

Abstract
Nox generated ROS, particularly those derived from Nox1, Nox2 and Nox4, have emerged as important regulators of the actin cytoskeleton and cytoskeleton-supported cell functions, such as migration and adhesion. The effects of Nox-derived ROS on cytoskeletal remodeling may be largely attributed to the ability of ROS to directly modify proteins that constitute or are associated with the cytoskeleton. Additionally, Nox-derived ROS may participate in signaling pathways governing cytoskeletal remodeling. In addition to these more extensively studied signaling pathways involving Nox-derived ROS, there also exist redox sensitive pathways for which the source of ROS is unclear. ROS from as of yet undetermined sources play a role in modifying, and thus regulating, the activity of several proteins critical for remodeling of the actin cytoskeleton. In this review we discuss ROS sensitive targets that are likely to affect cytoskeletal dynamics, as well as the potential involvement of Nox proteins.
Keywords: Cytoskeleton, NADPH oxidase, ROS, oxidation, signaling
CYTOSKELETON-RELATED STRUCTURES AND NADPH OXIDASES
The cytoskeleton is a network of filaments that helps cells maintain their shape and internal organization, as well as their mechanical properties. It is made up of three kinds of protein filaments: microtubules (about 25nm external diameter), intermediate filaments (about 10nm external diameter) and actin filaments (8nm external diameter) Fig. (1). Cytoskeletal dynamics enable cells to carry out essential functions such as migration, attachment, spreading and proliferation. Another important role of the cytoskeleton is to assist the movement of proteins, cargo-containing vesicles and organelles inside the cell. This is achieved by the association of structural proteins of the cytoskeleton with motor proteins such as kinesins, dyneins and myosins, which utilize ATP to generate movement. As expected of such energetically costly processes, cytoskeletal dynamics are highly regulated to suit the requirements of the cell as a whole. In this regard, there has been increasing interest in the regulation of cytoskeletal dynamics by reactive oxygen species, since many of the proteins participating in these processes are particularly sensitive to redox regulation [1].
Fig. 1. Main cytoskeleton components that can be affected by ROS.
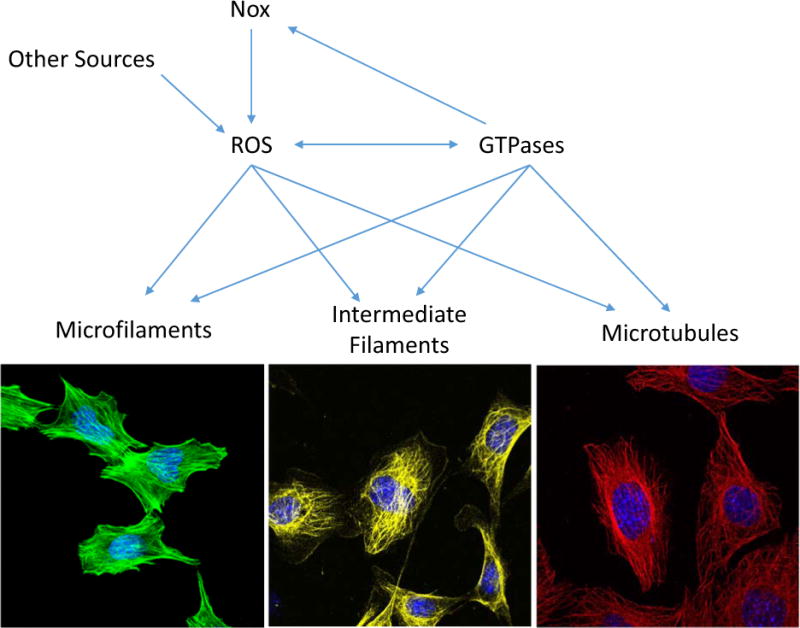
ROS generated by Nox (e.g. Nox1, Nox2, Nox4) and from other sources, has a direct effect on cytoskeleton by modifying directly microfilaments, intermediate filaments and microtubules. Additionally, ROS regulates the activity of proteins that are associated with cytoskeleton dynamic such as Rho GTPases affecting cell adhesion, migration and invasion. Moreover, Rac GTPase can activate Nox and increase the levels of ROS. Images show mouse embryonic fibroblasts (MEFs) stained for actin microfilaments (green, Phallodin-Alexa488), Vimentin as an example of intermediate filaments (yellow, α-Vimentin/α-mouse-Alexa568), α/β-tubulin (red, α-tubulin/α-rabbit-Alexa 568), and DAPI as a marker of nucleus (blue).
NADPH oxidases are a family of membrane-associated multisubunit enzymes that represent a major source of reactive oxygen species (ROS) in diverse systems [2–5]. Activation of members of the Nox family, which include Nox1-5 and Duox1/2, leads to production of O2•− and H2O2. These are released extracellularly and can reach the intracellular compartment by either simple diffusion, as in the case of H2O2, or via membrane-enclosed vesicles, in the case of superoxide [6]. Nox-derived ROS have been shown to modulate intracellular signaling pathways in a paracrine, autocrine, and even intracrine manner [7–10]. Although their activities have been linked to multiple disease states, today it is well accepted that under physiological conditions they are required for normal cell function. Indeed, NADPH oxidase-produced ROS participate in host defense [11, 12], cardiac repair [13], and cell differentiation [14–17].
Nox-based enzymes share a similar structure, containing at least six transmembrane domains and cytosolic flavin adenine dinucleotide (FAD) and NADPH binding domains [6]. Most of the NADPH oxidase complexes consist of a distinguishing catalytic subunit, and five variable regulatory phox subunits. The first member of this family to be identified and the best-studied is the Nox2-based NADPH oxidase, which mediates the respiratory burst of neutrophils [18]. This enzyme’s catalytic subunit is gp91phox (Nox2), which contains one FAD and two hemes, and catalyzes NADPH-dependent reduction of O2 to form O2•− [19, 20]. Nox proteins form a complex in the membrane with a stabilizing factor, p22phox [21–23] and, with the exception of Nox4 which is constitutively active, are dormant in resting cells [6]. A number of stimuli alter the expression and activity of Nox proteins including Angiotensin II (AngII) [24], platelet derived growth factor (PDGF) [24], and transforming growth factor beta (TGF-β) [25, 26]. Upon agonist stimulation, assembly of Nox proteins with cytosolic regulatory proteins and small GTPases results in their enzymatic activity [27]. A requirement for p47phox in Nox1 activation by AngII and PDGF has been clearly established both in vivo and in vitro [28, 29]. Nox1 can also interact with two novel homologues of p47phox and p67phox known as Noxo1 and Noxa1, respectively [30, 31], as well as the small GTPase Rac [32]. In contrast, no cytosolic subunits are required for ROS generation by Nox4 [33]; however, in vascular smooth muscle cells (VSMCs) the polymerase delta interacting protein 2 (Poldip2) binds to the C-terminal tail of p22phox and increases Nox4 activity [34].
Depending on the cell type, Nox proteins localize to different subcellular compartments. Interestingly, Nox proteins localize to subcellular compartments associated with the cytoskeleton. For example, in VSMCs, Nox4 localizes to stress fibers in differentiated cells and focal adhesions (FAs) in proliferating cells, while Nox1 is found in caveolae [17]. p47phox has been shown to colocalize with moesin and WAVE, two proteins closely related to lamellipodia [35], and is responsible for the production of H2O2 required to induce actin remodeling and directed ventral lamellipodia formation in endothelial cells [36]. In the same cell type, p47phox-dependent ROS production has also been shown to mediate vascular endothelial growth factor (VEGF)-induced membrane ruffle formation through its association with WAVE1, Rac1 and p21 associated kinase (PAK)1 [37]. In addition, Nox1 enzymatic activity is stimulated by growth-promoting agonists that induce membrane ruffling, such as PDGF and AngII [12, 13, 24, 38, 39], further implicating Nox proteins in growth factor stimulated actin remodeling. There is mounting evidence that Nox proteins may mediate growth factor signaling via ROS production in endosomal compartments, or “redoxosomes” [40, 41]. The colocalization of Nox proteins with specific receptors in distinct endosomal compartments provides another regulatory mechanism for generating stimuli-specific cellular responses (see [40, 41]).
During the following sections we will first review some of the most likely targets of redox regulation within the cytoskeleton, and then we will focus on how NADPH oxidase-mediated redox regulation may impact important cellular functions supported by the cytoskeleton such as migration and cell attachment.
CYTOSKELETON AND CYTOSKELETON-ASSOCIATED PROTEINS AS TARGETS OF REDOX REGULATION
Role of Actin Oxidation in Cytoskeletal Reorganization
Conceivably, ROS may participate in the remodeling of the cytoskeleton by modification of proteins and enzymes that regulate actin dynamics, or by direct oxidization of the cytoskeleton structural filaments. It is well understood that proteins containing thiols with a low pKa (capable of rendering thiolate anions at physiological pH) are targets of oxidation. Indeed, thiolates react with H2O2 to form sulfenic (SOH), sulfinic (SO2H) and sulfonic (SO3H) acids or protein disulfides (PrSSPr). Unlike the formation of sulfonic acid or sulfinic acid (the reduction of which is dependent on sulfite reductases), sulfenic acid is reversible, and has therefore been linked to cellular signalling [42]. Oxidized thiols can also react with glutathione (GSH) to form glutathiolated disulfides (PrSSG). Glutathiolation is reversible by reduction via glutathione peroxidase, thioredoxin or peroxiredoxins. A number of proteins involved in cytoskeletal reorganization are potential targets for oxidation or glutathiolation, but only a few have been confirmed, including Src [43], Csk [44], actin [45], and a number of phosphatases (PTP-PEST, LMW-PTP and SHP-2 [46, 47]). Of these, oxidation of β-actin has been extensively studied. Indeed, in the last few years it has been recognized that H2O2-mediated actin oxidation regulates actin dynamics. Direct treatment of β-actin with 10–20 mM of H2O2 has been shown to decrease the rate of actin polymerization. Although these doses are likely to be supraphysiological, these studies helped to identify several cysteines within the actin sequence that are redox sensitive. Indeed, mass spectrometry analysis shows that the C-terminal cysteine of α-actin (Cys376) or β-actin (Cys374) can be oxidized in either G-actin or F-actin [45, 48]. This high concentration of oxidants is capable of affecting most of the parameters that determine actin polymerization such as: the time required for half-maximum assembly, the elongation rate, the critical monomer concentration for polymerization, and the binding of the actin capping protein filamin [45, 48, 49]. Another report describes that the oxidation reaction in β-actin occurs at two main redox sensitive cysteine amino acid residues, Cys272 and Cys374 [50], and can be reversed by thioredoxin or glutathione. In particular, oxidation on Cys374 has been shown to cause actin depolymerization in part due to a decreased affinity for profilin [50]. Furthermore, Cys374 has been shown to be glutathiolated [51], which leads to a reduced rate of polymerization, instability of F-actin filaments, and enhancement of ATPase activity in vitro [52, 53]. As expected, actin-myosin-mediated contraction is decreased after treatment of permeabilized rabbit Psoas muscle fibers with 50 mM H2O2, as is actomyosin enzyme activity [54]. On the other hand, low concentrations of oxidants may have the opposite effect on actin dynamics. Treatment of P388D1 cells with 1–5 mM H2O2 increases stress fiber formation [55]. Moreover, mild oxidizing conditions that favor actin-actin disulfide bond formation induce a greater rate and extent of actin polymerization. Further evidence that oxidants promote actin polymerization is that treatment of migrating endothelial cells with the flavin-containing oxidase inhibitor DPI or the O2•− and peroxinitrite scavenger manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin (MnTMPyP) inhibits actin monomer incorporation at the barbed end [56]. Interestingly, modification of endogenous levels of glutaredoxin 1, with no addition of exogenous oxidants, increases actin glutathionylation and impaires its polymerization in neutrophils. Many of these pathways could be sensitive to low output enzymes such as NADPH oxidases, although more research is required to define the role of Nox-produced ROS in the described mechanism.
