

Abstract
Lantibiotics are potent antimicrobial peptides. Nisin is the most prominent member and contains five crucial lanthionine rings. Some clinically relevant bacteria express membrane-associated resistance proteins that proteolytically inactivate nisin. However, substrate recognition and specificity of these proteins is unknown. Here, we report the first three-dimensional structure of a nisin resistance protein from Streptococcus agalactiae (SaNSR) at 2.2 Å resolution. It contains an N-terminal helical bundle, and protease cap and core domains. The latter harbors the highly conserved TASSAEM region, which lies in a hydrophobic tunnel formed by all domains. By integrative modeling, mutagenesis studies, and genetic engineering of nisin variants, a model of the SaNSR/nisin complex is generated, revealing that SaNSR recognizes the last C-terminally located lanthionine ring of nisin. This determines the substrate specificity of SaNSR and ensures the exact coordination of the nisin cleavage site at the TASSAEM region.
Antibiotics provide a great advantage in the treatment of infections caused by bacteria such as Streptococcus pneumoniae and Streptococcus agalactiae. However, due to their widespread use, the number of resistant bacterial strains is increasing1, leading to an urgent need for the development of new antibiotics. Several approaches have been taken to identify new antibiotics where naturally occurring compounds are found to be the most promising ones2. Here, small antimicrobial peptides such as lantibiotics are excellent candidates because they exhibit high effectivity against various Gram-positive human pathogenic bacteria including Streptococcus pneumoniae and several methicillin-resistant Staphylococcus aureus (MRSA) strains3.
Lantibiotics display antimicrobial activities in the very low nanomolar range4,5. The anti-infective potency of lantibiotics such as nisin, mutacin, mersacidin and others has been recognized, and several are in the preclinical stages of medical application6,7. After translation, lantibiotics are modified and contain unusual amino acids such as dehydroalanine (Dha) and dehydrobutyrine (Dhb), which are covalently linked to the side chain of cysteine residues forming the so-called lanthionine rings8,9. The number as well as the exact location of the lanthionine rings vary within lantibiotics10. Lantibiotics have multiple modes of action, of which binding to lipid II, thereby inhibiting cell wall synthesis, and pore formation are the most predominant ones8,11. Nisin produced by Lactococcus lactis (L. lactis) is one of more than 50 lantibiotics discovered so far12 and is considered to be the role model. Active nisin consists of 34 amino acids and contains five lanthionine-based rings (Supplementary Fig. 1). The first three rings (A-C) are separated from the other two intertwined rings (D-E) by a flexible hinge region. The first two rings are able to bind lipid II13; the hinge region and the last two intertwined rings are able to flip into the membrane and create a pore14,15,16.
Due to their multiple modes of action, hardly any resistance against lantibiotics has developed over the past decades. However, some bacterial strains have been reported to be congenitally resistant against nisin17 via various mechanisms such as cell wall modifications, biofilm formation or the expression of resistance proteins18. For the latter case, a nsr gene was identified in the Streptococcus lactis subspecies diacetylactis (DRC3) that encodes the nisin resistance protein, NSR17,19. Similar genes were identified in other species17,20,21, including several human pathogenic strains22,23. NSR is a member of the S41 protease family, specifically the C-terminal processing peptidases (CTPs). NSR from L. lactis TS1640 has been shown to degrade nisin by cleaving the peptide bond between MeLan28 in ring E and Ser29. The resulting nisin1-28 fragment displays a significantly lower bactericidal efficacy and reduced affinity towards cellular membranes24. Furthermore, the NSR protein from S. agalactiae ATCC 13813 induced a 20-fold increased resistance towards nisin when expressed in L. lactis22.
NSR is localized within an operon comprising five genes, which encode for NSR, a two-component signaling system (NsrRK), and an ABC transporter (NsrFP). When expressed together, these proteins deliver full nisin resistance22. Interestingly, similar operon structures were also found to be associated with resistance against other lantibiotics18,23. These operons resemble (auto)-immunity systems found in lantibiotic producer10 strains. Structures of SpaI from B. subtilis25 conferring resistance against subtilin and MlbQ from the actinomycete Microbispora ATCC PTA-5024 conferring resistance against NAI-10726 were resolved by NMR. However, no significant sequence identity is found between NSR and SpaI or MlbQ, suggesting a different mechanism for the defense against lantibiotics. Furthermore, most (auto)-immunity proteins do not cleave or manipulate the lantibiotic but rather shield the host’s membrane from being harmed by its lantibiotic10,27.
The ability of NSR to cleave nisin is impressive because it has been shown for several lantibiotics that they are not easily accessible for protease cleavage14. Here, the lanthionine rings are likely causing steric hindrance within the active site of proteases, thereby inhibiting proteolysis. Thus, notwithstanding the recent advances in this field, we still structurally know relatively little about lantibiotic resistance. In particular, the lantibiotic binding site in NSR and the mechanism how substrate specificity is conferred remains elusive. In this study, we report the first structure of a nisin resistance protein, NSR from S. agalactiae COH1 (SaNSR). Mutagenesis studies guided by molecular dynamics (MD) simulations reveal that SaNSR recognizes the lanthionine ring closest to the C- terminus of nisin and that this ring binds at one end of the catalytic tunnel, thereby determining the substrate specificity and ensuring the exact coordination of the nisin cleavage site at the catalytic site region.
Results
Crystal structure of SaNSR
Nisin has been shown to be quite resistant against proteolytic digestion in general14, supposedly due to the presence of lanthionine rings. Therefore, it is intriguing to understand the proteolytic resistance mechanism mediated by NSRs. To obtain a molecular view on this mechanism, we solved the structure of SaNSR by X-ray crystallography.
Through sequence analyses, it was predicted that the first 30 amino acids encode for a transmembrane helix28. We deleted this N-terminal transmembrane helix and included a His8-tag for purification purposes, resulting in soluble expression of SaNSR. After over-expression, two-step purification yielded 5 mg of pure SaNSR protein per liter of cell culture (Supplementary Fig. 2a). SaNSR is a monomer in solution as determined by multiple angle light scattering (MALS) (Supplementary Fig. 2b). SaNSR protein was crystallized and cubic crystals were obtained that diffracted up to 2.2 Å resolution29. We solved the structure by Single Anomalous Dispersion (SAD) phasing, using crystals of selenomethionine-substituted protein (data and refinement statistics are shown in Table 1).
