

Abstract
The objective of the present work was to formulate and evaluate mucoadhesive in situ nasal gels of loratadine. This drug delivery system may overcome the first-pass metabolism and subsequently improve the bioavailability of the drug. A total of 16 formulations of in situ nasal gels were prepared using different polymeric ratios of hydroxypropyl methylcellulose (HPMC K-100) and xanthan gum. All formulations had a clear appearance in the sol form, with gelling temperature of the nasal gels ranging between 33.1 ± 0.43 and 34.8 ± 0.82 °C. The gelling time of all the formulations varied from 4.0 ± 0.21 to 11.3 ± 0.22 s; the drug content was >95%. The pH of the formulations ranged between 5.6 ± 0.004 and 6.0 ± 0.003, i.e. no mucosal irritation is expected as the pH was in the acceptable range. Mucoadhesive strength was adequate (3010.89 ± 1.21-6678.89 ± 0.45 dyne/cm2) to provide prolonged adhesion. In vitro drug release studies showed that the prepared formulations could release the drug for up to 10 h with all of them following Higuchi kinetics. The accelerated stability studies indicated that the gels were stable over the six months test period. The DSC and XRD analysis revealed that there was no drug-polymer interaction. From these findings it can be concluded that in situ nasal gels may be potential drug delivery systems for loratadine to overcome first-pass metabolism and thereby to improve the bioavailability.
Keywords: In situ nasal gel, Loratadine, Controlled drug release, Gelling temperature, Accelerated stability studies
INTRODUCTION
Allergic rhinitis (AR) is a heterogeneous disorder, which is often characterized by mucosal infiltration and action of eosinophils, plasma cells, and mast cells (1,2,3). It is a highly prevalent condition; however, its diagnosis and prevention is comparatively low. Many drug formulations have been used in AR; however, the associated constraints with drug delivery systems pose a major drawback. Some of the factors affecting the drug delivery system include capacity of the nasal cavity for the drug volume (<0.2 ml), mucociliary clearance, and anterior leakage (4).
Antihistamines are often employed to provide symptomatic relief to allergic symptoms due to histamine release. Antihistamine formulations used in the allergic responses are associated with a number of problems. Various studies have been conducted to explore the problems related to the formulation of antihistamines into hydrogels (5). Loratadine is a second-generation histamine H1 receptor antagonist most commonly used in the treatment of AR. Loratadine competes with free histamine and exhibits specific, selective peripheral H1 -antagonistic activity. It is rapidly absorbed following an oral administration, with a bioavailability of only 40% (6,7,8,9).
Intranasal drug delivery is an attractive option for drugs, such as oral antihistamine. Intranasal antihistamines are the most effective agents in the treatment of AR and nasal blockage owing to their efficacy over oral antihistamines. Factors that influence the drug delivery at nasal mucosa includes higher penetration of drugs for better onset of action, high vascularization and large absorption area, avoidance of degradation by gastrointestinal enzymes, potential to deliver the drug via olfactory region, and bypassing the blood-brain barrier. As a result of localized drug delivery, the nasal formulation provides a faster onset of action (around 30 min), with fewer systemic side-effects (10).
Relatively less residence time of the drug in nasal cavity affects its bioavailability. The possible strategy to improve the residence time is to decrease the rapid mucociliary clearance using mucoadhesive formulations. However, for ordinary gels and mucoadhesive powders, residence time cannot be enhanced due to the following drawbacks: accurate drug dose cannot be measured due to difficulty in administration and nasal mucosa irritation and a gritty appearance of the tissues respectively (11,12). The use of in situ nasal gels has been found to be an attractive alternative to overcome the drawbacks associated with ordinary gels and mucoadhesive powders.
Among the different nasal drug delivery systems, in situ gel formulations have been explored for both local and systemic drug delivery. These drug delivery systems exist in sol form before their administration; however, once administered, they undergo gelation to form a gel. The factors regulating the in situ gel formation process include microenvironment temperature, changes in pH, presence of ions, ultraviolet irradiation, and polymers. Rheological properties of gels, which are critical to their efficacy, are important in retaining the gel at the site of application or absorption (13). The present study aims at formulating loratadine into nasal in situ gel to circumvent the first-pass metabolism and improve the bioavailability.
MATERIALS AND METHODS
Materials
Loratadine was obtained as a gift sample from Caplin Point Laboratories Ltd, Chennai, India. Hydroxypropyl methylcellulose (HPMC K 100), xanthan gum, mannitol, polyethylene glycol (PEG), benzalkonium chloride and methanol were purchased from SD Fine Chemicals, Bangalore, India. All the chemicals and reagents used were of analytical grade.