Microtubules Oxidation
Microtubules are filamentous structures composed of a single type of globular protein, called tubulin. They are involved in cell division, organization of intracellular structures, intracellular transport, and ciliary and flagellar motility. Microtubules are formed by a dimer consisting of two closely related 55-kd polypeptides, α-tubulin and β-tubulin. In addition, a third type of tubulin (γ-tubulin) is specifically localized to the centrosome. When intracellular conditions favor assembly, tubulin heterodimers assemble into linear protofilaments, and later into microtubules. Though apparently stable, microtubular structures have an intrinsic in stability, which is normally recognized as a dynamic equilibrium, or steady state. Posttranslational modifications within tubulin seem to govern the degree of microtubule stability; these have been extensively studied, and the reader can find pertinent information in the literature [57, 58].
Recently, it has been recognized that cysteine oxidation may also contribute to the variety of posttranslational modifications found in tubulin. Indeed, it has been reported that oxidation of tubulin cysteines by peroxynitrite, 2-[(1-methylpropyl)dithio]-1H-imidazole or hypothiocyanous acid inhibits microtubule polymerization [59–61]. Since cysteine residues are not part of the protein domains responsible for the dimerization and polymerization of tubulin [62], more studies are needed to determine if this is a direct effect on filament stabilization, or an effect on the binding of proteins that regulate the polymerization process; in addition, more work is needed to determine the endogenous sources of ROS that may mediate these oxidations in vivo.
Intermediate Filament Oxidation
There are an increasing number of oxidative modifications of intermediate filaments reported in the literature, which we have summarized in Table 1. However, how these modifications impact cytoskeletal dynamics and cytoskeleton-supported functions have yet to be studied.
Table 1.
Oxidation of Intermediate Filaments.
| Oxidation Induced by: | Cell/Tissue | Relevance | Refs. | |
|---|---|---|---|---|
| Keratin 10 | Ultraviolet A irradiation, hypochlorite, and benzoyl peroxide | Human stratum corneum | Skin damage | [194] |
| Keratin 1 and 10 | Oxidative damage | Human skin | Potential biomarkers of oxidative skin damage | [195] |
| Cytokeratin 18 | Nox1 | Immortalized human epithelial | Prevents cytokeratin 18 degradation, Neoplasia | [196] |
| Vimentin | Hydrogen peroxide | Fibroblast-like (Type B) synoviocytes | n.d | [197] |
| Sodium arsenite | Human umbilical vein endothelial cells (HUVECs) | n.d | [198] | |
| Lamin A | Aging, hydrogen peroxide | Primary human dermal fibroblasts | Cellular senescence | [199] |
| Desmin | Punctual mutations | Muscle | Myofibrillar myopathies | [200, 201] |
| Hydrogen peroxide | Rat heart | Myocardial pathologies | [202] | |
| GFAP | Iron overload-induced ROS | Astrocytes | Aceruloplasminemia | [203] |
| Neurodegenerative disorder | Striatum | Neurodegenerative Tau-related pathology | [204] | |
| Illness related ROS | Spinal cord | Axonal pathologies such as autoimmune encephalomyelitis and multiple sclerosis | [205] | |
| ApoE−/− -induced ROS | Mouse Brain | Alzheimer’s disease | [206] | |
| Neurofilaments: NF-H, NF-M, NF-L | Illness related ROS | Spinal cord | Axonal pathologies such as autoimmune encephalomyelitis and multiple sclerosis | [205] |
| Serotonin/tryptamine-4,5-dione, Cytochrome C and Hydrogen peroxide | Neuroblastoma (SH-SY5Y) | Neurodegeneration processes | [207, 208] | |
| Hydrogen peroxide, Hdh140Q/140Q knock-in mice | Mouse striatal synaptosomes | Huntington’s disease | [209] | |
| Salsolinol (Dopamine metabolism), Hydrogen peroxide, Cu,Zn-SOD/H2O2 | In vitro | Neurodegenerative disorders | [210] [211] |
|
| α-Internexin | Kainic acid-induced status epilepticus | Rat hippocampus | Neurodegenerative disorders | [212] |
The table summarizes different intermediate filament proteins that can be oxidized in response to different stimuli. It is also shows the cell/tissue in which it was described and the possible relevance during physiological or pathological conditions. n.d: not determined.
GTPases as ROS Effectors
The Rho family of small GTPases is composed of GTP-binding proteins belonging to the Ras superfamily. There are 20 members in this family, the most studied of which are RhoA, Rac and Cdc42. The Rho GTPases act as molecular switches to control signal transduction by cycling between a GTP-bound state (active form) and a GDP-bound state (inactive form). When GTPases are active, they can bind to and signal through downstream effectors to regulate diverse cellular responses including cell spreading, adhesion, polarity and migration. The Rho GTPase activation cycle is tightly regulated by guanine exchange factors (GEFs), which promote the exchange of GDP for GTP to activate the GTPase; conversely, GTPase-activating proteins (GAPs) catalyze the GTPase-dependent hydrolysis of GTP to GDP, thus turning off the G protein’s activity. Additionally, another group of regulatory proteins known as guanine nucleotide dissociation inhibitors (GDIs) prevent the nucleotide exchange and bind to cytosolic GTPases, thereby protecting them from degradation [63, 64]. By controlling cytoskeletal dynamics, Rho GTPases play a pivotal role in physiological functions and pathological processes.
Rho GTPases can be either upstream regulators of ROS producing enzymes, or downstream effectors; in this section we will focus on their role as downstream effectors of ROS. In this regard, both ROS and reactive nitrogen species (RNS) can regulate the activity of GTPases by inducing the release of GDP. This redox-mediated regulation of Rho GTPases is specifically driven by O2•− and nitrogen dioxide (•NO2), which induce reversible oxidation of a cysteine residue (Cys18 in Rac and Cys20 in RhoA) located within the conserved sequence GXXXXGK(S/T)C in the phosphoryl-binding loop (p-loop) of several GTPases including RhoA, Rac and Cdc42 [65]. This cysteine oxidation disrupts the interaction between the GTPase and GDP, leaving the GTPase in a nucleotide free state. After the dissociation of GDP, if this oxidation is reduced back in an environment with a GTP:GDP of ~10:1 (as in the cytoplasm of the cell) it is more likely that the GTPase loads GTP and becomes active. Notably, this activation of GTPases by redox occurs in the absence of GEFs, suggesting that the redox-mediated nucleotide exchange occurs in parallel to classical enzymatic GEF activity.
Interestingly, RhoA and RhoB possess an additional cysteine residue (Cys16 in RhoA, GXXXCGK(S/T)C) that is associated with distinct redox properties compared to the ones present in Rac and Cdc42. Under high reduction potentials, as in the cell cytoplasm where physiological concentrations of GSH are present, H2O2 induces oxidation of cysteines in the p-loop of RhoA, potentially enhancing the rate of nucleotide dissociation and activation. Indeed, Aghajanian et al. have shown that H2O2-dependent RhoA activation and stress fiber formation is abolished by NAC or the mutation of the Cys16 and Cys20 of RhoA [66].
However, the oxidation state of a second cysteine can negatively regulate RhoA. In this case, •NO2 leads to thiyl radical formation at Cys20, which promotes intramolecular disulfide bond formation between Cys16 and Cys20, thereby obstructing nucleotide binding and inactivating RhoA. This disulfide bridge is not reversible by reduction in the cell cytoplasm, resulting in an irreversible inactivation of RhoA. This inactive form of RhoA is also unable to interact with the GEF Vav2 in in vitro studies [66, 67]. The inactivation of RhoA by this mechanism has also been observed after exposure to highly reactive compounds that cross-link vicinal thiol groups such as phenylarsine oxide (PAO) and platinated-chemotherapeutic agents such as cisplatin (Cis-Diamminedichloroplatinum (II)) or oxaliplatin ((trans-R,R)1,2-Diamino-cyclo-hexaneoxalato-platinum (II)) [68, 69].
Thus, the activation/inactivation of RhoA will depend on the ROS/RNS levels and redox state of the cell: RhoA activation occurs under elevated ROS/RNS in a high reducing potential environment. In this case, oxidation of Cys20 releases GDP, until the redox potential is restored and Cys20 is reduced, allowing RhoA to bind GTP; however, during higher ROS/RNS (oxidative stress, chemotherapeutic drugs or radical quenching) the oxidation allows the formation of a disulfide bridge between Cys16–Cys20, thus blocking the nucleotide binding pocket and leading to an irreversible inactivation of RhoA.
