

Abstract
Neutral sphingomyelinases (N-SMases) are major candidates for stress-induced ceramide production, but there is still limited knowledge of the regulatory mechanisms of the cloned N-SMase enzyme—nSMase2. We have reported that p38 mitogen-activated protein kinase (MAPK) was upstream of nSMase2 in tumor necrosis-α (TNF-α)-stimulated A549 cells (J Biol Chem 282:1384–1396, 2007). Here, we report a role for protein kinase C (PKC) in mediating TNF-induced translocation of nSMase2 from the Golgi to the plasma membrane (PM). Pharmacological inhibition of PKCs prevented TNF-stimulated nSMase2 translocation to the PM in A549 cells. Using phorbol 12-myristate 13-acetate (PMA) as a tool to dissect PKC responses, we found that PMA induced nSMase2 translocation to the PM in a time- and dose-dependent manner. Pharmacological inhibitors and specific siRNA implicated the novel PKCs, specifically PKC-δ, in both TNF and PMA-stimulated nSMase2 translocation. However, PMA did not increase in vitro N-SMase activity and PKC-δ did not regulate TNF-induced N-SMase activity. Furthermore, PKC-δ and nSMase2 did not coimmunoprecipitate, suggesting that other signaling proteins may be involved. PMA-stimulated nSMase2 translocation was independent of p38 MAPK, and neither PKC inhibitors nor small interfering RNA had significant effects on TNF-stimulated p38 MAPK activation, indicating that PKC-δ does not act through p38 MAPK in regulating nSMase2. Finally, down-regulation of PKC-δ inhibited induction of vascular cell and intercellular adhesion molecules, previously identified as downstream of nSMase2 in A549 cells. Taken together, these data implicate PKC-δ as a regulator of nSMase2 and, for the first time, identify nSMase2 as a point of cross-talk between the PKC and sphingolipid pathways.
Ceramide is well established as a bioactive lipid involved in the cellular responses to stress. The sphingomyelinase (SMase)-mediated hydrolysis of sphingomyelin has emerged as a major pathway of stress-induced ceramide production and the Mg2+-dependent neutral SMases (N-SMases) are considered strong candidates for mediating this pathway (Hannun and Obeid, 2002). Two cloned proteins with in vitro and in vivo N-SMase activity have been identified: nSMase2 and nSMase3 (Hofmann et al., 2000; Krut et al., 2007). Previous research has reported the translocation of nSMase2 to the plasma membrane (PM) in response to confluence (Marchesini et al., 2004), H2O2 (Levy et al., 2006), and TNF (Clarke et al., 2007). It is noteworthy that ceramide production in response to H2O2 or confluence and the up-regulation of adhesion proteins induced by TNF were all prevented by down-regulation of nSMase2 (Marchesini et al., 2004; Levy et al., 2006; Clarke et al., 2007). Moreover, in hepatocytes, nSMase2 is located constitutively at the PM, where it plays a role in interleukin-1β-stimulated activation of JNK (Karakashian et al., 2004). Taken together, these results suggest that nSMase2 localization or translocation to the PM is necessary for its signaling functions, but the regulation of this process is still poorly understood. We have reported that p38 MAPK was involved in regulation of nSMase2 translocation (Clarke et al., 2007). However, this regulation was indirect, suggesting that other signaling proteins could be involved.
Protein kinase C (PKC) is a family of serine/threonine kinases important in modulating a variety of biological responses, including cell growth, differentiation, and apoptosis. PKCs consist of 11 isoforms separated into three classes according to their regulation. The classic PKCs (α, βI and βII, γ) are activated by diacylglycerol (DAG), calcium, and phosphatidylserine (PS), the novel PKCs (δ, ε, η, θ) are activated by DAG and PS, and the atypical isoforms (ζ and λ/ι) are independent of both DAG and calcium but can be regulated by PS, arachidonic acid, and ceramide. The classic and novel PKCs are also robustly activated by phorbol 12-myristate 13-acetate (PMA), tumor-promoting compounds that mimic the action of DAG (for review, see Musashi and Shiroshita, 2000; Parker and Murray-Rust, 2004). There is accumulating evidence for isoform-specific roles of PKCs. For example, PKC-δ has been implicated in regulation of apoptosis of U937 cells (Jang et al., 2003), whereas PKC-ε is involved in UV-induced carcinoma development (Aziz et al., 2007). A role for PKCs in p38 MAPK activation has also been suggested in response to TNF and hyperglycemia (Woo et al., 2005; Nomiyama et al., 2007), and PMA induces p38 MAPK activation in many cell types, including MCF-7, A549, and A172 glioblastoma cells (Chang et al., 2005; Kitatani et al., 2006; Nomura et al., 2007). Thus, some of the downstream effects of PKC can be attributed to its subsequent activation of p38 MAPK.
Considerable evidence suggests cross-talk between the sphingolipid and PKC pathways. Ceramide is reported to have regulatory effects on activity and localization of PKC-α, -δ, -ε, and -ζ (Lee et al., 2000; Kajimoto et al., 2004; Fox et al., 2007). Moreover, PKC can act upstream of ceramide production because PMA activates acid SMase and the ceramide salvage pathway in MCF-7 cells (Kitatani et al., 2006; Zeidan and Hannun, 2007). PKCs can also regulate N-SMase activity. In mesangial cells, the cytokine-induced activation of N-SMase activity was inhibited by PMA (Kaszkin et al., 1998), and N-SMase activity stimulated by chemotherapeutic drugs in U937 cells was inhibited by PKC-ζ overexpression (Bezombes et al., 2002). In addition, both PKC-α and -δ were implicated in regulation of the Mg2+-independent cytosolic N-SMase in response to interferon-γ and vitamin D (Visnjic et al., 1999). Thus, PKC-mediated regulation of N-SMases may be an important point of cross-talk between the two pathways. However, no study has yet examined the regulation of the cloned N-SMases by PKC.
In this study, we investigated the role of PKC in nSMase2 regulation. We found that PKCs mediated the effects of TNF on nSMase2 translocation. In addition, PMA induced nSMase2 translocation to the PM in a time- and dose-dependent manner but did not increase endogenous N-SMase activity or overexpress nSMase2 activity. The use of PKC inhibitors and siRNA implicated PKC-δ in both PMA and TNF responses, but this was not through activation of p38 MAPK. Together, these data offer further insight into nSMase2 regulation and also implicate nSMase2 as a point of cross-talk between PKC and sphingolipid pathways in the cellular response to TNF-α.
Materials and Methods
Cell Culture and siRNA
A549 cells were maintained in 10% fetal bovine serum in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) at 37°C in 5% CO2. For siRNA experiments, cells were seeded in 60-mm dishes (50–75 × 103/dish) or 35-mm confocal dishes (15 × 103/dish). After 24 h, cells were transfected with scrambled (Scr) or specific siRNA (20 nM) using Oligofectamine according to the manufacturer’s protocol. Cells were allowed to grow for 48 to 60 h before experiments or transfection with 3′V5-tagged nSMase2. siRNA for PKC-δ was as described previously (Zeidan and Hannun, 2007); prevalidated siRNA for PKC-ε and -θ were from QIAGEN (Valencia, CA).
