

Abstract
Oral dictyoceratin-C (1) and A (2), hypoxia-selective growth inhibitors, showed potent in vivo antitumor effects in mice subcutaneously inoculated with sarcoma S180 cells. Structurally modified analogs were synthesized to assess the structure–activity relationship of the natural compounds 1 and 2 isolated from a marine sponge. Biological evaluation of these analogs showed that the exo-olefin and hydroxyl and methyl ester moieties were important for the hypoxia-selective growth inhibitory activities of 1 and 2. Thus far, only substitution of the methyl ester with propargyl amide in 1 was found to be effective for the synthesis of probe molecules for target identification.
Keywords: dictyoceratin-C, dictyoceratin-A, hypoxia-selective growth inhibitor, marine sponge, analog synthesis, structure-activity relationship, in vivo antitumor effect
1. Introduction
Marine natural products have attracted attention in recent years as a rich and promising source of drug candidates, especially in the field of anticancer drug discovery [1]. In the majority of cases, only small quantities of the compounds could be isolated from the extracts of marine organisms such as sponges and tunicates; therefore, sustainable supply is the major limitation for further evaluation of these compounds and drug development. One of the solutions to address this issue is chemical synthesis of the active compounds and their analogs. Structure-activity relationship studies and syntheses of the truncated natural products give us further opportunities to generate more promising drug leads with optimized activity, chemical stability, and accessibility [2,3].
It is widely accepted that hypoxia aggravates tumorigenesis by promoting tumor growth, angiogenesis, and metastasis, or by inducing resistance to chemotherapy and irradiation [4]. Therefore, compounds exhibiting hypoxia-selective growth inhibitory activity could be novel and promising drug leads for anticancer drug development [5], and the adaptation factors of tumor cells to hypoxia environment, with particular regard to hypoxia inducible factor-1 (HIF-1), have been extensively investigated as drug targets for cancer chemotherapy.
In our continuing search for bioactive compounds from marine organisms, we isolated dictyoceratin-C (1) [6] from the Indonesian marine sponge Dactylospongia elegans as a hypoxia-selective growth inhibitor, and found that dictyoceratin-A (2) [7] exhibited a similar biological activity (Figure 1). These two sesquiterpene phenols inhibited the proliferation of human prostate cancer DU145 cells selectively under hypoxic condition in a dose-dependent manner at concentrations ranging from 1.0 to 10 μM, by inhibiting the accumulation of HIF-1α under hypoxic condition [8].
Figure 1.
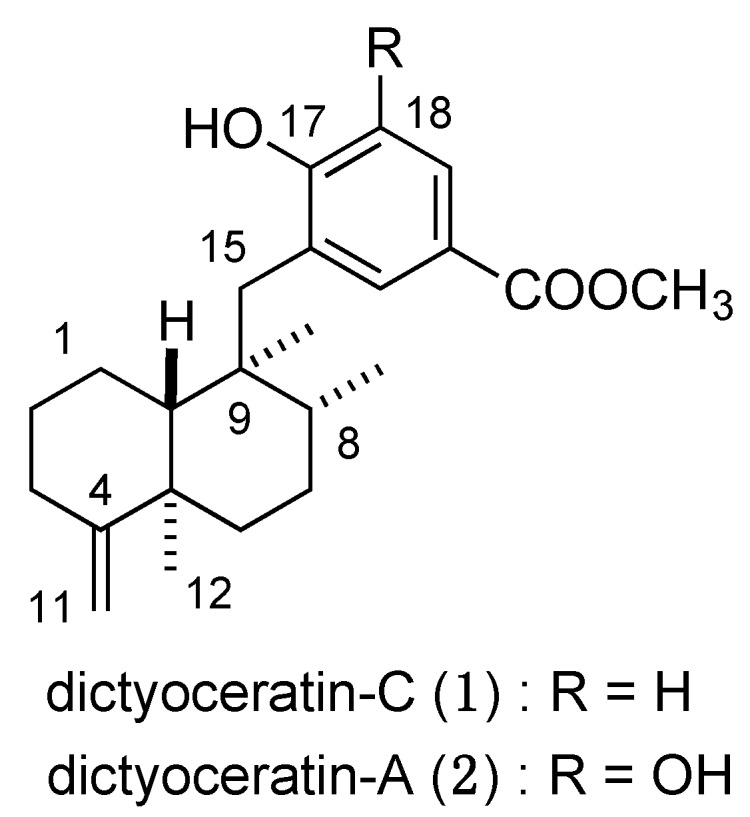
Chemical structures of (+)-dictyoceratin-C (1) and -A (2).
To obtain compounds in sufficient yields for further evaluation, we recently reported the enantioselective total synthesis of 1 and 2, with confirmation of absolute stereochemistry [9]. Moreover, we found that unnatural enantiomers of 1 and 2 also showed similar hypoxia-selective growth inhibitory activity against DU145 cells. It implies that the pharmacophore of these compounds is the para-hydroxybenzoyl ester moiety of the side chain and the chiral decalin moiety might not play a crucial role [9]. However, more precise structure-activity relationship (SAR) information is needed to develop promising anticancer drug leads based on these compounds. In this study, we investigated the in vivo antitumor effect of compounds 1 and 2 and analyzed their SAR through design and synthesis of various analog compounds.
2. Results and Discussion
2.1. In Vivo Antitumor Activity of 1 and 2
Enantioselective total synthesis of 1 and 2 yielded hundreds of milligrams of these compounds [9]. In order to verify the potential of these compounds as promising drug leads for cancer treatment, we examined their in vivo antitumor activity in mice subcutaneously inoculated with sarcoma S180 cells.
The compounds were orally administered every other day for two weeks, and the effectiveness of the compounds was determined by weighing the surged tumor on the day after final administration. Both compounds at 10–50 mg/kg inhibited the growth of implanted tumors, with ~90% reduction of the tumor weight at 50 mg/kg relative to respective control (Figure 2). Moreover, no significant acute toxicities, such as weight loss or diarrhea, were observed during the study period for both compounds. This result indicated that compounds 1 and 2 are potential anticancer drug leads.
Figure 2.

In vivo antitumor effect of dictyoceratin-C (1) and A (2). (a) Mean ± SD of tumor weight of each group. * p < 0.05; (b) Images of surged tumors after two weeks.
2.2. Design and Synthesis of Structure-Modified Analogs
SAR study of 1 or 2 was performed to identify the important moiety for their biological activities. In the initial SAR study of some natural sesquiterpene phenols/quinones isolated from sponge extracts, para-hydroxybenzoyl ester moiety was found to play a significant role in the hypoxia-selective growth inhibitory activity [8,9]. In order to examine the SAR in detail, we synthesized analogs of 1 or 2 by focusing on certain particular functional groups, such as methyl ester or hydroxyl group of the aromatic ring and exo-olefin or C-8 methyl groups in the decalin part (Figure 3).
Figure 3.

Modification of functional groups for structure-activity relationship (SAR).
2.2.1. Modification of Aromatic Ring
Involvement of the functionality of the aromatic ring was analyzed. To elucidate the functional group crucial for the bioactivity and to develop more potent compounds, modification of the ester group and hydroxyl group of 1 was executed (Scheme 1).
Scheme 1.