A different level of regulation of GTPases by ROS is by indirect modulation of GEFs and GAPs. Since the activity of multiple GEFs/GAPs are modulated by phosphorylation/dephosphorylation cycles, the enzymatic activity of their upstream kinases and phosphatases or the translocation of GEFs/GAPs to a specific subcellular location may represent another opportunity for redox regulation of GTPase activity. The net value of such regulation will depend on the balance between kinase/phosphatase activities after a specific stimulus. In this respect, ROS-dependent regulation of protein tyrosine phosphatases (PTPs) has been broadly studied [70, 71]. It is well-established that ROS induces a reversible (or even irreversible) oxidation of redox-sensitive cysteine residues located in or near the active site of PTPs, resulting in changes to the tertiary structure of the catalytic domain that inhibit or inactivate phosphatase activity [71–74]. In this context, it has been shown that ROS can inactivate the low-molecular weight protein tyrosine phosphatase (LMW-PTP), thereby increasing the phosphorylation and activation of its target p190Rho-GAP. Consequently, RhoA is inactivated, promoting Rac-induced formation of dorsal ruffles and cell spreading [75].
Non-Muscle Myosin Heavy Chain Oxidation
Myosins are motor proteins that hydrolyze ATP in order to move along actin filaments. There are at least 20 members of this family, one of which, the non-muscle myosin heavy chain type II (NMII), plays an important role in cytokinesis, endocytosis, cell shape and cell migration [76–79]. There are at least three different NMII isoforms in humans termed NMIIA (MyH9), NMIIB (MyH10) and NMIIC (MyH14). In particular, NMIIA is highly expressed in platelets, lymphocytes and neutrophil granulocytes. One of NMIIA’s most notable functions is to participate in the disengagement of integrin lymphocyte function-associated antigen (LFA)-1 at the rear end to allow tail retraction during T lymphocyte polarization [80, 81]. Fiaschi and collaborators have demonstrated that NMIIA is a target of ROS generated by integrin engagement during cell adhesion [82]. It is notable that the same group previously described that Nox4 produces ROS after integrin engagement (see integrin section for more details). They also showed that after cell spreading, oxidized NMIIA can interact with β-actin, and that this interaction is abolished using an antioxidant. Since functionality of myosin relies on its association with β-actin, these results suggest that NMIIA oxidation is integral for its contractile and structural role during cell migration.
Cortactin Oxidation
Cortactin is a nucleation-promoting factor that binds F-actin at the cell periphery of most cell types [83]. Its activity can be modulated by phosphorylation at tyrosine residues by kinases activated downstream of cell adhesion receptors such as integrins and cadherin [84–86]. After activation, cortactin induces rearrangement of the cortical actin by binding the Arp2/3 complex, facilitating actin branching and promoting the formation of structures such as lamellipodia and invadopodia during cell migration, or by facilitating endocytosis [83].
In addition to the canonical activation, there are also several non-receptor tyrosine kinases that can phosphorylate cortactin such as: Src, Abl, Syk and Fer [87–90]. Of these, Fer kinase has been linked to cortactin phosphorylation after integrin activation [90]. In this case, mouse embryonic fibroblast (MEF) adhesion on fibronectin induces Fer and cortactin phosphorylation, which is inhibited by pre-treatment with N-acetyl cysteine (NAC), the ROS scavenger Tiron or DPI [90]. Treatment with exogenous H2O2 induces both Fer and cortactin phosphorylation [90, 91], and cortactin phosphorylation is blunted in MEFs derived from mice harboring a catalytically inactive form of Fer (ferDR/DR) [90]. The remaining cortactin phosphorylation in ferDR/DR MEFs [90] was abrogated after the treatment with the Src family (Src/Yes/Fyn) kinase inhibitor PP2, suggesting that Src also acts in parallel with Fer during the phosphorylation of cortactin [90]. It is also noteworthy that Src directly binds cortactin through a disulfide bond formed between the Cys185 in the SH2 domain of Src and the Cys112/246 in the repeats domain of cortactin. This interaction is independent of tyrosine phosphorylation of cortactin, but is required for Src-mediated cortactin phosphorylation and cell migration [92].
Additionally, it has been shown that the mechanism by which ROS induces Fer activation is not direct, as autophosphorylation of Fer or GST-cortactin phosphorylation is not observed in vitro after H2O2 treatment of immunoprecipitated Fer [90]. Taken together, these results suggest that ROS can indirectly increase Fer phosphorylation levels by the inactivation of a phosphatase, and at the same time directly mediate the binding of Src to cortactin. In both cases the result is an increase in phospho-cortactin leading to actin reorganization.
Furthermore, it has been shown that p47phox binds to cortactin [93], an association that suggests new possible levels of regulation. First, the subunit p47phox can have a regulatory effect on cortactin that may not be dependent on ROS. Accordingly, in recent years Patel et al have shown that the effect of p47phox on cytoskeletal remodeling seems to be independent of Nox2 in hearts isolated from both p47phox and Nox2 KO mice subjected to transverse aortic constriction-induced pressure overload and heart failure. In this case, after mechanical stress the hearts from p47phox KO animals exhibited an upregulation of N-cadherin and β-catenin, but the cortactin/N-cadherin interaction was abolished, leading to impaired actin filament rearrangement [94]. In contrast, Nox2 did not interact with cortactin, and Nox2 KO hearts were protected against biomechanical stress-induced remodeling [94]. Similarly, the p47phoxcortactin interaction may modulate the trafficking and localization of the subunits p47phox/p67phox/p40phox to mediate NADPH oxidase activation at a particular subcellular location; it may also be possible that cortactin modulates Nox2 activity by influencing the signaling pathways implicated in the activation of p47phox [93, 95, 96].
Histone Deacetylase (HDAC) Oxidation
HDACs are enzymes that remove acetyl groups from N-acetyl lysine residues within histones and other proteins such as tubulin, cortactin, Smad7 and HSP90. Class IIa HDACs include HDAC4, HDAC5, HDAC7, HDAC9 and a truncated splice variant of HDAC9 known as myocyte enhancer factor (MEF)2-interacting transcriptional repressor (MITR) or HDAC-related protein (HDRP). They are distinguished from the other HDAC classes because they shuttle between the nucleus and the cytoplasm by a redox-dependent mechanism. One of the most studied effects of HDAC nuclear-cytoplasm export is the activation of gene transcription observed during hypertrophy, development and the inflammatory response [97]. In this context, HDAC4 and HDAC5 are in the nucleus under basal conditions and accumulate in the cytoplasm after pro-hypertrophic stimuli such as phenylephrine (PE) and Endothelin-1 (ET-1). In cardiomyocytes, the nuclear export of HDAC4 is independent of phosphorylation and completely driven by Nox4 [98]. Although the mechanisms of redox-induced nuclear export are still unknown, it has been proposed that the formation of a disulfide bond between the Cys667 and Cys669 in HDAC leads to a conformational change that exposes the nuclear export signal [99]. In the case of HDAC5, its nuclear export also seems to be regulated by redox, since it is abrogated by the antioxidant NAC or overexpression of the disulfide oxido-reductase (thioredoxin-1) [99–102]. However, in skeletal muscle fibers, the export of HDAC5 to the cytoplasm in response to stretch has been shown to be dependent on Nox2 (Fig. 2) [100]. Whether the oxidation of HDAC5 is a direct consequence of O2-derived H2O2 diffusion to the nucleus, or Nox2-dependent changes in other redox enzymes is unclear and requires further study.
Fig. 2. Nox-mediated regulation of the cytoskeleton.
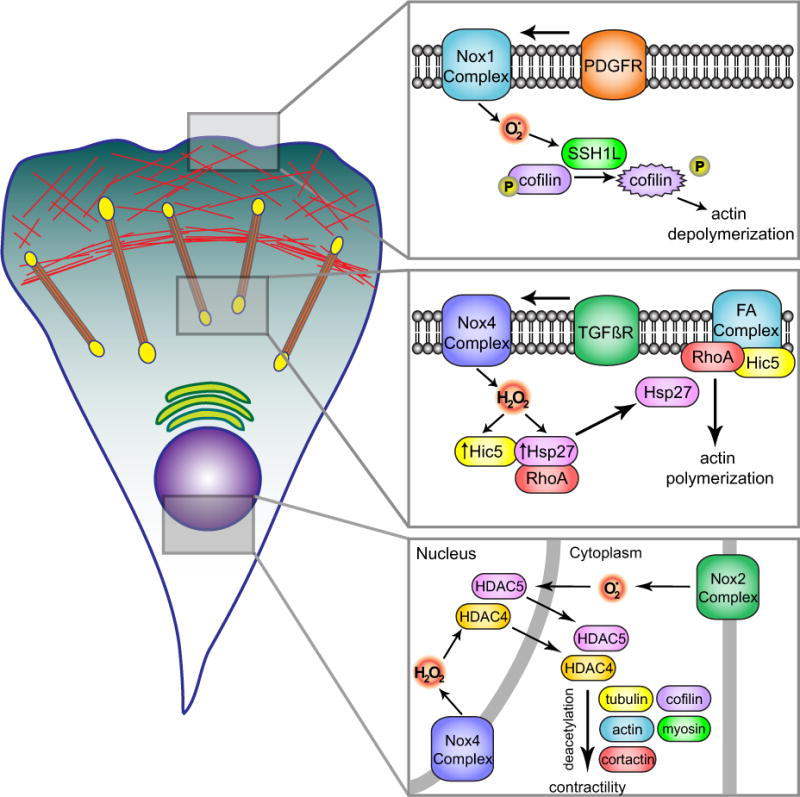
Different cytoskeletal pathways are directly influenced by Nox-produced ROS. Top insert: PDGF-induced actin polymerization uses Nox1-derived superoxide that after being dismutased to H2O2, activates SSH1L phosphatase by oxidation of inhibitory 14-3-3 proteins. SSH1L phosphatase induces dephosphorylation and activation of the actin binding protein cofilin stimulating actin depolymerization. Middle insert: Participation of Nox4 in TGFβ-induced FA formation. After TGFßR activation, Nox4 produces H2O2, which mediates the expression of Hic-5 and the chaperone protein Hsp27. Hic-5 localizes to FA and mediates TGFβ induced adhesive forces while Hsp27 aids Hic-5 localization to FA and RhoA activation-induced actin polymerization. Bottom insert: Nox complexes participate in the redox-depending shuttling of HDACs between nucleus and cytosol. Nox2 and Nox4 oxidize HDACs leading to their translocation to the cytosol and the consequent deacetylation of cytoskeleton-related proteins which leads to changes in cellular contractility.