Table 1. Data collection, phasing and refinement statistics for SaNSR.
| Native SaNSR | SeMet SaNSR | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P432 |
| Cell dimensions | ||
| a, b, c (Å) | 58.8, 137.2, 164.0 | 186.1, 186.1, 186.1 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 0.87260 | 0.97625 |
| Resolution (Å) | 100.0–2.21 (2.29–2.21) | 100.0–2.80 (2.9-2.8) |
| Rmerge | 11.5 (63.8) | 29.3 (110.5) |
| <I / σ (I)> | 8.67 (1.79) | 20.33 (1.72) |
| Completeness (%) | 99.6 (99.3) | 99.8 (98.7) |
| Redundancy | 4.5 (4.2) | 75.6 (70.5) |
| Refinement | ||
| Resolution (Å) | 55.36-2.21 (2.28-2.21) | |
| No. reflections | 303208 (27954) | |
| Rwork/Rfree | 0.19 (0.27)/ 0.24 (0.31) | |
| No. of atoms | 9588 | |
| Protein | 9017 | |
| Ligand/ion | 48 | |
| Water | 523 | |
| B-factors (Å3) | 40.5 | |
| Protein | 40.3 | |
| Ligand/ion | 68.1 | |
| Water | 41.2 | |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 1.09 | |
*Values in parentheses are for highest-resolution shell.
The asymmetric unit (Supplementary Fig. 2c) contained four copies of SaNSR that were virtually identical (root mean square deviation (RMSD) between the monomers = 0.15–0.5 Å over 300 amino acids). Therefore, the overall structure is described only for monomer A. The entire sequence of SaNSR could be fitted into the electron density, with the exception of the N-terminal His8-tag that was disordered. The Rwork and Rfree values after refinement were 0.19 and 0.24, respectively.
A SaNSR monomer (Fig. 1) consists of eleven helices (α1-α11) and eleven β-strands (βa-βk), which form three domains: an N-terminal helical bundle and two protease subdomains. Altogether, these domains form a hydrophobic tunnel of ~10 Å width (Fig. 1b), which could very well harbor the nisin molecule. The N-terminal helical bundle (Fig. 1b, represented in green) comprises 65 amino acid residues (Lys31-Gly96), which form helices α1-α3. This domain ends in a triple glycine motif (94GGG96) before entering the protease cap domain (Fig. 1b, represented in red). The protease cap domain consists of helix α4 and a β-hairpin structure formed by strands βi-j. The protease cap forms a lid-like structure above the tunnel. The third domain is the so-called protease core domain (Fig. 1b, represented in grey), which adopts a ‘protease fold’ domain as observed in other S41 peptidases30,31,32. The protease core domain is formed by six strands βb-βg and five helices α5-α9. It contains the highly conserved TASSAEM region that harbors the previously identified catalytically active serine at position 23622 (Fig. 1, represented in blue; Supplementary Fig. 3). The TASSAEM region lies in the tunnel between the two protease subdomains (Fig. 2a).
Figure 1. Structural architecture of the SaNSR monomer.
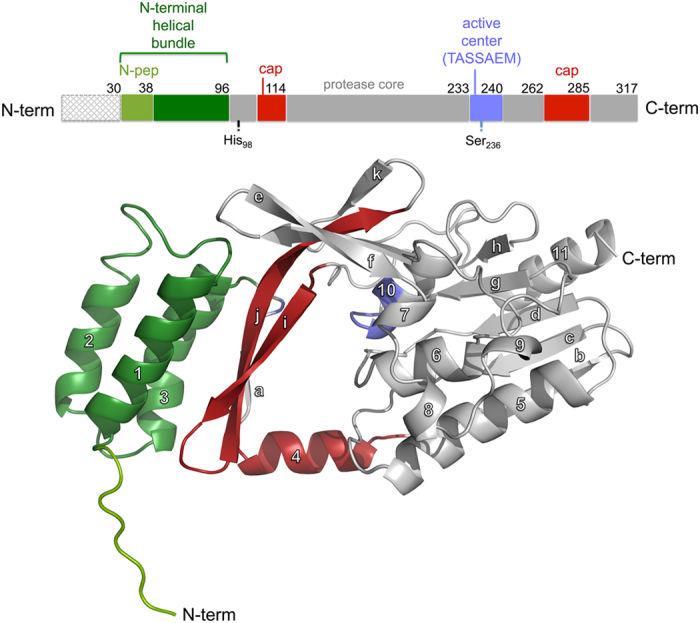
(a) Schematic illustration of the domain organization of SaNSR indicating the domain borders and catalytically important residues (His98 and Ser236). (b) The overall structure of a SaNSR monomer in a cartoon representation. The N-terminal helical bundle is depicted in green where the light green region represents the N-pep. The protease cap and core domains are highlighted in red and grey, respectively. The catalytically important residues and the highly conserved “TASSAEM” region are depicted in blue.
Figure 2. Surface representation of SaNSR.

(a) The surface representation of SaNSR in white, highlighting the tunnel localized in between the protease cap and the core domain. The TASSAEM motif is colored in blue. (b) Surface representation of SaNSR with bound N-pep (colored in orange). (c) Stereo view on the active site architecture of SaNSR highlighting the N-pep that is bound within the tunnel as ball and stick representation. The corresponding 2FoFc omit electron density map is calculated at 2.2 Å and contoured at 1.0 σ. The water mediated interactions of N-pep (colored in orange) with residues of the protease cap (depicted in red) and the direct interactions with the residues of the protease core (grey color) are shown.
N-terminal helical bundle
A comparison of the N-terminal helical bundle with all available entries in the Protein Data Bank was performed using the Dali server33. The Dali server identifies similarities in 3D structures irrespective of sequence similarities. A structurally similar helical bundle has been identified in the human Factor H (Z-score of 5.2), which is responsible for tight binding of the pneumococcal protein virulence factor CbpA (choline-binding protein A)34. Furthermore, a similar helical bundle is present at the C-terminus of the 70 kDa human heat shock protein (HSP70) (Z-score of 5.0). This region is responsible for causing a structural switch during HSP70 allosteric activation, which is important for maintaining a proper conformation of the protein for binding to the J-domain and ATPase activity purposes35. Finally, a dynamic helical region is present at the N-termini of staphylococcal complement inhibitors (SCINs) (Z-score of 4.7), which is responsible for binding to the substrate C3b and is also necessary for the formation of higher order complexes of C3b, which blocks phagocytosis36. While these findings suggest that a certain degree of mobility of the found helical bundles is required for function, in three replicates of molecular dynamics (MD) simulations of a monomer of SaNSR (termed NSRApo; see online methods for details), each of 500 ns length, the N-terminal helical bundle is rather immobile with respect to the protease core and cap domain (backbone root mean square fluctuations (RMSF) <2.5 Å; Supplementary Fig. 4a).
N-pep bound to SaNSR
In the crystal structure, the hydrophobic tunnel is filled with the N-terminal residues 31KNIYLLPP38 of a neighboring SaNSR molecule (termed N-pep; Fig. 2b; shown in light green in Fig. 1). N-pep is predominantly bound to SaNSR via direct backbone interactions to amino acids 167NNTGGN172 of β-strand βd, which is part of the protease core domain and is structurally located on the opposite site of the TASSAEM sequence motif (Fig. 2c). In addition, N-pep is stabilized via water-mediated hydrogen bonds between backbone atoms of Asn32, Tyr34, Leu35 and residues Asn265 and Thr267 of the protease cap domain (Fig. 2c). The presence of N-pep within the tunnel is clearly an induced artifact of the crystallization procedure, since in the full-length SaNSR protein, another 30 amino acids are attached at the N-terminus of the N-pep sequence that form a transmembrane helix. Yet, during MD simulations of 500 ns length of a SaNSR monomer complexed with N-pep (termed NSRTail; see online methods for details), N-pep remains stably bound within the hydrophobic tunnel (mean backbone RMSD < 1.6 Å; Supplementary Fig. 4b). The predominance of backbone interactions of N-pep with the protease core and cap domains could explain why N-pep binds into the putative binding region of nisin despite its sequence being very dissimilar to the one of nisin.