Synthesis of aromatic ring-modified analogs. Reagents and conditions: (a) Ph3PCH3Br, KHMDS, THF, 94%; (b) SOCl2, octanol, 50 °C, 13%; (c) EDCI·HCl, HOBt, propargylamine, DMF, 58%; (d) MeI, K2CO3, DMF, quant.; (e) propargyl bromide, K2CO3, DMF, 8: 35%, 9: 13%; (f) n-nonyl bromide, K2CO3, DMF, 10: 21%, 11: 14%.
First, octyl ester analog 5 and propargyl amide analog 6 were synthesized from compound 3, an intermediate in the total synthesis of 1 [9], through Wittig olefination and subsequent condensation with n-octanol or propargylamine, respectively. Further, the phenolic hydroxyl group of 1 was alkylated with MeI and K2CO3 to provide a methyl ether analog 7.
Second, participation of the respective hydroxyl groups of 2 was examined. In this analysis, propargyl and nonyl ether were selected to determine the relationship between lipophilicity/steric bulkiness and bioactivity. Alkylation of the diol group of 2 via a general method (alkyl bromide and K2CO3 in DMF) yielded a mixture of two mono-alkylated compounds, together with di-alkylated compound as a major product; a selective mono-alkylation condition could not be achieved. Fortunately, each mono-alkylated compound could be separated by open column chromatography to yield 17-O-alkylated analogs (8,10) and 18-O-alkylated analogs (9,11), respectively.
2.2.2. Modification of Decalin Part
Next, modification of the decalin part was examined. Many reaction steps were needed to elaborate the decalin part in a chemo- and stereoselective manner, mainly for the construction of 8α-methyl group and relatively unstable exo-olefin at C-4. Therefore, synthetic study of the truncated natural compounds might be effective for obtaining analogs with improved accessibility or stability or both.
First, modifications of the exo-olefin at C-4 of 1 were attempted (Scheme 2). Hydrogenation of 1 yielded 3,4-dihydro analog (12) as a diastereomixture (1:1), which was separated by HPLC to provide 12a (4S) and 12b (4R). Stereochemistry of each compound was determined by NOE experiment. Isomerization of the exo-olefin of 1 was accomplished by treatment with RhCl3·H2O in refluxing ethanol [10] to yield compound 13 having an internal olefin.
Scheme 2.

Synthesis of exo-methylene-modified analogs. Reagents and conditions: (a) H2, Pd-C, MeOH (12a: 51%, 12b: 47%); (b) RhCl3·H2O, EtOH, reflux, 61%.
Next, transformation of the 8-methyl group of 1 was also examined (Scheme 3). We designed and synthesized 8-hydroxyl and 8-desmethyl analogs from 8-keto compound 14, an intermediate in the total synthesis of 1. Reduction of the ketone of 14 by NaBH4 yielded compound 15 as a single diastereomer. Multiplicity of the newly formed oxymethine proton signal in the 1H-NMR spectrum of 15 was ddd (J = 10.3, 5.2, 3.4 Hz), indicating that the reduction proceeded selectively from the β-side of the molecule. Removal of all protecting groups of 15 yielded a benzoic acid 16, and subsequent treatment with SOCl2 in MeOH followed by Wittig olefination provided an 8-hydroxy analog 18. Conversely, removal of the hydroxyl group of 15 was achieved with the Barton–McCombie deoxygenation reaction [11] to yield 20, which was converted in the same manner as analog 18 to yield an 8-desmethyl analog 23.
Scheme 3.

Synthesis of 8-methyl group-modified analogs. Reagents and conditions: (a) NaBH4, CeCl3·7H2O, MeOH, 94%; (b) 80% TFA, THF, 50 °C, 16: 97%, 21: 93%; (c) SOCl2, MeOH, 50 °C, 17: 98%, 22: 87%; (d) Ph3PCH3Br, KHMDS, THF, 18: 39%, 23: 95%; (e) NaH, CS2, THF, rt, then MeI, 50 °C, 94%; (f) nBu3SnH, AIBN, toluene, 80 °C, 86%.
2.3. Biological Evaluation of Synthetic Analogs
Following the synthesis of the desired analogs, growth inhibitory activities of the analogs against DU145 cells were evaluated under normoxic and hypoxic conditions. As briefly summarized in Figure 4, the majority of the analogs lost the hypoxia-selective growth inhibitory activity of the parent natural product. Thus, methyl ether analog 7, 17-O-propargyl analog 8, and 18-O-propargyl analog 9 did not possess hypoxia-selectivity. These results suggested that the 17-hydroxyl group might be necessary for hypoxia selectivity and the 18-hydroxyl group was partially concerned with growth inhibitory activity. In contrast, octyl ester analog 5, 17-O-nonyl ether analog 10, and 18-O-nonyl analog 11 showed only weak hypoxia-selective growth inhibitory activity. Because these analogs were highly lipophilic, this significant change in their physical properties might affect their biological activity. Furthermore, all the exo-olefin-modified analogs (12a,12b,13) and 8-hydroxyl analog (18) lost hypoxic/normoxic selectivity, while 8-desmethyl analog 23 showed moderate hypoxia-selective growth inhibitory activity. These results indicated that the exo-olefin might be an essential moiety for hypoxia-selective growth inhibitory activity and the 8-methyl group also affected growth inhibition. Taken together, not only the para-hydroxybenzoyl moiety but also the decalin part, including 4-exo-olefin and 8-methyl groups, were essential for the hypoxia-selective growth inhibitory activity of 1 and 2. Of the synthesized analogs, only the propargyl amide analog 6 showed excellent hypoxia-selective growth inhibitory activity. In particular, potent and hypoxia-selective growth inhibitory activity was observed at 30 µM relative to the parent compound (Figure 5).
Figure 4.

Growth inhibitory activity of dictyoceratin analogs against DU145 cells. * p < 0.05.
Figure 5.

Growth inhibitory activity of dictyoceratin-C (1) and propargyl amide analog (6) against DU145 cells. * p < 0.05.
3. Experimental Section
3.1. General
The following instruments were used to obtain physical data: a JASCO P-2200 digital polarimeter (L = 50 mm) for specific rotations; a JEOL ECS-300 (1H-NMR: 300 MHz, 13C-NMR: 75 MHz), ECA-500 (1H-NMR: 500 MHz, 13C-NMR: 125 MHz) and a Varian NMR system (1H-NMR: 600 MHz, 13C-NMR: 150 MHz) spectrometer for 1H and 13C NMR data using tetramethylsilane or residual solvent peak as internal standards; a JASCO FT/IR-5300 infrared spectrometer for IR spectra; a Waters Q-Tof Ultima API mass spectrometer for ESI-TOF MS; HPLC was performed using a Hitachi L-6000 pump equipped with Hitachi L-4000H UV detector. Silica gel (Kanto, 40–100 μm) and pre-coated thin layer chromatography (TLC) plates (Merck, 60F254) were used for column chromatography and TLC. Spots on TLC plates were detected by spraying acidic p-anisaldehyde solution (p-anisaldehyde: 25 mL, c-H2SO4: 25 mL, AcOH: 5 mL, EtOH: 425 mL) or phosphomolybdic acid solution (phosphomolybdic acid: 25 g, EtOH: 500 mL) with subsequent heating. Unless otherwise noted, all the reaction was performed under N2 atmosphere. After workup, the organic layer was dried over Na2SO4. 1H and 13C NMR spectra of compounds 4–23 are available in the supplementary information.