Preparation of loratadine in situ nasal gels
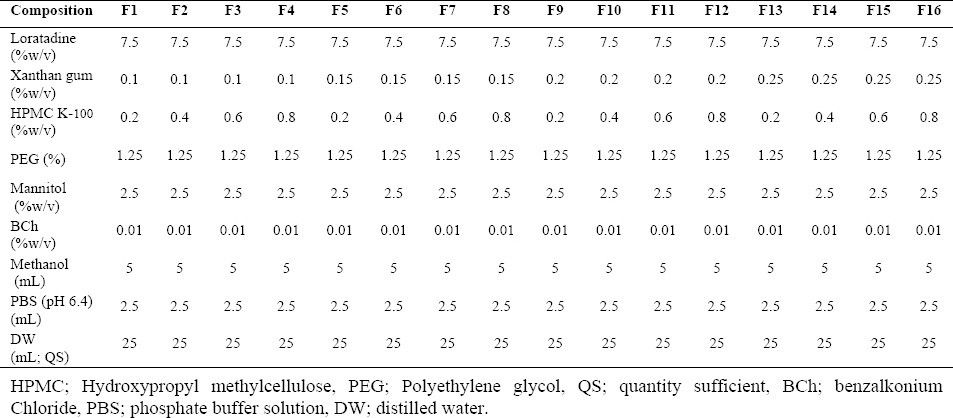
A 24 factorial design was used for optimization of the process parameters. The composition of different formulations of loratadine in situ nasal gels is shown in Table 1. The drug was dissolved in methanol and 10 ml of distilled water (Milli-Q) was added. The solution was mixed by constant stirring. To the above drug solution, mannitol, PEG, and benzalkonium chloride were added. The polymeric solution of HPMC K 100 and xanthan gum were prepared separately in distilled water and thoroughly mixed with the above mixture. The resultant mixture was stirred for 15 min on a magnetic stirrer and phosphate buffer solution was added. The final volume was made up to the desired quantity with distilled water (14).
Table 1.
Composition of different in situ nasal gels of loratadine.
Gelling temperature and gelling time
Gelling temperature refers to the temperature when the meniscus of the formulation would no longer move upon slanting the test tubes at 90 ° (15). Miller and Donovan's technique (15) was used to determine the gelation studies. The gelling temperature was determined by placing the test tube, containing sufficient quantity of the prepared solutions, in a water bath at 4 °C. The temperature of water bath was increased slowly at a constant rate of 1 °C every 2 min.
Gelling time of formulations was determined using the procedures described by Miller and Donovan (15). The delivery systems exist in sol form before administration, however, once they are administered; they undergo gelation to form a gel. Gelling time was recorded as the time for first detection of gelation. The sol-gel transition temperature (Tsol-gel) of the prepared in situ gel formulations was evaluated by transferring 2 ml of the prepared formulation to a test tube (10 ml), with a diameter of 1.0 cm. After sealing with a parafilm, the tube was kept in a circulation water bath at 37 °C. Following each temperature setting, equilibration was allowed for 10 min. Finally, the test tube was placed horizontally to observe the state of the sample and to examine the gelation.
Viscosity of solution
Viscosity of the in situ gel systems was determined using Brookfield viscometer DV-II+Pro coupled with S-94 spindle (Brookfield Engineering Laboratories Inc., MA, USA). The prepared gel formulations were transferred to the beaker. The spindle was lowered perpendicularly into the gel at l00 rpm and temperature was maintained at 37 ± 0.5 °C. The viscosity was determined during the cooling of the system (12). All the measurements were performed in triplicates.
Rheological properties of in situ gels
The rheological properties of in situ gel formulations were investigated using Brookfield LVDV-E Viscometer (Brookfield Engineering Laboratories, Inc, USA). The temperature was initially maintained above 40 °C. The rheological properties were measured by increasing the spindle rotational speed from 0.3 to 100 rpm, and the shear rate (g), shear stress (t), and viscosity (h) were recorded. All the measurements were performed in triplicates.
Determination of pH
One ml of the prepared gels was transferred to a 10 ml volumetric flask, and the solution was diluted with distilled water. The pH of resulting solution was determined using a digital pH meter (Shambhavi Impex, India), which was previously calibrated using phosphate buffers at pH 4 and pH 7 (16).
Drug content assay
One ml of the prepared formulation was dispersed in 10 ml of methanol for 2–3 min with occasional shaking. The resulting solution was filtered through a 0.45 µm filter paper and was diluted with methanol. The amount of loratadine in the formulation was determined spectrophotometrically at 280 nm (Shimadzu 1800, Japan).