The redox dependency of HDAC shuttling has implications for cardiac hypertrophy. When HDAC is in the nucleus it suppresses the activity of pro-hypertrophic transcription factors and recruitment of epigenetic regulators such as histone methyl-transferases and class I HDAC [99, 103]. Moreover, the inhibition of nuclear export has been shown to prevent cardiomyocyte hypertrophy, suggesting an important role for the cytoplasmic localization of HDAC in the hypertrophic response [98, 102–104]. Besides the effect of HDAC on gene transcription after its trafficking to the cytoplasm, little is known about the direct effect of HDAC shuttling on cell morphology and cytoskeletal dynamics during hypertrophy. Because the oxidation of HDAC4 does not affect the deacetylase activity of the enzyme, and considering that other HDACs belonging to class IIb that are normally present in the cytoplasm affect the acetylation status of cytoskeletal proteins such as tubulin, actin, cortactin, cofilin and myosin, it is likely that redox-controlled cytosolic shuttling of HDAC4 and HDAC5 also play a role in cytoskeletal rearrangement.
Acetylation/deacetylation of cytoskeletal components play a pivotal role in a plethora of cell processes. For example, HDAC3, HDAC4 and the acetylase PCAF have been shown to localize at cardiac sarcomeres and play a role in regulating myofilament contractile activity by regulating α- and β-myosin heavy chain (MHC) activity [105, 106]. The same group showed that the levels of myosin acetylation increased progressively after hypertrophy, indicating that a balance between acetylases/deacetylases may be essential for the contractile performance of the heart [106]. Moreover, HDAC8, which has been accepted as a cytosolic marker of smooth muscle cell differentiation, can also associate with α-smooth muscle actin and may regulate the contractile capacity of smooth muscle cells [107]. Similarly, α-tubulin acetylation at the C-terminus affects the binding of microtubule-associated proteins (MAPs) or motors [108], and HDAC6 can deacetylate α-tubulin and cortactin to regulate microtubule-dependent cell movement.
Integrin Activation as a Redox Sensitive Process
Integrins are transmembrane receptors that mediate cell-cell and cell-extracellular matrix interactions. They are heterodimers composed of α and β subunits that transduce signaling in the cell to control diverse processes such as cell transcription, cell cycle, cell shape, endocytosis and motility. There are about 18 α and 8 β subunits, which are differentially expressed in diverse cell types and combine in specific α/β pairs that recognize and interact with specific ligands to elicit precise cellular responses.
ROS have emerged as important mediators of both integrin function and integrin-triggered signal transduction. For example, the platelet α2bβ3 integrin contains several unpaired cysteine residues, the redox status of which can modulate the activation of this integrin [109, 110]. It has also been shown that reducing agents, such as dithiothreitol (DTT), can induce activation of α2bβ3 integrin [111]. This observation was corroborated with in vitro assays which demonstrate that DTT reduces two disulfide bonds in the cysteine rich domain of the integrin; this reduction induces a conformational change on both α2b and β3 subunits, thereby opening the integrin RGD and fibrinogen binding sites [109]. The α2bβ3 integrins are the primary mediators of platelet aggregation. Accordingly, a “reducing extracellular environment” (produced by application of DTT) activates integrins, and consequently increases platelet aggregation (activation of fibrinogen receptors), as opposed to higher concentrations of H2O2 which inhibit platelet aggregation by inducing an “oxidizing extracellular environment” [111–113]. Nevertheless, it is still not clear how lower concentrations of H2O2 induce platelet aggregation.
The mechanisms of ROS-mediated integrin activation have been studied in more detail for the integrin β3 subunit. This protein contains four epidermal growth factor (EGF)-like domains that each harbor a disulfide bond (Cys437-Cys457 in EGF-1, Cys473-Cys503 in EGF-2, Cys523-Cys544 in EGF-3, and Cys560-Cys583 in EGF-4). Mutagenesis of these cysteine residues differentially regulates α2bβ3 and αvβ3 integrins. Further in silico analyses suggest that Cys560 within EGF-4 of the β3 subunit reacts with one of the cysteines in the Cys567–Cys581 bonded pair, thus producing a disulfide exchange reaction leading to integrin activation [114]. The Cys560–Cys583 bond is integrin-specific, and the naturally occurring mutation C560R was identified as the cause for a bleeding disorder associated with a constitutively active state of integrin α2bβ3 [115].
Redox-mediated integrin activation has also been described for the α4 subunit in Jurkat cells [116] and for α7β1 in VSMCs [117], which increases cell adhesion on fibronectin or laminin respectively. Moreover, in the case of α7β1 they also showed that there is a Nox4-dependent cysteine oxidation during early stage adhesion that could be responsible for concomitant integrin activation [117]. Another result suggests that the cysteine rich region of integrins can change the affinity of the receptors for their substrates and also help constrain the activation of integrins, since the replacement of the cysteine rich region of the β2 integrin with the corresponding region of β1 (β2NV1) allows the formation of a constitutively active form of αLβ2NV1 [118, 119].
Integrins are not only a target for ROS modification, but can also produce ROS following their engagement after cell adhesion. This ROS production has been shown to be derived from NADPH oxidases, 5-LOX and mitochondria. Fibroblast adhesion results from integrin-dependent ROS production, mainly by 5-LOX and mitochondria [120, 121]. Further assays show that adhesion on fibronectin results in ROS release from the mitochondria during early adhesion (first 10 min) and a more robust 5-LOX-derived ROS production during late adhesion (after 45 min) [47]. It is well known that integrins can activate Rac, and at the same time Rac activity regulates Nox1, Nox2 and Nox3. In addition, other proteins such as Hic-5, TRF4, IQGAP and WAVE can target Nox4 or Nox regulatory subunits (Poldip2 and p22phox) to integrin sites to induce localized ROS production [122, 123]. However, the exact mechanisms by which integrins activate Nox complexes and the regulatory feedback loop by which ROS regulate integrin activity are not fully understood. Until now, it has only been shown that α2β1 integrin adhesion to type IV collagen induces Nox1 activation in the human adenocarcinoma cell line (Caco-2) [90, 124].
Nox Proteins in Cell Migration and Adhesion
The study of platelet derived growth factor-induced signaling and migration has shed light on the role of ROS in cell migration. Since the seminal study by Sundaresan et al. [125] showing that incubation of cells with catalase prevents PDGF-induced signaling, the important role of Nox enzymes in cellular signaling under normal physiological conditions has become well accepted. With respect to cellular migration, it has been shown that Nox1 participates in endothelial cell (EC) migration during angiogenesis, as Nox1 silencing decreases EC migration and tube-like structure formation through the inhibition of PPARα, a regulator of NF-κB [126]. Similarly, VSMCs from Nox1 deficient mice display inhibited PDGF or bFGF-induced migration [127, 128], while PDGF-induced migration is increased in VSMCs from Nox1 transgenic mice [13]. Additionally, studies have implicated Nox1 activity in the response to agonists that stimulate growth in cell culture such as AngII or LDL [129, 130] and in vascular pathologies with migratory components such as restenosis following balloon injury [131]. Indeed, neointimal formation following femoral artery injury is significantly reduced in Nox1 knockout mice [13].
The role of Nox proteins in migration is neither agonist nor cell type restricted. Phenylephrine- and VEGF-induced VSMC migration are prevented by catalase treatment and the antioxidants N-acetyl cysteine (NAC) and pyrrolidine dithiocarbamate, respectively [132, 133]. Meng et al. [134] found a role for ROS in IGF-1 induced VSMC migration that is derived from a Rac-dependent oxidase (presumably Nox1) as well as Nox4. Haurani et al. [135] showed that overexpression of Nox4 impaired AngII-induced migration in fibroblasts. In addition, Nox1 and Nox4 are required for a full migratory response to PDGF [13, 34]. Thrombin-stimulated migration is blocked by DPI and the putative NADPH oxidase inhibitor apocynin, implicating NADPH oxidase-derived ROS in this response as well [136]. Similarly, PDGF-induced migration of VSMCs is attenuated by the Nox inhibitor VAS2870 [137]. Endothelial cell migration is also reduced by antioxidant treatment [56] and inhibition of the NADPH oxidases [138].
Nox proteins participate at multiple levels in the remodeling of the actin cytoskeleton, a process that is integral to migration. Upon sensing a chemoattractant, the cell initiates migration via the formation of F-actin rich membrane protrusions called lamellipodia [139]. This lamellipodia formation is driven by actin reorganization, which in turn is regulated by actin modifications, actin binding proteins, and small GTPases. Actin polymerization occurs at the leading edge of the cell [140], while depolymerization predominates at the interface of the lamellipodium with the cell body (the lamella) [141]. Nox1 regulates the activity of Slingshot 1L (SSH1L), a member of a recently described family of protein phosphatases [142] that dephosphorylates and activates cofilin, an actin binding protein that is absolutely required for cell migration in VSMCs [143] (Fig. 2). Furthermore, we and others have recently shown that the mechanism of PDGF-induced SSH1L is redox sensitive and uses Nox1-derived ROS to disrupt an inhibitory complex with 14-3-3 proteins (Fig. 2) [12, 13, 144, 145]. Conversely, cofilin activity is negatively regulated by the LIM kinase (LIMK) family of serine/threonine kinases through phosphorylation of cofilin at Ser-3 [146–148]. LIMK may also be regulated by NADPH-produced ROS downstream of PAK or Src [149]. We should mention that the oxidative modification of cofilin has emerged as an important node of redox-mediated cytoskeleton regulation, as has been recently reviewed in the literature [150].
REGULATION OF CELL ATTACHMENT BY NOX PROTEINS
In addition to regulating the polymerization state of actin, Nox proteins may also support migration by regulating the adhesive forces responsible for the generation of cell traction [151, 152]. Focal adhesions (FAs), which are integrin-containing macromolecular complexes that serve as mechanical links between the actin cytoskeleton and the extracellular matrix (ECM), mediate these adhesive forces; at the same time, their dissolution in the rear allows the cell to contract and move forward. FAs are localized at the ventral surface of cells, where they concentrate and regulate numerous signaling proteins in response to integrin activation during cell adhesion [153]. Efficient cell migration involves the formation of focal complexes at the front of the cell, which are subsequently converted to FAs. It is important to note that both FAs formation and turnover are required for cell motility, such that focal adhesions must form and dissolve properly for normal migration. These processes are primarily controlled by FAK, a non-receptor tyrosine kinase that is activated by integrin receptors. FAK mediated translocation of paxillin and p130Cas to FAs enhances their formation [154], while autophosphorylation of FAK on Y397 is essential for FAK-induced FA disassembly [155].