3.2. In Vivo Anti-Tumor Effect of Dictyoceratin-C (1) and -A (2)
All procedures of animal experiment were approved as No. DOUYAKU24-8 by the Committee on Animal Experimentation of Osaka University. Murine sarcoma S180 cells (1 × 106 cells/body), suspended in the RPMI1640 without FBS and kanamycin, were subcutaneously implanted into the right ventral flank of female ddY mice of 6 weeks old. After one week from implantation, dictyoceratin-C (1) or -A (2) was orally administered on every other day for two week (total seven times) as a suspension in 1% sodium carboxymethyl cellulose (CMC-Na). Then, tumor was isolated and weighed to calculate the ratio of inhibition. The control group and the groups treated with 1 or 2 consisted of six mice each in this study.
3.3. Design and Synthesis of Structure-Modified Analogues
3.3.1. 4-Hydroxy-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoic acid (4)
Under an Ar atmosphere, KHMDS (5.9 mL, of a 0.5 M solution in toluene, 3.0 mmol) was added to a solution of Ph3PCH3Br (1.05 g, 3.0 mmol) in anhydrous THF (1.9 mL), and the whole mixture was stirred for 1 h at rt. The above solution (7.2 mL, about 2.7 mmol) was added dropwise to a solution of 3 (93.5 mg, 0.27 mmol) in anhydrous THF (0.9 mL) at 0 °C, and the whole mixture was stirred for 20 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 3:2) to give 4 (87.3 mg, 94%) as a white solid.
+33 (c = 1.22, MeOH). 1H-NMR (500 MHz, CDCl3) δ: 7.85–7.83 (2H, m), 6.76 (1H, d, J = 8.6 Hz), 4.42 (1H, s), 4.37 (1H, s), 2.70 (1H, d, J = 14.4 Hz), 2.63 (1H, d, J = 14.4 Hz), 2.34 (1H, td, J = 13.6, 5.1 Hz), 2.12–2.06 (2H, m), 1.95–1.91 (1H, m), 1.64–1.56 (1H, m), 1.48 (1H, dt, J = 12.4, 3.1 Hz), 1.44–1.19 (5H, m), 1.07 (3H, s), 1.04 (3H, d, J = 6.7 Hz), 0.95 (1H, dd, J = 12.2, 1.8 Hz), 0.89 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 171.9, 159.9, 159.4, 135.7, 130.1, 125.2, 121.2, 115.5, 102.9, 48.1, 42.1, 40.2, 37.1, 36.5, 36.3, 32.9, 27.9, 27.7, 23.3, 20.5, 17.62, 17.57. MS (ESI-TOF) m/z: 365 [M + Na]+. HRMS (ESI-TOF) m/z: 365.2093 calcd for C22H30O3Na; Found: 365.2083.
3.3.2. Octyl 4-hydroxy-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (5)
The components SOCl2 (6.0 µL, 0.083 mmol) and DMF (1 drop) were added to a solution of 4 (4.8 mg, 0.014 mmol) in anhydrous THF (0.2 mL), and the whole mixture was stirred for 1 h at rt. n-Octanol (6.0 µL, 0.083 mmol) was added to the mixture, and the whole mixture was stirred for 7 h at 50 °C. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 200:3:1, lower phase) to give 5 (0.8 mg, 13%) as a white solid.
+14 (c = 0.31, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.78 (1H, dd, J = 8.2, 1.9 Hz), 7.75 (1H, d, J = 1.9 Hz), 6.73 (1H, d, J = 8.2 Hz), 5.17 (1H, s), 4.41 (1H, s), 4.37 (1H, s), 4.31–4.20 (2H, m), 2.67 (1H, d, J = 14.3 Hz), 2.62 (1H, d, J = 14.3 Hz), 2.34 (1H, td, J = 13.7, 5.2 Hz), 2.08 (2H, t, J = 11.7 Hz), 1.94–1.91 (1H, m), 1.76–1.70 (2H, m), 1.60 (2H, qd, J = 13.2, 6.6 Hz), 1.47–1.41 (5H, m), 1.31–1.23 (10H, m), 1.07 (3H, s), 1.03 (3H, d, J = 6.9 Hz), 0.96 (1H, dd, J = 12.0, 1.7 Hz), 0.88 (3H, t, J = 6.9 Hz), 0.88 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 166.6, 159.9, 158.5, 134.8, 129.3, 124.8, 122.5, 115.3, 102.9, 64.8, 47.9, 42.0, 40.2, 37.0, 36.5, 36.2, 33.0, 31.8, 29.3, 29.2, 28.8, 27.9, 27.7, 26.1, 23.3, 22.7, 20.5, 17.63, 17.60, 14.1. IR (KBr) : 2926, 1672, 1599, 1279 cm−1. MS (ESI-TOF) m/z: 477 [M + Na]+. HRMS (ESI-TOF) m/z: 477.3345 calcd for C30H46O3Na; Found: 477.3348.
3.3.3. 4-Hydroxy-N-(prop-2-yn-1-yl)-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzamide (6)
Propargylamine (7.0 µL, 0.10 mmol), EDCI·HCl (18.0 mg, 0.094 mmol) and HOBt (14.2 mg, 0.11 mmol) were added to a solution of 4 (10.8 mg, 0.032 mmol) in anhydrous DMF (0.3 mL), and the whole mixture was stirred for 6 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 30:3:1, lower phase) to give 6 (7.2 mg, 60%) as a white solid.
+22.0 (c = 0.58, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 8.89 (1H, d, J = 4.0 Hz), 7.79 (1H, brs), 7.71 (1H, d, J = 2.3 Hz), 7.61 (1H, dd, J = 8.6, 2.3 Hz), 6.86 (1H, d, J = 8.6 Hz), 4.37 (1H, s), 4.33 (1H, s), 4.20–4.09 (2H, m), 2.72 (1H, d, J = 14.3 Hz), 2.68 (1H, d, J = 14.3 Hz), 2.63 (1H, t, J = 2.6 Hz), 2.35 (1H, td, J = 13.7, 5.2 Hz), 2.22 (1H, d, J = 13.2 Hz), 2.08–2.04 (1H, m), 1.90–1.85 (1H, m), 1.62–1.54 (1H, m), 1.51–1.36 (4H, m), 1.21–1.15 (1H, m), 1.07 (3H, s), 1.05 (3H, d, J = 5.7 Hz), 1.01 (1H, dd, J = 11.7, 2.0 Hz), 0.89 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 166.8, 160.7, 160.0, 133.6, 127.0, 126.0, 125.8, 115.4, 103.2, 81.8, 71.6, 48.9, 42.7, 40.9, 37.6, 37.5, 37.1, 33.7, 29.2, 28.8, 28.4, 23.7, 20.9, 18.1, 18.0. IR (KBr): 3299, 2921, 1630, 1588, 1490, 1268 cm−1. MS (ESI-TOF) m/z: 402 [M + Na]+. HRMS (ESI-TOF) m/z: 402.2409 calcd for C25H33NO2Na; Found: 402.2418.
3.3.4. Methyl 4-methoxy-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (7)
K2CO3 (7.5 mg, 0.054 mmol) and MeI (6.0 µL, 0.096 mmol) were added to a solution of 1 (6.8 mg, 0.018 mmol) in anhydrous DMF (0.2 mL), and the whole mixture was stirred for 12.5 h at rt. Sat. Na2S2O3 aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 8:1) to give 7 (7.6 mg, quantitative yield) as a white solid.