Gel strength
Sample (50 g) was placed in a 100 ml graduated cylinder. Gelation was carried out by placing the formulations in a thermostat at 37 °C. The strength of the gel was determined by measuring the time taken by a weight of 35 g to sink 5 cm in the gel (17).
Spreadability
Spreadability was determined using a 10 × 4 cm rectangular glass slide. The sheep nasal mucosa from serosal side was tied on the surface of slide with a thread. The slide was kept in a hot air oven (Navyug Udyog, India), at 37 °C and one drop of gel was placed on the mucosa at an angle of 120 °. Spreadability was determined relative to the distance travelled by the drop of gel (liquid) before its gelation. Average of three readings was recorded (17).
Mucoadhesive strength
Ex vivo mucoadhesive strength was determined using fresh sheep nasal mucosa. The mucosal membrane was separated by removing the underlying fat and loose tissues. The membrane was washed thrice with distilled water and phosphate buffer (pH 6.4). Modified balance method was used to design the experiment.
The balance was equilibrated on both sides by placing one beaker on the left pan and a weight (5 g) on the opposite pan. The sheep nasal mucosa was cleaved into 1 cm2 and glued with cyanoacrylate over the glass support so as to allow the smooth surface of nasal mucosa face the upper side of the glass. The glued sheep nasal mucosa was wetted with phosphate buffer (pH 6.4) by filling the beaker with the buffer on the right hand side of the balance by lowering the glass support. The above setup was placed below the right side of the pan.
A thin film of the prepared gel (1 g) was spread on the lower surface of the right pan. The right pan was lowered and was spread with gel by removing the beaker from the left pan. The pan was left undisturbed for 2 min to ensure proper contact between the nasal mucosa and the gel. Following this, water was slowly added to the left pan using a burette until the nasal mucosa was separated from the gel film. The mucoadhesive force was calculated by determining the weight required to separate the mucosa. The force was expressed in dynes per square centimetre (dyne/cm2) (18).
In vitro drug release
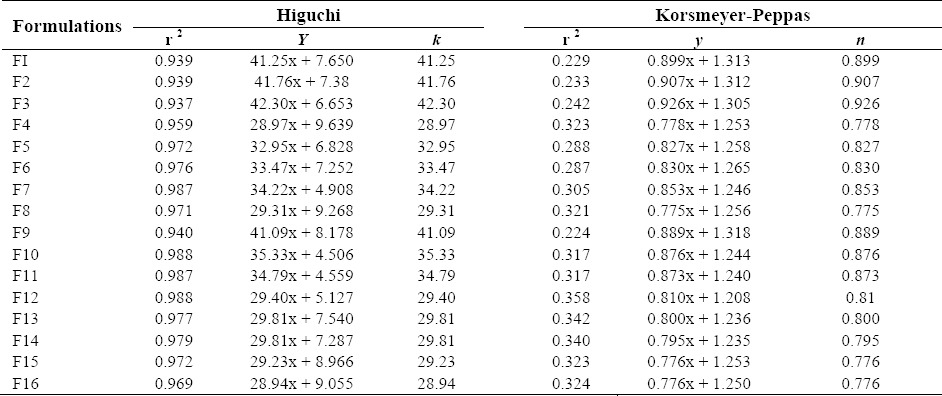
In vitro drug release of loratadine was determined using Franz-diffusion cell (Orchid Scientific & Innovative India Pvt Ltd, India). The sheep nasal mucosa was prepared as described in the above procedures. The prepared nasal mucosa was mounted between the donor and receptor compartments. The receptor compartment was filled with phosphate buffer (pH 6.4) at 37 °C. The solution was stirred at 100 rpm. The gel (10 mg) was placed on the nasal mucosa and the compartments were clamped together. One ml of the sample was withdrawn at predetermined time intervals (0, 0.5, 1, 2, 4, 6, 8, 10, and 12 h) from receptor compartments and immediately replaced using phosphate buffer (pH 6.4). After filtering through 0.45 μm filter and appropriate dilution, the samples were analyzed for drug content at 280 nm (Shimadzu 1800, Japan) (19). The mechanism of drug release from the in situ nasal was determined by plotting the best fit of the release data in Higuchi and Korsmeyer-Peppas plots. The release rate constants k and n of each model were calculated by linear regression analysis using Microsoft Excel 2003 software. Coefficients of determination (R2) were used to evaluate the accuracy of the fit (20,21).
