type 2 in stable chronic heart failure: workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI) Contrib Nephrol. 2013;182:117–136. doi: 10.1159/000349968. [DOI] [PubMed] [Google Scholar]
- 5.McCullough PA. Prevention of cardiorenal syndromes. Contrib Nephrol. 2010;165:101–111. doi: 10.1159/000313749. [DOI] [PubMed] [Google Scholar]
- 6.Kishimoto T, Maekawa M, Abe Y, Yamamoto K. Intrarenal distribution of blood flow and renin release during renal venous pressure elevation. Kidney Int. 1973;4:259–266. doi: 10.1038/ki.1973.112. [DOI] [PubMed] [Google Scholar]
- 7.Colombo PC, Onat D, Sabbah HN. Acute heart failure as ‘acute endothelitis’ – interaction of fluid overload and endothelial dysfunction. Eur J Heart Fail. 2008;10:170–175. doi: 10.1016/j.ejheart.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 8.Ronco C, McCullough PA, Anker SD, Anand I, Aspromonte N, Bagshaw SM, Bellomo R, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, Hillege H, House A, Katz NM, Maisel A, Mankad S, Zanco P, Mebazaa A, Palazzuoli A, Ronco F, Shaw A, Sheinfeld G, Soni S, Vescovo G, Zamperetti N, Ponikowski P. Cardiorenal syndromes: an executive summary from the consensus conference of the Acute Dialysis Quality Initiative (ADQI) Contrib Nephrol. 2010;165:54–67. doi: 10.1159/000313745. [DOI] [PubMed] [Google Scholar]
- 9.Cuoco L, Vescovo G, Castaman R, Ravara B, Cammarota G, Angelini A, Salvagnini M, Dalla Libera L. Skeletal muscle wastage in Crohn's disease: a pathway shared with heart failure? Int J Cardiol. 2008;127:219–227. doi: 10.1016/j.ijcard.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Niebauer J, Volk HD, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–1842. doi: 10.1016/S0140-6736(98)09286-1. [DOI] [PubMed] [Google Scholar]
- 11.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 12.Dalla Libera L, Ravara B, Angelini A, Rossini K, Sandri M, Thiene G, Battista Ambrosio G, Vescovo G. Beneficial effects on skeletal muscle of the angiotensin II type 1 receptor blocker irbesartan in experimental heart failure. Circulation. 2001;103:2195–2200. doi: 10.1161/01.cir.103.17.2195. [DOI] [PubMed] [Google Scholar]
- 13.Vescovo G, Ceconi C, Bernocchi P, Ferrari R, Carraro U, Ambrosio GB, Libera LD. Skeletal muscle myosin heavy chain expression in rats with monocrotaline-induced cardiac hypertrophy and failure. Relation to blood flow and degree of muscle atrophy. Cardiovasc Res. 1998;39:233–241. doi: 10.1016/s0008-6363(98)00041-8. [DOI] [PubMed] [Google Scholar]
- 14.Angelini A, Castellani C, Ravara B, Franzin C, Pozzobon M, Tavano R, Libera LD, Papini E, Vettor R, De Coppi P, Thiene G, Vescovo G. Stem-cell therapy in an experimental model of pulmonary hypertension and right heart failure: role of paracrine and neurohormonal milieu in the remodeling process. J Heart Lung Transplant. 2011;30:1281–1293. doi: 10.1016/j.healun.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Castellani C, Vescovo G, Ravara B, Franzin C, Pozzobon M, Tavano R, Gorza L, Papini E, Vettor R, De Coppi P, Thiene G, Angelini A. The contribution of stem cell therapy to skeletal muscle remodeling in heart failure. Int J Cardiol. 2013;168:2014–2021. doi: 10.1016/j.ijcard.2013.01.168. [DOI] [PubMed] [Google Scholar]