+23 (c = 0.68, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.87 (1H, dd, J = 8.9, 1.7 Hz), 7.74 (1H, d, J = 1.7 Hz), 6.82 (1H, d, J = 8.9 Hz), 4.40 (1H, s), 4.35 (1H, s), 3.87 (3H, s), 3.81 (3H, s), 2.68 (1H, d, J = 14.3 Hz), 2.65 (1H, d, J = 14.3 Hz), 2.33 (1H, td, J = 13.5, 5.2 Hz), 2.11–2.08 (2H, m), 1.95–1.92 (1H, m), 1.58–1.53 (2H, m), 1.45 (1H, dt, J = 12.4, 3.3 Hz), 1.41–1.30 (3H, m), 1.25–1.15 (1H, m), 1.06 (3H, s), 1.01 (3H, d, J = 6.3 Hz), 0.88 (1H, d, J = 12.6 Hz), 0.86 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.2, 162.1, 160.1, 134.1, 129.4, 127.4, 121.5, 109.6, 102.7, 55.1, 51.8, 48.0, 42.1, 40.2, 36.7, 36.6, 36.3, 33.1, 27.9, 27.7, 23.2, 20.6, 17.6, 17.5. IR (KBr): 2927, 1719, 1437, 1298, 1269 cm−1. MS (ESI-TOF) m/z: 393 [M + Na]+. HRMS (ESI-TOF) m/z: 393.2406 calcd for C24H34O3Na; Found: 393.2414.
3.3.5. Methyl 3-hydroxy-4-(prop-2-yn-1-yloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (8); Methyl 4-hydroxy-3-(prop-2-yn-1-yloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (9)
K2CO3 (13.3 mg, 0.096 mmol) and propargyl bromide (9.0 µL, of a 80 wt% in toluene, 0.10 mmol) were added to a solution of 2 (30.6 mg, 0.082 mmol) in anhydrous DMF (1.65 mL), and the whole mixture was stirred for 3 h at 0 °C and for 13 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 5:1) to give 8 (11.9 mg, 35%) as a colorless amorphous and 9 (4.3 mg, 13%) as a colorless amorphous.
8: +22 (c = 0.94, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.48 (1H, d, J = 2.0 Hz), 7.38 (1H, d, J = 2.0 Hz), 5.74 (1H, s), 4.59 (1H, dd, J = 15.5, 2.5 Hz), 4.53 (1H, dd, J = 15.5, 2.5 Hz), 4.40 (1H, s), 4.35 (1H, s), 3.87 (3H, s), 2.66 (1H, d, J = 13.7 Hz), 2.62 (1H, d, J = 13.7 Hz), 2.58 (1H, t, J = 2.5 Hz), 2.37–2.28 (1H, m), 2.10–2.07 (1H, m), 1.99 (1H, d, J = 13.2 Hz), 1.94–1.88 (1H, m), 1.57 (1H, qd, J = 13.0, 3.3 Hz), 1.45 (1H, dt, J = 12.4, 3.3 Hz), 1.42–1.33 (2H, m), 1.27–1.13 (3H, m), 1.05 (3H, s), 1.00 (3H, d, J = 6.3 Hz), 0.87 (3H, s), 0.84 (1H, dd, J = 12.0, 1.7 Hz). 13C-NMR (150 MHz, CDCl3) δ: 166.7, 159.8, 149.0, 148.8, 132.8, 126.2, 125.9, 115.2, 102.9, 78.2, 76.8, 61.0, 52.1, 47.8, 42.4, 40.1, 37.7, 36.4, 36.1, 32.9, 27.7, 27.5, 23.2, 20.6, 17.8, 17.6. IR (KBr): 3302, 2928, 1718, 1591, 1437, 1262 cm−1. MS (ESI-TOF) m/z: 433 [M + Na]+. HRMS (ESI-TOF) m/z: 433.2355 calcd for C26H34O4Na; Found: 433.2372.
9: +14 (c = 0.36, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.493 (1H, d, J = 2.0 Hz), 7.486 (1H, d, J = 2.0 Hz), 6.12 (1H, s), 4.78 (2H, d, J = 2.2 Hz), 4.41 (1H, s), 4.36 (1H, s), 3.87 (3H, s), 2.68 (2H, s), 2.58 (1H, t, J = 2.3 Hz), 2.34 (1H, td, J = 13.7, 5.2 Hz), 2.14–2.07 (2H, m), 1.92 (1H, m), 1.66–1.52 (1H, m), 1.48–1.36 (3H, m), 1.32–1.17 (3H, m), 1.06 (3H, s), 1.03 (3H, d, J = 6.9 Hz), 0.94 (1H, dd, J = 11.7, 2.0 Hz), 0.87 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.0, 160.2, 149.7, 143.9, 128.5, 125.0, 120.4, 111.1, 102.6, 77.6, 76.5, 57.3, 51.9, 48.0, 42.1, 40.2, 36.9, 36.6, 36.3, 33.0, 27.9, 27.7, 23.1, 20.6, 17.7, 17.6. IR (KBr): 3295, 2926, 1713, 1437, 1300 cm−1. MS (ESI-TOF) m/z: 433 [M + Na]+. HRMS (ESI-TOF) m/z: 433.2355 calcd for C26H34O4Na; Found: 433.2364.
3.3.6. Methyl 3-hydroxy-4-(nonyloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (10); Methyl 4-hydroxy-3-(nonyloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (11)
K2CO3 (9.2 mg, 0.067 mmol) and n-nonyl bromide (8.0 µL, 0.042 mmol) were added to a solution of 2 (12.6 mg, 0.034 mmol) in anhydrous DMF (0.34 mL), and the whole mixture was stirred for 3 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 30:1 to 5:1) to give 10 (3.6 mg, 21%) as a colorless amorphous and 11 (2.3 mg, 14%) as a colorless amorphous.
10: +15 (c = 0.20, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.43 (1H, d, J = 1.7 Hz), 7.36 (1H, d, J = 1.7 Hz), 6.16 (1H, s), 4.41 (1H, s), 4.36 (1H, s), 4.07 (2H, t, J = 6.6 Hz), 3.86 (3H, s), 2.69 (1H, d, J = 14.0 Hz), 2.66 (1H, d, J = 14.0 Hz), 2.34 (1H, td, J = 13.5, 5.2 Hz), 2.13–2.07 (2H, m), 1.94–1.90 (1H, m), 1.85–1.79 (2H, m), 1.62–1.52 (3H, m), 1.48–1.19 (16H, m), 1.06 (3H, s), 1.03 (3H, d, J = 6.3 Hz), 0.96 (1H, dd, J = 12.0, 1.7 Hz), 0.89 (3H, t, J = 6.9 Hz), 0.87 (3H, s). 13C-NMR (150 MHz, CDCl3) δ: 167.2, 160.3, 149.4, 145.3, 127.4, 124.2, 120.3, 109.9, 102.6, 69.2, 51.9, 48.0, 42.1, 40.2, 36.9, 36.6, 36.4, 33.1, 31.9, 29.5, 29.4, 29.2, 29.1, 27.9, 27.7, 26.0, 23.1, 22.7, 20.7, 17.7, 17.6, 14.1. IR (KBr): 2925, 1716, 1437, 1301 cm−1. MS (ESI-TOF) m/z: 521 [M + Na]+. HRMS (ESI-TOF) m/z: 521.3607 calcd for C32H50O4Na; Found: 521.3616.