Accelerated stability studies
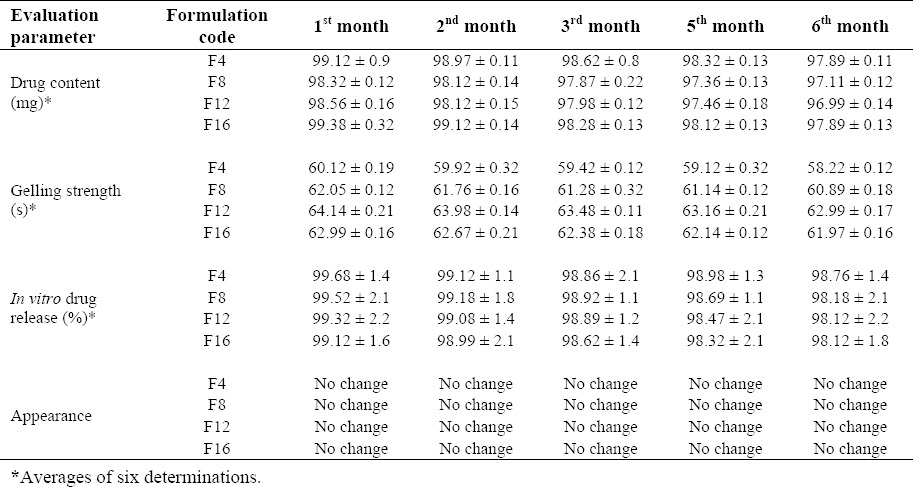
Stability studies were carried on in situ gel formulation according to International Conference on Harmonization guidelines. A sufficient quantity of in situ gel in nasal spray bottles was stored in desiccator (Sabar Scientific, India), containing saturated solution of sodium chloride (relative humidity (RH) of 75 ± 5%). The desiccator was placed in hot air oven maintained at 40 ± 2 °C, and the samples were withdrawn at 1, 2, 3, 5, and 6 months. Changes in the appearance, drug content, gelling strength, and in vitro drug release of the stored formulations were investigated. Mean values from the three determinations were recorded.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) thermograms of selected formulation (F4) (stored at 40 ± 2 °C/ 75 ± 5% RH for 2 months) were recorded. The samples were placed in sealed aluminium pans and scanned at heating rate of 10 °C min-1 over the temperature range of 30–200 °C (22–23).
X-Ray diffraction studies
X-Ray diffraction studies (XRD) pattern of selected formulation (F4) was recorded on Jeol JDX 8030 X-ray diffractometer (MTI Corporation, USA), for a specified quantity of pure drug using copper target at a voltage of 40 kV and a current of 30 mA. The scanning was done over °2 θ range of 10–80 ° (22,23).
RESULTS
Gelling temperature, gelling time, viscosity of solution, drug content and gel strength
In the present study, in situ nasal gels (16 formulations) were prepared using 24 factorial design. HPMC K-100 and xanthan gum were used as the base polymers. The gelling temperature, gelling time, viscosity of solution, drug content and gel strength of the prepared formulation are shown in Table 2.
Table 2.
Gelling temperature, gelling time, viscosity of solution, drug content, and gel strength of in situ nasal gel of loratadine.
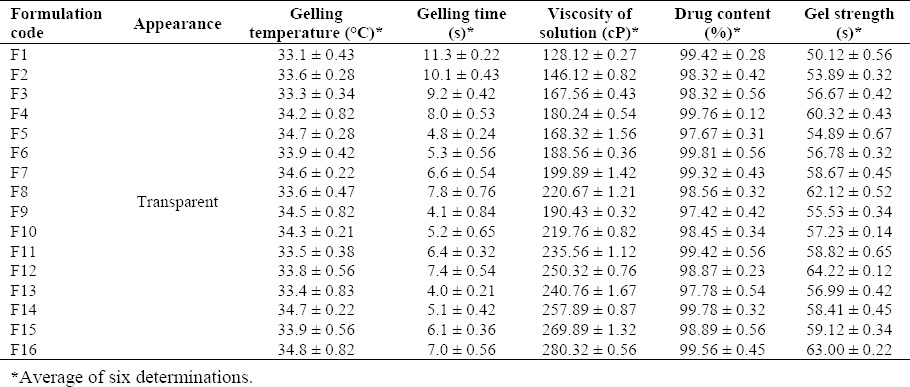
All the formulations showed a clear appearance in the sol form. The gelling temperature of the prepared in situ nasal gel ranged between 33.1 ± 0.43 °C and 34.8 ± 0.82 °C. The gelling temperature suitable for thermoreversible nasal gel ranges between 30–36 °C. The gelation point refers to the temperature when the meniscus of the formulation would no longer move upon slanting the test tubes at 90 °, with gradual increase in the temperature (24,25). In the present study, all the formulations showed a gelling temperature within the range. However, if the temperature is increased, gelation does not occur at the nasal mucosal region, thereby leading to immediate nasal clearance. The gelling time (s) of the formulation ranged between 4.0 ± 0.21 s and 11.3 ± 0.22 s. The formulations F5, F9, and F13 showed an adequate gelling time. The drug content of the developed formulation ranged between 97.42 ± 0.42% and 99.81 ± 0.56%.