Substructures of nisin determining its molecular recognition
To investigate the substrate specificity of SaNSR and determine substructures of nisin important for its recognition by the protein, we used different nisin variants. In order to test the influence of rings D and E located next to the cleavage site of nisin, we genetically replaced the last or the last two cysteine(s) in nisin by alanine, resulting in the expression of active nisin containing only rings A-D (termed CCCCA) or A-C (termed CCCAA), respectively27. Similarly, we removed the last six amino acids of nisin (termed nisin1-28), resulting in the product of the proteolysis reaction mediated by SaNSR. Furthermore, a truncated variant (nisin1-22) was expressed, which contained the rings A-C but lacked the rest of the C-terminus of nisin27. Since all variants show a different activity against the nisin sensitive L. lactis NZ9000Erm strain (Supplementary Table 1a), we analyzed them with respect to the fold of resistance mediated by the expression of the SaNSR protein in the NZ9000SaNSR strain. From current and previous work, it is known that SaNSR confers a 20-fold increased resistance against wildtype nisin (Fig. 3a)22.
Figure 3. Influence of wild type SaNSR and its mutations against nisin and its variants.

(a) Growth inhibition experiment of SaNSR with nisin. The activity of SaNSR is determined using the L. lactis NZ9000 strain, where the plasmid encoding the SaNSR wildtype and the mutations were transformed, and the IC50 against nisin was determined. As a control, the empty vector was transformed and used in the IC50 study (termed NZ9000Erm). Black lines represent the NZ9000Erm (filled Δ) and NZ9000-SaNSR (♦) strains, respectively. The black dotted lines represent the NZ9000-SaNSR-His98Ala (☐) and NZ9000-SaNSR-Ser236Ala (Ο) strains. The data were fitted and evaluated as described in ref. 51. The difference in the growth exhibited by the strains was used to calculate the percentage of activity. Each experiment was performed at least in triplicates. (b) Graphical representation of the fold of resistance exhibited by SaNSR with nisin and different nisin variants (CCCCA, CCCAA, nisin1-22 and nisin1-28). The NZ9000Erm and NZ9000SaNSR strains were used to determine the activity of all the nisin variants. The error bars indicate the standard error of at least three independent experiments. (c) The activity of SaNSR and its mutations is determined using the L. lactis NZ9000 strain. A normalization of the IC50 values were done by setting the values exhibited by the empty vector (NZ9000Erm) and NZ9000SaNSR to 0% and 100%, respectively. The error bars indicate the standard error of at least three independent experiments.
For CCCCA as well as CCCAA, the resistance mediated by SaNSR decreased to roughly 1.4–1.7 fold when comparing the IC50 values of the different strains (Fig. 3b; Supplementary Table 1a). For the truncated variants, nisin1-22 and nisin1-28, no resistance was observed anymore as the IC50 values dropped to the levels observed for the NZ9000Erm strain. Thus, the lanthionine ring E is clearly important for the recognition by SaNSR.
Structural model of nisin binding to SaNSR
Despite intensive trials, we were not successful in obtaining a crystal structure of a SaNSR/nisin complex. Thus, we resorted to generating a structural model by integrative modeling and validating it by mutagenesis studies. Initially, we structurally aligned the backbone of residues 31–36 of nisin to the backbone of N-pep such that the nisin cleavage site between ring E and Ser29 was oriented towards the catalytically active Ser236 in SaNSR. Rings D and E were then manually placed in three orientations at the tunnel entrances such that they showed good complementarity with the SaNSR surface. This resulted in three structural models of SaNSR/nisin complexes, two (termed NSRNisin,1, NSRNisin,2) where rings D and E are located close to Asn172, Met173, and Ile174, and one (termed NSRNisin,3) where nisin is oriented oppositely with respect to the tunnel axis such that Tyr261 stacks onto rings D and E (Supplementary Fig. 5a). The three models were subjected to MD simulations37 of 500 ns length, with three replicate simulations each.
The average distances between the side chain oxygen of Ser236, previously identified as the catalytically active serine22, and the carbonyl carbon of Ser29 at the nisin cleavage site24 are 3.71 Å, 4.13 Å, and 7.74 Å for NSRNisin,1, NSRNisin,2 and NSRNisin,3, respectively (Fig. 4a). This strongly indicates that a nucleophilic attack of the side chain of Ser236 at the nisin cleavage site as a first step in the catalytic mechanism24 is possible for the first two models but not for the third, suggesting that NSRNisin,1 and NSRNisin,2 represent the most likely orientation of nisin within the SaNSR tunnel. Thus, we focused further analyses on the NSRNisin,1 and NSRNisin,2 models.
Figure 4. Structural and energetic analysis of MD simulations of SaNSR/nisin model complexes.

(a) Distance between the side chain oxygen of Ser236 and the carbonyl carbon of ring E at the nisin cleavage site (black dotted line in the upper right panel) in NSRNisin, {1, 2, 3} during 500 ns of MD simulations; lines were smoothed by cubic splines. Mean values and standard error of the mean (SEM; in parentheses) are shown in the legend. The mean distance over all three MD simulations is shown in the lower right panel (SEM < 0.1 Å and not shown). (b) Mean backbone RMSF (SEM indicated as error bars) for NSRNisin, {1, 2} models over three trajectories each of 500 ns length. Rings D and E, Ser29, and Ile30 compose the NisinCore. (c) Mean effective binding energy per residue for NSRNisin,{1, 2} models. Error bars indicate SEM over three trajectories. (d) Superimposition of six close-to-average structures (based on the backbone RMSD) of nisin (ball-and-stick models each colored differently), extracted from three independent MD simulations each of NSRNisin,1 and NSRNisin,2, within the tunnel of SaNSR (white surface representation). Ser236 of the catalytic dyad is colored in blue. For clarity, the N-terminal helical bundle and part of the cap region of SaNSR have been omitted. (e) Representative nisin model (orange and green ball-and-stick model) within the tunnel of SaNSR (white cartoon representation with transparent surface). Residues Asn172, Met173, and Ile174 that bind to rings D and E are colored in magenta.