Immunofluorescence and Confocal Microscopy
A549 cells (2.5 × 104) were seeded in 35-mm confocal dishes; 48 h later, cells were transiently transfected with 0.3 μg of 3′V5-tagged-nSMase2 using Effectene (QIAGEN) according to the manufacturer’s protocol. After 12 to 15 h, media were replaced and cells were allowed to grow for a further 3 to 6 h before stimulation with PMA (100 nM) or TNF-α (50 ng/ml) as indicated. For PKC inhibitor experiments, A549 cells were preincubated with 2 μM bisindolylmalemide, 3 μM Go6976, or 10 μM rottlerin for 1 h before stimulation. Cells were fixed, permeablized, and stained as described previously (Clarke et al., 2007) and viewed on an LSM 510 Meta Confocal Microscope (Zeiss, Welwyn Garden City, UK).
Total Cell Lysate Preparation
Cells were washed in ice-cold PBS, scraped in 1 ml of PBS and phosphatase inhibitors (Halt Cocktail; Pierce, Rockford, IL), and pelleted (5 min, 1000g). Cells were resuspended in 100 μl of PBS, and two 3-μl aliquots were removed for protein estimation by Bradford assay (Bradford, 1976). Forty microliters of 2× Laemmli buffer was added to an equal volume of cell suspension and vortexed 3 times for 10 s to lyse cells. Samples were boiled for 10 min for immediate use or storage at −20°C.
Immunoblotting
Protein samples were separated on 4 to 20% gradient gels (Criterion; BioRad Laboratories, Hercules, CA) at 60 to 100 V before transfer to nitrocellulose membrane in tris/glycine buffer (100 V, 30 min, 4°C). Membranes were blocked (5% milk, >30 min) and probed with primary antibody overnight (4°C). Membranes were washed (three times with 0.1% Tris-buffered saline/Tween 20), probed with horseradish peroxidase-conjugated secondary antibody (1:5000 mouse or rabbit in 5% nonfat dry milk) for 30 to 45 min at room temperature, and washed (three times with 0.1% Tris-buffered saline/Tween 20). Proteins were visualized by enhanced chemiluminescence (Pierce).
In Vitro Sphingomyelinase Assay
Neutral sphingomyelinase assay was assessed in vitro using [choline-methyl-14C]sphingomyelin as described previously (Marchesini et al., 2004; Clarke et al., 2007)
Immunoprecipitation
A549 cells were mock- or transiently transfected with 3′V5-nSMase2 before stimulation with PMA (100 nM) or TNF-α (50 ng/ml) as indicated in text and figures. Cells were washed (twice with ice-cold PBS) and lysed in immunoprecipitation buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 5 mM NaF, 5 mM Na3VO4, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitors) by sonication (four times for 10 s each). V5 and PKC-δ were immunoprecipitated from 100 μg of lysate using protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) as described previously (Clarke et al., 2007)
Statistical Analysis
Comparisons between two groups were analyzed by Student’s t test. P < 0.05 was considered statistically significant; n = number of experiments, as indicated.
Materials
Monoclonal anti-V5 antibody was from Invitrogen; polyclonal giantin was from Covance Research Products (Princeton, NJ); polyclonal phospho-p38 MAPK was from Promega (Madison, WI); polyclonal anti-p38 MAPK (clone C-20), polyclonal PKC-δ (C-20), monoclonal ICAM-1 (G-5), and polyclonal VCAM-1 (H-276) antibodies were from Santa Cruz Biotechnology; monoclonal PKC-ε, PKC-θ, PKC-η, and polyclonal p230 antibodies were from BD Biosciences (San Jose, CA). Anti phospho-PKC-δ, -ε, and -θ antibodies were from Cell Signaling Technology Inc. (Danvers, MA). Polyclonal anti TGN46 came from Novus Biologicals (Littleton, CO). Fluorescent secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). PMA, bisindolylmalemide, Go6976, and rottlerin were from Calbiochem (San Diego, CA). Human recombinant TNF was purchased from Peprotech Inc. (Rocky Hill, NJ). [choline-methyl-14C]-Sphingomyelin was provided by Dr. Alicja Bielawska (Medical University of South Carolina, Charleston, SC). All lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Scintillation mixture Safety Solve was from Research Products International (Mt. Prospect, IL). All other products were from Sigma Aldrich (St. Louis, MO) unless stated otherwise.
Results
PKCs Are Involved in TNF-Stimulated Translocation of nSMase2 to the Plasma Membrane
Previous studies have suggested cross-talk between PKC and N-SMase pathways (Kaszkin et al., 1998; Visnjic et al., 1999; Bezombes et al., 2002). Because nSMase2 translocation to the PM is important for nSMase2-mediated signaling (Marchesini et al., 2004; Levy et al., 2006; Clarke et al., 2007), the role of PKCs in this process was investigated initially by a pharmacological approach. A549 cells overexpressing V5-nSMase2 were preincubated with the PKC inhibitors bisindolylamide (2 μM) or Go6976 (3 μM) for 1 h before stimulation with TNF (50 ng/ml, 30 min). Localization of nSMase2 was investigated by immunofluorescence and confocal microscopy as described under Materials and Methods. Preincubation of A549 cells with either inhibitor had no significant effect on basal nSMase2 localization with V5-nSMase2 colocalizing with the Golgi marker giantin consistent with our previous studies (Clarke et al., 2007). However, bisindolylamide inhibited the TNF-stimulated nSMase2 translocation, whereas Go6976 had no effect (Fig. 1A). Taken together, this indicates a role for PKC in nSMase2 translocation with the differential effects of bisindolylamide (classic and novel PKC inhibitor) and Go6976 (classic PKC inhibitor) suggesting that the novel PKCs are involved.
Fig. 1.

PKC is involved in regulation of nSMase2 translocation in response to TNF and PMA. A549 cells (20–25 ×n 35-mm confocal dishes and 48 h later were transiently transfected with 3′V5-tagged nSMase2 (0.25 μg/dish) for 12 h, media were changed, and cells were allowed to grow for 6 to 12 h before stimulation as indicated. Cells were fixed and stained with anti-V5 (green) and anti-giantin (red) as described under Materials and Methods. A, effect of PKC inhibitors on TNF-stimulated nSMase2 translocation. Cells were preincubated in bisindolylamide I (2 μM) or Go6976 (3 μM) for 1 h before stimulation with TNF (50 ng/ml) for 30 min. B, time course of PMA stimulation (100 nM) from 0 to 60 min. C, dose response of PMA stimulation: 0 to 200 nM for 30 min. D, effect of PKC inhibitors on PMA-stimulated nSMase2 translocation. Cells were preincubated as in A, before stimulation with PMA (100 nM) for 30 min. Pictures are representative of more than five fields taken from at least four independent experiments.
PMA Induces nSMase2 Translocation to the Plasma Membrane
To further define the role of PKCs in nSMase2 translocation, PMA (a direct activator of novel and classic PKCs) was used. As before, V5-nSMase2 colocalized with giantin in unstimulated cells. Stimulation with PMA (100 nM) induced a rapid (10–20 min) and sustained translocation of nSMase2 to the PM (Fig. 1B). To further characterize this effect, a dose response at 30 min of stimulation was performed. At concentrations of 1 and 10 nM PMA, nSMase2 was intracellular, mainly localizing to the Golgi. However, concentrations of PMA ≥ 50 nM were sufficient to induce PM translocation of nSMase2 (Fig. 1C).