11: +20 (c = 0.61, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.46 (1H, d, J = 2.0 Hz), 7.36 (1H, d, J = 2.0 Hz), 5.57 (1H, s), 4.40 (1H, s), 4.35 (1H, s), 3.87 (3H, s), 3.84–3.74 (2H, m), 2.66 (1H, d, J = 14.0 Hz), 2.58 (1H, d, J = 14.0 Hz), 2.31 (1H, td, J = 13.6, 5.5 Hz), 2.08 (1H, dd, J = 14.0, 3.2 Hz), 2.01 (1H, d, J = 12.6 Hz), 1.91–1.87 (1H, m), 1.83–1.77 (2H, m), 1.69–1.51 (3H, m), 1.47–1.11 (16H, m), 1.05 (3H, s), 0.99 (3H, d, J = 6.9 Hz), 0.89 (3H, t, J = 7.2 Hz), 0.87 (3H, s), 0.84 (1H, dd, J = 12.0, 1.7 Hz). 13C-NMR (150 MHz, CDCl3) δ: 166.8, 159.8, 149.9, 148.9, 132.2, 125.8, 125.5, 114.4, 102.9, 74.6, 52.0, 47.9, 42.3, 40.1, 37.3, 36.5, 36.1, 33.0, 31.8, 30.2, 29.5, 29.4, 29.2, 27.9, 27.6, 26.0, 23.1, 22.7, 20.6, 17.8, 17.5, 14.1. IR (KBr): 3394, 2926, 1722, 1437, 1335 cm−1. MS (ESI-TOF) m/z: 521 [M + Na]+. HRMS (ESI-TOF) m/z: 521.3607 calcd for C32H50O4Na; Found: 521.3630.
3.3.7. Methyl 4-hydroxy-3-(((1S,2R,4aS,5S,8aS)-1,2,4a,5-tetramethyldecahydronaphthalen-1-yl)methyl)benzoate (12a); Methyl 4-hydroxy-3-(((1S,2R,4aS,5R,8aS)-1,2,4a,5-tetramethyldecahydronaphthalen-1-yl)methyl)benzoate (12b)
An amount of 10% Pd-C (6.8 mg) was added to a solution of 1 (15.4 mg, 0.041 mmol) in anhydrous MeOH (2.0 mL), and the whole mixture was stirred for 16 h under H2 atmosphere (baloon). The mixture was filtered through a short pad of Celite. Removal of the solvent from the filtrate under reduced pressure gave a crude product, which was purified by reversed-phase HPLC (Cosmosil AR-II, 85% aq. CH3CN, flow rate: 3 mL/min, UV: 220 nm) to give 12a (7.5 mg, 51%) and 12b (7.0 mg, 47%) as a white amorphous solid.
12a: −3.8 (c = 0.66, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.83 (1H, d, J = 2.2 Hz), 7.79 (1H, dd, J = 8.5, 2.2 Hz), 6.78 (1H, d, J = 8.5 Hz), 5.22 (1H, s), 3.88 (3H, s), 2.63 (2H, s), 1.91–1.83 (2H, m), 1.69–1.55 (3H, m), 1.48–1.31 (3H, m), 1.28–1.21 (2H, m), 1.17–1.11 (2H, m), 1.04 (3H, s), 0.99 (3H, d, J = 6.3 Hz), 0.88 (1H, dt, J = 13.0, 3.2 Hz), 0.82 (3H, s), 0.70 (3H, d, J = 6.9 Hz). 13C-NMR (150 MHz, CDCl3) δ: 167.2, 158.8, 135.0, 129.2, 125.1, 121.8, 115.3, 51.8, 42.2, 41.1, 38.8, 36.8, 36.51, 36.48, 36.4, 29.1, 27.6, 23.5, 22.1, 21.2, 17.7, 17.5, 14.6. IR (KBr): 3291, 2934, 1680, 1601, 1420, 1285 cm−1. MS (ESI-TOF) m/z: 381 [M + Na]+. HRMS (ESI-TOF) m/z: 381.2406 calcd for C23H34O3Na; Found: 381.2413.
12b: −6.5 (c = 0.67, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.80 (1H, d, J = 2.0 Hz), 7.78 (1H, dd, J = 8.4, 2.0 Hz), 6.76 (1H, d, J = 8.4 Hz), 5.25 (1H, s), 3.88 (3H, s), 2.63 (2H, s), 1.93 (1H, d, J = 12.0 Hz), 1.84–1.80 (1H, m), 1.67–1.42 (5H, m), 1.40–1.13 (5H, m), 1.01 (3H, d, J = 6.3 Hz), 0.90 (1H, dd, J = 12.3, 2.6 Hz), 0.84 (3H, s), 0.80 (3H, s), 0.65 (3H, d, J = 6.9 Hz). 13C-NMR (125 MHz, CDCl3) δ: 167.1, 158.7, 135.1, 129.2, 125.0, 122.0, 115.3, 51.8, 48.9, 45.4, 41.6, 38.3, 37.3, 36.9, 36.1, 30.8, 27.5, 26.4, 23.1, 17.73, 17.68, 15.0, 13.2. IR (KBr): 2928, 1717, 1692, 1283 cm−1. MS (ESI-TOF) m/z: 381 [M + Na]+. HRMS (ESI-TOF) m/z: 381.2406 calcd for C23H34O3Na; Found: 381.2413.
3.3.8. Methyl 4-hydroxy-3-(((1S,2R,4aR,8aR)-1,2,4a,5-tetramethyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl)methyl)benzoate (13)
Compound 1 (7.6 mg, 0.020 mmol) and RhCl3·3H2O (2.6 mg, 0.010 mmol) were mixed and dried. EtOH (1.0 mL) was added to the mixture, and the whole mixture was stirred for 12 h under reflux. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 5:1) and reversed-phase HPLC (Cosmosil AR-II, 85% aq. CH3CN, flow rate: 3 mL/min, UV: 220 nm) to give 13 (4.0 mg, 61%) as a white amorphous solid.
−27 (c = 0.39, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.80 (1H, d, J = 2.0 Hz), 7.77 (1H, dd, J = 8.6, 2.0 Hz), 6.74 (1H, d, J = 8.6 Hz), 5.27 (1H, s), 5.15 (1H, s), 3.87 (3H, s), 2.72 (1H, d, J = 14.6 Hz), 2.68 (1H, d, J = 14.6 Hz), 2.27–2.19 (1H, m), 2.13–2.08 (1H, m), 2.04 (1H, dd, J = 13.5, 6.6 Hz), 1.66–1.55 (2H, m), 1.50 (3H, d-like, J = 1.7 Hz), 1.38–1.25 (3H, m), 1.18 (1H, dd, J = 12.0, 1.1 Hz), 1.03 (3H, d, J = 6.3 Hz), 1.02 (3H, s), 0.93–0.88 (1H, m), 0.88 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.1, 158.7, 144.1, 135.2, 129.3, 125.0, 122.1, 120.6, 115.3, 51.8, 45.5, 41.7, 38.3, 37.2, 36.0, 35.7, 27.7, 26.0, 20.0, 19.9, 18.1, 17.7, 17.6. IR (KBr): 2944, 1688, 1603, 1433, 1285 cm−1. MS (ESI-TOF) m/z: 379 [M + Na]+. HRMS (ESI-TOF) m/z: 379.2249 calcd for C23H32O3Na; Found: 379.2252.
