

Abstract
Reduced intensity conditioning (RIC) is desirable for hematopoietic stem cell (HSC) targeted gene therapy; however, RIC may be insufficient for efficient engraftment and inducing immunological tolerance to transgenes. We previously established long-term gene marking in our rhesus macaque autologous HSC transplantation model following 10 Gy total body irradiation (TBI). In this study, we evaluated RIC transplantation with 4 Gy TBI in two rhesus macaques that received equal parts of CD34+ cells transduced with green fluorescent protein (GFP)-expressing lentiviral vector and empty vector not expressing transgenes. In both animals, equivalently low gene marking between GFP and empty vectors was observed 6 months post-transplantation, even with efficient transduction of CD34+ cells in vitro. Autologous lymphocyte infusion with GFP marking resulted in an increase of gene marking in lymphocytes in a control animal with GFP tolerance, but not in the two RIC-transplanted animals. In vitro assays revealed strong cellular and humoral immune responses to GFP protein in the two RIC-transplanted animals, but this was not observed in controls. In summary, 4 Gy TBI is insufficient to permit engraftment of genetically modified HSCs and induce immunological tolerance to transgenes. Our findings should help in the design of conditioning regimens in gene therapy trials.
Keywords: lentiviral vector, CD34+ cells, large animal model, hematopoietic stem cell transplantation, lymphocyte infusion
INTRODUCTION
Reduced intensity conditioning (RIC) regimens are thought to be appropriate for hematopoietic stem cell (HSC)-targeted gene therapy, as autologous HSCs are utilized in gene therapy and most gene therapy patients have potential complications, such as chronic infection due to the underlying immunodeficiency and organ damage as a result of long-standing sickle cell disease (SCD). However, previous gene therapy trials revealed low gene marking in peripheral blood cells after transplantation using RIC or no conditioning, even when high gene marking was observed in transduced HSCs in vitro before transplantation.1–6 One trial showed relatively high marking levels in vivo, but these patients developed a hematological malignancy later.6 More efficient engraftment of genetically modified HSCs thus remains crucial for development of successful gene therapy.
Conditioning therapy in HSC transplantation involves chemotherapy and/or irradiation before infusion of HSCs.7 In HSC gene therapy, autologous HSCs are genetically modified with a therapeutic gene and infused into patients after conditioning therapy. Myeloablative conditioning regimens have been historically utilized for both autologous and allogeneic transplantation for hematological malignancies; however, this conditioning carries a relatively high risk of treatment-related mortality and is not tolerable for older patients and patients with co-morbidities, such as adult SCD patients.8,9 Recently, RIC regimens were developed in the allogeneic transplantation setting to reduce mortality and extend application.9,10 Our group recently developed an allogeneic HSC transplantation protocol for adult SCD patients using a RIC regimen,8 in which 3–4 Gy total body irradiation (TBI) proved effective for adult SCD patients. The low-dose TBI conditioning resulted in a mean donor chimerism of ~80% among granulocytes following transplantation.
Conditioning therapy for allogeneic transplantation has two main purposes: (1) myeloablation to open niches for engraftment of HSCs and (2) immunosuppression to permit immunological tolerance to allogeneic antigens.7 We hypothesized that RIC regimens not incorporating tolerance induction strategies might be insufficient in the autologous gene therapy setting because autologous cells are modified to express therapeutic genes that are potentially immunogenic foreign proteins. Previously, we established a rhesus autologous HSC transplantation model, in which cytokine-mobilized CD34+ cells were transduced with a chimeric human immunodeficiency virus type 1 (HIV-1)-based vector (χHIV vector) and transplanted following a high-dose TBI at 10 Gy.11–13 Using this model, stable high gene marking in peripheral blood cells was observed in the circulation long term. In the current study, we evaluated both engraftment and tolerance for genetically modified cells using this rhesus gene therapy model following a RIC regimen of 4 Gy TBI.
RESULTS
RIC HSC transplantation with lentiviral transduction in rhesus macaques
Previously we established long-term gene marking in rhesus HSC transplantation following a split dose of 10 Gy TBI.12,13 To evaluate whether a RIC regimen is sufficient for engraftment and tolerance induction for genetically modified cells, two rhesus macaques (DCFC and 07E083) were conditioned with 4 Gy TBI, as we have recently demonstrated that 3–4 Gy TBI was sufficient for engraftment of sibling donor cells.8 Half of the autologous CD34+ cells were transduced with an enhanced green fluorescent protein (GFP)-expressing vector (GFP vector), whereas the other half was transduced with a control vector not expressing the GFP transgene (empty vector), as we also sought to evaluate the specific immunological reaction to the GFP transgene in the transplanted animals (Figure 1a). The autologous transduced cells (5.5–10.3×10e6 cells kg−1) were transplanted into the two animals following a RIC of 4 Gy TBI. We evaluated both GFP-positive rates (%GFP) in peripheral blood cells by flow cytometry and average vector copy number (VCN) per cell by real-time polymerase chain reaction (PCR) for 6–8 months after transplantation.
Figure 1.