The viscosity of the formulation ranged between 128.12 ± 0.27 and 280.32 ± 0.56 cP. The viscosity was increased when the formulations were prepared with 0.8% of HPMC K-100 along with a concentration of 0.1%, 0.15%, 0.2%, and 0.25% of xanthan gum. However, a variation was observed in the viscosity of the formulations when prepared with 0.2%, 0.4% and 0.6% of HPMC K-100. The mucoadhesive strength ranged between 3010.89 ± 1.21 and 6678.89 ± 0.45. Mucoadhesive strength was directly proportional to the concentration of HPMC K-100. The relationship between the polymer ratios (xanthan gum and HPMC ratios) and the viscosity of the gels are presented in Fig. 1.
Fig. 1.
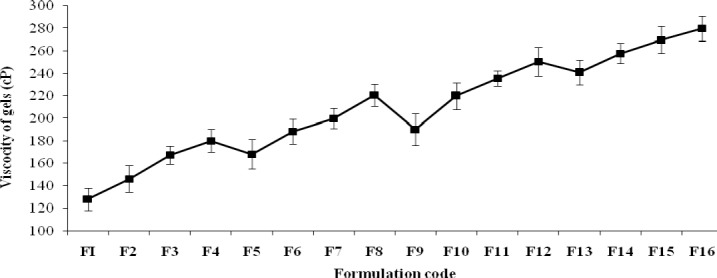
The relationship between the polymer ratios and the viscosity of the formulations (F1-F16).
Spreadability, pH, and mucoadhesive strength
The physicochemical properties, such as spreadability, pH, and mucoadhesive strength are shown in Table 3. The spreadability of the formulation ranged between 10.2 ± 0.11 to 9.2 ± 0.39 cm. The pH of the formulation ranged between 5.6 ± 0.004 and 6.0 ± 0.003. These findings indicated that the formulations showed pH in the acceptable range. The mucoadhesive strength of the formulations varied between 3010.89 ± 1.21 and 6678.89 ± 0.45 dyne/cm2.
Table 3.
Mucoadhesive strength, spreadability, and pH of different formulations of in situ nasal gel of loratidine.
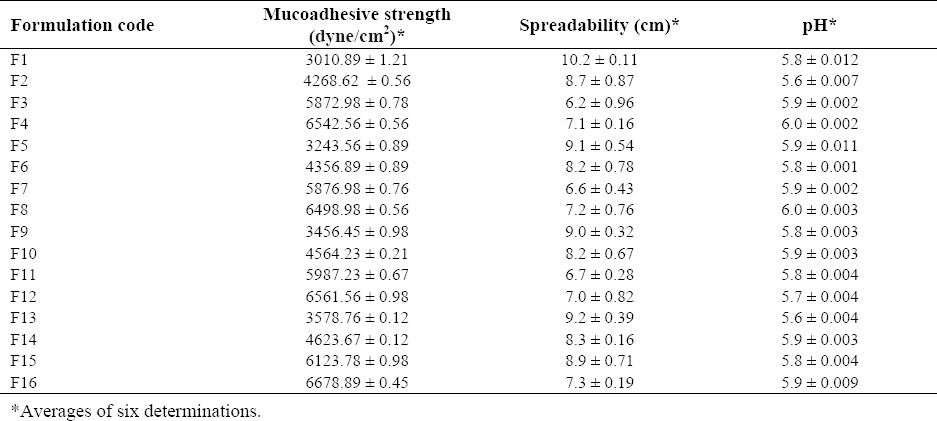
Mucoadhesive drug delivery systems allows rapid dissipation of drug in the circulatory system, thereby preventing the first-pass metabolism and prolonging the residence time of the dosage at the site of application or absorption. In the present study, the formulations prepared with high concentration of HPMC K-100 exhibited more mucoadhesion strength as compared to other formulations. Therefore, the high concentration of HPMC K-100 is more critical for the formulation than xanthan gum.
In vitro drug release
The in vitro drug release of the prepared formulations is shown in (Fig. 2a, b, c, d). The formulations F1 (99.95%), F2 (99.96%), F3 (99.93%), and F9 (99.99%) showed maximum drug release after 6 h, followed by the formulations F5 (99.78%), F6 (99.89%), F7 (99.98%), F10 (99.89%), and F11 (99.99%) after 8 h. The remaining formulations F4 (99.87%), F8 (99.76%), F12 (99.48%), F13 (99.98%), F14 (99.89%), F15 (99.98%), and F16 (99.33%) showed drug release after 10 h. In vitro drug release data indicated that the prepared nasal gels can maintain the drug in the matrix network and prevent early release of drug, thereby maintaining their integrity during the study period. The R2, k and n values are given in Table 4.