3.3.9. tert-Butyl 3-(((4a′R,5′R,6′R,8a′R)-6′-hydroxy-5′,8a′-dimethyloctahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalen]-5′-yl)methyl)-4-(methoxymethoxy)benzoate (15)
CeCl3·7H2O (545 mg, 1.46 mmol) was added to a solution of 14 (285 mg, 0.58 mmol) [9] in MeOH (5.8 mL), and the whole mixture was stirred for 5 min. NaBH4 (110 mg, 2.9 mmol) was added to the mixture at 0 °C, and the whole mixture was stirred for 2.5 h at 0 °C. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 4:1) to give 15 (270 mg, 94%) as a white amorphous solid.
−8 (c = 1.07, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.87 (1H, dd, J = 8.9, 2.0 Hz), 7.68 (1H, d, J = 2.0 Hz), 7.14 (1H, d, J = 8.9 Hz), 5.28 (2H, s), 3.86 (1H, q, J = 6.1 Hz), 3.79–3.76 (2H, m), 3.71 (1H, q, J = 6.5 Hz), 3.52 (3H, s), 3.37 (1H, d, J = 3.4 Hz), 3.00 (1H, ddd, J = 10.3, 5.2, 3.4 Hz), 2.82 (1H, d, J = 14.3 Hz), 2.59 (1H, d, J = 14.3 Hz), 1.93 (1H, d, J = 13.2 Hz), 1.76–1.61 (2H, m), 1.58 (9H, s), 1.57–1.45 (5H, m), 1.31–1.22 (2H, m), 1.10 (3H, s), 0.97 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 165.6, 158.9, 133.6, 129.4, 126.7, 125.5, 113.2, 113.0, 95.1, 80.7, 73.0, 65.0, 64.7, 56.8, 43.7, 43.1, 41.4, 35.8, 30.4, 28.3, 28.2, 25.0, 22.6, 21.3, 17.2, 16.6. MS (ESI-TOF) m/z: 513 [M + Na]+. HRMS (ESI-TOF) m/z: 513.2828 calcd for C28H42O7Na; Found: 513.2829.
3.3.10. 4-Hydroxy-3-(((1R,2R,4aR,8aR)-2-hydroxy-1,4a-dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)benzoic acid (16)
An amount of 80% TFA (3.6 mL) was added to a solution of 15 (51.5 mg, 0.11 mmol) in THF (1.2 mL) at 0 °C, and the whole mixture was stirred for 3.5 h at 50 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 15:3:1, lower phase) to give 16 (36.6 mg, 97%) as a white amorphous solid.
+14 (c = 1.02, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 7.77 (1H, dd, J = 8.6, 2.0 Hz), 7.69 (1H, d, J = 2.0 Hz), 6.88 (1H, d, J = 8.6 Hz), 3.06–3.03 (1H, m), 2.81 (1H, d, J = 14.3 Hz), 2.76 (1H, d, J = 14.3 Hz), 2.65 (1H, td, J = 14.0, 7.1 Hz), 2.27 (1H, dd, J = 14.0, 2.0 Hz), 2.18–2.13 (1H, m), 2.08–2.07 (1H, m), 1.96 (1H, qd, J = 13.0, 3.6 Hz), 1.76–1.59 (3H, m), 1.44 (1H, dt, J = 14.1, 3.3 Hz), 1.35–1.25 (2H, m), 1.20 (3H, s), 1.15 (1H, dd, J = 12.3, 2.0 Hz), 1.10 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 213.7, 167.5, 162.0, 134.8, 130.7, 124.5, 122.6, 117.1, 73.9, 49.0, 47.2, 44.3, 37.7, 37.3, 32.0, 26.1, 26.0, 21.7, 19.6, 17.2. MS (ESI-TOF) m/z: 369 [M + Na]+. HRMS (ESI-TOF) m/z: 369.1678 calcd for C20H26O5Na; Found: 369.1680.
3.3.11. Methyl 4-hydroxy-3-(((1R,2R,4aR,8aR)-2-hydroxy-1,4a-dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)benzoate (17)
SOCl2 (0.02 mL, 0.28 mmol) was added to a solution of 16 (23.6 mg, 0.068 mmol) in anhydrous MeOH (0.7 mL), and the whole mixture was stirred for 6.5 h at 45 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 60:3:1, lower phase) to give 17 (23.9 mg, 98%) as a white solid.
+12 (c = 0.82, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 7.74 (1H, dd, J = 8.2, 1.9 Hz), 7.67 (1H, d, J = 1.9 Hz), 6.89 (1H, d, J = 8.2 Hz), 3.80 (3H, s), 3.04 (1H, dd, J = 10.3, 5.7 Hz), 2.88 (1H, br), 2.82 (1H, d, J = 14.9 Hz), 2.77 (1H, d, J = 14.9 Hz), 2.66 (1H, td, J = 14.0, 7.0 Hz), 2.27 (1H, dd, J = 14.0, 2.0 Hz), 2.22–2.17 (1H, m), 2.10–2.06 (1H, m), 2.03–1.97 (1H, m), 1.75–1.55 (3H, m), 1.44 (1H, dt, J = 14.3, 3.4 Hz), 1.33–1.23 (2H, m), 1.21 (3H, s), 1.16–1.13 (1H, dd, J = 12.3, 2.3 Hz), 1.11 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 213.6, 167.0, 162.1, 134.5, 130.3, 124.6, 122.3, 117.1, 73.9, 51.9, 49.0, 47.2, 44.3, 37.7, 37.3, 32.0, 26.1, 26.0, 21.7, 19.6, 17.2. MS (ESI-TOF) m/z: 383 [M + Na]+. HRMS (ESI-TOF) m/z: 383.1834 calcd for C21H28O5Na; Found: 383.1847.
3.3.12. Methyl 4-hydroxy-3-(((1R,2R,4aR,8aS)-2-hydroxy-1,4a-dimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (18)
Under an Ar atmosphere, KHMDS (0.9 mL, of a 0.5 M solution in toluene, 0.45 mmol) was added to a solution of Ph3PCH3Br (165 mg, 0.46 mmol) in anhydrous THF (0.22 mL), and the whole mixture was stirred for 1 h at rt. The above solution was added dropwise to a solution of 17 (10.9 mg, 0.030 mmol) in anhydrous THF (0.1 mL) at 0 °C, and the whole mixture was stirred for 1 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 3:1) to give 18 (5.6 mg, 39%) as a white solid.
−32 (c = 0.55, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.81 (1H, dd, J = 8.5, 1.9 Hz), 7.63 (1H, d, J = 1.9 Hz), 6.89 (1H, d, J = 8.5 Hz), 4.45 (1H, s), 4.37 (1H, s), 3.85 (3H, s), 3.12 (1H, dd, J = 11.7, 4.3 Hz), 2.71 (2H, s), 2.31 (1H, td, J = 13.7, 5.7 Hz), 2.16–2.09 (2H, m), 2.04–2.00 (1H, m), 1.78 (1H, qd, J = 12.6, 3.2 Hz), 1.71–1.56 (4H, m), 1.48–1.40 (1H, m), 1.36 (1H, td, J = 13.3, 3.8 Hz), 1.11 (3H, s), 1.03 (3H, s), 1.00 (1H, dd, J = 12.6, 2.3 Hz). 13C-NMR (150 MHz, CDCl3) δ: 167.1, 160.9, 158.5, 133.9, 129.8, 123.2, 121.7, 116.6, 103.2, 74.7, 51.8, 46.2, 43.1, 39.5, 36.6, 34.8, 32.7, 27.4, 26.9, 22.1, 21.2, 16.3. IR (KBr): 3210, 2938, 1721, 1435, 1289 cm−1. MS (ESI-TOF) m/z: 381 [M + Na]+. HRMS (ESI-TOF) m/z: 381.2042 calcd for C22H30O4Na; Found: 381.2032.