Cell contraction, and hence movement of the cell, occurs via engagement of actin-myosin interactions. This is facilitated by the Rho signaling pathway, leading to increased Rho kinase activity and inhibition of myosin light chain phosphatase [156]. As the body of the cell moves forward, the newly formed FAs become stronger and arrive at the rear of the cell where they are disassembled, allowing their components to recycle to the leading edge of the cell for the next wave of migration [151].
One major mechanism by which ROS regulate FAs is through inactivation of protein tyrosine phosphatases (PTPs). PTPs have been shown to localize to FAs and regulate their stability [46]. Furthermore, inactivation of PTPs has also been shown to be a major mechanism by which Nox proteins regulate various signaling pathways [157]. Shinohara et al. [158] found that in Ras-transformed cells, Nox1-generated ROS mediate down-regulation of Rho activity via oxidative inactivation of the low molecular weight protein tyrosine phosphatase (LMW-PTP). Similarly, ROS production induced by integrin mediated cell attachment also inhibits LMW-PTP, thus preventing inactivation of focal adhesion associated protein FAK [121]. Another PTP, PTP-PEST, has been strongly associated with FA turnover, and like other phosphatases, is known to be inactivated by ROS [46]. Overexpression or ablation of PTP-PEST results in impaired migration, suggesting that tight regulation of PTP-PEST is necessary for efficient migration [159–161]. In PTP-PEST overexpressing fibroblasts, p130Cas phosphorylation and motility are inhibited [160]. Similarly, PTP-PEST null fibroblasts exhibit impaired motility, but in this case exhibit hyperphosphorylation of p130Cas, paxillin and FAK, and also display an enhanced rate of spreading and an increase in the size and number of FAs [159].
Another important cytoskeletal protein that stabilizes FAs and F-actin networks is cortactin [162]. Cortactin localizes to specialized microdomains at the focal adhesion/stress fiber interface and cooperates with p190RhoGAP in the control of FA turnover [163]. Knockdown of cortactin causes a reduction of FA formation in fibrosarcoma cells [164]. A role in cell migration can be inferred from the observation that the cortactin gene is upregulated after PDGF treatment in VSMCs [165]. Of importance, as we described in detail previously in this review, cortactin is a main target of direct oxidation. Not surprisingly, ROS have been shown to participate in cortactin regulation during integrin-mediated cell adhesion [90].
Focal Adhesion Kinase
FAK is a non-receptor tyrosine kinase that is activated by integrin receptors and phosphorylates paxillin and p130Cas, thereby regulating their translocation to FAs [154] and enhancing FA formation. At the same time, autophosphorylation of FAK on Y397 is essential for FAK-induced FA disassembly [155]. It is important to note that FA turnover is required for cell motility, so FAs must form and dissolve properly for normal migration. Indeed, depletion of FAK in fibroblasts results in enhanced FAs and impaired migration [166]. FAK has been shown to either activate RhoA by activating p190RhoGEF [167], or inhibit it by activating p190RhoGAP [168]. The precise role of FAK in regulating cell-substrate interactions is therefore context-specific, and may evolve temporally during migration. In this sense, local changes in ROS can indirectly regulate FAK activity after cell adhesion. Indeed, Chiarugi et al. have shown that in fibroblasts, Rac activity generates ROS after integrin engagement. At the same time they showed that ROS is necessary for cell adhesion since the incubation with DPI, a 5-lypoxigenase (LOX) inhibitor (nordihydroguaiaretic acid or NDGA) or an antioxidant N-acetylcysteine (NAC) dramatically delays the adhesion of cell to fibronectin [121]. Normally, LMW-PTP localizes to focal adhesions and dephosphorylates FAK; thus, ROS produced downstream of integrins after cell spreading induce the oxidation and inactivation of LMW-PTP, thereby increasing the phosphorylation of FAK and other downstream effectors such as Src and MAPK [121, 169, 170]. Although LOX-mediated ROS seem to play a main role in the phosphorylation of FAK in this study [121], further work is necessary to estimate the contribution of NADPH oxidases in FAK activation during attachment.
In agreement with these results, H2O2 mediated increases in phospho-FAK levels correlate with an increase in stress fiber formation [171] and cell migration [172]. Moreover, in a model of oxidative stress-induced disruption of tight junctions in Caco-2 monolayers, FAK activation is mediated by Src and PI3K. Likewise, H2O2 induced migration is dependent on Src and PI3K activities [172]. A high migration rate after redox-induced FAK activation can be achieved after growth factor stimulation. Specifically, VEGF causes S-glutathionylation of LMW-PTP and subsequent FAK activation. Furthermore, the effect of VEGF by itself or in combination with low concentrations of peroxynitrite (0.1–0.2 mM) increase cell migration; however, VEGF in combination with higher concentrations of peroxynitrite (5mM) inhibit migration [173]. Interestingly, VEGF can induce ROS in leukemic cells by NADPH oxidases [174]. These results suggest that tight control of the redoxstate in the cell is necessary to control VEGF-driven migration.
Nox4 Regulation of FA Dynamics
In particular, Nox4 may play a critical role in FA dynamics as it has been shown to co-localize with vinculin and paxillin at FAs in VSMCs and monocytes respectively [175, 176]. Silencing either Nox4 or its activator, Poldip2, disorganizes FAs and stress fibers. Moreover, overexpression of Poldip2 results in an increase of both FA and stress fiber formation through a RhoA-dependent pathway and a decrease in cell migration in response to PDGF [34, 123]. By using a nocodazol-induced microtubule-dependent FA turnover assay, the authors demonstrate that Poldip2 overexpression impairs FA dissolution. At the same time, local levels of H2O2 in FAs remain high in Poldip2 overexpressing cells, which is consistent with the fact that an intracellular increase in H2O2 can activate FAK and subsequently stimulate cell adhesion. Indeed, the levels of FAK phosphorylated at Y397 decrease after the silencing of Poldip2 and Nox4 [123].
Regarding the mechanism by which Nox4 regulates FAs, recent work from our lab implicate the hydrogen peroxide-inducible clone-5 (Hic-5) and the chaperone protein Hsp27 [177]. Hic-5, first identified as a H2O2 and TGF-β-inducible gene [178], is a member of the paxillin family of proteins and localizes to FAs, where it reportedly binds to GIT1 [179], FAK [180], PTP-PEST [180] and Pyk2 [181]. For example, loss of Hic-5 impairs, and overexpression of Hic-5 increases, lysophosphatidic acid-induced migration of endothelial cells [182]. Overexpression of Hic-5 in mammary gland epithelial cells also stimulates migration [183]. Furthermore, we have recently shown that in VSMCs exposed to TGF-β, Hic-5 mediates cell adhesives forces and migration by a mechanism that is dependent on the production of H2O2 by Nox4 [177] (Fig. 2).
The precise mechanisms by which Hic-5 regulates FAs are unclear. As previously mentioned, Hic-5 forms a complex with Pyk-2 [181] in a Src-dependent manner [184] and becomes phosphorylated. This phosphorylation has been reported to release the Hic-5-mediated inhibition of Rac [185]. Another GTPase regulated by Hic-5 is RhoA. Indeed, Hic-5 downregulation by siRNA inhibits RhoA activation by TGF-β in epithelial cells, which is accompanied by inhibition of stress fiber and FA formation [186]. Likewise, overexpression of Hic-5 in these cells leads to increased formation of stress fibers in a Rho kinase-dependent manner.
As previously indicated, another redox sensitive pathway that is implicated in Nox4 regulation of FAs involves the microfilament capping and actin polymerization inhibiting protein Hsp27 [187]. We have reported that in TGF-β-treated VSMCs Hsp27 is upregulated by a Nox4-dependent mechanism. Furthermore, we demonstrated that Hsp27 is indispensable for the localization of Hic-5 (but not paxillin) to FAs. Additionally, Hsp27 phosphorylation has been linked to its increased association with RhoA [188, 189] and to enhanced actin polymerization [188, 189]. Importantly, this pathway is redox-sensitive, as evidenced by its activation by H2O2 in endothelial cells [190], and is required for endothelial cell migration [191], PDGF-induced migration of hepatic myofibroblasts [192] and migration of tracheal smooth muscle cells induced by PDGF, IL-1β and TGF-β [193].
CONCLUSION
Given the important roles of Nox proteins in signaling pathways that contribute to normal cell physiology [6], it is not surprising that they also have a predominant role in the regulation of cytoskeletal dynamics. In fact, some well-known inhibitors of NADPH oxidases impact cell physiology via alterations of the cytoskeleton (Table 2). Cytoskeletal and cytoskeleton-associated proteins have a remarkable susceptibility to oxidization [1], based on the presence of reactive cysteine residues within their sequence [1]. At the molecular level, Cys modification is likely to impact protein structure and function, as well as interactions with other macromolecules.
Table 2.
Nox inhibitors that target cytoskeletal-dependent structures and functions.
| Inhibitor | Mechanism of action | Target | Refs. |
|---|---|---|---|
| ML171 | Inhibits Nox1 (IC50 of 0.25 μM) and have a lower effect over other NADPH oxidases (IC50 >3 μM). Mechanism of action is not established. | -Impairs the formation of functional invadopodia in Colon cancer. | [213] |
| GKT136901 | Inhibits Nox1 and Nox4 by an unknown mechanism of action. Structural similarity with NADPH suggests that it may act as a competitive inhibitor enzymatic inhibitor. | -Inhibits endothelial cell migration and tumor angiogenesis. | [126] |
| PR-39 | Binds to SH3 domains of the p47phox subunit and prevents binding to p22phox. | -Inhibits endothelial cell migration/angiogenesis. -Alters invasive activity and actin structure of human hepatocellular carcinoma cells. |
[214] [215] |
| Nebivolol | Induces the dissociation of p67phox and Rac1. | -Controls vascular tone through the Inhibition of RhoA activity. | [216] |
| Gliotoxin | Inhibits the phosphorylation of p47phox. | -Modulates of immune response by affecting actin cytoskeleton organization-mediated phagocytosis in human neutrophil. -Inhibits neointimal formation after vascular injury in rats. |
[217] [218] [219] [220] |
| VAS2870 | Inhibits NOX2 and NOX4 containing oxidases by an undefined mechanism of action. | -Reduces retrograde F-actin flow and neurite outgrowth. -Attenuates PDGF-dependent smooth muscle cell chemotaxis. -Reverses the over contraction of diabetic rat aorta. |
[221] [137] [222] |
Of the numerous oxidative modifications described, only a fraction of them have been linked to specific signaling pathways (Fig. 2). Similarly, most of the functional consequences of these modifications have yet to be determined. Nonetheless, it is clear that these effects are likely dependent on the source and localization of ROS, as well as the concentration. In this manner, ROS have the ability to elicit finely tuned temporal and spatial cellular responses to stimuli. It is the coordinated activity of ROS producing enzymes, in conjunction with other signaling molecules, which bring about the precise cytoskeletal changes required for complex cellular responses such as migration. Advances in understanding the structural changes induced by many of these modifications will be transcendental to improve the mechanistic understanding of redox signaling within the cytoskeleton, and is likely to impact a variety of aspects of human health and disease.