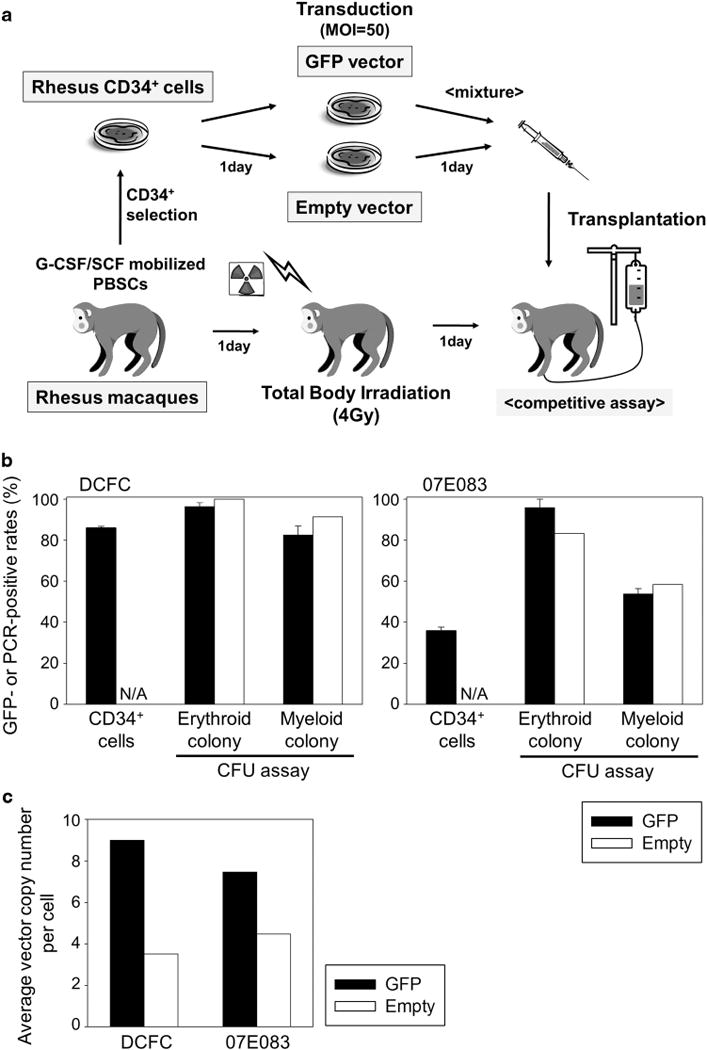
RIC rhesus HSC transplantation with efficient lentiviral transduction. (a) We previously established a rhesus HSC transplantation model with long-term gene marking, in which mobilized CD34+ cells were transduced with a chimeric HIV-1 vector (χHIV vector) and transplanted following 10 Gy TBI.13 In the current study, two rhesus macaques (DCFC and 07E083) were conditioned with a RIC regimen of 4 Gy TBI, and they received autologous mobilized CD34+ cells split for transduction with a GFP-expressing vector (GFP vector) or a control vector not expressing a transgene (empty vector). (b and c) We observed efficient transduction in liquid culture of transduced CD34+ cells, which was evaluated by GFP-positive rates in flow cytometry and VCN per cell in real-time PCR. CFU assay of transduced CD34+ cells also revealed efficient transduction, evaluated by GFP-positive rates in fluorescent microscopy and PCR-positive rates in empty vector-transduced cells. Abbreviations: PBSC, peripheral blood stem cell; G-CSF, granulocyte colony-stimulating factor; SCF, stem cell factor; MOI, multiplicity of infection; and N/A, not applicable.
In vitro transduction efficiency was evaluated from small aliquots of rhesus CD34+ cells before transplantation. In vitro %GFP was 36–86% (Figure 1b) at 4 days after transduction, and in vitro VCN was 7.5–9.0 for GFP-transduced cells and 3.5–4.5 for empty vector-transduced cells (Figure 1c) at 6 days after transduction. In addition, we performed colony-forming unit (CFU) assay using the transduced CD34+ cells (Figure 1b), which demonstrated efficient transduction as evaluated by %GFP in GFP-transduced cells using fluorescent microscopy (erythroid: 96% and myeloid: 54–82%) and PCR-positive rates in empty vector-transduced cells (erythroid: 53–100% and myeloid: 58–91%).
After transplantation following 4 Gy TBI, mild bone marrow suppression was followed by relatively slow reconstitution of peripheral blood cells in the two animals (Figure 2 and Supplementary Tables 1 and 2). Recovery dates for DCFC were day 27 for white blood cells (WBC ⩾1000 μl−1), day 34 for granulocytes (Gr ⩾500 μl−1) and day 20 for platelets (PLT ⩾50 000 μl−1). In 07E083, baseline blood counts never fell below the thresholds; however, this animal also required ~1 month until the blood counts returned to normal. DCFC received a platelet transfusion at day 16 after transplantation, whereas 07E083 required neither red blood cell (RBC) nor platelet transfusion. No complications were observed in the two transplanted animals. After peripheral blood reconstitution, similar blood counts continued for the 1.0–1.2 years of follow-up (data not shown).
Figure 2.
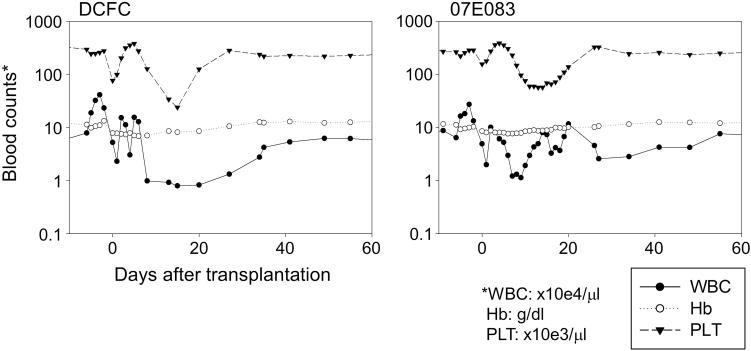
Delayed recovery of complete blood counts following RIC HSC transplantation. After transplantation with 4 Gy TBI, mild bone marrow suppression followed by relatively slow reconstitution of peripheral blood cells was observed in the two animals (DCFC and 07E083). The recovery dates of each blood cell lineage in DCFC were day 27 for white blood cells (WBC ⩾1000 μl−1), day 34 for granulocytes (Gr ⩾500 μl−1) and day 20 for platelets (PLT ⩾50 000 μl−1). In 07E083, baseline blood counts never reached these thresholds; however, this animal also required around 1 month until the blood counts plateaued. Abbreviation: Hb, hemoglobin.
At 2 weeks after transplantation in both animals, we observed low %GFP (1.6–2.1%) in RBCs, and %GFP was undetectable in granulocytes, lymphocytes, monocytes and platelets (Figure 3a). %GFP in RBCs gradually decreased and became undetectable by 3 months. Interestingly, both animals displayed very high %GFP in both granulocytes (97–99%) and monocytes (81–84%) starting at 1–2 months after transplantation (Figure 3a), which later declined to undetectable levels by 3–4 months after transplantation. Despite high %GFP, relatively low GFP intensity was observed in both granulocytes and monocytes, compared with intensity of the GFP-positive RBCs (Supplementary Figure 1). Using lineage marker-specific analysis (CD3, CD20, CD33, CD41a and RBC-specific antibodies), similar patterns were observed for both animals (Figure 3b). Equivalent VCN between GFP and empty vectors was observed in both granulocytes and lymphocytes for 6–8 months after transplantation (Figure 4). Analysis of bone marrow cells in both animals revealed <1% GFP-positivity in various lineage cells at 1 month after transplantation (Figure 5a). CFU assay using these bone marrow mononuclear cells resulted in no GFP-positive CFUs under fluorescent microscopy and 0–6% empty vector-positive CFUs when evaluated by PCR (Figure 5b). Equivalent VCN between GFP and empty vectors was observed in the bone marrow mononuclear cells from both animals (Figure 5c). These data suggest that 4 Gy TBI is insufficient to support engraftment of genetically modified hematopoietic repopulating cells.