3.3.13. tert-Butyl 3-(((4a′R,5′R,6′R,8a′R)-5′,8a′-dimethyl-6′-(((methylthio)carbonothioyl)oxy)octahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalen]-5′-yl)methyl)-4-(methoxymethoxy)benzoate (19)
Under an Ar atmosphere, NaH (91.0 mg, 60% suspension in oil, 2.3 mmol) was added to a solution of 15 (136 mg, 0.28 mmol) in anhydrous THF (2.8 mL) at 0 °C, and the whole mixture was stirred for 30 min at rt. CS2 (0.17 mL, 2.8 mmol) was added dropwise to the mixture, and the whole mixture was stirred for 50 min at rt. MeI (0.21 mL, 3.4 mmol) was added to the mixture, and the whole mixture was stirred for 5 h at 50 °C. Sat. Na2S2O3 aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 5:1) to give 19 (152 mg, 94%) as a white amorphous solid.
−18 (c = 1.17, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.79 (1H, dd, J = 8.6, 2.3 Hz), 7.68 (1H, d, J = 2.3 Hz), 7.02 (1H, d, J = 8.6 Hz), 5.18 (2H, dd, J = 8.0, 6.9 Hz), 5.13 (1H, dd, J = 11.2, 4.3 Hz), 3.92–3.88 (3H, m), 3.79–3.77 (1H, m), 3.43 (3H, s), 2.96 (1H, d, J = 14.0 Hz), 2.64 (1H, d, J = 14.0 Hz), 2.28–2.24 (1H, m), 2.19 (3H, s), 1.88 (1H, d, J = 12.6 Hz), 1.80 (1H, dd, J = 12.0, 2.3 Hz), 1.77–1.61 (3H, m), 1.58 (9H, s), 1.54–1.44 (4H, m), 1.39–1.35 (1H, m), 1.17 (3H, s), 1.15 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 213.4, 165.8, 158.8, 133.6, 129.3, 127.1, 124.6, 113.0, 112.9, 93.9, 88.7, 80.4, 65.2, 64.9, 56.2, 45.7, 43.2, 38.9, 30.4, 28.2 (3C), 27.7, 22.8, 21.6, 21.0, 17.8, 17.0, 16.9. MS (ESI-TOF) m/z: 603 [M + Na]+. HRMS (ESI-TOF) m/z: 603.2426 calcd for C30H44O7S2Na; Found: 603.2449.
3.3.14. tert-Butyl 3-(((4a′R,5′R,8a′R)-5′,8a′-dimethyloctahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalen]-5′-yl)methyl)-4-(methoxymethoxy)benzoate (20)
Under an Ar atmosphere, nBu3SnH (0.21 mL, 0.80 mmol) and AIBN (34.5 mg, 0.21 mmol) were added to a solution of 19 (138 mg, 0.24 mmol) in anhydrous toluene (3.4 mL), and the whole mixture was stirred for 3 h at 80 °C. H2O was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 8:1) to give 20 (96.6 mg, 86%) as a white amorphous solid.
+21 (c = 1.26, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.79 (1H, dd, J = 8.6, 2.3 Hz), 7.70 (1H, d, J = 2.3 Hz), 7.07 (1H, d, J = 8.6 Hz), 5.21 (1H, d, J = 6.9 Hz), 5.18 (1H, d, J = 6.9 Hz), 3.94–3.89 (3H, m), 3.83–3.79 (1H, m), 3.46 (3H, s), 2.88 (1H, d, J = 13.0 Hz), 2.40 (1H, d, J = 13.0 Hz), 1.81 (1H, d, J = 13.2 Hz), 1.75–1.64 (2H, m), 1.61–1.58 (1H, m), 1.57 (9H, s), 1.52–1.50 (1H, m), 1.44–1.21 (8H, m), 1.10 (3H, s), 0.85 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 165.9, 159.5, 134.1, 128.9, 127.9, 124.3, 113.5, 112.7, 94.2, 80.4, 65.2, 64.8, 56.2, 47.6, 43.8, 42.3, 37.9, 37.6, 30.6, 30.2, 28.2 (3C), 23.0, 20.9, 19.3, 17.8, 17.1. MS (ESI-TOF) m/z: 497 [M + Na]+. HRMS (ESI-TOF) m/z: 497.2879 calcd for C28H42O6Na; Found: 497.2897.
3.3.15. 3-(((1R,4aR,8aR)-1,4a-Dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)-4-hydroxybenzoic acid (21)
An amount of 80% TFA (4.0 mL) was added to a solution of 20 (74.9 mg, 0.16 mmol) in THF (2.0 mL), and the whole mixture was stirred for 2.5 h at 50 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 30:3:1, lower phase) to give 21 (50.0 mg, 93%) as a white amorphous solid.
+46 (c = 1.13, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 7.75 (1H, dd, J = 8.6, 2.3 Hz), 7.74 (1H, d, J = 2.3 Hz), 6.92 (1H, d, J = 8.6 Hz), 2.77 (1H, d, J = 13.2 Hz), 2.70–2.60 (1H, m), 2.61 (1H, d, J = 13.2 Hz), 2.16–2.07 (2H, m), 1.83 (1H, qd, J = 12.9, 3.7 Hz), 1.69–1.40 (4H, m), 1.37–1.26 (5H, m), 1.20 (3H, s), 1.05 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 214.3, 161.1, 135.6, 130.3, 125.8, 122.0, 115.7, 78.1, 51.1, 49.7, 42.5, 39.6, 37.88, 37.86, 33.8, 26.6, 21.8, 21.4, 19.4, 18.2. MS (ESI-TOF) m/z: 353 [M + Na]+. HRMS (ESI-TOF) m/z: 353.1729 calcd for C20H26O4Na; Found: 353.1730.
3.3.16. Methyl 3-(((1R,4aR,8aR)-1,4a-dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)-4-hydroxybenzoate (22)
SOCl2 (0.03 mL, 0.413 mmol) was added to a solution of 21 (30.3 mg, 0.092 mmol) in anhydrous MeOH (0.92 mL), and the whole mixture was stirred for 15 h at 50 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 3:1) to give 22 (27.5 mg, 87%) as a white solid.
+46 (c = 0.73, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 9.10 (1H, br s), 7.72 (1H, dd, J = 8.6, 1.7 Hz), 7.70 (1H, d, J = 1.7 Hz), 6.91 (1H, d, J = 8.6 Hz), 3.81 (3H, s), 2.77 (1H, d, J = 13.2 Hz), 2.66 (1H, td, J = 13.7, 6.9 Hz), 2.60 (1H, d, J = 13.2 Hz), 2.16–2.08 (3H, m), 1.83 (1H, qd, J = 13.2, 3.7 Hz), 1.68–1.25 (7H, m), 1.20 (3H, s), 1.11 (1H, t, J = 7.2 Hz), 1.05 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 214.2, 167.1, 161.1, 135.2, 130.0, 125.9, 121.8, 115.8, 51.8, 51.1, 49.7, 42.4, 39.6, 37.89, 37.86, 33.8, 26.6, 21.8, 21.4, 19.4, 18.2. MS (ESI-TOF) m/z: 367 [M + Na]+. HRMS (ESI-TOF) m/z: 367.1885 calcd for C21H28O4Na; Found: 367.1875.