Acknowledgments
Declared none.
SOURCES OF FUNDING
The National Institutes of Health, through awards R01HL113167 and T32HL007745, supports our research.
Biography

Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Go YM, Jones DP. The redox proteome. J Biol Chem. 2013;288:26512–20. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zafari AM, Ushio-Fukai M, Akers M, et al. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–95. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 3.Rajagopalan S, Kurz S, Münzel T, et al. Angiotensin II mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohazzab KM, Wolin MS. Sites of superoxide anion production detected by lucigenin in calf pulmonary artery smooth muscle. Am J Physiol. 1994;267:L815–22. doi: 10.1152/ajplung.1994.267.6.L815. [DOI] [PubMed] [Google Scholar]
- 5.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–8. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 6.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–90. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Investig. 1996;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tummala PE, Chen XL, Sundell CL, et al. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation. 1999;100:1223–9. doi: 10.1161/01.cir.100.11.1223. [DOI] [PubMed] [Google Scholar]
- 9.Wang HD, Pagano PJ, Du Y, et al. Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ Res. 1998;82:810–8. doi: 10.1161/01.res.82.7.810. [DOI] [PubMed] [Google Scholar]
- 10.Wang HD, Xu S, Johns DG, et al. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice [see comment] Circ Res. 2001;88:947–53. doi: 10.1161/hh0901.089987. [DOI] [PubMed] [Google Scholar]
- 11.Lassegue B, Sorescu D, Szocs K, et al. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–94. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 12.Maheswaranathan M, Gole HK, Fernandez I, et al. Platelet-derived growth factor (PDGF) regulates Slingshot phosphatase activity via Nox1-dependent auto-dephosphorylation of serine 834 in vascular smooth muscle cells. J Biol Chem. 2011;286:35430–7. doi: 10.1074/jbc.M111.268284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MY, San Martin A, Mehta PK, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–7. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao Q, Luo Z, Pepe AE, Margariti A, Zeng L, Xu Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am J Physiol Cell Physiol. 2009;296:C711–23. doi: 10.1152/ajpcell.00442.2008. [DOI] [PubMed] [Google Scholar]
- 15.Mouche S, Mkaddem SB, Wang W, et al. Reduced expression of the NADPH oxidase NOX4 is a hallmark of adipocyte differentiation. Biochim Biophys Acta. 2007;1773:1015–27. doi: 10.1016/j.bbamcr.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Stouffs M, Serrander L, et al. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell. 2006;17:3978–88. doi: 10.1091/mbc.E05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clempus RE, Sorescu D, Dikalova AE, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–8. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dang PM, Cross AR, Quinn MT, Babior BM. Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67PHOX and cytochrome b558 II. Proc Natl Acad Sci USA. 2002;99:4262–5. doi: 10.1073/pnas.072345299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babior BM. The respiratory burst oxidase. Curr Opin Hematol. 1995;2:55–60. doi: 10.1097/00062752-199502010-00008. [DOI] [PubMed] [Google Scholar]
- 20.Babior BM. The respiratory burst oxidase. Curr Opin Hematol. 1995;2:55–60. doi: 10.1097/00062752-199502010-00008. [DOI] [PubMed] [Google Scholar]
- 21.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–41. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 22.Hanna IR, Dikalova A, Hilenski L, Quinn MT, Griendling KK. Nox 1 binds p22phox to form a functional oxidase in vascular smooth muscle cells (VSMCs) Circulation. 2002;106:II-164. [Google Scholar]
- 23.Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280:31859–69. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- 24.Lassegue B, Sorescu D, Szocs K, et al. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways [see comment] Circ Res. 2001;88:888–94. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 25.Martin-Garrido A, Brown DI, Lyle AN, et al. NADPH oxidase 4 mediates TGF-beta-induced smooth muscle alpha-actin via p38MAPK and serum response factor. Free Radic Biol Med. 2011;50:354–62. doi: 10.1016/j.freeradbiomed.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–51. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambeth JD. Regulation of the phagocyte respiratory burst oxidase by protein interactions. J Biochem Mol Biol. 2000;33:427–439. [Google Scholar]
- 28.Landmesser U, Cai H, Dikalov S, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–5. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavigne MC, Malech HL, Holland SM, Leto TL. Genetic demonstration of p47phox-dependent superoxide anion production in murine vascular smooth muscle cells. Circulation. 2001;104:79–84. doi: 10.1161/01.cir.104.1.79. [DOI] [PubMed] [Google Scholar]
- 30.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 31.Takeya R, Ueno N, Kami K, et al. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem. 2003;278:25234–46. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 32.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 33.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 34.Lyle AN, Deshpande NN, Taniyama Y, et al. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–59. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wientjes FB, Reeves EP, Soskic V, Furthmayr H, Segal AW. The NADPH oxidase components p47(phox) and p40(phox) bind to moesin through their PX domain. Biochem Biophys Res Commun. 2001;289:382–8. doi: 10.1006/bbrc.2001.5982. [DOI] [PubMed] [Google Scholar]
- 36.Martinelli R, Kamei M, Sage PT, et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol. 2013;201:449–65. doi: 10.1083/jcb.201209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu RF, Gu Y, Xu YC, Nwariaku FE, Terada LS. Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. J Biol Chem. 2003;278:36830–40. doi: 10.1074/jbc.M302251200. [DOI] [PubMed] [Google Scholar]
- 38.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45(9):1340–51. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dikalova A, Clempus R, Lassegue B, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–76. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 40.Klomsiri C, Rogers LC, Soito L, et al. Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radic Biol Med. 2014;71:49–60. doi: 10.1016/j.freeradbiomed.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11:1313–33. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113:4633–79. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intra-cellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mills JE, Whitford PC, Shaffer J, Onuchic JN, Adams JA, Jennings PA. A novel disulfide bond in the SH2 Domain of the C-terminal Src kinase controls catalytic activity. J Mol Biol. 2007;365:1460–8. doi: 10.1016/j.jmb.2006.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DalleDonne I, Milzani A, Colombo R. H2O2-treated actin: assembly and polymer interactions with cross-linking proteins. Biophys J. 1995;69:2710–9. doi: 10.1016/S0006-3495(95)80142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heneberg P, Draber P. Regulation of cys-based protein tyrosine phosphatases via reactive oxygen and nitrogen species in mast cells and basophils. Curr Med Chem. 2005;12:1859–71. doi: 10.2174/0929867054546636. [DOI] [PubMed] [Google Scholar]
- 47.Taddei ML, Parri M, Mello T, et al. Integrin-mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxid Redox Signal. 2007;9:469–81. doi: 10.1089/ars.2006.1392. [DOI] [PubMed] [Google Scholar]
- 48.DalleDonne I, Milzani A, Colombo R. The tert-butyl hydroperoxide-induced oxidation of actin Cys-374 is coupled with structural changes in distant regions of the protein. Biochemistry. 1999;38:12471–80. doi: 10.1021/bi990367k. [DOI] [PubMed] [Google Scholar]
- 49.Milzani A, DalleDonne I, Colombo R. Prolonged oxidative stress on actin. Arch Biochem Biophys. 1997;339:267–74. doi: 10.1006/abbi.1996.9847. [DOI] [PubMed] [Google Scholar]
- 50.Lassing I, Schmitzberger F, Bjornstedt M, et al. Molecular and structural basis for redox regulation of beta-actin. J Mol Biol. 2007;370:331–48. doi: 10.1016/j.jmb.2007.04.056. [DOI] [PubMed] [Google Scholar]
- 51.Johansson M, Lundberg M. Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. BMC Biochem. 2007;8:26. doi: 10.1186/1471-2091-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drewes G, Faulstich H. The enhanced ATPase activity of glutathione-substituted actin provides a quantitative approach to filament stabilization. J Biol Chem. 1990;265:3017–21. [PubMed] [Google Scholar]
- 53.Stournaras C, Drewes G, Blackholm H, Merkler I, Faulstich H. Glutathionyl(cysteine-374) actin forms filaments of low mechanical stability. Biochim Biophys Acta. 1990;1037:86–91. doi: 10.1016/0167-4838(90)90105-o. [DOI] [PubMed] [Google Scholar]
- 54.Prochniewicz E, Lowe DA, Spakowicz DJ, et al. Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am J Physiol Cell Physiol. 2008;294:C613–26. doi: 10.1152/ajpcell.00232.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Omann GM, Harter JM, Burger JM, Hinshaw DB. H2O2-induced increases in cellular F-actin occur without increases in actin nucleation activity. Arch Biochem Biophys. 1994;308:407–12. doi: 10.1006/abbi.1994.1057. [DOI] [PubMed] [Google Scholar]
- 56.Moldovan L, Moldovan NI, Sohn RH, Parikh SA, Goldschmidt-Clermont PJ. Redox changes of cultured endothelial cells and actin dynamics. Circ Res. 2000;86:549–57. doi: 10.1161/01.res.86.5.549. [DOI] [PubMed] [Google Scholar]
- 57.Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12:773–86. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- 58.Wloga D, Gaertig J. Post-translational modifications of microtubules. J Cell Sci. 2010;123:3447–55. doi: 10.1242/jcs.063727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber K, Patel P, Zhang L, et al. 2-[(1-methylpropyl)dithio]-1H-imidazole inhibits tubulin polymerization through cysteine oxidation. Mol Cancer Ther. 2008;7:143–51. doi: 10.1158/1535-7163.MCT-07-0486. [DOI] [PubMed] [Google Scholar]
- 60.Landino LM, Hasan R, McGaw A, et al. Peroxynitrite oxidation of tubulin sulfhydryls inhibits microtubule polymerization. Arch Biochem Biophys. 2002;398:213–20. doi: 10.1006/abbi.2001.2729. [DOI] [PubMed] [Google Scholar]