Figure 3.
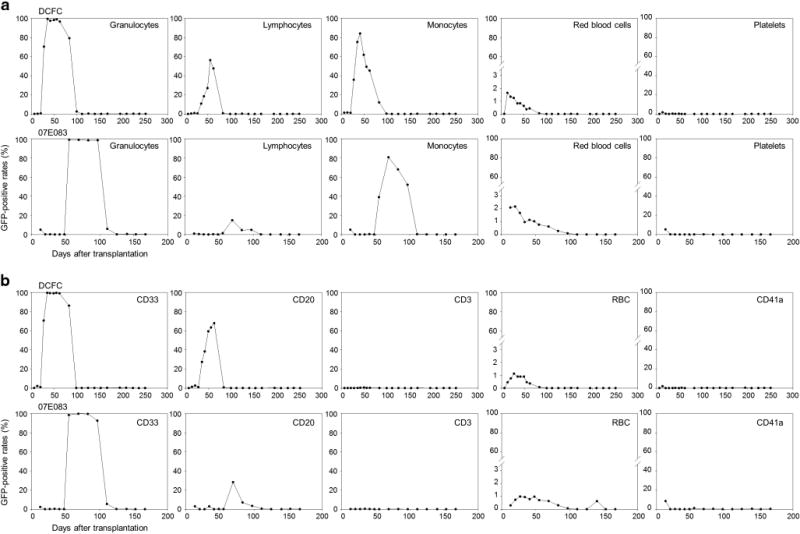
High GFP-positive rates in granulocytes and monocytes at 1–2 months after RIC HSC transplantation. (a) At 2 weeks after transplantation in both animals, we observed ~2% of GFP-positive rates (%GFP) in RBCs, but undetectable in granulocytes, lymphocytes, monocytes or platelets. %GFP in RBCs gradually decreased and was undetectable by 3 months. Interestingly, %GFP in granulocytes and monocytes were unexpectedly high (80–99%) starting at 1–2 months after transplantation, and declined to undetectable levels by 3–4 months. (b) In lineage marker specific analysis (CD3, CD20, CD33, CD41a and RBC-specific antibodies), similar patterns were observed.
Figure 4.
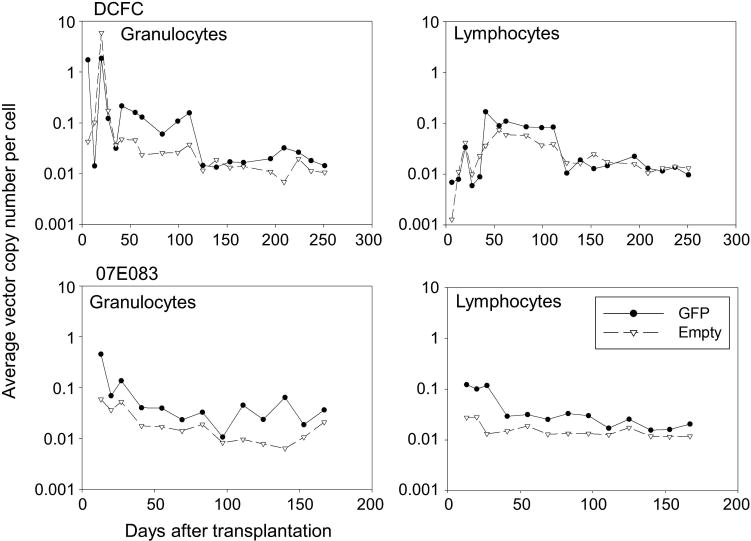
Equivalent gene marking levels between GFP and empty vectors in peripheral blood cells in the RIC-transplanted animals. We evaluated average VCN per cell in peripheral blood cells in the two transplanted animals with 4 Gy TBI (DCFC and 07E083). Equivalent VCNs between GFP and empty vectors were observed in both granulocytes and lymphocytes for 6–8 months after transplantation in both animals.
Figure 5.
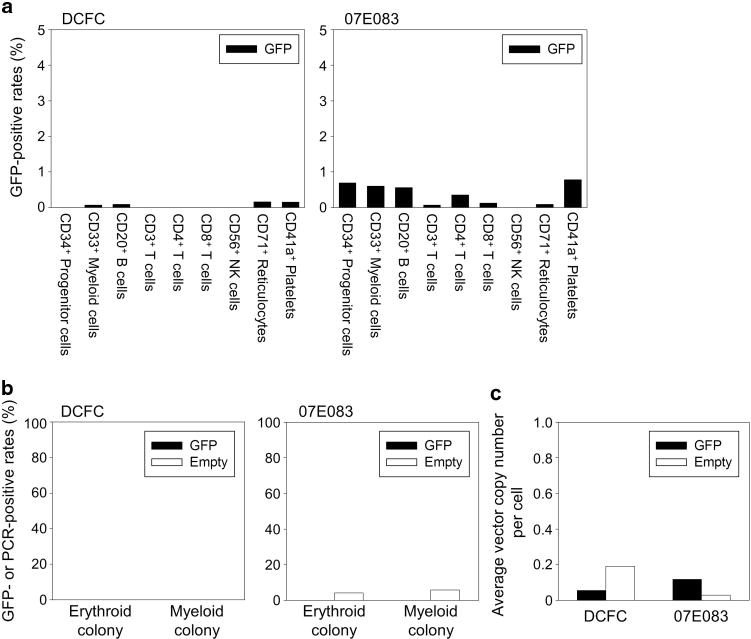
Low gene making levels in bone marrow cells in the RIC-transplanted animals. (a) Lineage-specific cell surface analysis in various types of bone marrow cells revealed <1% of GFP-positive rates at 1 month after transplantation in both animals. (b) In CFU assay using bone marrow cells in the transplanted animals, we observed no GFP-positive CFUs evaluated by fluorescent microscopy and 0–6% PCR-positive evaluated specific for the empty vector. (c) Equivalent average VCN per cell between GFP and empty vectors were observed in bone marrow cells at 1 month after transplantation in both animals.