3.3.17. Methyl 3-(((1R,4aR,8aR)-1,4a-dimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)-4-hydroxybenzoate (23)
Under an Ar atmosphere, KHMDS (0.57 mL, of a 0.5 M solution in toluene, 0.29 mmol) was added to Ph3PCH3Br (114 mg, 0.32 mmol), and the whole mixture was stirred for 1 h at rt. The above solution was added dropwise to a solution of 22 (14.5 mg, 0.042 mmol) in anhydrous THF (0.21 mL) at 0 °C, and the whole mixture was stirred for 3.5 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 4:1) to give 23 (13.7 mg, 95%) as a white solid.
+60 (c = 0.89, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.78 (1H, dd, J = 8.3, 2.3 Hz), 7.73 (1H, d, J = 2.3 Hz), 6.78 (1H, d, J = 8.3 Hz), 5.28 (1H, br s), 4.47 (1H, s), 4.46 (1H, s), 3.87 (3H, s), 2.68 (1H, d, J = 13.5 Hz), 2.44 (1H, d, J = 13.5 Hz), 2.36 (1H, td, J = 13.7, 5.2 Hz), 2.15–2.11 (1H, m), 1.97–1.92 (2H, m), 1.62–1.46 (4H, m), 1.44–1.23 (4H, m), 1.11 (3H, s), 1.09 (1H, d, J = 12.0 Hz), 0.95 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.0, 160.2, 158.4, 134.9, 129.3, 124.9, 122.1, 115.2, 102.5, 51.9, 42.5, 40.2, 38.7, 38.1, 37.0, 33.0, 29.7, 28.4, 22.3, 20.8, 20.3, 18.4. IR (KBr): 2928, 1688, 1603, 1429, 1283 cm−1. MS (ESI-TOF) m/z: 365 [M + Na]+. HRMS (ESI-TOF) m/z: 365.2093 calcd for C22H30O3Na; Found: 365.2089.
3.4. Biological Evaluation of Analogues
3.4.1. Cell Cultures
Human prostate cancer cell line DU145 was cultured in RPMI 1640 supplemented with heat-inactivated 10% fetal bovine serum (FBS) and kanamycin (50 μg/mL) in a humidified atmosphere of 5% CO2 at 37 °C.
3.4.2. Assay for Anti-Proliferative Activity under Hypoxic Condition
The DU145 cells in the culture medium were plated into each well of 96-well plates (1 × 104 cells/well/200 μL) in a humidified atmosphere of 5% CO2 at 37 °C (normoxic condition). After 4 h, the plates were incubated for 12 h in the hypoxic condition (94% nitrogen, 5% CO2, and 1% O2) inducing hypoxia-related genes such as HIF-1α. Then, testing compounds were added, and the plates were incubated for additional 24 h in the hypoxic condition. The cell proliferation was detected by MTT method as previously described [8]. The growth inhibition rate was calculated as percentage of parallel negative controls. The anti-proliferative activities of the testing compounds under normoxic condition were also evaluated by the MTT method.
4. Conclusions
In summary, we demonstrated the potential of dictyoceratin-C (1) and A (2) as anticancer drug leads by evaluating their in vivo antitumor activity. In addition, we investigated the SAR of 1 and 2 by using our synthetic methodology and concluded that the characteristic moieties of 1 were important for hypoxia-selective growth inhibition. This evidence conflicts with a previous study, which showed that the unnatural enantiomers of 1 and 2 also exhibited similar hypoxia-selective growth inhibitory activity. In order to clarify this contradiction, we focused on determining the target molecules of 1 and 2. Substitution of the methyl ester of 1 with the more physiologically stable propargyl amide was found to be effective. Moreover, the propargyl group could be a useful tag for further functionalization through click chemistry, leading to the synthesis of probe molecules for target identification of 1 and 2. These studies are now underway.
Acknowledgments
The authors thank Dr. Kazutake Tsujikawa (Osaka University) for providing DU145 cells. The authors are grateful to JSPS KAKENHI (Grant No. 23102005, No. 26242074 and No. 26860065), the Hoansha Foundation, Uehara Memorial Foundation, Naito Foundation, and Osaka University Project MEET for financial support. This study was also supported by the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and Japan Agency for Medical Research and Development (AMED).
Supplementary Files
Author Contributions
Naoyuki Kotoku conceived and designed the experiments; Yuji Sumii and Akinori Fukuda performed the synthetic experiments; Takashi Kawachi and Masayoshi Arai performed the biological experiments; Naoyuki Kotoku, Masayoshi Arai and Motomasa Kobayashi analyzed the data; Yuji Sumii, Naoyuki Kotoku and Motomasa Kobayashi wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Molinski T.F., Dalisay D.S., Lievens S.L., Saludes J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 2.Szychowski J., Truchon J.-F., Bennani Y.L. Natural products in medicine: Transformational outcome of synthetic chemistry. J. Med. Chem. 2014;57:9292–9308. doi: 10.1021/jm500941m. [DOI] [PubMed] [Google Scholar]
- 3.Wach J.-Y., Gademann K. Reduce to the maximum: Truncated natural products as powerful modulators of biological processes. Synlett. 2012;23:163–170. [Google Scholar]
- 4.Dewhirst M.W., Cao Y., Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer. 2008;8:425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson W.R., Hay M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 6.Kushlan D.M., Faulkner D.J., Parkanyi L., Clardy J. Metabolites of the Palauan sponge Dactylospongia sp. Tetrahedron. 1989;45:3307–3312. doi: 10.1016/S0040-4020(01)81009-7. [DOI] [Google Scholar]
- 7.Nakamura H., Deng S., Kobayashi J., Ohizumi Y., Hirata Y. Dictyoceratin-A and -B, novel antimicrobial terpenoids from the Okinawan marine sponge Hippospongia sp. Tetrahedron. 1986;42:4197–4201. doi: 10.1016/S0040-4020(01)87643-2. [DOI] [Google Scholar]
- 8.Arai M., Kawachi T., Sato H., Setiawan A., Kobayashi M. Marine spongian sesquiterpene phenols, dictyoceratin-C and smenospondiol, display hypoxia-selective growth inhibition against cancer cells. Bioorg. Med. Chem. Lett. 2014;24:3155–3157. doi: 10.1016/j.bmcl.2014.04.116. [DOI] [PubMed] [Google Scholar]
- 9.Sumii Y., Kotoku N., Fukuda A., Kawachi T., Sumii Y., Arai M., Kobayashi M. Enantioselective synthesis of dictyoceratin-A (smenospondiol) and -C, hypoxia-selective growth inhibitors from marine sponge. Bioorg. Med. Chem. 2015;23:966–975. doi: 10.1016/j.bmc.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Stahl P., Kissau L., Mazitschek R., Huwe A., Furet P., Giannis A., Waldmann H. Total synthesis and biological evaluation of the nakijiquinones. J. Am. Chem. Soc. 2001;123:11586–11593. doi: 10.1021/ja011413i. [DOI] [PubMed] [Google Scholar]
- 11.Barton D.H.R., McCombie S.W. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc. Perkin Trans. 1975;1:1574–1585. doi: 10.1039/p19750001574. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.