The higher doses of PMA required to induce nSMase2 translocation again suggested that the novel PKCs are involved, as they are reported to require higher PMA concentrations for activation (Kazanietz et al., 1993). Accordingly, the inhibitors bisindolylamide and Go6976 were used. Consistent with the effects seen with TNF, preincubation with both inhibitors had no effect on basal nSMase2 localization. However, whereas bisindolylamide inhibited nSMase2 translocation to the PM, Go6976 had no effect (Fig. 1D), supporting the evolving hypothesis that novel PKCs play a role in regulating nSMase2.
Investigation of the Novel PKC Isoform Involved in nSMase2 Translocation
To identify the novel PKC involved in regulation of nSMase2, it was first necessary to confirm which novel PKCs were detectable in A549 cells. Accordingly, total cell lysate was immunoblotted for PKC-δ, -ε, -η, and -θ. As can be seen, the isoforms PKC-δ, -ε, and -θ were readily detected (Fig. 2A). However, we were unable to detect PKC-η.
Fig. 2.
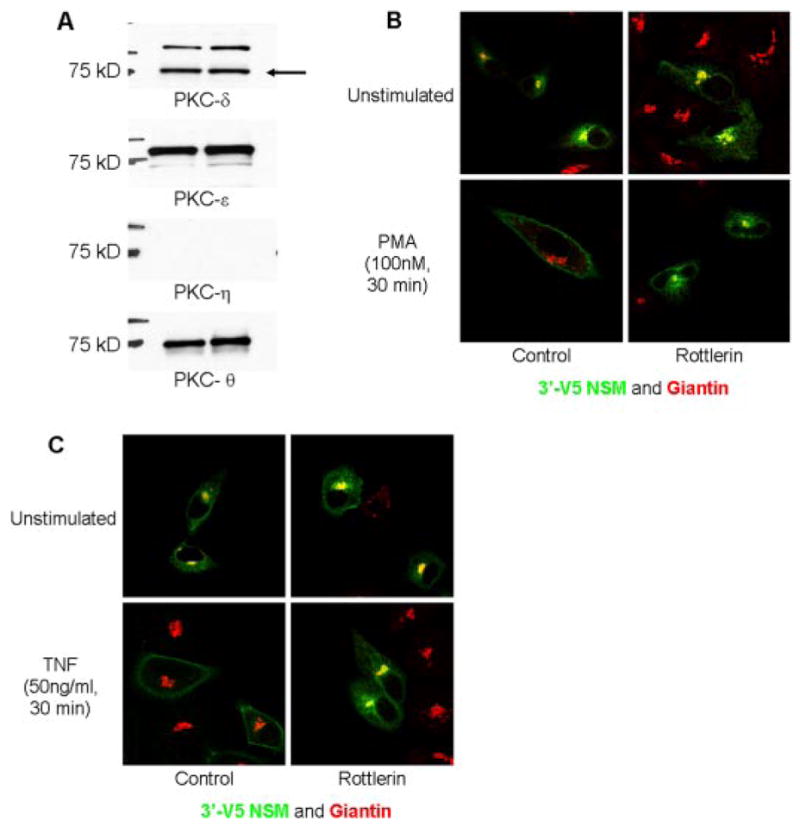
Rottlerin inhibits nSMase2 translocation in response to TNF and PMA. A, total lysate was prepared from A549 cells and immunoblotted for novel PKCs as described under Materials and Methods. B and C, A549 cells (20–25 × 103) were seeded in 35-mm confocal dishes and 48 h later were transiently transfected with 3′V5-tagged nSMase2 (0.25 μg/dish) for 12 h, media were changed and cells were allowed to grow for 6 to 12 h before stimulation as indicated. Cells were preincubated with rottlerin (10 μM) for 1 h before stimulation as indicated. Cells were fixed and stained with anti-V5 (green) and anti-giantin (red) as described under Materials and Methods. B, effect of rottlerin on PMA-stimulated nSMase2 translocation. After preincubation, cells were stimulated with PMA (100 nM) for 30 min. C, effect of rotterlin on TNF-stimulated nSMase2 translocation. After preincubation, cells were stimulated with TNF (50 ng/ml) for 30 min. Pictures are representative of more than five fields taken from at least four independent experiments.
To further explore the novel PKC role in nSMase2 translocation, we initially used rottlerin, an inhibitor shown to have some specificity for the novel PKC-δ (Geschwendt et al., 1994). Whereas preincubation of cells with rottlerin (10 μM, 1 h) had no effect on basal nSMase2 localization, translocation of nSMase2 in response to both PMA (Fig. 2B) and TNF (Fig. 2C) was prevented, further consistent with a role for the novel PKCs in regulating nSMase2 translocation.
To consolidate this data and avoid nonspecificity issues of pharmacological inhibitors, siRNA for each novel isoform detected was used. To validate the efficacy and specificity of each siRNA, A549 cells were transfected with 20 nM PKC-δ, -ε, -θ, or a nonspecific targeting (Scr) siRNA as control. After 48 to 60 h, total protein was extracted and immunoblotted for all novel PKCs. Each siRNA was found to strongly and specifically down-regulate the respective isoform with a minimal effect on the levels of the other novel PKC isoforms (Fig. 3A). Using these siRNAs, the involvement of each novel PKC in regulation of nSMase2 was investigated. A549 cells were treated with siRNA for 48 h before transient transfection with V5-nSMase2. Cells were stimulated with PMA (100 nM, 30 min) or TNF (50 ng/ml, 30 min), and nSMase2 localization was investigated by immunofluorescence and confocal microscopy. In all cases, basal localization of nSMase2 was unaffected by siRNA treatment (Fig. 3B). However, on PMA stimulation, PKC-ε siRNA had no effect on nSMase2 translocation. In contrast, PKC-δ down-regulation markedly inhibited nSMase2 translocation with a predominantly intra-cellular localization of V5-nSMase2 observed (Fig. 3B). It is noteworthy that PKC-θ siRNA had a small effect such that both PM and intracellular localization of nSMase2 could be seen.
Fig. 3.

siRNA down-regulation implicates PKC-δ in PMA- and TNF-stimulated nSMase2 translocation. A, validation of novel PKC siRNA. A549 cells were seeded in 60-mm dishes (65 × 103) and 24 h later were transfected with negative control (Scr) or novel PKC siRNA (20 nM). After 48 h, total protein was extracted and immunoblotted for novel PKCs as described under Materials and Methods. B and C, cells were seeded in 35-mm confocal dishes and, 24 h later, were transfected with Scr, PKC-δ, -ε, or -θ siRNA. After 48 h, cells were transfected with 3′V5-tagged nSMase2 (0.25 μg/dish) for 12 h, media were changed, and cells were allowed to grow for 3 to 6 h before stimulation as indicated. Cells were fixed and stained with anti-V5 (green) and anti-giantin (red) as described under Materials and Methods. B, effect of PKC siRNA on PMA-stimulated nSMase2 translocation. After siRNA down-regulation and nSMase2 transfection, cells were stimulated with PMA (100 nM) for 30 min. C, effect of PKC siRNA on TNF-stimulated nSMase2 translocation. After siRNA down-regulation and V5-nSMase2 transfection, cells were stimulated with TNF (50 ng/ml) for 30 min. Pictures are representative of more than five fields taken from at least three independent experiments performed in duplicate.