Evaluation of immunological reaction for the GFP transgene in the transplanted animals
To evaluate whether the introduction of transduced HSCs after 4 Gy TBI could establish immunological tolerance to the GFP transgene, we performed autologous lymphocyte infusion after transduction with a woodchuck hepatitis virus post-transcriptional regulatory element (WPRE)-containing GFP-expressing χHIV vector (GFP/WPRE vector) in these animals (DCFC and 07E083). As a control animal with GFP tolerance, we utilized a rhesus (RQ7277) that had stable GFP expression at 5–15% among peripheral blood cells for 2 years after HSC transplantation following conditioning with a split dose of 10 Gy TBI.11 Cells collected by leukapheresis from the three animals were cultured on CD3/28 antibody-coated plates. After 1 day of culture, these cells were transduced with the GFP/WPRE vector (Figure 6a), on the following day, the transduced cells were infused into the animals without conditioning (5.4–11.7×10e7 cells kg−1). The reinfused cells mainly contained lymphocytes but not granulocytes as determined by surface marker analysis (CD3: 35–60%, CD20: 14–40% and CD33: 0.5–0.8%, Figure 6b). The GFP/WPRE-VCN was evaluated by real-time PCR using WPRE-specific probe/primers, which can detect the infused lymphocytes with GFP/WPRE marking (Figure 6a) but not reconstituted blood cells from the transplanted HSCs with GFP marking (Figure 1a).
Figure 6.
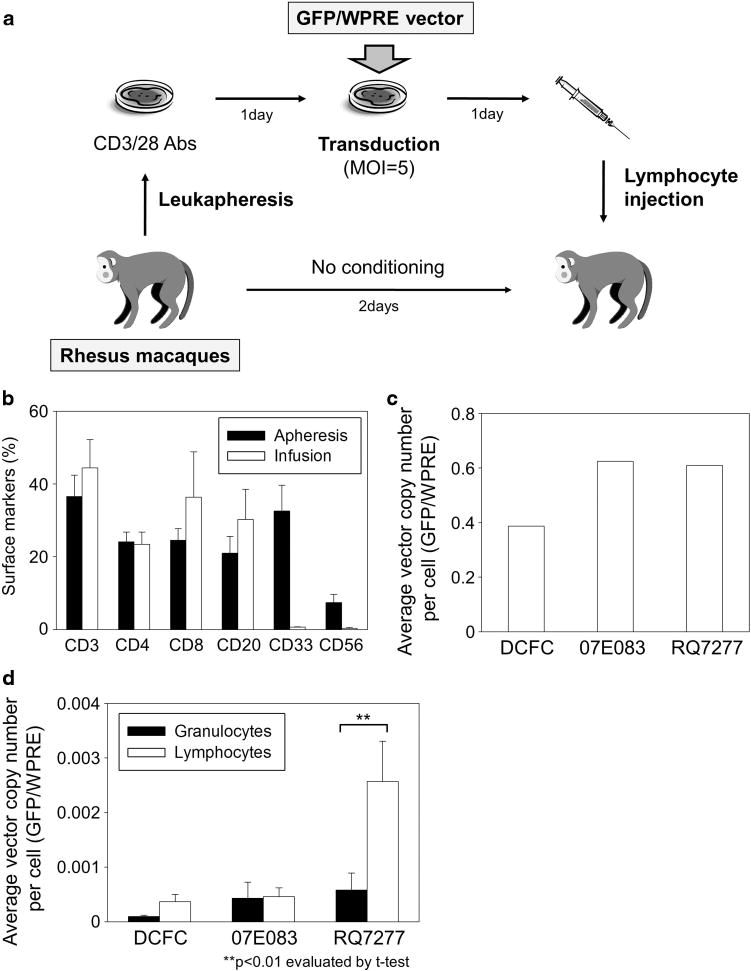
Lymphocyte infusion with GFP/WPRE marking into the transplanted animals. (a) To evaluate whether the introduction of transduced HSCs after 4 Gy TBI could establish immunological tolerance for the GFP transgene, we performed autologous lymphocyte infusion after transduction with a WPRE-containing GFP-expressing vector (GFP/WPRE vector) in the two RIC-transplanted animals (DCFC and 07E083). As a control animal with GFP tolerance, we utilized a rhesus (RQ7277) that had 5–10% of GFP-positive rates in peripheral blood cells for 2 years after HSC transplantation with 10 Gy TBI. Cells collected by leukapheresis from the three animals were cultured on CD3/28 antibody-coated plates and transduced with the WPRE/GFP vector at MOI 5. The cultured cells were infused into the animals without conditioning. (b) The cultured cells mainly contained lymphocytes but not granulocytes. (c) We observed efficient GFP/WPRE marking in the transduced lymphocytes in vitro, which was evaluated by average VCN per cell in real-time PCR using WPRE-specific probe/primers. (d) In the control animal (RQ7277), lymphocytes had higher GFP/WPRE marking levels in vivo for 12 weeks after infusion, compared with granulocytes (Po0.01), whereas no difference in GFP/WPRE signals between lymphocytes and granulocytes was observed in the two RIC-transplanted animals (n.s.). Abbreviation: CD3/28 Abs, CD3 antibody and CD28 antibody.
We observed efficient transduction with the GFP/WPRE vector in the cultured cells in vitro, as evaluated by %GFP (29.5–49.1%) at 3 days following transduction (Supplementary Figure 2a) and GFP/WPRE-VCN (0.39–0.61) at 6 days after transduction (Figure 6c). In the control animal (RQ7277), in vivo GFP/WPRE-VCN in lymphocytes increased for 12 weeks after infusion, compared with that of granulocytes (P<0.01), whereas no difference in GFP/WPRE-VCN between lymphocytes and granulocytes was observed in the two RIC-transplanted animals (n.s.) (Figure 6d and Supplementary Figure 2b), which had no remaining detectable GFP expression in peripheral blood cells (Supplementary Figures 3a and b).
In addition, we performed mixed lymphocyte culture to evaluate in vitro proliferation of autologous T lymphocytes after stimulation with irradiated lymphocytes transduced with a GFP-expressing vector (Figure 7a). In lymphocyte cultures from the two RIC-transplanted animals, significant proliferation was observed after stimulation with GFP-positive autologous lymphocytes compared with no stimulation control (P<0.01), whereas no significant proliferation of T cells was observed in the control animal (n.s.) (Figure 7b). In addition, T-cell proliferation was observed after stimulation with empty vector-transduced lymphocytes in DCFC (P<0.01).