- 61.Clark HM, Hagedorn TD, Landino LM. Hypothiocyanous acid oxidation of tubulin cysteines inhibits microtubule polymerization. Arch Biochem Biophys. 2014;541:67–73. doi: 10.1016/j.abb.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirchner K, Mandelkow EM. Tubulin domains responsible for assembly of dimers and protofilaments. EMBO J. 1985;4:2397–402. doi: 10.1002/j.1460-2075.1985.tb03945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 64.Boulter E, Garcia-Mata R, Guilluy C, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–83. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–10. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 66.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heo J, Raines KW, Mocanu V, Campbell SL. Redox regulation of RhoA. Biochemistry. 2006;45:14481–9. doi: 10.1021/bi0610101. [DOI] [PubMed] [Google Scholar]
- 68.Mordovanakis AG, Easter J, Naumova N, et al. Quasimonoenergetic electron beams with relativistic energies and ultrashort duration from laser-solid interactions at 0.5 kHz. Phys Rev Lett. 2009;103:235001. doi: 10.1103/PhysRevLett.103.235001. [DOI] [PubMed] [Google Scholar]
- 69.Matasova LV, Semenikhina AV, Popova TN, et al. Effect of N-[Imino(4-morpholyl)methyl]guanidine on the oxidative status in rats with toxic hepatitis. Bull Exp Biol Med. 2009;148:619–22. doi: 10.1007/s10517-010-0779-5. [DOI] [PubMed] [Google Scholar]
- 70.Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251–2. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 71.Frijhoff J, Dagnell M, Godfrey R, Ostman A. Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid Redox Signal. 2014;20:1994–2010. doi: 10.1089/ars.2013.5643. [DOI] [PubMed] [Google Scholar]
- 72.Maremmani I, Pacini M, Pani PP, et al. Use of street methadone in italian heroin addicts presenting for opioid agonist treatment. J Addict Dis. 2009;28:382–8. doi: 10.1080/10550880903183000. [DOI] [PubMed] [Google Scholar]
- 73.Peremyslova LM, Kostiuchenko VA, Popova I, Safronova NG. Radioecological situation in the Iset riverside settlements. Radiats Biol Radioecol. 2009;49:714–20. [PubMed] [Google Scholar]
- 74.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–99. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 75.Formanovskii AA, Popova IS, Mikhura IV. The total large-scale synthesis of argiopine. Bioorg Khim. 2009;35:837–44. [PubMed] [Google Scholar]
- 76.Chandrasekar I, Goeckeler ZM, Turney SG, et al. Nonmuscle myosin II is a critical regulator of clathrin-mediated endocytosis. Traffic. 2014;15:418–32. doi: 10.1111/tra.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Recuenco MC, Ohmori T, Tanigawa S, et al. Nonmuscle Myosin II Regulates the Morphogenesis of Metanephric Mesenchyme-Derived Immature Nephrons. J Am Soc Nephrol. 2015;26(5):1081–91. doi: 10.1681/ASN.2014030281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shutova M, Yang C, Vasiliev JM, Svitkina T. Functions of non-muscle myosin II in assembly of the cellular contractile system. PLoS One. 2012;7:e40814. doi: 10.1371/journal.pone.0040814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee MY, Lee SH, Kim YH, et al. Effect of EGF on [3H]-thymidine incorporation and cell cycle regulatory proteins in primary cultured chicken hepatocytes: Involvement of Ca2+/PKC and MAPKs. J Cell Biochem. 2006;99:1677–87. doi: 10.1002/jcb.21026. [DOI] [PubMed] [Google Scholar]
- 80.Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121:11–8. doi: 10.1242/jcs.007112. [DOI] [PubMed] [Google Scholar]
- 81.Morin NA, Oakes PW, Hyun YM, et al. Nonmuscle myosin heavy chain IIA mediates integrin LFA-1 de-adhesion during T lymphocyte migration. J Exp Med. 2008;205:195–205. doi: 10.1084/jem.20071543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fiaschi T, Cozzi G, Chiarugi P. Redox regulation of nonmuscle myosin heavy chain during integrin engagement. J Signal Transduct. 2012;2012:754964. doi: 10.1155/2012/754964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacGrath SM, Koleske AJ. Cortactin in cell migration and cancer at a glance. J Cell Sci. 2012;125:1621–6. doi: 10.1242/jcs.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Helwani FM, Kovacs EM, Paterson AD, et al. Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization. J Cell Biol. 2004;164:899–910. doi: 10.1083/jcb.200309034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lapetina S, Mader CC, Machida K, Mayer BJ, Koleske AJ. Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J Cell Biol. 2009;185:503–19. doi: 10.1083/jcb.200809085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vuori K, Ruoslahti E. Tyrosine phosphorylation of p130Cas and cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J Biol Chem. 1995;270:22259–62. doi: 10.1074/jbc.270.38.22259. [DOI] [PubMed] [Google Scholar]
- 87.Fan L, Di Ciano-Oliveira C, Weed SA, et al. Actin depolymerization-induced tyrosine phosphorylation of cortactin: the role of Fer kinase. Biochem J. 2004;380:581–91. doi: 10.1042/BJ20040178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maruyama S, Kurosaki T, Sada K, Yamanashi Y, Yamamoto T, Yamamura H. Physical and functional association of cortactin with Syk in human leukemic cell line K562. J Biol Chem. 1996;271:6631–5. doi: 10.1074/jbc.271.12.6631. [DOI] [PubMed] [Google Scholar]
- 89.Boyle SN, Michaud GA, Schweitzer B, Predki PF, Koleske AJ. A critical role for cortactin phosphorylation by Abl-family kinases in PDGF-induced dorsal-wave formation. Curr Biol. 2007;17:445–51. doi: 10.1016/j.cub.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 90.Sangrar W, Gao Y, Scott M, Truesdell P, Greer PA. Fer-mediated cortactin phosphorylation is associated with efficient fibroblast migration and is dependent on reactive oxygen species generation during integrin-mediated cell adhesion. Mol Cell Biol. 2007;27:6140–52. doi: 10.1128/MCB.01744-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Liu J, Zhan X. Tyrosine phosphorylation of cortactin is required for H2O2-mediated injury of human endothelial cells. J Biol Chem. 2000;275:37187–93. doi: 10.1074/jbc.M005301200. [DOI] [PubMed] [Google Scholar]
- 92.Evans JV, Ammer AG, Jett JE, et al. Src binds cortactin through an SH2 domain cystine-mediated linkage. J Cell Sci. 2012;125:6185–97. doi: 10.1242/jcs.121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:512–8. doi: 10.1161/01.ATV.0000154141.66879.98. [DOI] [PubMed] [Google Scholar]
- 94.Patel VB, Wang Z, Fan D, et al. Loss of p47phox subunit enhances susceptibility to biomechanical stress and heart failure because of dysregulation of cortactin and actin filaments. Circ Res. 2013;112:1542–56. doi: 10.1161/CIRCRESAHA.111.300299. [DOI] [PubMed] [Google Scholar]
- 95.Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:981–7. doi: 10.1161/01.ATV.0000069236.27911.68. [DOI] [PubMed] [Google Scholar]
- 96.Usatyuk PV, Gorshkova IA, He D, et al. Phospholipase D-mediated activation of IQGAP1 through Rac1 regulates hyperoxia-induced p47phox translocation and reactive oxygen species generation in lung endothelial cells. J Biol Chem. 2009;284:15339–52. doi: 10.1074/jbc.M109.005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. 2007;150:829–31. doi: 10.1038/sj.bjp.0707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matsushima S, Kuroda J, Ago T, et al. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res. 2013;112:651–63. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ago T, Liu T, Zhai P, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–93. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 100.Liu Y, Hernandez-Ochoa EO, Randall WR, Schneider MF. NOX2-dependent ROS is required for HDAC5 nuclear efflux and contributes to HDAC4 nuclear efflux during intense repetitive activity of fast skeletal muscle fibers. Am J Physiol Cell Physiol. 2012;303:C334–47. doi: 10.1152/ajpcell.00152.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Haworth RS, Stathopoulou K, Candasamy AJ, Avkiran M. Neuro-hormonal regulation of cardiac histone deacetylase 5 nuclear localization by phosphorylation-dependent and phosphorylation-independent mechanisms. Circ Res. 2012;110:1585–95. doi: 10.1161/CIRCRESAHA.111.263665. [DOI] [PubMed] [Google Scholar]
- 102.Vega RB, Harrison BC, Meadows E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–85. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oka S, Ago T, Kitazono T, Zablocki D, Sadoshima J. The role of redox modulation of class II histone deacetylases in mediating pathological cardiac hypertrophy. J Mol Med (Berl) 2009;87:785–91. doi: 10.1007/s00109-009-0471-2. [DOI] [PubMed] [Google Scholar]
- 104.Monovich L, Vega RB, Meredith E, et al. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010;584:631–7. doi: 10.1016/j.febslet.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 105.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283:10135–46. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Samant SA, Courson DS, Sundaresan NR, et al. HDAC3-dependent reversible lysine acetylation of cardiac myosin heavy chain isoforms modulates their enzymatic and motor activity. J Biol Chem. 2011;286:5567–77. doi: 10.1074/jbc.M110.163865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 107.Waltregny D, Glenisson W, Tran SL, et al. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–8. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 108.Perdiz D, Mackeh R, Pous C, Baillet A. The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell Signal. 2011;23:763–71. doi: 10.1016/j.cellsig.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 109.Yan B, Smith JW. Mechanism of integrin activation by disulfide bond reduction. Biochemistry. 2001;40:8861–7. doi: 10.1021/bi002902i. [DOI] [PubMed] [Google Scholar]
- 110.Yan B, Smith JW. A redox site involved in integrin activation. J Biol Chem. 2000;275:39964–72. doi: 10.1074/jbc.M007041200. [DOI] [PubMed] [Google Scholar]
- 111.Peerschke EI. Regulation of platelet aggregation by post-fibrinogen binding events. Insights provided by dithiothreitol-treated platelets. Thromb Haemost. 1995;73:862–7. [PubMed] [Google Scholar]
- 112.Pratico D, Iuliano L, Alessandri C, Camastra C, Violi F. Polymor-phonuclear leukocyte-derived O2-reactive species activate primed platelets in human whole blood. Am J Physiol. 1993;264:H1582–7. doi: 10.1152/ajpheart.1993.264.5.H1582. [DOI] [PubMed] [Google Scholar]
- 113.Iuliano L, Colavita AR, Leo R, Pratico D, Violi F. Oxygen free radicals and platelet activation. Free Radic Biol Med. 1997;22:999–1006. doi: 10.1016/s0891-5849(96)00488-1. [DOI] [PubMed] [Google Scholar]