Given the above results, it became critical to determine whether PKC-δ plays a key role in mediating the effects of TNF on nSMase2. Therefore, the effects of the siRNAs were evaluated as above. As before, no effect of the siRNA on basal nSMase2 localization was observed (Fig. 3C). However, as with PMA, PKC-δ down-regulation markedly prevented relocalization of nSMase2 to the PM, whereas PKC-ε and -θ siRNA were without significant effect (Fig. 3C). As an additional control, PKC-α siRNA was used and with both TNF and PMA stimulation, no significant effect on nSMase2 localization was observed (data not shown). These studies reveal a critical role for PKC-δ in mediating the effects of TNF on nSMase2 translocation.
The Role of PKC-δ in Regulation of N-SMase Activity
In addition to inducing PM translocation of nSMase2, we have also reported that TNF rapidly and transiently increases nSMase2 activity, peaking at 5 min of stimulation (Clarke et al., 2007). Therefore, we were interested to determine whether PMA also stimulated nSMase2 activity in A549 cells. Accordingly, N-SMase activity both endogenously and in cells overexpressing nSMase2 was measured in vitro as described above (Fig. 4A). Results indicated that, unlike with TNF, PMA stimulation had no significant effect on endogenous N-SMase activity between 0 and 60 min of stimulation. In contrast, PMA decreased activity of overexpressed nSMase2 at 30 and 60 min of stimulation. This is consistent with previous studies reporting a PMA-induced reduction of N-SMase activity (Kaszkin et al., 1998) and suggests that PKCs may not be involved in post-translational activation of nSMase2 (see Discussion).
Fig. 4.

PKC does not regulate in vitro nSMase2 activity in A549 cells. A, A549 cells with (■) and without (○) nSMase2 overexpression were stimulated with PMA (100 nM) for 0 to 60 min (*, P < 0.05, n = 5). B, A549 cells were transfected with negative control (Scr), PKC-δ or -ε siRNA (20 nM) for 48 to 60 h before stimulation with TNF for 5 min (*, P < 0.05, n = 4). In vitro N-SMase activity was measured as described under Materials and Methods.
To further explore this in response to TNF, the effects of PKC-δ and PKC-ε siRNA on TNF-stimulated endogenous N-SMase activity were investigated (Fig. 4B). A time point of 5 min was used because this was shown previously to be the peak stimulation time in A549 cells (Clarke et al., 2007). As can be seen, neither PKC-δ nor PKC-ε siRNA had effects on basal activity. Moreover, neither siRNA prevented TNF stimulation of N-SMase activity. This is consistent with the lack of effect of PMA on endogenous N-SMase activity in A549 cells. Furthermore, this suggests that PKC-δ is important for nSMase2 translocation, but not activation in A549 cells.
Coimmunoprecipitation Studies of nSMase2 and PKC-δ
Translocation and regulation of proteins can often occur through direct interactions with other proteins at the membrane. To determine whether PKC-δ and nSMase2 directly interact with each other in cells, coimmunoprecipitation studies were performed. Mock- or V5-nSMase2-transfected A549 cells were stimulated with either PMA (100 nM; 0, 10, or 30 min) or TNF (3 nM; 0, 10, or 30 min), lysates were immunoprecipitated with V5 or PKC-δ antibody and immunoblotted for PKC-δ and V5-nSMase2, respectively. With both TNF (Fig. 5A) and PMA (Fig. 5B), there was no coimmunoprecipitation of V5-nSMase2 and PKC-δ under either basal or stimulated conditions. Together, this suggests that PKC-δ may function as an indirect regulator of nSMase2 translocation in both PMA and TNF responses.
Fig. 5.

Coimmunoprecipitation studies of PKC-δ and nSMase2. A549 cells (1.5–2 × 105) were seeded in 60-mm dishes and 48 h later were either mock-transfected (UTF) or transiently transfected with 3′V5-tagged nSMase2 (2 μg/dish) for 12 h, media were changed, and cells were allowed to grow for 6 to 12 h before TNF (50 ng/ml) (A) and PMA (100 nM) (B) stimulation as indicated. V5-nSMase2 and PKC-δ were immunoprecipitated from cell lysates as described under Materials and Methods. Immunoprecipitates (IP) and supernatants (SUP) were analyzed for V5-nSMase2 and PKC-δ content by SDS-PAGE and immunoblotting. Shown are immunoblots of V5-nSMase2 and PKC-δ immunoprecipitates and supernatants, representative of at least three independent experiments. N, negative control.
p38 MAPK Does Not Play a Role in PMA-Stimulated nSMase2 Translocation
In a previous study, we identified p38 MAPK as an indirect regulator of nSMase2 translocation and activation in response to TNF (Clarke et al., 2007). Previous studies have shown that PMA can induce p38 activation (Chang et al., 2005; Kitatani et al., 2006; Nomura et al., 2007) and implicated PKC upstream of p38 MAPK (Woo et al., 2005; Nomiyama et al., 2007). Given that PKC-δ and nSMase2 did not seem to interact, it became important to determine the role of PKC with respect to p38 MAPK in regulating nSMase2 translocation. To investigate this with PMA, the p38 MAPK inhibitor SB202190 was used. A549 cells overexpressing V5-nSMase2 were pretreated with 10 μM SB202190 for 1 h before stimulation with PMA (100 nM, 30 min) and the effects on nSMase2 localization investigated. Preincubation with the inhibitor alone had no effect on basal nSMase2 localization consistent with our previous study (Clarke et al., 2007). Furthermore, inhibition of p38 MAPK had no effect on the translocation of nSMase2 stimulated by PMA but inhibited the TNF response on nSMase2 (Fig. 6A). These results suggest that p38 MAPK does not act downstream of PKC-δ in inducing translocation of nSMase2.
Fig. 6.

p38 MAPK is not involved in PMA-stimulated nSMase2 translocation. A, A549 cells (200K) were seeded in 35-mm confocal dishes and 48 h later were transiently transfected with 3′V5-tagged nSMase2 (0.25 μg/dish) for 12 h, media were changed, and cells were allowed to grow for 3 to 6 h before preincubation with SB202190 (10 μM). Subsequently, cells were stimulated with PMA (100 nM) or TNF (3 nM) for 30 min, fixed, and stained with anti-V5 (green) and anti-giantin (red) as described under Materials and Methods. B and C, A549 cells (2 × 105) were seeded in 60-mm dishes; 48 h later, media were changed for 1 to 2 h. Cells were preincubated with bisindolylamide I (2 μM) or Go6976 (B) or rottlerin (10 μM) for 1 h before stimulation with PMA (100 nM) for 30 min (C), total protein was extracted and immunoblotted for phospho-p38 MAPK and p38 MAPK as described under Experimental Procedures. (*, P < 0.05, n = 4).
To consolidate these results, the effects of PKC inhibitors on PMA-stimulated p38 MAPK activation, as assessed by phosphorylation, were investigated. As can be seen, both bisindolylamide I and Go6976 inhibited p38 MAPK phosphorylation in response to PMA (Fig. 6B), suggesting a classic PKC-mediated activation of p38 MAPK. Consistent with this, rottlerin also had no effect on PMA-stimulated p38 phosphorylation (Fig. 6C). Taken together, these results suggest that activation of p38 MAPK by PMA is independent of the novel PKC-mediated regulation of nSMase2.