Figure 7.
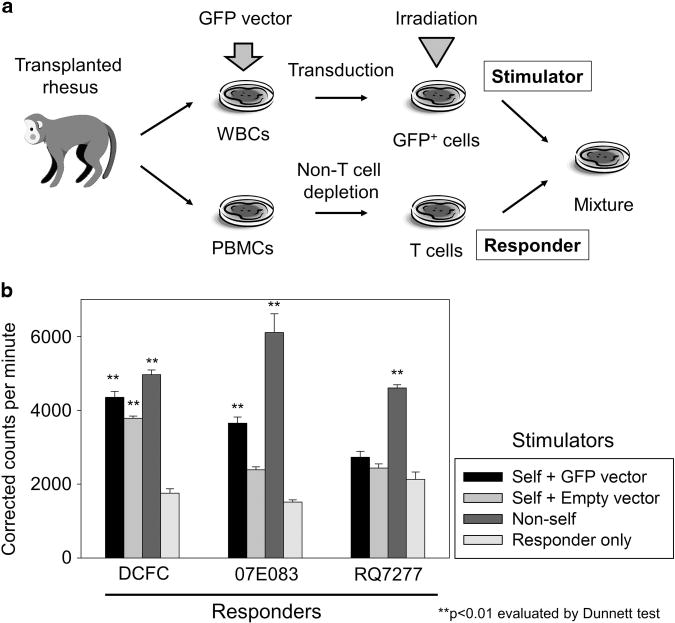
T-cell proliferation by GFP stimulation in the RIC-transplanted animals. (a) We performed mixed lymphocyte culture, to evaluate in vitro proliferation of autologous T lymphocytes after stimulation with irradiated lymphocytes transduced with a GFP-expressing vector (GFP vector). (b) In the two RIC-transplanted animals (DCFC and 07E083), significant proliferation was observed after stimulation with GFP-positive autologous lymphocytes compared with no stimulation control (P<0.01), whereas no significant proliferation of T cells was observed in the control animal RQ7277 (n.s.). Abbreviations: PBMCs, peripheral blood mononuclear cells; WBCs, white blood cells.
To investigate humoral immune response, we measured anti-GFP antibody production using serum obtained from the RIC-transplanted animals (DCFC and 07E083), the control animal with GFP tolerance (RQ7277) and an untransplanted animal (Figure 8). We observed high titers of GFP-specific immunoglobulin G (IgG) in the RIC-transplanted animals, detectable to a dilution of 1500–3900-fold. Anti-GFP IgG titers in the tolerant and untransplanted animals were dramatically lower in comparison and there was relatively little difference (threefold) between them. These data suggest that 4 Gy TBI alone is insufficient for inducing tolerance to the transgene and that the two RIC-transplanted animals have established cellular and humoral immunological memory to the GFP transgene. In contrast, animals transplanted with ablative doses of TBI with stable engraftment of genetically modified cells have tolerance to the transgene product.
Figure 8.
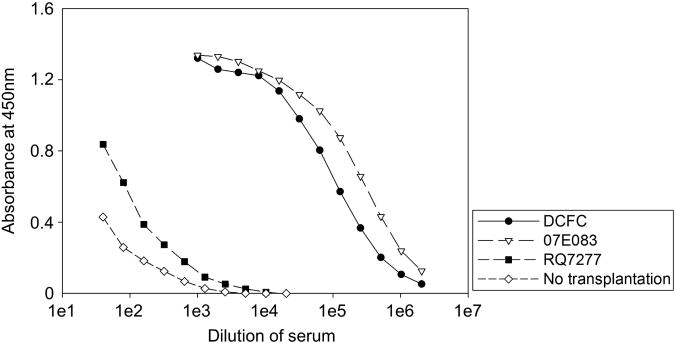
Anti-GFP antibody production in the RIC-transplanted animals. To evaluate humoral immune response in rhesus macaques, we performed ELISA that detected anti-GFP antibody in the sera of the RIC-transplanted animals (DCFC and 07E083), the control animal with GFP tolerance (RQ7277) and an untransplanted animal. We observed anti-GFP antibody in the RIC-transplanted animals, out to dilution of 1500–3900-fold. On the other hand, there was little difference (threefold) of anti-GFP antibody titers between the control animal with GFP tolerance (RQ7277) and the untransplanted animal.
DISCUSSION
RIC regimens are thought to be suitable for HSC gene therapy due to their presumed superior safety profile; however, previous clinical gene therapy trials demonstrated low gene marking in peripheral blood cells, even with efficient gene marking in vitro.1–6 To investigate the effects of a RIC regimen in a gene therapy setting using optimized gene-transfer methods, we utilized a RIC regimen of 4 Gy TBI in our rhesus HSC transplantation model with lentiviral transduction (Figure 1a),11–13 as we previously demonstrated long-term engraftment of sibling donor cells in SCD patients following 3–4 Gy TBI.8 In the two rhesus macaques receiving 4 Gy TBI, we observed no detectable GFP expression by flow cytometry (Figures 3 and 5) and low vector signals by real-time PCR (Figures 4 and 5), while we have previously demonstrated high long-term gene marking following 10 Gy TBI in several animals.11–13 In addition, in these two RIC-transplanted animals, no difference in VCN between GFP and empty vectors was observed in peripheral blood cells (Figure 4). These data suggest that the lack of GFP expression in peripheral blood cells is due to poor engraftment of transduced CD34+ cells instead of gene silencing. TBI conditioning (4 Gy) does not provide sufficient niche space for engraftment of transduced CD34+ cells.
In the two RIC-transplanted animals, we observed mild bone marrow suppression and relatively slow recovery to baseline in peripheral blood cell counts (around 1 month, Figure 2). In rhesus HSC transplantation following 10 Gy TBI, GFP-positive peripheral blood cells routinely recovered 1–2 weeks after transplantation.11–13 This slower recovery of GFP-negative peripheral blood cells also suggests reconstitution from autologous HSCs which survived conditioning with 4 Gy TBI rather than from engrafted transplanted CD34+ cells. In addition, the 4 Gy TBI conditioning produced mild bone marrow suppression with one animal not falling below either WBC 1000 μl−1 or PLT 50 000 μl−1.