Visual inspection of the MD trajectories and computations of the backbone RMSF identified residues Lys22, His31, Val32, Dha33, and Lys34 of nisin as highly mobile (RMSF values up to 6.39 Å ± 0.49 Å) (Fig. 4b). In contrast, the core region (NisinCore) composed of the rings D and E, and residues Ser29 and Ile30 revealed RMSF values <1.85 Å ± 0.24 Å (Fig. 4b) suggesting a tightly bound NisinCore region. This was corroborated by a per-residue decomposition of effective binding energies computed by the MM-PBSA approach38. Here, rings D and E (treated as one residue in the energy decomposition) and Ile30 are identified as essential for nisin binding (residue-wise effective binding energies in the range from −4.26 kcal mol−1 to -8.63 kcal mol−1) (Fig. 4c). In contrast, for Ser29, a smaller contribution to the effective binding energy of -1.64 kcal mol−1 (−0.70 kcal mol−1) for SaNSRNisin,1 (SaNSRNisin,2) was found (Fig. 4c). Overall, this suggests that the rings D and E as well as Ile30 form a binding motive, that way ensuring that also Ser29 at the nisin cleavage site is correctly positioned within the catalytic site.
In Figure 4d, a representative set of six nisin structures within the SaNSR tunnel is shown. For this, the structure with the smallest backbone RMSD to the average structure was extracted from each of the NSRNisin,1 and NSRNisin,2 MD trajectories. The set shows that the location and orientation of rings D and E, Ser29, and Ile30 agree well in all cases, with RMSD values with respect to the average structure for the NisinCore ranging from 0.80 Å to 2.27 Å (Supplementary Fig. 5b). Thus, both NSRNisin,1 and NSRNisin,2 models were considered equivalent and used to identify residues in SaNSR important for catalysis and nisin binding for mutagenesis studies. The remaining residues of nisin show large structural deviations, in agreement with the above analyses (Supplementary Fig. 5b, Fig. 4b,c).
The TASSAEM region and His98 form the active site
The NSR superfamily contains a highly conserved sequence motif “TASSAEM” (Supplementary Fig. 3) located at the rear end of the protease core domain. Within this TASSAEM region, Ser236 has been previously identified as the catalytically active serine22. This serine is in close proximity to the strictly conserved His98 residue, which is localized at the end of the N-terminal helical bundle directly next to the 94GGG96 motif (Fig. 1) and is in hydrogen bonds distance with the side chain of Ser236. In the NSRNisin,1 and NSRNisin,2 MD simulations, hydrogen bonds were found in up to ~23% of all conformations (Supplementary Fig. 6a), which indicates that both residues likely interact also in the nisin-bound state. Based on the interactions of Ser236 and His98 and the absence of any other lysine or aspartate residue localized nearby, we presume that SaNSR acts via a catalytic dyad mechanism as observed for some other serine proteases39,40. The NZ9000SaNSR-Ser236Ala strain displayed a low background activity as observed by an IC50 value of 12.6 ± 0.7 nM (Supplementary Table 1b), which relates to a SaNSR residual activity of 14% (Fig. 3c). The His98Ala mutation displayed a similar IC50 value of 12.3 ± 1.5 nM and a residual activity of 14% (Fig. 3c, Supplementary Table 1a). The residual activity displayed by both variants is likely due to the binding of nisin to that particular SaNSR variant such that a higher concentration of nisin is required to kill the corresponding nisin sensitive NZ9000Erm L. lactis strain.
Within the TASSAEM sequence, a second serine residue, Ser237, is present. In the NSRNisin,1 and NSRNisin,2 MD simulations, the mean distance between the side chain oxygen and the carbonyl carbon of ring E is <5.7 Å (Supplementary Fig. 6b). However, the distance to the δ-nitrogen of His98 is >9 Å (Supplementary Fig. 6c), and no hydrogen bonds were detected between both residues, making a proton shift between Ser237 and His98 unlikely. Instead, we observed hydrogen bond formation between the side chain of Ser237 and the backbone of Gly171 of the protease core in at least 46% of the conformations that may be relevant for nisin recognition (see section “Residues involved in nisin recognition and SaNSR specificity”) but not for catalytic activity. Thus, Ser237 is not expected to be involved in the catalytic mechanism. In accordance, a Ser237Ala mutation does not have a pronounced effect on the activity of SaNSR (residual activity 74%; see Fig. 3c, Supplementary Table 1b).
The next residue in the TASSAEM motif is Glu239, which is pointing away from the active site. In the crystal structure, the Glu239 side chain interacts with backbone atoms of Gly260 and Tyr261 via hydrogen bonds, and during the NSRNisin,1 and NSRNisin,2 MD simulations this interaction is present in at least 82% of all conformations (Supplementary Fig. 6a). Additionally, we found hydrogen bond interactions between Glu239 and Ser236 in at least 25% of the cases (Supplementary Fig. 6a). These interactions are likely important for the correct positioning of the TASSAEM region. This is in line with the drastically lowered activity of the Glu239Ala mutant (IC50 value of 17.1 ± 0.7 nM; residual activity of 22%; Fig. 3c, Supplementary Table 1b). Furthermore, we found stabilizing hydrogen bonds between Thr263 and His98 in up to ~28%, and between Asn265 and His98 in up to ~20% of all cases (Supplementary Fig. 6a). These interactions likely ensure a correct orientation of His98 as the mutations Thr263Ala and Asn265Ala decreased the residual activities of SaNSR to 20% and 30%, respectively, (Fig. 3c) with associated IC50 values of 16.0 ± 0.3 and 22.1 ± 1.1 nM (Supplementary Table 1b). Taken together, the TASSAEM sequence is crucial for the activity of SaNSR and contains the catalytically active serine as well as a glutamic acid residue, which is likely responsible for a correct positioning of the TASSAEM helix.
Residues involved in nisin recognition and SaNSR specificity
Next, we investigated nisin recognition by SaNSR. Residue-wise effective binding energies were computed for both the NSRNisin,1 and NSRNisin,2 MD trajectories to identify SaNSR residues likely to be important for nisin binding (Supplementary Fig. 7a). Considering energies < −0.8 kcal mol−1 resulted in seven candidates (Leu102, Leu137, Asn172, Met173, Ile174, Glu266, Ala277). Our model (Fig. 4d) suggests that the hydrophobic residues Leu102, Leu137, Met173, Ile174, Ala277 and the polar/charged ones Asn172 and Glu266 bind to rings D and E in nisin. Asn172, Met173, and Ile174 form a pocket that harbors rings D and E in our model (Fig. 4e). The Asn172Ala mutant displayed an activity of 47% (IC50 value of 33.5 ± 2.9 nM) (Fig. 3c, Supplementary Table 1b). Furthermore, when mutating the strictly conserved Met173 residue (Supplementary Fig. 3), a reduced activity of 42% compared to the wild type value was observed (IC50 value of 30.3 ± 1.4 nM). Additionally, the Ile174Ala mutant exhibited an activity of 33% (IC50 value of 24.1 ± 2.2 nM) (Fig. 3c, Supplementary Table 1b).