Fig. 2.

In vitro drug release of in situ formulations. a; F1-F4, b; F5-F8, c; F9-12, d; F13-F16.
Table 4.
Coefficient values of different formulations of nasal in situ gel of loratadine.
Accelerated stability studies
Data from the accelerated stability studies of the selected in situ gel formulations is shown in Table 5. During and at the end of the accelerated stability study, the selected in situ gel formulations showed drug content similar to what was observed at the beginning of the study. These formulations exhibited satisfactory gelling strength and in vitro drug release during and at the end of the accelerated study period. No color changes or unexpected change in the texture were observed.
Table 5.
Accelerated stability studies of various formulations of in situ gel of loratadine.
Differential scanning calorimetry
The thermal curves of selected formulation (F4) are illustrated in Fig. 3. The DSC provides useful information about the physical properties (crystalline or amorphous nature) of the sample and demonstrates a possible interaction between the drug and polymers in the formulations. Since, the melting point of xanthan gum and HPMC K-100 is molecular-dependent, its determination becomes difficult. Further, the thermograms of xanthan gum and HPMC did not show any endothermic and exothermic peaks between 50–280 °C. The thermal curve of loratadine exhibited a profile typical of a pure, crystalline, anhydrous drug, with a sharp exothermic peak (T peak=137.31 °C) due to its melting process. Whereas, the thermal profile of mannitol showed the exothermic and endothermic peaks at 172.50 °C and 114.90 °C respectively.
Fig. 3.

Differential scanning calorimetry of selected formulation (F4).
X-Ray diffraction studies
The XRD pattern of selected formulation (F4) is shown in Fig. 4. The XRD patterns were detected using X-Ray diffractometer with Cu at an interval of 10-800/2θ. The degree of diffraction was measured at a scanning speed of 40/min, voltage 40.0 (kV), and current 30.0 (mA).
Fig. 4.

X-ray diffraction pattern of selected formulation (F4).
DISCUSSION
the current study, a total of 16 formulations of in situ nasal gels of loratadine was developed and evaluated for gelling temperature and time, viscosity, drug content, gel strength, spreadability, pH, mucoadhesive strength, in vitro drug release, differential scanning calorimetry and X-Ray diffraction studies. 24 factorial designs were used to design the experiment and HPMC K-100 and xanthan gum were used as the base polymers. The nature of the resulting gel depended on the concentration of polymers. The temperature at which the liquid phase is transitioned into gel is referred to as the gelling temperature (15). A gel can be formed at room temperature, if the gelling temperature is lower than the referred period. However, if the temperature is increased, gelation does not occur at the nasal mucosal region, thereby leading to immediate nasal clearance. However, all the formulations exhibited integrity for an extended period of time for sustained release of the drug and showed immediate gelation when contacted with the solution. The time taken by a sol form to form a gel is referred as the gelling time.
The viscosity of the test gels was increased with the increase in the concentration of HPMC K-100. The viscosity was directly dependent on the polymeric content of the formulations (13). When the in situ gels were mixed with phosphate buffer solution (pH 6.4), an elastic and viscous gel was instantaneously formed. The cellulose derivatives can markedly prolong the residence time of the drugs in the nasal cavity because of their desirable mucoadhesive property. Additionally, sustained release of the drug can be maintained due to the high viscosity of the cellulose following hydration in the nasal cavity (26). Thus, cellulose is effective in increasing the intranasal bioavailability of both small hydrophobic and hydrophilic macromolecular drugs. However, using the combination of the cellulose with other absorption enhancers, such as aminoglycoside antibiotics and bisphosphonates, results in enhanced bioavailability of the drug than polymer alone. Moreover, the presence of combination of polymers significantly increased the viscosity as well as gel strength. However, the formulations prepared with high concentration of HPMC K-100 were associated less spreadability. This was attributed to the high viscous nature of HPMC K-100. The results of pH of the formulations did not show any mucosal irritation because all the formulations pH was within in the acceptable range.
Mucoadhesion involves 3 stages: wetting; interpenetration; and mechanical interlocking between mucin and polymer. Owing to the characteristic anatomy and physiology of the nasal passage, i.e., large surface area, highly vascularized epithelium, porous endothelial membrane, nasal drug delivery has emerged as a promising route of drug administration for the systemic therapy (27). The nasal mucus layer can be divided into lower-, low-viscosity layer, and the more viscous upper layer (28). The viscoelastic properties of the nasal mucus is due to the presence of glycoproteins (2%) (29). In addition, nasal mucus is also composed of water (95%), albumin, immunoglobulins, lysozyme, lactoferrin and other proteins (1%), inorganic salts (1%), and lipids (<1%).