Novel PKCs Do Not Act Upstream of p38 MAPK in Response to TNF
Having found that p38 MAPK and novel PKCs seemed to act independently in response to PMA, it was important to further explore this relationship in response to TNF. Initially, A549 cells were treated with bisindolylamide 1 or Go6976, and p38 MAPK phosphorylation was assessed. As with PMA, neither inhibitor had a significant effect on basal p38 MAPK phosphorylation. However, neither inhibitor had a significant effect on TNF-stimulated p38 MAPK phosphorylation (Fig. 7A), suggesting that, unlike with PMA, classic PKC is not upstream of p38 MAPK activation in response to TNF. To consolidate these data, the effect of rottlerin was determined. Although a small effect of rottlerin on TNF-stimulated p38 MAPK phosphorylation was observed (20%), this was not statistically significant (p > 0.1) (Fig. 7B). Thus, as with PMA, the TNF-stimulated activation of p38 MAPK is independent of novel PKCs, but unlike PMA, it is also independent of classic PKCs.
Fig. 7.

PKC is not upstream of p38 MAPK in response to TNF. A and B, A549 cells (200 × 103) were seeded in 60-mm dishes; 48 h later, they were preincubated with bisindolylamide I (2 μM) or Go6976 (3 μM) (A) or rottlerin (10 μM) for 1 h before stimulation with TNF (50 ng/ml, 10 min) (B). C, A549 cells (65 × 103) were seeded and, 24 h later, transfected with negative control (Scr) or novel PKC siRNA. After 48 to 60 h, media were changed and cells were incubated for 1 to 2 h and stimulated with TNF (3 nM, 10 min). Total protein samples were prepared and analyzed for phospho-p38 MAPK and p38 MAPK as described under Materials and Methods. (*, P < 0.05 versus unstimulated, n = 5).
To further support inhibitor data, avoid inhibitor specificity issues, and clarify individual roles of novel PKCs in p38 MAPK activation or not, the effects of novel PKC (δ, ε, θ) siRNA on p38 MAPK phosphorylation were determined. As can be seen, siRNA down-regulation of novel PKCs had no significant effect on basal or TNF-stimulated p38 MAPK phosphorylation (Fig. 7C), confirming that PKC-δ is not upstream of p38 MAPK in A549 cells. It is noteworthy that these data are also consistent with the lack of a role for PKC-δ in regulating endogenous N-SMase activity in A549 cells, because we have shown previously that this activity is p38 MAPK-dependent (Clarke et al., 2007). Taken together, this confirms that novel PKCs do not act upstream of p38 MAPK in regulating nSMase2 in response to TNF.
Regulation of Novel PKCs by TNF in A549 Cells
Although previous studies have reported that TNF increases PKC activity as early as 10 min in A549 cells (Chen et al., 2001), there is currently little information as to the PKC isoforms regulated by TNF in A549 cells. Therefore, the specific novel PKCs regulated by TNF in our system were investigated. To this end, antibodies specific to phosphorylated forms of novel PKCs were used. These sites, previously reported to correspond to active forms of the enzymes, were Thr-505 for PKC-δ (Le Good et al., 1998), Ser-729 for PKC-ε (Cenni et al., 2002), and Thr-538 for PKC-θ (Liu et al., 2002). A549 cells were stimulated with TNF for various time between 0 and 60 min, and phosphorylation state was probed by immunoblot (Fig. 8). For normalization, blots were reprobed for total PKC levels. As can be seen, PKC-δ was basally phosphorylated to some extent at Thr-505, as reported previously (Le Good et al., 1998). Stimulation with TNF increased this phosphorylation, peaking at 30 min at 1.47 ± 0.12-fold over basal, with levels returning toward that of unstimulated cells by 60 min (Fig. 8A). In contrast to this, phosphorylation of PKC-δ at Tyr-311, known to be regulated by H2O2 (Konishi et al., 2001) was not observed at any time point (data not shown). It is noteworthy that, phosphorylation of PKC-ε at Ser-729 was detected neither basally nor on stimulation (Fig. 8B, top). In addition, whereas basal phosphorylation of PKC-θ at Thr-538 was observed, this was not significantly affected by TNF at any of the time points investigated (Fig. 8B, lower). Taken together, these data suggest that PKC-δ is the major novel PKC regulated by TNF in A549 cells, consistent with its role in regulating nSMase2.
Fig. 8.

PKC-δ is the major novel PKC regulated by TNF in A549 cells. A549 cells (200K) were seeded in 60-mm dishes and, 48 h later, media were changed. Cells were incubated for 1 to 2 h and then stimulated with TNF (50 ng/ml) for 0 to 60 min as indicated. Total protein samples were prepared in the presence of phosphatase inhibitors and analyzed for phospho-PKC-δ (Thr-505) (A), phospho-PKC-ε (Ser-729) and phospho-PKC-θ (Thr-538) (B) as described under Materials and Methods. Blots were reprobed with total PKC antibodies as loading controls.
PKC-δ Is Upstream of VCAM and ICAM Induction in A549 Cells
The results above suggest that PKC-δ is an upstream regulator of nSMase2. Previously, nSMase2 was reported as upstream of TNF-stimulated VCAM and ICAM induction in A549 cells (Clarke et al., 2007). Therefore, as a measurable functional consequence, the effects of PKC siRNA on VCAM and ICAM induction were investigated (Fig. 9). A549 cells treated with siRNA to PKC-δ and -ε for 48 to 60 h were stimulated with TNF (3 nM, 3 h), and VCAM and ICAM levels were analyzed by immunoblot (Fig. 9A). As can be seen, siRNA to PKC-δ resulted in significant reduction of VCAM and ICAM induction at 3 h of TNF stimulation (41 ± 2% for ICAM, p < 0.04; 31 ± 5% for VCAM, p < 0.01). This is consistent with the effects of nSMase2 reported previously (Clarke et al., 2007) and further supports a role for PKC-δ as an upstream regulator of nSMase2. In contrast, PKC-ε siRNA had no effect on ICAM levels but did have a modest effect on VCAM levels (25 ± 4%, p < 0.05). Because PKC-ε did not seem to be activated at early time points (Fig. 8B), it is possible that this isoform may play a regulatory role at later time points of TNF stimulation.
Fig. 9.

PKC-δ is upstream of TNF-stimulated VCAM and ICAM induction in A549 cells. A549 cells were seeded in 60-mm dishes (70 × 103) and 24 h later were transfected with negative control (Scr), PKC-δ, or -ε siRNA (20 nM). After 48 to 60 h, cells were stimulated with vehicle or TNF (3 nM) for 3 h, and total protein was extracted and analyzed for VCAM and ICAM levels by immunoblot. A, representative immunoblot; B, quantification of effect of PKC siRNA on ICAM (*, P < 0.05, n = 4); C, quantification of effect of PKC siRNA on VCAM (*, P < 0.05, n = 4).
Discussion
In this study, PKC-δ was implicated as an upstream regulator of nSMase2 translocation to the PM in PMA- and TNF-stimulated A549 cells. Moreover, this regulation does not occur through activation of p38 MAPK, previously identified as a regulator of nSMase2 (Clarke et al., 2007). A schematic of the major conclusions from the results obtained is shown (Fig. 10). Taken together, these data shed light on the regulation of nSMase2 in TNF responses and implicate nSMase2 as a point of cross-talk between the PKC and sphingolipid signaling pathways.