Moreover, we found hydrogen bonds between backbone atoms of Thr169 and Gly171 with the NisinCore residues (Supplementary Fig. 7b). These interactions are reminiscent to those found for N-pep (Fig. 2c) and likely ensure a proper placement of the NisinCore within the binding site. Additional stabilizing hydrogen bonds were observed between Asn168 and Gly170 (Supplementary Fig. 7b), which could contribute to nisin binding indirectly. A similar indirect effect was found for Glu266, for which we observed salt-bridge formation with Arg54 from the N-terminal helical bundle (Supplementary Fig. 7c; mean distance <3.4 Å). We also found water-mediated hydrogen bonds between backbone atoms of rings D and E, and Asn265 and Thr267, respectively (Supplementary Fig. 7d), again mimicking what was observed for the bound N-pep (Fig. 2c). Accordingly, the mutations Asn265Ala (see above) and Thr267Ala decreased the residual activity of SaNSR to 30% and 71%, respectively, (Fig. 3c) with associated IC50 values of 22.1 ± 1.1 and 48.5 ± 0.6 nM (Supplementary Table 1b).
Role of the protease cap domain in SaNSR
Other S41 peptidases also contain a protease cap domain comprising a helix and a β-hairpin structure, where the helix appears to open and close depending on the presence of the peptide substrate: once a peptide is bound, the cap closes and seals the active site. As such, the protease CtpB from Bacillus subtilis has been crystallized in an open and closed state with the helix of the protease cap moving by 10–15 Å towards the active site once the peptide was bound30. In SaNSR, helix α4 (103SKETVRRDTLDS114) was identified as the protease cap helix, localized directly after the N-terminal helical bundle. Out of all residues of this helix, only the side chain of Asp110 is intruding into the tunnel, which neither forms an interaction to N-pep in the crystal structure nor in the NSRTail MD simulations. This suggests that the protease cap is not adopting a fully closed state, rather an intermediate state. MD simulations show a salt-bridge formation between Asp110 and Arg275 of the protease cap domain for both NSRNisin1,2 models (Supplementary Fig. 8a). In those cases where the salt-bridge formation is weak (mean distance is > 10 Å), a loss of the secondary structures of helix α4 is observed (Supplementary Fig. 8b). The Asp110Ala mutant of SaNSR is still active although with a lower IC50 value of 32.8 ± 2.1 nM (residual activity of 46%; Fig. 3c, Supplementary Table 1b). The Arg275Ala mutant revealed an identical IC50 value of 33.6 ± 2.3 nM (residual activity of 48%). Taken together, this suggests that a proper secondary structure of helix α4 is required for SaNSR function, and that Asp110 contributes to the stability of the secondary structure.
Discussion
The present study reveals that the lanthionine ring E of nisin determines substrate specificity of the nisin resistance protein (NSR) and contributes to the coordination of the nisin cleavage site at the catalytic center. These results are based on the first structure of a nisin resistance protein from S. agalactiae COH1 (SaNSR) at 2.2 Å resolution and subsequent integrative modeling and mutagenesis studies. The SaNSR structure consists of an N-terminal helical bundle, a protease cap domain, and a protease core domain (Fig. 1). The core domain harbors the highly conserved TASSAEM motif, which contains the catalytically important Ser236 residue, in a hydrophobic tunnel formed by all three domains. In this tunnel, an N-terminal peptide from another SaNSR protomer (N-pep) in the asymmetric unit is bound predominantly by direct and water-mediated backbone hydrogen bonds (Fig. 2). A very similar binding pattern is found for the C-terminal lanthionine rings D and E, and residues Ser29, and Ile30 of nisin in our model of the SaNSR/nisin complex (Fig. 5a,b; Supplementary Fig. 7d). According to this model, lanthionine ring E binds at one end of the hydrophobic tunnel (Fig. 5a,b) and ensures the exact coordination of the nisin cleavage site at the highly conserved TASSAEM region (Fig. 5a,b).
Figure 5. Nisin/SaNSR binding model.

(a) Representative structure of nisin (residues 22-34; extracted from the NSRNisin,1 model) bound to the crystal structure of SaNSR (cartoon representation with transparent surface; each domain is colored differently). Orange spheres with one-letter/three letter amino acid code indicate nisin residues 1–21 not considered for modeling studies (abbreviations: abu = aminobutyric acid; dha = dehydroalanine; dhb = dehydrobutyrine; ala-S-X = lanthionine derivatives). (b) Close up view of nisin binding to SaNSR residues important for nisin recognition (left), residues with catalytic function (middle), and residues with a regulatory function (right). Amino acids of interest are depicted as ball-and-stick model; residues for which experimental data is reported in this study are, additionally, shown in transparent surface representation. (c) Schematic representation of the Nisincore bound to SaNSR residues (residue numbers according to the crystal structure described here). Residues that compose the catalytic site are colored in blue, residues that contribute to nisin binding in magenta, residues that have an indirect effect on binding in black-magenta, and residues with a supposedly regulatory function in SaNSR in red. For residues with colored background, SaNSR activity information for alanine mutants is available (see Fig. 3c). Residues marked with a star form the catalytic dyad. In panels a, b, and c, the nisin structure is depicted as orange ball-and-stick model.
In contrast to some other C-terminal processing proteases30,32, the active center of SaNSR consists of a catalytic dyad formed by residues Ser23622, which is part of the TASSAEM motif, and His98 as determined by mutational analysis and also described for some other proteases41 (Fig. 5a–c). Mutational analysis and geometric parameters in the crystal structure and during MD simulations exclude that the neighboring Ser237 participates in the catalytic step. Residues Glu239, Gly260, Tyr261 and Thr263, form hydrogen bonds with either Ser236 or His98 during all-atom MD simulations of the SaNSR/nisin complexes (Supplementary Fig. 6a, Figure 5a–c) and, thus, likely stabilize the catalytic residues, as also indicated by alanine mutations of these residues that lead to a decrease in SaNSR activity (Fig. 3c).
Since all our efforts to obtain crystals of SaNSR with bound nisin were unsuccessful, we generated a model (Fig. 5a–c) of the SaNSR/nisin complex by integrative modeling and subsequent site-directed mutagenesis studies and activity measurements for validation. The modeling step was guided by exploiting the knowledge on the location of N-pep in the SaNSR crystal structure as well as on the substructures of nisin determining its molecular recognition. As to the latter, we focused on the C-terminus of nisin (nisin22-34) where NSR from L. lactis TS1640 has been shown to cleave24. As a result, nisin variants in which the bulky lanthionine rings D and E, or only E, were replaced by a linear sequence (CCCCA, CCCAA) showed a large drop in the fold of resistance comparable to those exhibited when the last 12 or 6 residues of nisin (nisin1-22, nisin1-28) were missing (Fig. 3b). These results demonstrated that ring E is essential for nisin recognition by SaNSR.