Intriguingly, at 1–2 months after transplantation, we observed very high %GFP positivity (80–90%) with low GFP intensity in both granulocytes and monocytes which declined to undetectable levels 3–4 months after transplantation (Figure 3). The majority GFP-positive cells could not be easily explained by reconstitution from GFP-transduced HSCs, because only half of CD34+ cells were transduced with GFP vector, whereas the other half of CD34+ cells were transduced with empty vector not expressing GFP. It is also unusual that GFP intensity in granulocytes and monocytes is lower than RBCs (Supplementary Figure 1). Interestingly, %GFP in RBCs (~2%) also declined to undetectable levels in the same time period (3–4 months). It is possible that the GFP protein expressed in RBCs was recognized and phagocytized by granulocytes and monocytes. This hypothesis is also supported by the real-time PCR data of low gene marking in peripheral blood cells, even when high %GFP in granulocytes and monocytes was observed.
We then evaluated immunological reactions to the GFP transgene in the two RIC-transplanted animals. As a control animal with GFP tolerance, we utilized a rhesus that has 5–10% GFP-positive in peripheral blood cells 2 years after HSC transplantation, as we previously demonstrated that stable engraftment of gene-modified HSCs induces tolerance to the transgene products.14,15 Strikingly, autologous lymphocyte injection with GFP/WPRE marking resulted in no increase of GFP/WPRE-positive lymphocytes in peripheral blood cells in the RIC-transplanted animals (Figure 6 and Supplementary Figures 2 and 3), and in vitro T-cell proliferation was observed after GFP stimulation in mixed lymphocyte culture in the two RIC-transplanted animals (Figure 7). In addition, substantial anti-GFP antibody production was detected exclusively in the RIC-transplanted animals (Figure 8). Much lower titers were observed in the previously transplanted control animal (RQ7277) with GFP tolerance and the untransplanted control. These data suggest that the two RIC-transplanted animals acquired immunological memory for the GFP transgene rather than immunological tolerance. This specific immunological reaction to the GFP protein may be the result of GFP epitopes frequently being presented to newly emerging lymphocytes by granulocytes and monocytes when these cells had high %GFP 1–2 months after transplantation. Interestingly, the mixed lymphocyte culture in one RIC animal (DCFC) revealed T-cell proliferation after stimulation with empty vector-transduced lymphocytes as well as GFP-transduced lymphocytes (Figure 7). The empty vector should not stably express any transgene, but it contains viral component proteins which were packaged when viral vectors were prepared. Immunological reactions might therefore be induced by not only transgenes but also vector components in the gene therapy setting.
Generally, HSC transplantation follows conditioning therapy, which has two main effects to open niche space for efficient engraftment (myeloablation) and to induce immunological tolerance for donor cells (immunosuppression).7 Traditionally, RIC in allogeneic transplantation focused on immunosuppression, as reconstituted donor cells can recognize and deplete recipient cells remaining in the bone marrow, often producing a desirable graft-versus-leukemia effect which can be further enhanced by donor lymphocyte infusion.9,10 However, in our gene therapy setting, genetically modified blood cells should not recognize non-modified cells, as autologous HSCs are utilized for gene modification. On the other hand, genetically modified cells can be recognized by non-modified cells due to immunoreaction to the transgene, which was confirmed by our lymphocyte infusion and mixed lymphocyte culture in the RIC-transplanted animal (Figures 6 and 7). Immunological tolerance to the transgene should be required for long-term gene marking in peripheral blood cells after gene therapy. We also demonstrated immunological tolerance to the GFP protein in an HSC-transplanted rhesus macaque, RQ7277 (Figures 6 and 7) that had long-term GFP marking in peripheral blood cells.11 Theoretically, conventional myeloablative conditioning should be sufficient for engraftment and immunological tolerance for genetically modified cells, as our rhesus model has revealed long-term gene marking after a split dose of 10 Gy TBI.11–13 Our data suggest that RIC regimens for gene therapy should focus not only on creating open niches for engraftment but also on immunomodulatory therapy to achieve immunological tolerance to the transgene-encoded product.
In clinical gene therapy settings for various diseases, preexisting immunity may be present in patients. For example, the β-globin gene can be used for a therapeutic application in SCD gene therapy,16,17 whereas the RBCs in patients include sickle-globin which is caused by a point mutation in β-globin that replaces the glutamic acid with valine at the sixth position.18 β-globin may be tolerable in SCD patients, as almost all amino acids except one at the sixth position are same as the sickle-globin. Clinically, RBCs from healthy donors (with β-globin) can be infused into sickle cell patients (with sickle-globin) without clinically relevant immunological reactions. On the other hand, antibodies in a pig model were reported to discriminate between β-globin and sickle-globin.19 As it is controversial whether the one amino-acid difference between β-globin and sickle-globin is immunogenic, we may still need to consider a possibility of immunological reaction to β-globin in SCD gene therapy.
In summary, 4 Gy TBI allows transient existence of GFP-positive RBCs in peripheral blood in transplanted animals; however, it is incapable to support efficient engraftment of genetically modified hematopoietic repopulating cells. This low dose TBI-only approach is insufficient to induce immunological tolerance. Our findings on RIC in a rhesus HSC transplantation model will inform the design of future conditioning regimens for gene therapy trials.
METHODS
Lentiviral vector preparation
We previously developed a chimeric HIV-1-based lentiviral vector system (χHIV vector), in which the HIV-1 genomes were packaged into the simian immunodeficiency virus capsid to efficiently transduce rhesus hematopoietic cells.11–13 The χHIV vectors were prepared by using self-inactivating HIV-1 vector, gag/pol, rev/tat and vesicular stomatitis virus G glycoprotein envelope plasmids.20,21 The GFP-expressing vector contained a GFP expression cassette under the control of a murine stem cell virus promoter (pCL20cMpGFP).22,23 The empty vector contained a multiple cloning site instead of the GFP gene, which did not express a transgene. The GFP/WPRE vector contained a WPRE element between GFP and 3′LTR. The HIV-1 vector system was kindly provided from Dr Arthur Nienhuis (St Jude Children’s Research Hospital, Memphis, TN, USA).22,24 The viral titers in GFP-expressing vectors were evaluated by %GFP in transduced HeLa cells, whereas the viral titers in the empty vector were calculated by directly comparing relative VCN in empty vector-transduced HeLa cells to that of GFP-transduced cells.13,25
RIC HSC transplantation with lentiviral transduction in rhesus macaques
We previously developed rhesus HSC transplantation with lentiviral transduction following a split dose (2×5 Gy) of 10 Gy TBI, which demonstrated efficient long-term gene marking in peripheral blood cells.11–13 In this study, we utilized 4 Gy TBI as a RIC regimen, instead of 10 Gy TBI. Briefly, granulocyte colony-stimulating factor (Amgen, Thousand Oaks, CA, USA) and stem cell factor (Amgen)-mobilized CD34+ cells were cultured in serum-free X-VIVO10 media (Lonza, Allendale, NJ, USA) containing stem cell factor, FMS-related tyrosine kinase 3 ligand (FLT3L) and thrombopoietin (all 100 ng ml−1; R&D Systems, Minneapolis, MN, USA). After 1 day prestimulation, one half of CD34+ cells were transduced with GFP vector at multiplicity of infection (MOI) 50, and the other half of cells were transduced with empty vector on the identical conditions. One day later, these cells were infused into rhesus macaques following one dose of 4 Gy TBI.