Fig. 10.
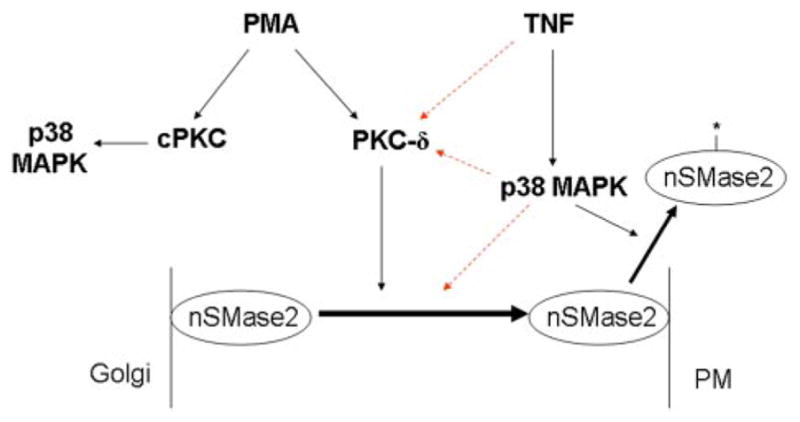
PKC-δ is upstream of nSMase2 but is not upstream of p38 MAPK in TNF and PMA responses. TNF-induced translocation of nSMase2 from the Golgi to the PM requires both p38 MAPK and PKC-δ. However, although p38 MAPK is upstream of nSMase2 activation by post-translational modification (*, nSMase2), PKC-δ does not play a role. The colored dashed lines represent three possibilities: 1) TNF activates PKC-δ independently of p38 MAPK, 2) p38 MAPK may regulate nSMase2 through activation of PKC-δ, or 3) p38 MAPK regulates nSMase2 independently of PKC-δ activation. With PMA, nSMase2 translocation is independent of p38 MAPK, and activation of p38 MAPK is downstream of a classic PKC (cPKC) isoform in a pathway distinct from the PKC-δ-mediated regulation of nSMase2.
Using a combination of pharmacological inhibitors and direct activation of PKC by PMA, nSMase2 was identified as a PKC-regulated enzyme. The effects of specific inhibitors together with the higher doses of PMA required for nSMase2 translocation suggested that this was mediated by a novel PKC. Upon further investigation with specific siRNA, PKC-δ was identified as the novel PKC isoform involved in regulating nSMase2 translocation to the PM in response to both PMA and TNF. Previous research has suggested that nSMase2 translocation to the PM is important for its signaling functions (Karakashian et al., 2004; Marchesini et al., 2004; Levy et al., 2006; Clarke et al., 2007). Consistent with this, siRNA down-regulation of PKC-δ decreased TNF-stimulated induction of VCAM and ICAM in A549 cells, matching what we have previously reported for nSMase2 (Clarke et al., 2007). Taken together, these data implicate PKC-δ as a positive regulator of nSMase2 in A549 cells.
Although previous studies have reported regulation of N-SMase by PKC, these have focused solely on endogenous N-SMase activities without explicit identification of the specific enzyme involved owing to a lack of molecular tools. It is noteworthy that PMA-sensitive PKC isoforms inhibited N-SMase activation by interleukin-1β in mesangial cells (Kaszkin et al., 1998), and PKC-δ negatively regulated N-SMase activity in HL-60 cells. In that study, pretreatment of cytosol extracts with PMA suppressed vitamin D3 stimulation of N-SMase activity, but depletion of the novel PKC-δ from the cytosol using specific antibodies prevented this. It is noteworthy that similar depletion of the classic PKC-α prevented vitamin D3 stimulation of N-SMase activity (Visnjic et al., 1999), suggesting it functions upstream of N-SMase. These differences could be attributed to both cell type and stimuli used in these studies as well as possible divergent roles of PKC isoenzymes. Moreover, because both studies investigated endogenous (generic) N-SMase activities and the latter study in HL-60 cells focused on the cytosolic Mg2+-independent N-SMase activity, the contribution of nSMase2 to these effects is not clear.
The current study also clarifies the mutual roles of p38 MAPK and PKC-δ in regulation of nSMase2. As we have previously reported, p38 MAPK acted upstream of nSMase2 translocation in TNF responses (Clarke et al., 2007), so the relationship between PKC, p38 MAPK, and nSMase2 was further explored. It is noteworthy that PKC-δ did not seem to regulate nSMase2 through activation of p38 MAPK, as indicated by a number of lines of evidence: 1) p38 MAPK was not required for PMA-induced nSMase2 translocation of nSMase2; 2) classic PKCs act upstream of p38 MAPK in PMA-stimulated A549 cells, consistent with the idea that this pathway is distinct from the novel PKC-mediated regulation of nSMase2; 3) neither inhibition nor siRNA down-regulation of novel PKCs has significant effects on basal or TNF-stimulated p38 MAPK activation; and 4) TNF stimulation of N-SMase activity was PKC-δ-independent but was previously shown to require p38 MAPK (Clarke et al., 2007). Taken together, these points constitute strong evidence that p38 MAPK activation in TNF-stimulated A549 cells is independent of both novel and classic PKCs, agreeing with studies in neutrophils (Konishi et al., 2001) and monocytes (Nguyen et al., 2006). However, this is contrary to previous research in A549 cells, where inhibitors and antisense oligonucleotides implicated PKC-δ upstream of p38 MAPK in TNF responses (Woo et al., 2005). This might be due to the different TNF concentrations used (10 ng/ml there versus 50 ng/ml here). Although these results suggested that PKC-δ and p38 MAPK may regulate nSMase2 independently, the possibility that p38 MAPK may be upstream of PKC-δ in regulating nSMase2 cannot be ruled out.
PKC-δ was the major novel PKC regulated by TNF in A549 cells, as evidenced by increased phosphorylation at Thr-505. This agrees with previous work in A549 cells (Woo et al., 2005) and in bronchial epithelial cells, where TNF increased PKC-δ activity (Page et al., 2003). In addition, the lack of effect of TNF on both PKC-ε and -θ provides additional evidence against a role of these isoforms in regulating nSMase2. Because phosphorylation of these residues on -ε and -θ is reported to correlate with the active enzyme (Cenni et al., 2002; Liu et al., 2002), this also suggests that TNF does not activate either isoform in A549 cells.