The formulations prepared with high viscous polymers prolonged the drug release during the study period. The drug release from the formulations was correlated with the concentration and the viscosity of the polymer used. At fixed drug concentrations, the release rate was dependent on the concentration of HPMC K-100. The drug release was lower when HPMC K-100 concentration was high. The results of stability studies indicated no influence on the chemical and physical stability of the formulations during the test period.
The thermogram of the selected formulation showed no interactions between loratadine and other excipients because the drug melting peak was observed at135.62 °C. However, in terms of crystalline nature, there was no noticeable reduction in heat of fusion (ΔH; 81.7856 J/g)), in the selected formulation (78.9968 J/g). These findings suggested that the selected excipients were able to maintain the crystalline nature of loratadine. However, it should be noted that the transformation of the physical state of the drug to amorphous or partially amorphous state results in high energy state and high disorder, thereby resulting in enhanced solubility and faster dissolution. The number of intense peaks formed from the XRD pattern was almost similar in selected formulation and thereby indicating reduced conversion of crystalline form of the drug to amorphous form. Furthermore, the occurrence of peak the physical mixture indicated no interactions between selected excipients and drug.
CONCLUSION
Novel mucoadhesive in situ gels of loratadine was developed to overcome the first-pass metabolism and enhance the subsequent low bioavailability of the drug. In vitro studies have shown that in situ gels act as potential drug delivery system for loratadine, with better stability and release profile. However, future in vivo studies are warranted to confirm these results.
ACKNOWLEDGEMENTS
The authors like to thank SciWrite Global, a medical communications and scientific writing company (www.sciwriteglobal.com), for their expert review and editing assistance.
REFERENCES
- 1.Togias AG. Systemic immunologic and inflammatory aspects of allergic rhinitis. J Allergy Clin Immunol. 2000;106:S247–S250. doi: 10.1067/mai.2000.110157. [DOI] [PubMed] [Google Scholar]
- 2.Druce HM. Allergic and nonallergic rhinitis. In: Middleton EM Jr, Reed CE, Ellis EF, Adkinson NF Jr, Yunginger JW, Busse WW, editors. Middleton allergy: principles and practice. 5th ed. St. Louis, Mo: Mosby Year-Book; 1998. pp. 1005–1016. [Google Scholar]
- 3.Skoner DP. Allergic rhinitis: definition, epidemiology, pathophysiology, detection, and diagnosis. J Allergy Clin Immunol. 2001;108:S2–S8. doi: 10.1067/mai.2001.115569. [DOI] [PubMed] [Google Scholar]
- 4.Capkova Z, Vitkova Z, Subova M. Formulation of loratadine into hydrogels. Acta Facult Pharm Univ Comenianae. 2005;52:73–78. [Google Scholar]
- 5.Kisan RJ, Manoj NG, Ishaque MS, Vilarsrao JK, Sambjahi, SP Nasal drug delivery system-factors affecting and applicatins. Curr Drug Ther. 2007;2:27–38. [Google Scholar]
- 6.Casale TB, Blaiss MS, Gelfand E. First do no harm: managing antihistamine impairment in patients with allergic rhinitis, antihistamine impairment roundtable. J Allergy Clin Immunol. 2003;111:S835–S842. doi: 10.1067/mai.2003.1550. [DOI] [PubMed] [Google Scholar]
- 7.Sharma A, Hamelin BA. Classic histamine H1 receptor antagonists: a critical review of their metabolic and pharmacokinetic fate from a bird's eye view. Curr Drug Metab. 2003;4:105–129. doi: 10.2174/1389200033489523. [DOI] [PubMed] [Google Scholar]
- 8.Oppenheimer JJ, Casale TB. Next generation antihistamines: therapeutic rationale, accomplishments and advances. Expert Opin Investig Drugs. 2002;11:807–817. doi: 10.1517/13543784.11.6.807. [DOI] [PubMed] [Google Scholar]
- 9.Assanasen P, Naclerio RM. Antiallergic anti-inflammatory effects of H1-antihistamines in humans. Clin Allergy Immunol. 2002;17:101–139. [PubMed] [Google Scholar]