We evaluated both %GFP and mean fluorescence intensity of GFP in GFP-positive fractions in peripheral blood cells by flow cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA). Lineage-specific surface marker analysis was performed as previously described.11 The VCN was evaluated by real-time PCR using GFP-specific probe/primers13 and empty vector-specific probe/primers (forward primer: 5′-CTG TTC GCG CGC TTC TGC TC-3′, Reverse primer: 5′-CGA TGC GGC CGC GAA TTC TG-3′, Probe: 5′-AGT CCT CCG ATA GAC TGC GTC GCC CT-3′) for 40 cycles of 30 s at 95 °C, 30 s at 60 °C and 15 s at 72 °C. The CFU assay was performed as previously described.13,26,27
Autologous lymphocyte infusion with lentiviral transduction in rhesus macaques
We performed rhesus lymphocyte infusion with lentiviral transduction using a modified protocol, which we previously developed for retroviral vectors.14,28 Non-tissue-culture-treated flasks were coated by both anti-monkey CD3 antibody (Clone FN-18; Life Technologies, Grand Island, NY, USA) and anti-human CD28 antibody with cross-reactivity against rhesus (Clone CD28.2; BD Biosciences).29–31 Leukapheresis cells (1–2×10e8) were cultured on CD3/28 antibody-coated 75 cm2 flasks including 25 ml T-cell expansion SFM media (Life Technologies). After 1 day prestimulation, the cultured leukapheresis cells were transduced with the GFP/WPRE vector at MOI 5.32,33 The next day, the transduced cells were infused into rhesus macaques without conditioning. The transduction rates were evaluated by %GFP in flow cytometry and VCN in real-time PCR using WPRE-specific probe/primers,34 which can detect the GFP/WPRE-transduced lymphocytes (from lymphocyte infusion), but not blood cells from GFP-transduced HSCs (from stem cell transplantation).
Mixed lymphocyte culture
We performed mixed lymphocyte culture in rhesus macaques using a modified standard protocol. As a stimulator, autologous white blood cells were cultured on CD3/28-coated dishes and transduced with the GFP-expressing vector. The transduced cells were then subjected to 25 Gy irradiation to prevent proliferation. In parallel, autologous responder T lymphocytes were negatively selected from rhesus peripheral blood mononuclear cells using the Pan T Cell Isolation Kit, non-human primate (Miltenyi Biotec Inc., Auburn, CA, USA). In total, 1×105 responder T lymphocytes and 1×105 stimulator GFP-positive irradiated lymphocytes were co-cultured at a 1:1 ratio in triplicate wells of a 96-well plate for 4 days before adding 25 μl media containing 1 μCi 3H-thymidine per well. Cells were then cultured for another 24 h before harvesting and evaluating counts per minute (cpm) using a PerkinElmer (Waltham, MA, USA) MicroBeta microplate scintillation counter.
Enzyme-linked immunosorbent assay
We performed ELISA (enzyme-linked immunosorbent assay) to detect anti-GFP antibody in the serum among the transplanted rhesus macaques, as previously described.35 We used frozen serum from DCFC and 07E083 obtained 1 year after RIC transplantation, serum from the control animal with GFP tolerance (RQ7277) obtained 2 years after transplantation and serum from an untransplanted animal. Briefly, we coated a 96-well plate with a recombinant GFP protein (Clontech, Mountain View, CA, USA), and added serial dilutions of monkey serum or control anti-GFP IgG (Abcam, Cambridge, MA, USA) into these wells. After incubation and washing, anti-monkey IgG secondary antibody conjugated to horseradish peroxidase (Abcam) was added to detect GFP-bound immunoglobulin, and tetramethylbenzidine reaction signals were detected by absorbance at 450 nm.
Statistical analysis
Statistical analyses were performed using the JMP 9 software (SAS Institute Inc., Cary, NC, USA). Two averages were evaluated by the Student’s t-test. The averages in various conditions were evaluated by Dunnett’s test (one-way ANOVA for one control). A P-value of <0.01 was deemed significant. Standard errors of the mean are shown as error bars in figures.