Despite the identification of PKC-δ (here) and p38 MAPK (Clarke et al., 2007) as regulators of nSMase2 translocation, the mechanism by which nSMase2 translocates remains unclear. However, nSMase2 is a membrane-associated protein with two hydrophobic segments; thus, fundamental principles indicate that it must move as part of membrane trafficking. Moreover, nSMase2 is palmitoylated, as previously reported by our laboratory (Tani and Hannun, 2007), but the palmitoylation status of nSMase2 does not change on PMA or TNF stimulation (Tani and Hannun, unpublished observation). Therefore, regulated palmitoylation does not play a role in nSMase2 translocation. The basal localization of nSMase2 to the Golgi complex suggests a role for Golgi-derived vesicles in an exocytic pathway. Consistent with this, V5-nSMase2 colocalizes with both p230 and TGN46 (Supplemental Fig. 1), markers of the trans-Golgi network and trans-Golgi network-derived vesicles. Furthermore, p38 MAPK has been implicated in Golgi to PM transport of the bile salt export pump in HepG2 cells and rat hepatocytes (Kubitz et al., 2004). Although PKCs have yet to be implicated in transport of proteins from Golgi to PM, studies have suggested they play a regulatory role in sphingolipid transport to the apical PM of hepatoma cells (Zegers and Hoekstra, 1997). Thus, we speculate that PM translocation of nSMase2 occurs through trans-Golgi network-derived vesicles. It is noteworthy that nSMase2 also contains two putative recycling endosome motifs (N. Marchesini, T.-G. Truong, and Y.A. Hannun, unpublished observations), suggesting that the recycling endosomes may be important in regulating nSMase2 localization. Current studies in our laboratory are investigating this mechanism further.
It is noteworthy that although implicated in nSMase2 translocation, PKCs were not necessary for the observed increase in N-SMase activity in A549 cells. Direct activation of PKC by PMA had no effect on endogenous N-SMase activity, which is consistent with previous research in MCF-7 cells (Zeidan and Hannun, 2007) but conflicts with results of studies in mesangial cells (Kaszkin et al., 1998). This is probably due to cell specific differences, either in PKC isoenzymes or in N-SMase isoforms present. We were surprised to find that persistent PMA stimulation decreased activity of overexpressed nSMase2. Because the observed decrease (30–60 min) is temporally distinct from PMA-induced translocation (5–10 min), this suggests that nSMase2 translocation and activation are distinct processes. This hypothesis is further supported by the role of PKC-δ in TNF-induced translocation, but not activation, of nSMase2. However, it should be noted that “activation” in this case most likely denotes a post-translational modification, because it persists after cell lysis (which would usually dissociate allosteric regulators). This “activation” by TNF might not be necessary for nSMase2 activation in cells. Indeed, translocation itself may be a sufficient mechanism of cellular activation by relocating the enzyme to contact and act on substrate.
Cross-talk among different signaling pathways is often important for amplification and specificity of agonist responses, and there has been considerable study on the crosstalk between PKC and sphingolipid pathways. This study provides further evidence that PKC-δ is a primary point of cross-talk between these two pathways. Previous studies have found roles for PKC-δ in regulation of acid SMase activation and subsequent ceramide production in MCF-7 cells (Zeidan and Hannun, 2007). In addition, ceramide is also known to have effects on PKC-δ. In leukemia cells, ceramide treatment or generation by SMase caused PKC-δ translocation to the cytosol, whereas in HeLa cells, ceramide caused PKC-δ activation subsequent to Golgi translocation (Lee et al., 2000; Kajimoto et al., 2004). It is noteworthy that membrane translocation of PKC-δ in interferon-γ-stimulated HeLa cells and on chemotherapeutic stimulation of C6 glioma cells required N-SMase activation (Kajimoto et al., 2001; Peng et al., 2006). When considered with data presented here, this raises the interesting possibility that N-SMases might function both upstream and downstream of PKC-δ; this is currently the subject of ongoing studies. Finally, by identifying the connection between PKC-δ and nSMase2, this offers insight into the mechanisms by which some PKC-δ-mediated functions may occur.
In conclusion, this study has identified a role for PKC-δ as a regulator of nSMase2 translocation to the PM in TNF-stimulated A549 cells. This offers further insight into the regulation of nSMase2 and, for the first time, identifies a specific N-SMase as a point of cross-talk between PKC and sphingolipid pathways.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grant GM43825 (Y.A.H.) and a Mid-Atlantic Affiliate Postdoctoral Fellowship from the American Heart Association (C.J.C.)
We thank Drs. Nana Bartke, Ashley Snider, and Jolanta Idkowiak-Baldys for careful reading and advice on the manuscript. We are also grateful to Dr. Lina Obeid for advice. We also thank Hollings Cancer Center Molecular Imaging Facility for use of the confocal microscope.
ABBREVIATIONS
- SMase
sphingomyelinase
- N-SMase
neutral sphingomyelinase
- PM
plasma membrane
- TNF
tumor necrosis factor-α
- PKC
protein kinase C
- DAG
diacylglycerol
- PS
phosphatidylserine
- PMA
phorbol 12-myristate 13-acetate
- MAPK
mitogen-activated protein kinase
- siRNA
small interfering RNA
- Scr
scrambled
- PBS
phosphate-buffered saline
- ICAM
intercellular adhesion molecule
- VCAM
vascular cell adhesion molecule
- Go6976
12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole
- SB202190
4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole
Footnotes
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
References
- Aziz MH, Manoharan HT, Verma AK. Protein kinase Cε, which sensitizes skin to sun’s UV radiation-induced cutaneous damage and development of squamous cell carcinomas, associates with Stat3. Cancer Res. 2007;67:1385–1394. doi: 10.1158/0008-5472.CAN-06-3350. [DOI] [PubMed] [Google Scholar]
- Bezombes C, de Thonel A, Apostolou A, Louat T, Jaffrézou JP, Laurent G, Quillet-Mary A. Overexpression of protein kinase Cζ confers protection against antileukemic drugs by inhibiting the redox-dependent sphingomyelinase activation. Mol Pharmacol. 2002;62:1446–1455. doi: 10.1124/mol.62.6.1446. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cenni V, Döppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A. Regulation of novel protein kinase C ε by phosphorylation. Biochem J. 2002;363:537–545. doi: 10.1042/0264-6021:3630537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MS, Chen BC, Yu MT, Sheu JR, Chen TF, Lin CH. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell Signal. 2005;17:299–310. doi: 10.1016/j.cellsig.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Chen C, Chou C, Sun Y, Huang W. Tumor necrosis factor α-induced activation of downstream NF-kB site of the promoter mediate epithelial ICAM-1 expression and monocyte adhesion: involvement of PKCα, tyrosine kinase, and IKK2, but not MAPKs, pathway. Cell Signal. 2001;13:543–553. doi: 10.1016/s0898-6568(01)00171-1. [DOI] [PubMed] [Google Scholar]
- Clarke CJ, Truong TG, Hannun YA. Role for neutral sphingomyelinasee-2 in tumor necrosis factor α-stimulated expression of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in lung epithelial cells: p38 MAPK is an upstream regulator of nSMase2. J Biol Chem. 2007;282:1384–1396. doi: 10.1074/jbc.M609216200. [DOI] [PubMed] [Google Scholar]
- Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M. Ceramide recruits and activates protein kinase C ζ (PKC ζ) within structured membrane microdomains. J Biol Chem. 2007;282:12450–12457. doi: 10.1074/jbc.M700082200. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Müller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–25850. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Tomiuk S, Wolff G, Stoffel W. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc Natl Acad Sci U S A. 2000;97:5895–5900. doi: 10.1073/pnas.97.11.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang BC, Choi ES, Im KJ, Baek WK, Kwon TK, Suh MH, Kim SP, Park JW, Suh SI. Induction of apoptosis by Se-MSC in U937 human leukemia cells through release of cytochrome c and activation of caspases and PKC-delta: mutual regulation between caspases and PKC-delta via a positive feedback mechanism. Int J Mol Med. 2003;12:733–739. [PubMed] [Google Scholar]
- Kajimoto T, Ohmori S, Shirai Y, Sakai N, Saito N. Subtype-specific translocation of the δ subtype of protein kinase C and its activation by tyrosine phosphorylation induced by ceramide in HeLa cells. Mol Cell Biol. 2001;21:1769–1783. doi: 10.1128/MCB.21.5.1769-1783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto T, Shirai Y, Sakai N, Yamamoto T, Matsuzaki H, Kikkawa U, Saito N. Ceramide-induced apoptosis by translocation, phosphorylation and activation of protein kinase C-δ in the Golgi complex. J Biol Chem. 2004;279:12668–12676. doi: 10.1074/jbc.M312350200. [DOI] [PubMed] [Google Scholar]
- Karakashian AA, Giltiay NV, Smith GM, Nikolova-Karakashian MN. Expression of neutral sphingomyelinase-2 (NSMase-2) in primary rat hepatocytes modulates IL-beta-induced JNK activation. FASEB J. 2004;18:968–970. doi: 10.1096/fj.03-0875fje. [DOI] [PubMed] [Google Scholar]
- Kaszkin M, Huwiler A, Scholz K, van den Bosch H, Pfeilschifter J. Negative regulation of interleukin-1β-activated neutral sphingomyelinase by protein kinase C in rat mesangial cells. FEBS Lett. 1998;440:163–166. doi: 10.1016/s0014-5793(98)01445-8. [DOI] [PubMed] [Google Scholar]
- Kazanietz MG, Areces LB, Bahador A, Mischak H, Goodnight J, Mushinski JF, Blumberg PM. Characterization of ligand and substrate specificity for the calcium-dependent and calcium-independent protein kinase C isozymes. Mol Pharmacol. 1993;44:298–307. [PubMed] [Google Scholar]
- Kilpatrick LE, Sun S, Mackie D, Baik F, Li H, Korchak HM. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: role of delta-PKC and ERK1/2. J Leukocyte Biol. 2006;80:1512–1521. doi: 10.1189/jlb.0406284. [DOI] [PubMed] [Google Scholar]
- Kitatani K, Idkowiak-Baldys J, Bielawski J, Taha TA, Jenkins RW, Senkal CE, Ogretmen B, Obeid LM, Hannun YA. Protein kinase C-induced activation of a ceramide/protein phosphatase 1 pathway leading to dephosphorylation of p38 MAPK. J Biol Chem. 2006;281:36793–36802. doi: 10.1074/jbc.M608137200. [DOI] [PubMed] [Google Scholar]
- Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, Ohmae K, Kikkawa U, Nishizuka Y. Phosphorylation sites of protein kinase C δ in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci U S A. 2001;98:6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krut O, Wiegmann K, Kashkar H, Yazdanpanah B, Kronke M. Novel tumor necrosis factor-responsive mammalian neutral sphingomyelinase-3 is a C-tail-anchored protein. J Biol Chem. 2006;81:13784–13793. doi: 10.1074/jbc.M511306200. [DOI] [PubMed] [Google Scholar]
- Kubitz R, Sütfels G, Kühlkamp T, Kölling R, Häussinger D. Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology. 2004;126:541–553. doi: 10.1053/j.gastro.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Lee JY, Hannun YA, Obeid LM. Functional dichotomy of protein kinase C in tumor necrosis factor-α (TNF-α) signal transduction in L929 cells. J Biol Chem. 2000;275:29290–29298. doi: 10.1074/jbc.M000170200. [DOI] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Levy M, Castillo SS, Goldkorn T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem Biophys Res Commun. 2006;344:900–905. doi: 10.1016/j.bbrc.2006.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Graham C, Li A, Fisher RJ, Shaw S. Phosphorylation of the protein kinase C-theta activation loop and hydrophobic motif regulates its kinase activity, but only activation loop phosphorylation is critical to in vivo nuclear-factor-kB induction. Biochem J. 2002;361:255–265. doi: 10.1042/bj3610255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini N, Osta W, Bielawski J, Luberto C, Obeid LM, Hannun YA. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J Biol Chem. 2004;279:25101–25111. doi: 10.1074/jbc.M313662200. [DOI] [PubMed] [Google Scholar]
- Musashi M, Ota S, Shiroshita N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int J Hematol. 2000;72:12–19. [PubMed] [Google Scholar]
- Nguyen J, Gogusev J, Knapnougel P, Bauvois B. Protein tyrosine kinase and p38 MAP kinase pathways are involved in stimulation of matrix metallopro-teinase-9 by TNF-alpha in human monocytes. Immunol Lett. 2006;106:34–41. doi: 10.1016/j.imlet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Nomiyama Y, Tashiro M, Yamaguchi T, Watanabe S, Taguchi M, Asaumi H, Nakamura H, Otsuki M. High glucose activates rat pancreatic stellate cells through protein kinase C and p38 mitogen-activated protein kinase pathway. Pancreas. 2007;34:364–372. doi: 10.1097/MPA.0b013e31802f0531. [DOI] [PubMed] [Google Scholar]
- Nomura N, Nomura M, Sugiyama K, Hamada JI. Phorbol 12-myristate 13-acetate (PMA)-induced migration of glioblastoma cells is mediated via p38MAPK/Hsp27 pathway. Biochem Pharmacol. 2007;74:690–701. doi: 10.1016/j.bcp.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB, et al. Regulation of airway epithelial cells NF-kB-dependent gene expression by protein kinase Cδ. J Immunol. 2003;170:5681–5689. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- Parker PJ, Murray-Rust J. PKC at a glance. J Cell Sci. 2004;117:131–132. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- Peng CH, Huang CN, Hsu SP, Wang CJ. Penta-acetyl geniposide induced apoptosis in C6 glioma cells by modulating the activation of neutral sphingomyelinase-induced p75 never growth factor receptor and protein kinase Cδ pathway. Mol Pharmacol. 2006;70:997–1004. doi: 10.1124/mol.106.022178. [DOI] [PubMed] [Google Scholar]
- Tani M, Hannun YA. Neutral sphingomyelinase 2 is palmitoylated on multiple cysteine residues. Role of palmitoylation in subcellular localization. J Biol Chem. 2007;282:10047–10056. doi: 10.1074/jbc.M611249200. [DOI] [PubMed] [Google Scholar]
- Visnjic D, Batinić D, Banfić H. Different roles of protein kinase C alpha and delta isoforms in the regulation of neutral sphingomyelinase activity in HL-60 cells. Biochem J. 1999;344:921–928. doi: 10.1042/0264-6021:3440921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CH, Lim JH, Kim JH. VCAM-1 upregulation via PKCδ-p38 kinase-linked cascade mediates the TNF-α-induced leukocyte adhesion and emigration in the lung airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2005;288:L307–L316. doi: 10.1152/ajplung.00105.2004. [DOI] [PubMed] [Google Scholar]
- Zegers MM, Hoekstra D. Sphingolipid transport to the apical plasma membrane domain in human hepatoma cells is controlled by PKC and PKA activity: a correlation with cell polarity in HepG2 cells. J Cell Biol. 1997;138:307–321. doi: 10.1083/jcb.138.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidan YH, Hannun YA. Activation of acid sphingomyelinase by protein kinase Cδ-mediated phosphorylation. J Biol Chem. 2007;282:11549–11561. doi: 10.1074/jbc.M609424200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.