Initial models of SaNSR/nisin complexes were generated in which the linear, C-terminal sequence (sequence Lys22 - Lys34) were placed at the location of the backbone trace of N-pep and where rings D and E showed a good complementarity with the SaNSR surface at the tunnel entrance. We considered that no a priori knowledge on the direction of nisin with respect to the tunnel axis was available by generating models with both possible directions. By subsequent all-atom MD simulations, we could exclude one of the possibilities (NSRNisin,3) as in this case the distance between Ser236 and the nisin cleavage site was too large as to allow for a nucleophilic attack of the serine side chain (Fig. 4a). In contrast, for the other direction (NSRNisin,1, NSRNisin,2), such an attack is very likely according to distances that are only slightly larger than the sum of van der Waals radii of oxygen and carbon. This model of a SaNSR/nisin complex is further supported by rather immobile residues of the core region of nisin (rings D and E, Ser29 and Ile30), which is considered to facilitate a nucleophilic attack, in contrast to the more mobile C-terminal residues 31-34 (Fig. 4b), and by a residue-wise decomposition of the effective binding energy, which identified rings D and E as well as Ile30 as major contributors to the binding affinity (Fig. 4c).
The model (Fig. 5a–c) reveals that SaNSR binding to rings D and E of nisin is dominated by hydrophobic interactions (Fig. 5b,c). Within the protease core Asn172, Met173, and Ile174 form a pocket that harbors both rings D and E (Figs 4e,5b). In agreement with this model, mutation of these residues reduces the activity of SaNSR. Furthermore, water-mediated hydrogen bonds between backbone atoms of rings D and E and side chains of Asn265 and Thr267, respectively, were identified, mimicking interactions with N-pep. Asn265Ala and Thr267Ala mutations decreased the residual activity of SaNSR (Fig. 3c). Finally, along the tunnel, hydrogen bonds between backbone atoms of Thr169 and Gly171 of SaNSR with Ser29 and Ile30 of nisin were found (Fig. 5b,c; Supplementary Fig. 7b), which likely contribute towards the correct orientation of the nisin cleavage site at the catalytic center and are again reminiscent of interactions observed for N-pep in the crystal structure.
N-pep and the C-terminus of nisin are not similar on the amino acid level. Together with the above findings of similar interactions along the tunnel between backbone atoms of SaNSR and the two peptides, respectively, this suggests that the tunnel’s role in peptide binding is not to confer substrate specificity but rather to “rope in” the peptide while establishing these interactions. In the case of nisin, this “roping in” is stopped when the lanthionine ring E starts interacting with SaNSR, thereby acting as a plug on the tunnel (Fig. 5c). These interactions are highly relevant for the molecular recognition of nisin and the substrate specificity of SaNSR, as shown by a decrease in the fold of resistance for the nisin variants CCCCA and CCCAA (Fig. 3b) and a decrease in the activity of SaNSR mutants Asn172Ala, Met173Ala, and Ile174Ala (Fig. 3c). In addition, rings D and E are highly likely relevant for a proper placement of the nisin cleavage site with respect to the catalytic Ser236, as only with nisin a distance to this residue compatible with a nucleophilic attack and, simultaneously, hydrogen bonds with His98 are found in the MD simulations. In contrast, during MD simulations of NSRTail, no hydrogen bond formation between Ser236 and His98 was detected. This may explain why N-pep binds to SaNSR but is not cleaved.
Previously, an “inhibiting role” of lanthionine rings has been recognized in that they protect lantibiotics from degradation by standard proteases42, likely because of their bulky 3D structure which prevents a proper placement in the substrate binding regions of proteases evolved to cleave linear peptides. In turn, the findings in this study for the first time reveal a significant “fostering role” of the lanthionine rings D and E in nisin for the highly specific cleavage of this lantibiotic by SaNSR. These findings and our structural model of the SaNSR/nisin complex open up a new avenue in the understanding of lantibiotic resistance by human pathogens. They may also facilitate the development of therapeutics to overcome nisin resistance.
Methods
Cloning, expression and purification of SaNSR
The nsr gene from Streptococcus agalactiae COH1 was cloned into pET28b and purified as previously described. For details see Supplementary Information.
Multiple angle light scattering
For HPLC-MALS analysis, a Bio SEC-5 HPLC column (Agilent Technologies Deutschland GmbH, Böblingen, Germany) with a pore size of 300 Å was equilibrated with 25 mM MES pH 6.0, 150 mM NaCl for HPLC using a system from Agilent Technologies connected to a triple-angle light-scattering detector (miniDAWN TREOS, Wyatt Technology Europe GmbH, Dernbach, Germany) followed by a differential refractive index detector (OPTILab T-rEX, Wyatt Technology). Typically, 100 μl of purified SaNSR (2.0 mg/ml) was loaded onto the Bio SEC-5 HPLC column, and the obtained data were analyzed with the ASTRA software package (Wyatt Technology).
Crystallization, data collection and structure determination of SaNSR
Crystals were obtained and optimized as described in the Supplementary Information. X-ray diffraction data were collected at the ID23eh2 or ID29 beamlines of the European Synchrotron Radiation Facility (ESRF), Grenoble. All the data sets were processed and scaled using XDS and XSCALE software package43. Data sets from native crystals were collected at a wavelength of 0.872 Å at 100 K. For selenomethionine-substituted crystals, the ID29 beamline (ESRF Synchrotron, Grenoble)44 was used for anomalous diffraction data collection, done at 100 K. The structure was solved by single-wavelength anomalous dispersion (SAD) from a single selenomethionine derivative crystal measured at 0.976 Å, which diffracted up to 2.7 Å. The Auto-Rickshaw program45 was then used to phase the protein and build an initial model, which was further manually build and refined using COOT46 and phenix.refine from the Phenix package47. This model was then used to phase the native data set at a resolution of 2.2 Å. After molecular replacement, automatic model building was performed with the program ARP/wARP48, followed by manual iterative cycles of model refinement using the program phenix.refine47. Manual adjustments between the refinement cycles were done with the program Coot46 and Ramachandran validation was done using MolProbity49. Almost all residues (96.3%) were in the preferred regions of the Ramachandran plot, and the remaining 3.7% were in the additionally allowed regions. The data collection and refinement statistics are listed in Table 1. The images of the models were prepared using MacPyMOL50.
IC50 determination of nisin and its variants
Cells from the different expressing strains were grown overnight in GM17 media supplemented with 5 μgml−1 erythromycin in the presence of 1 ngml−1 nisin. The diluted cells (final OD600 of 0.1) were incubated with a serial dilution of nisin or its variants in a 96-well plate. The total volume in each well was 200 μl, consisting of 50 μl nisin or its variants and 150 μl GM17 containing the corresponding L. lactis strain. The plate was then incubated at 30 °C and after 5 hours, the optical density was measured at 600 nm via 96-well plate reader BMG. The IC50 value was determined as previously described51.