- 16.Vescovo G, Volterrani M, Zennaro R, Sandri M, Ceconi C, Lorusso R, Ferrari R, Ambrosio GB, Dalla Libera L. Apoptosis in the skeletal muscle of patients with heart failure: investigation of clinical and biochemical changes. Heart. 2000;84:431–437. doi: 10.1136/heart.84.4.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavano R, Franzoso S, Cecchini P, Cartocci E, Oriente F, Aricò B, Papini E. The membrane expression of Neisseria meningitidis adhesin A (NadA) increases the proimmune effects of MenB OMVs on human macrophages, compared with NadA- OMVs, without further stimulating their proinflammatory activity on circulating monocytes. J Leukoc Biol. 2009;86:143–153. doi: 10.1189/jlb.0109030. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Bakky MS, Hammad MA, Walker LA, Ashfaq MK. Silencing of tissue factor by antisense deoxyoligonucleotide prevents monocrotaline/LPS renal injury in mice. Arch Toxicol. 2001;85:1245–1256. doi: 10.1007/s00204-011-0663-8. [DOI] [PubMed] [Google Scholar]
- 19.Carstens LA, Allen BS, Allen JR. Arterial degeneration and glomerular hyalinization in the kidney of monocrotaline-intoxicated rats. Am J Pathol. 1970;60:75–92. [PMC free article] [PubMed] [Google Scholar]
- 20.Maisel AS, Katz N, Hillege HL, Shaw A, Zanco P, Bellomo R, Anand I, Anker SD, Aspromonte N, Bagshaw SM, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, House AA, Mankad S, McCullough P, Mebazaa A, Palazzuoli A, Ponikowski P, Ronco F, Sheinfeld G, Soni S, Vescovo G, Zamperetti N, Ronco C. Biomarkers in kidney and heart disease. Nephrol Dial Transplant. 2011;26:62–74. doi: 10.1093/ndt/gfq647. [DOI] [PubMed] [Google Scholar]
- 21.Vescovo G, Ravara B, Gobbo V, Sandri M, Angelini A, Della Barbera M, Dona M, Peluso G, Calvani M, Mosconi L, Dalla Libera L. L-Carnitine: a potential treatment for blocking apoptosis and preventing skeletal muscle myopathy in heart failure. Am J Physiol Cell Physiol. 2002;283:C802–C810. doi: 10.1152/ajpcell.00046.2002. [DOI] [PubMed] [Google Scholar]
- 22.Lekawanvijit S, Kompa AR, Zhang Y, Wang BH, Kelly DJ, Krum H. Myocardial infarction impairs renal function, induces renal interstitial fibrosis, and increases renal KIM-1 expression: implications for cardiorenal syndrome. Am J Physiol Heart Circ Physiol. 2012;302:H1884–H1893. doi: 10.1152/ajpheart.00967.2011. [DOI] [PubMed] [Google Scholar]
- 23.Lu J, Wang X, Wang W, Muniyappa H, Deshmukh A, Hu C, Das K, Mehta JL. Abrogation of lectin-like oxidized LDL receptor-1 attenuates acute myocardial ischemia-induced renal dysfunction by modulating systemic and local inflammation. Kidney Int. 2012;82:436–444. doi: 10.1038/ki.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Virzi GM, Torregrossa R, Cruz DN, et al. Cardiorenal syndrome type 1 may be immunologically mediated: a pilot evaluation of monocyte apoptosis. Cardiorenal Med. 2012;2:33–42. doi: 10.1159/000335499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AH, Suzuki F, Nagai Y, Nakano D, Hitomi H, Kitada K, Urushihara M, Kobori H, Kohno M, Nishiyama A. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation. 2012;125:1402–1413. doi: 10.1161/CIRCULATIONAHA.111.064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakraborty S, Kaur S, Guhsa S. The multifaced roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 2012;1826:129–169. doi: 10.1016/j.bbcan.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 28.Martensson J, Xu S, Bell M, Martling CR, Venge P. Immunoassays distinguishing between HNL/NGAL released in urine from kidney epithelial cells and neutrophils. Clin Chim Acta. 2012;413:1661–1667. doi: 10.1016/j.cca.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP9 and neutrophil gelatinase-associated lipocalin (NGAL) J Biol Chem. 2001;276:37258–37265. doi: 10.1074/jbc.M106089200. [DOI] [PubMed] [Google Scholar]
- 30.Jessup M, Fox KA, Komajda M, McMurray JJ, Packer M. PARADIGM-HF – the experts' discussion. N Engl J Med. 2014;371:e15. doi: 10.1056/NEJMp1410203. [DOI] [PubMed] [Google Scholar]