- 10.Cingil C, Kayabasoglu G, Nacar A. Update on the medical treatment of allergic rhinitis. Inflam Allergy Drug Targets. 2009;8:96–103. doi: 10.2174/187152809788462653. [DOI] [PubMed] [Google Scholar]
- 11.Romeo VD, deMeireles J, Sileno AP, Pimplaskar HK, Behl CR. Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev. 1998;29:89–116. doi: 10.1016/s0169-409x(97)00063-x. [DOI] [PubMed] [Google Scholar]
- 12.Shinde JS, Mali KK, Dias RJ, Havaldar VD, Mahajan NS. In situ mucoadhesive nasal gels of metoclopramide hydrochloride: preformulation and formulation studies. J Pharm Res. 2008;1:88–96. [Google Scholar]
- 13.Shah RA, Mehta MR, Patel DM. Design and optimization of mucoadhesive nasal in situ gel containing sodium cromoglycate using factorial design. Asian J Pharm. 2011;5:65–74. [Google Scholar]
- 14.Nandgude T, Thube R, Jaiswal N, Deshmukh P, Chatap V, Hire N. Formulation and evaluation of pH induced in-situ nasal gel of salbutamol sulphate. Int J Pharm Sci Nanotechnol. 2008;1:177–182. [Google Scholar]
- 15.Miller SC, Donovan MD. Effect of poloxamer 407 gels on the miotic activity of pilocarpine nitrate in rabbits. Int J Pharm. 1982;12:142–152. [Google Scholar]
- 16.Kumar MV, Aravindram AS, Rohitash K, Gowda DV, Parjanya K. Formulation and evaluation of in-situ gel of bromhexine hydrochloride for nasal delivery. Der Pharmacia Sinica. 2012;3:699–707. [Google Scholar]
- 17.Choi HG, Shim CK, Kim DD. Development of in situ gelling and mucoadhesive acetaminophen liquid suppository. Int J Pharm. 1998;165:33–44. [Google Scholar]
- 18.Singh RM, Kumar A, Pathak K. Thermally triggered mucoadhesive in situ gel of loratadine: ß-cyclodextrin complex for nasal delivery. AAPS Pharm Sci Tech. 2013;14:412–424. doi: 10.1208/s12249-013-9921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahajan HS, Gattani SG. Gellan gum based microparticles of metoclopromide hydrochloride for intranasal delivery: development and evaluation. Chem Pharm Bull. 2009;57:388–392. doi: 10.1248/cpb.57.388. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50:874–875. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 21.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanism of potassium chloride release from compressed, hydrophilic, polymeric matrices: Effect of entrapped air. J Pharm Sci. 1983;72:1189–1191. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- 22.Kohda Y, Kobayashi H, Baba Y, Yuasa H, Ozeki T, Kanaya Y. Controlled release of lidocaine hydrochloride from buccal mucosa adhesive films with solid dispersion. Int J Pharm. 1997;158:147–155. [Google Scholar]
- 23.Jug M, Bećirević-Laćan M. Influence of hydroxypropyl-ß-cyclodextrin complexation on piroxicam release from buccoadhesive tablets. Eur J Pharm Sci. 2004;21:251–260. doi: 10.1016/j.ejps.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 24.Majithiya RJ, Ghosh PK, Umrethia ML, Murthy RSR. Thermoreversible- mucoadhesive gel for nasal delivery of sumatriptan. AAPS Pharm Sci Tech. 2006;7:E80–E86. doi: 10.1208/pt070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulsson M, Helene HH, Edsman K. Rheological studies of the gelation of deacetylated gellan gum (Gelrite®) in physiological conditions. Eur J Pharmaceut Sci. 1999;9:99–105. doi: 10.1016/s0928-0987(99)00051-2. [DOI] [PubMed] [Google Scholar]
- 26.Chaturvedi M, Kumar M, Pathak K. A review on mucoadhesive polymer used in nasal drug delivery system. J Adv Technol Res. 2011;2:215–222. doi: 10.4103/2231-4040.90876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mainardes RM, Urban MC, Cinto PO, Chaud MV, Evangelista RC, Gremiao MP. Liposomes and micro/nanoparticles as colloidal carriers for nasal drug delivery. Curr Drug Deliv. 2006;3:275–285. doi: 10.2174/156720106777731019. [DOI] [PubMed] [Google Scholar]
- 28.Merkus FW, Verhoef JC, Schipper NG, Marttin E. Nasal mucociliary clearance as a factor in nasal drug delivery. Adv Drug Del Rev. 1998;29:13–38. doi: 10.1016/s0169-409x(97)00059-8. [DOI] [PubMed] [Google Scholar]
- 29.Kaliner M, Marom Z, Patow C, Shelhamer J. Human respiratory mucus. J Allergy Clin Immunol. 1984;73:318–323. doi: 10.1016/0091-6749(84)90403-2. [DOI] [PubMed] [Google Scholar]