Molecular dynamics simulations
In order to investigate nisin recognition by SaNSR we performed molecular dynamics (MD) simulations of an unbound SaNSR monomer (NSRApo), a SaNSR monomer bound to the N-terminal part of SaNSR (residues 31–36; in the following named “Tail”) from an adjacent subunit (NSRTail) in the crystal structure (see Fig. 1), and a SaNSR monomer bound to the C-terminal part (residues 22–34; Supplementary Fig. 2c) of nisin (NSRNisin). Initial coordinates for NSRApo and NSRTail were taken from the crystal structure described here. Since no structural information is available for nisin bound to SaNSR, we generated models as starting structures for MD simulations by structurally aligning the nisin part to the Tail using the program Moloc. The nisin cleavage site between ring E and Ser29 was oriented towards the catalytically active Ser236 in SaNSR22. Rings D and E were manually placed in three orientations within the binding site such that they showed good complementarity with the SaNSR surface, resulting in three different models of SaNSR/nisin complexes (NSRNisin,1, NSRNisin,2, and NSRNisin,3, Supplementary Fig. 4a).
For the MD simulations, structures of NSRApo, NSRTail, and NSRNisin,1-3 were prepared, relaxed, and thermalized as described in detail in the Supporting Information. Three independent production runs of MD simulations of 500 ns length in the canonical (NVT) ensemble at 300 K were then conducted for each of the five systems, leading to a total simulation time of 5 × 3 × 500 ns = 7.5 μs; see Supporting Information for details.
The trajectories were analyzed with respect to distances, root mean square fluctuations (RMSF) and deviations (RMSD) as a measure for mobility and structural similarity, respectively, and hydrogen bonds defined by a distance between the two donor and acceptor atoms <3.2 Å and an angle (donor atom, H, acceptor atom) between 120° and 180° using cpptraj52. Salt-bridge interactions are defined by a distance <4.0 Å between the center of mass of both charged groups. The set of structural models binding to SaNSR (see section “Structural model of nisin binding to SaNSR) was generated by structurally aligning SaNSR and subsequent RMSD calculations for the nisin peptide.
Calculation of the effective binding energy
In order to identify amino acids in SaNSR that contribute most to nisin binding, we computed the residue-wise contribution to binding effective energies by the “single trajectory” molecular mechanics Poisson-Boltzman area (MM-PBSA) approach53,54,55. To determine the per-residue contribution, the decomposition scheme56 as implemented in the mm_pbsa.pl script in Amber 1437 was applied. The calculations were performed with the ff99SB force field57,58. The polar part of the solvation free energy was determined by applying the PBSA solver using a dielectric constant of 1 (solute) and 80 (solvent) together with Parse radii59. The conformational ensemble consists of 10,000 snapshots and was extracted from the 1–200 ns interval of each of the NSRNisin,1-2 trajectories. Prior to the MM-PBSA computations, counter ions and water molecules were stripped from the snapshots. For the computations, we considered the SaNSR protein the receptor, whereas the nisin C-terminus was considered the ligand. All residues in SaNSR and nisin were considered for per-residue decomposition. Rings D and E in nisin were treated as one residue.
PDB Deposition
The final model has been deposited in the PDB database under the accession code: 4Y68.
Additional Information
How to cite this article: Khosa, S. et al. Structural basis of lantibiotic recognition by the nisin resistance protein from Streptococcus agalactiae. Sci. Rep. 6, 18679; doi: 10.1038/srep18679 (2016).
Supplementary Material
Acknowledgments
We thank Lutz Schmitt for fruitful discussions, encouragement, support and invaluable advice. We are grateful to Philipp Ellinger for initiating the project, André Abts for stimulating discussions, Michael Lenders and Iris Fey for technical assistance. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities and are grateful to the staff of ESRF ID23-2 and ID29 for support during crystal screening and data collection, especially Christoph Mueller-Dieckmann for his enormous patience, assistance and support. We are thankful to Heinrich Heine International Graduate School of Protein Science and Technology (iGRASPseed) for providing a scholarship to S.K and to the International NRW Research School BioStruct, granted by the Ministry of Innovation, Science and Research of the State North Rhine-Westphalia, the Heinrich Heine University Düsseldorf, and the Entrepreneur Foundation at the Heinrich Heine University Düsseldorf for a scholarship to B.F. We are also grateful to the “Zentrum für Informations- und Medientechnologie” (ZIM) at the Heinrich-Heine-University Düsseldorf for providing computational support.
Footnotes
Author Contributions S.K., A.H., D.K. performed the biochemical and structural experiments. B.F., D.M. performed molecular modeling and M.D. simulations. S.K., B.F., H.G., S.S. designed the experiments, evaluated the data, and wrote the manuscript. All authors reviewed the manuscript.
References
- Levy S. B. Antibiotic Resistance: Consequences of Inaction. Clin. Infect. Dis. 33, S124–S129 (2001). [DOI] [PubMed] [Google Scholar]
- Ling L. L. et al. A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl H.-G. & Bierbaum G. Lantibiotics: biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 52, 41–79 (1998). [DOI] [PubMed] [Google Scholar]
- Willey J. M. & van der Donk W. A. Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501 (2007). [DOI] [PubMed] [Google Scholar]
- Breukink E. et al. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Sci 286, 2361–2364 (1999). [DOI] [PubMed] [Google Scholar]
- Breukink E. & de Kruijff B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 5, 321–323 (2006). [DOI] [PubMed] [Google Scholar]
- Hancock R. E. & Sahl H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 (2006). [DOI] [PubMed] [Google Scholar]
- Bierbaum G. & Sahl H. G. Lantibiotics: Mode of Action, Biosynthesis and Bioengineering. Curr. Pharm. Biotechnol. 10, 2–18 (2009). [DOI] [PubMed] [Google Scholar]
- Xie L. & Van Der Donk W. A. Post-translational modifications during lantibiotic biosynthesis. Curr. Opin. Chem. Biol. 8, 498–507 (2004). [DOI] [PubMed] [Google Scholar]
- Chatterjee C., Paul M., Xie L. L. & van der Donk W. A. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 105, 633–683 (2005). [DOI] [PubMed] [Google Scholar]
- Héchard Y. & Sahl H.-G. Mode of action of modified and unmodified bacteriocins from Gram-positive bacteria. Biochimie 84, 545–557 (2002). [DOI] [PubMed] [Google Scholar]
- Dischinger J., Basi Chipalu S. & Bierbaum G. Lantibiotics: promising candidates for future applications in health care. Int. J. Med. Microbiol. 304, 51–62 (2014). [DOI] [PubMed] [Google Scholar]
- Hsu S. T. et al. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol. 11, 963–7 (2004). [DOI] [PubMed] [Google Scholar]
- Wiedemann I. et al. Specific Binding of Nisin to the Peptidoglycan Precursor Lipid II Combines Pore Formation and Inhibition of Cell Wall Biosynthesis for Potent Antibiotic Activity. J. Biol. Chem. 276, 1772–1779 (2001). [DOI] [PubMed] [Google Scholar]
- van Heusden H. E., de Kruijff B. & Breukink E. Lipid II induces a transmembrane orientation of the pore-forming peptide lantibiotic nisin. Biochemistry 41, 12171–12178 (2002). [DOI] [PubMed] [Google Scholar]