Supplementary Material
Acknowledgments
This work was supported by the intramural research program of the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute of Diabetes, Digestive, and Kidney Diseases (NIDDK) at the National Institutes of Health (NIH). We thank the animal care staff and technicians at 5 Research Court for their excellent care and handling of the animals. We would also like to thank William DeGraff and Dr Jim Mitchell of the Radiation Biology Branch, National Cancer Institute (NCI) for the use of the Eldorado Cobalt-60 irradiator.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on Gene Therapy website (http://www.nature.com/gt)
References
- 1.Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 2.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 3.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 5.Schwarzwaelder K, Howe SJ, Schmidt M, Brugman MH, Deichmann A, Glimm H, et al. Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo. J Clin Invest. 2007;117:2241–2249. doi: 10.1172/JCI31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 7.Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh MM, Kang EM, Fitzhugh CD, Link MB, Bolan CD, Kurlander R, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Storb RF, Champlin R, Riddell SR, Murata M, Bryant S, Warren EH. Non-myeloablative transplants for malignant disease. Hematology Am Soc Hematol Educ Program. 2001:375–391. doi: 10.1182/asheducation-2001.1.375. [DOI] [PubMed] [Google Scholar]
- 10.Antin JH. Reduced-intensity stem cell transplantation: ‘…whereof a little More than a little is by much too much.’ King Henry IV, part 1, I, 2. Hematology Am Soc Hematol Educ Program. 2007:47–54. doi: 10.1182/asheducation-2007.1.47. [DOI] [PubMed] [Google Scholar]
- 11.Uchida N, Bonifacino A, Krouse AE, Metzger ME, Csako G, Lee-Stroka A, et al. Accelerated lymphocyte reconstitution and long-term recovery after transplantation of lentiviral-transduced rhesus CD34(+) cells mobilized by G-CSF and plerixafor. Exp Hematol. 2011;39:795–805. doi: 10.1016/j.exphem.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uchida N, Hargrove PW, Lap CJ, Evans ME, Phang O, Bonifacino AC, et al. High-efficiency transduction of rhesus hematopoietic repopulating cells by a modified HIV1-based lentiviral vector. Mol Ther. 2012;20:1882–1892. doi: 10.1038/mt.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchida N, Washington KN, Hayakawa J, Hsieh MM, Bonifacino AC, Krouse AE, et al. Development of a human immunodeficiency virus type 1-based lentiviral vector that allows efficient transduction of both human and rhesus blood cells. J Virol. 2009;83:9854–9862. doi: 10.1128/JVI.00357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heim DA, Hanazono Y, Giri N, Wu T, Childs R, Sellers SE, et al. Introduction of a xenogeneic gene via hematopoietic stem cells leads to specific tolerance in a rhesus monkey model. Mol Ther. 2000;1:533–544. doi: 10.1006/mthe.2000.0072. [DOI] [PubMed] [Google Scholar]
- 15.Kung SK, An DS, Bonifacino A, Metzger ME, Ringpis GE, Mao SH, et al. Induction of transgene-specific immunological tolerance in myeloablated nonhuman primates using lentivirally transduced CD34 + progenitor cells. Mol Ther. 2003;8:981–991. doi: 10.1016/j.ymthe.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 16.Nienhuis AW. Development of gene therapy for blood disorders. Blood. 2008;111:4431–4444. doi: 10.1182/blood-2007-11-078121. [DOI] [PubMed] [Google Scholar]
- 17.Persons DA. Hematopoietic stem cell gene transfer for the treatment of hemoglobin disorders. Hematology Am Soc Hematol Educ Program. 2009;1:690–697. doi: 10.1182/asheducation-2009.1.690. [DOI] [PubMed] [Google Scholar]
- 18.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 19.Otieno S, Bloor A, Karol R, Reichlin M, Noble RW. Specific antibodies to hemoglobin A1 (anti-Glu) and hemoglobin S (anti-Val) in the guinea pig: immunologic and structural correlations. J Immunol. 1978;121:2458–2462. [PubMed] [Google Scholar]
- 20.Uchida N, Hsieh MM, Hayakawa J, Madison C, Washington KN, Tisdale JF. Optimal conditions for lentiviral transduction of engrafting human CD34(+) cells. Gene Therapy. 2011;18:1078–1086. doi: 10.1038/gt.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida N, Washington KN, Lap CJ, Hsieh MM, Tisdale JF. Chicken HS4 insulators have minimal barrier function among progeny of human hematopoietic cells transduced with an HIV1-based lentiviral vector. Mol Ther. 2010;19:133–139. doi: 10.1038/mt.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanawa H, Persons DA, Nienhuis AW. Mobilization and mechanism of transcription of integrated self-inactivating lentiviral vectors. J Virol. 2005;79:8410–8421. doi: 10.1128/JVI.79.13.8410-8421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uchida N, Hanawa H, Yamamoto M, Shimada T. The chicken HS4 core insulator blocks promoter interference in lentiviral vectors. Hum Gene Ther Methods. 2013;24:117–124. doi: 10.1089/hgtb.2012.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanawa H, Hematti P, Keyvanfar K, Metzger ME, Krouse A, Donahue RE, et al. Efficient gene transfer into rhesus repopulating hematopoietic stem cells using a simian immunodeficiency virus-based lentiviral vector system. Blood. 2004;103:4062–4069. doi: 10.1182/blood-2004-01-0045. [DOI] [PubMed] [Google Scholar]
- 25.Uchida N, Hanawa H, Dan K, Inokuchi K, Shimada T. Leukemogenesis of b2a2-type p210 BCR/ABL in a bone marrow transplantation mouse model using a lentiviral vector. J Nippon Med Sch. 2009;76:134–147. doi: 10.1272/jnms.76.134. [DOI] [PubMed] [Google Scholar]
- 26.Tisdale JF, Hanazono Y, Sellers SE, Agricola BA, Metzger ME, Donahue RE, et al. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood. 1998;92:1131–1141. [PubMed] [Google Scholar]
- 27.Uchida N, Hsieh MM, Washington KN, Tisdale JF. Efficient transduction of human hematopoietic repopulating cells with a chimeric HIV1-based vector including SIV capsid. Exp Hematol. 2013;41:779–788. doi: 10.1016/j.exphem.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanazono Y, Brown KE, Handa A, Metzger ME, Heim D, Kurtzman GJ, et al. In vivo marking of rhesus monkey lymphocytes by adeno-associated viral vectors: direct comparison with retroviral vectors. Blood. 1999;94:2263–2270. [PubMed] [Google Scholar]
- 29.Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- 30.Pollok KE, Hanenberg H, Noblitt TW, Schroeder WL, Kato I, Emanuel D, et al. High-efficiency gene transfer into normal and adenosine deaminase-deficient T lymphocytes is mediated by transduction on recombinant fibronectin fragments. J Virol. 1998;72:4882–4892. doi: 10.1128/jvi.72.6.4882-4892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou P, Lee J, Moore P, Brasky KM. High-efficiency gene transfer into rhesus macaque primary T lymphocytes by combining 32 degrees C centrifugation and CH-296-coated plates: effect of gene transfer protocol on T cell homing receptor expression. Hum Gene Ther. 2001;12:1843–1855. doi: 10.1089/104303401753153901. [DOI] [PubMed] [Google Scholar]
- 32.Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 33.Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. 2008;14:1390–1395. doi: 10.1038/nm.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geraerts M, Willems S, Baekelandt V, Debyser Z, Gijsbers R. Comparison of lentiviral vector titration methods. BMC Biotechnol. 2006;6:34. doi: 10.1186/1472-6750-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houter JVKD. Antibody response induced after intraocular viral gene addition therapy using adeno-associated virus, lentivirus, and adenovirus vectors with the GFP Transgene in dogs. ProQuest LLC; Ann Arbor, MI, USA: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.