- Hasper H. E., de Kruijff B. & Breukink E. Assembly and stability of nisin-lipid II pores. Biochemistry 43, 11567–75 (2004). [DOI] [PubMed] [Google Scholar]
- Froseth B. R. & Mckay L. L. Molecular characterization of the nisin resistance region of Lactococcus lactis subsp. lactis biovar diacetylactis DRC3. Appl. Environ. Microbiol. 57, 804–811 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper L. A., Cotter P. D., Hill C. & Ross R. P. Lantibiotic Resistance. Microbiol. Mol. Biol. Rev. 79, 171–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay L. L. & Baldwin K. A. Conjugative 40-megadalton plasmid in Streptococcus lactis subsp. diacetylactis DRC3 is associated with resistance to nisin and bacteriophage. Appl. .Environ. Microbiol. 47, 68–74 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan K., Harvey M., Liu C. Q. & Dunn N. Identification and characterization of a mobilizing plasmid, pND300, in Lactococcus lactis M189 and its encoded nisin resistance determinant. J. Appl. Bacteriol. 81, 493–500 (1996). [DOI] [PubMed] [Google Scholar]
- Klaenhammer T. R. & Sanozky R. B. Conjugal transfer from Streptococcus lactis ME2 of plasmids encoding phage resistance, nisin resistance and lactose-fermenting ability: evidence for a high-frequency conjugative plasmid responsible for abortive infection of virulent bacteriophage. J. Gen. Microbiol. 131, 1531–1541 (1985). [DOI] [PubMed] [Google Scholar]
- Khosa S., Alkhatib Z. & Smits S. H. NSR from Streptococcus agalactiae confers resistance against nisin and is encoded by a conserved nsr operon. Biol. Chem. 394, 1543–1549 (2013). [DOI] [PubMed] [Google Scholar]
- Kawada-Matsuo M. et al. Three distinct two-component systems are involved in resistance to the class I bacteriocins, nukacin ISK-1 and nisin A, in Staphylococcus aureus. PLoS One 8, e69455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z. et al. Novel mechanism for nisin resistance via proteolytic degradation of nisin by the nisin resistance protein NSR. Antimicrob. Agents Chemother. 53, 1964–1973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ N. A. et al. The First structure of a lantibiotic immunity protein, SpaI from Bacillus subtilis, reveals a novel fold. J. Biol. Chem. 287, 35286–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi R. et al. Distinct mechanisms contribute to immunity in the lantibiotic NAI-107 producer strain Microbispora ATCC PTA-5024. Environ. Microbiol. (2015). [DOI] [PubMed] [Google Scholar]
- Alkhatib Z. et al. The C-terminus of nisin is important for the ABC transporter NisFEG to confer immunity in Lactococcus lactis. MicrobiologyOpen 3, 752–763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernsel A., Viklund H., Hennerdal A. & Elofsson A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. gkp363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosa S., Hoeppner A., Kleinschrodt D. & Smits S. Overexpression, purification, crystallization and preliminary X-ray diffraction of the nisin resistance protein from Streptococcus agalactiae. Acta Crystallogr. F Biol. Crystallogr. 71, 671–675 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastny M. et al. CtpB assembles a gated protease tunnel regulating cell-cell signaling during spore formation in Bacillus subtilis. Cell 155, 647–658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-S. et al. Navigation inside a protease: substrate selection and product exit in the tricorn protease from Thermoplasma acidophilum. J. Mol. Biol. 324, 1041–1050 (2002). [DOI] [PubMed] [Google Scholar]
- Liao D.-I., Qian J., Chisholm D. A., Jordan D. B. & Diner B. A. Crystal structures of the photosystem II D1 C-terminal processing protease. Nat. Struct. Mol. Biol. 7, 749–753 (2000). [DOI] [PubMed] [Google Scholar]
- Holm L. & Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- David A. et al. Structural determinants of host specificity of complement Factor H recruitment by Streptococcus pneumoniae. Biochem. J 465, 325–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.-C. et al. The C-terminal Helices of Heat Shock Protein 70 Are Essential for J-domain Binding and ATPase Activation. J. Biol. Chem. 287, 6044–6052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B. L. et al. A structurally dynamic N-terminal helix is a key functional determinant in staphylococcal complement inhibitor (SCIN) proteins. J. Biol. Chem. 288, 2870–2881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A. et al. AMBER 14. University of California, San Francisco. (2014). [Google Scholar]
- Metz A. et al. Hotspots and transient pockets: predicting the determinants of small-molecule binding to a protein-protein interface. J Chem. Inf. Model 52, 120–133 (2012). [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang Y. & Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature 444, 179–180 (2006). [DOI] [PubMed] [Google Scholar]
- Hodel A. E. et al. The Three-Dimensional Structure of the Autoproteolytic, Nuclear Pore-Targeting Domain of the Human Nucleoporin Nup98. Mol. Cell 10, 347–358 (2002). [DOI] [PubMed] [Google Scholar]
- Page M. & Di Cera E. Serine peptidases: classification, structure and function. Cell. Mol. Life Sci. 65, 1220–1236 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum G. et al. Engineering of a novel thioether bridge and role of modified residues in the lantibiotic Pep5. Appl. Environ. Microbiol. 62, 385–392 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sanctis D. et al. ID29: a high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J. Synchrotron Radiat. 19, 455–461 (2012). [DOI] [PubMed] [Google Scholar]
- Panjikar S., Parthasarathy V., Lamzin V. S., Weiss M. S. & Tucker P. A. On the combination of molecular replacement and single-wavelength anomalous diffraction phasing for automated structure determination. Acta Crystallogr. D Biol. Crystallogr. 65, 1089–97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. & Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G., Cohen S. X., Lamzin V. S. & Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano W. L. The PyMOL molecular graphics system. (2002).
- Abts A. et al. Easy and Rapid Purification of Highly Active Nisin. International Journal of Peptides 2011, 9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D. R. & Cheatham T. E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
- Srinivasan J., Cheatham I., T. E., Cieplak P., Kollman P. A. & Case D. A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate - DNA helices. J. Am. Chem. Soc. 120, 9401–9409 (1998). [Google Scholar]
- Gohlke H. & Case D. A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 25, 238–50 (2004). [DOI] [PubMed] [Google Scholar]
- Homeyer N. & Gohlke H. Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Molecular Informatics 31, 114–122 (2012). [DOI] [PubMed] [Google Scholar]
- Gohlke H., Kiel C. & Case D. A. Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras–Raf and Ras–RalGDS complexes. J. Mol. Biol. 330, 891–913 (2003). [DOI] [PubMed] [Google Scholar]
- Cornell W. D. et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 118, 2309–2309 (1996). [Google Scholar]
- Hornak V. et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Bioinf. 65, 712–725 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitkoff D., Sharp K. A. & Honig B. Accurate Calculation of Hydration Free-Energies Using Macroscopic Solvent Models. J. Phys. Chem. 98, 1978–1988 (1994). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.