- 114.Levin L, Zelzion E, Nachliel E, Gutman M, Tsfadia Y, Einav Y. A single disulfide bond disruption in the beta3 integrin subunit promotes thiol/disulfide exchange, a molecular dynamics study. PLoS One. 2013;8:e59175. doi: 10.1371/journal.pone.0059175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fang J, Nurden P, North P, et al. C560Rbeta3 caused platelet integrin alphaII b beta3 to bind fibrinogen continuously, but resulted in a severe bleeding syndrome and increased murine mortality. J Thromb Haemost. 2013;11:1163–71. doi: 10.1111/jth.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Laragione T, Bonetto V, Casoni F, et al. Redox regulation of surface protein thiols: identification of integrin alpha-4 as a molecular target by using redox proteomics. Proc Natl Acad Sci USA. 2003;100:14737–41. doi: 10.1073/pnas.2434516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de Rezende FF, Martins Lima A, Niland S, et al. Integrin alpha7beta1 is a redox-regulated target of hydrogen peroxide in vascular smooth muscle cell adhesion. Free Radic Biol Med. 2012;53:521–31. doi: 10.1016/j.freeradbiomed.2012.05.032. [DOI] [PubMed] [Google Scholar]
- 118.Douglass WA, Hyland RH, Buckley CD, et al. The role of the cysteine-rich region of the beta2 integrin subunit in the leukocyte function-associated antigen-1 (LFA-1, alphaLbeta2, CD11a/CD18) heterodimer formation and ligand binding. FEBS Lett. 1998;440:414–8. doi: 10.1016/s0014-5793(98)01498-7. [DOI] [PubMed] [Google Scholar]
- 119.Gofer-Dadosh N, Klepfish A, Schmilowitz H, Shaklai M, Lahav J. Affinity modulation in platel et al pha 2 beta 1 following ligand binding. Biochem Biophys Res Commun. 1997;232:724–7. doi: 10.1006/bbrc.1997.6201. [DOI] [PubMed] [Google Scholar]
- 120.Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;158:357–68. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chiarugi P, Pani G, Giannoni E, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–44. doi: 10.1083/jcb.200211118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA, Jr, Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol. 2005;171:893–904. doi: 10.1083/jcb.200507004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Datla SR, McGrail DJ, Vukelic S, et al. Poldip2 controls vascular smooth muscle cell migration by regulating focal adhesion turnover and force polarization. Am J Physiol Heart Circ Physiol. 2014;307:H945–57. doi: 10.1152/ajpheart.00918.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Honore S, Kovacic H, Pichard V, Briand C, Rognoni JB. Alpha2beta1-integrin signaling by itself controls G1/S transition in a human adenocarcinoma cell line (Caco-2): implication of NADPH oxidase-dependent production of ROS. Exp Cell Res. 2003;285:59–71. doi: 10.1016/s0014-4827(02)00038-1. [DOI] [PubMed] [Google Scholar]
- 125.Sundaresan M, Zu-Xi Y, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 126.Garrido-Urbani S, Jemelin S, Deffert C, et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS One. 2011;6:e14665. doi: 10.1371/journal.pone.0014665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schroder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1736–43. doi: 10.1161/ATVBAHA.107.142117. [DOI] [PubMed] [Google Scholar]
- 128.Lee MY, San Martin A, Mehta PK, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009 doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med. 2001;31:1456–64. doi: 10.1016/s0891-5849(01)00727-4. [DOI] [PubMed] [Google Scholar]
- 130.Yin CC, Huang KT. H2O2 but not O2- elevated by oxidized LDL enhances human aortic smooth muscle cell proliferation. J Biomed Sci. 2007;14:245–54. doi: 10.1007/s11373-006-9132-4. [DOI] [PubMed] [Google Scholar]
- 131.Szöcs K, Lassègue B, Sorescu D, et al. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. 2002;22:21–7. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 132.Nishio E, Watanabe Y. The involvement of reactive oxygen species and arachidonic acid in alpha 1-adrenoceptor-induced smooth muscle cell proliferation and migration. Br J Pharmacol. 1997;121:665–70. doi: 10.1038/sj.bjp.0701171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang Z, Castresana MR, Newman WH. Reactive oxygen and NF-kappaB in VEGF-induced migration of human vascular smooth muscle cells. Biochem Biophys Res Commun. 2001;285:669–74. doi: 10.1006/bbrc.2001.5232. [DOI] [PubMed] [Google Scholar]
- 134.Meng D, Lv DD, Fang J. Insulin-like growth factor-I induces reactive oxygen species production and cell migration through Nox4 and Rac1 in vascular smooth muscle cells. Cardiovasc Res. 2008;80(2):299–308. doi: 10.1093/cvr/cvn173. [DOI] [PubMed] [Google Scholar]
- 135.Haurani MJ, Cifuentes ME, Shepard AD, Pagano PJ. Nox4 oxidase overexpression specifically decreases endogenous Nox4 mRNA and inhibits angiotensin II-induced adventitial myofibroblast migration. Hypertension. 2008;52:143–9. doi: 10.1161/HYPERTENSIONAHA.107.101667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang Z, Castresana MR, Newman WH. Reactive oxygen species-sensitive p38 MAPK controls thrombin-induced migration of vascular smooth muscle cells. J Mol Cell Cardiol. 2004;36:49–56. doi: 10.1016/j.yjmcc.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 137.ten Freyhaus H, Huntgeburth M, Wingler K, et al. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–41. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 138.Abid MR, Kachra Z, Spokes KC, Aird WC. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000;486:252–6. doi: 10.1016/s0014-5793(00)02305-x. [DOI] [PubMed] [Google Scholar]
- 139.Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–20. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 140.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–9. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- 141.Vallotton P, Small JV. Shifting views on the leading role of the lamellipodium in cell migration: speckle tracking revisited. J Cell Sci. 2009;122:1955–8. doi: 10.1242/jcs.042036. [DOI] [PubMed] [Google Scholar]
- 142.Kaji N, Ohashi K, Shuin M, Niwa R, Uemura T, Mizuno K. Cell cycle-associated changes in Slingshot phosphatase activity and roles in cytokinesis in animal cells. J Biol Chem. 2003;278:33450–5. doi: 10.1074/jbc.M305802200. [DOI] [PubMed] [Google Scholar]
- 143.San Martin A, Lee MY, Williams HC, Mizuno K, Lassegue B, Griendling KK. Dual regulation of cofilin activity by LIM kinase and Slingshot-1L phosphatase controls platelet-derived growth factor-induced migration of human aortic smooth muscle cells. Circ Res. 2008;102:432–8. doi: 10.1161/CIRCRESAHA.107.158923. [DOI] [PubMed] [Google Scholar]
- 144.Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell. 2009;20:2650–60. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.San Martin A, Lee MY, Griendling KK. Novel Nox1-Mediated Mechanism of SSH1L Activation in VSMC: Role in Cell Migration. Atheroscler Thromb Vasc Biol. 2008;28:e 109. [Google Scholar]
- 146.Arber S, Barbayannis FA, Hanser H, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–9. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 147.Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell. 2001;12:1131–45. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yang N, Higuchi O, Ohashi K, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–12. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 149.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006;21:269–80. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- 150.Samstag Y, John I, Wabnitz GH. Cofilin: a redox sensitive mediator of actin dynamics during T-cell activation and migration. Immunol Rev. 2013;256:30–47. doi: 10.1111/imr.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ballestrem C, Hinz B, Imhof BA, Wehrle-Haller B. Marching at the front and dragging behind: differential alphaVbeta3-integrin turnover regulates focal adhesion behavior. J Cell Biol. 2001;155:1319–32. doi: 10.1083/jcb.200107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Carpenter CL. Actin cytoskeleton and cell signaling. Crit Care Med. 2000;28:N94–9. doi: 10.1097/00003246-200004001-00011. [DOI] [PubMed] [Google Scholar]
- 153.Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol. 2011;3(5):a005033. doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 155.Hamadi A, Bouali M, Dontenwill M, Stoeckel H, Takeda K, Ronde P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci. 2005;118:4415–25. doi: 10.1242/jcs.02565. [DOI] [PubMed] [Google Scholar]
- 156.Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 157.Chiarugi P, Cirri P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem Sci. 2003;28:509–14. doi: 10.1016/S0968-0004(03)00174-9. [DOI] [PubMed] [Google Scholar]
- 158.Shinohara M, Shang WH, Kubodera M, et al. Nox1 redox-signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007 doi: 10.1074/jbc.M609450200. [DOI] [PubMed] [Google Scholar]
- 159.Angers-Loustau A, Cote JF, Charest A, et al. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J Cell Biol. 1999;144:1019–31. doi: 10.1083/jcb.144.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Garton AJ, Tonks NK. Regulation of fibroblast motility by the protein tyrosine phosphatase PTP-PEST. J Biol Chem. 1999;274:3811–8. doi: 10.1074/jbc.274.6.3811. [DOI] [PubMed] [Google Scholar]
- 161.Sastry SK, Lyons PD, Schaller MD, Burridge K. PTP-PEST controls motility through regulation of Rac1. J Cell Sci. 2002;115:4305–16. doi: 10.1242/jcs.00105. [DOI] [PubMed] [Google Scholar]
- 162.Cai L, Makhov AM, Schafer DA, Bear JE. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell. 2008;134:828–42. doi: 10.1016/j.cell.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Burgstaller G, Gimona M. Actin cytoskeleton remodelling via local inhibition of contractility at discrete microdomains. J Cell Sci. 2004;117:223–31. doi: 10.1242/jcs.00839. [DOI] [PubMed] [Google Scholar]
- 164.Bryce NS, Clark ES, Leysath JL, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol. 2005;15:1276–85. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 165.Minta JO, Yun JJ, Kabiawu O, Jones J. mRNA differential display identification of vascular smooth muscle early response genes regulated by PDGF. Mol Cell Biochem. 2006;281:63–75. doi: 10.1007/s11010-006-0524-6. [DOI] [PubMed] [Google Scholar]
- 166.Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112(Pt 16):2677–91. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- 167.Zhai J, Lin H, Nie Z, et al. Direct interaction of focal adhesion kinase with p190RhoGEF. J Biol Chem. 2003;278:24865–73. doi: 10.1074/jbc.M302381200. [DOI] [PubMed] [Google Scholar]
- 168.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem. 2006;281:2296–305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- 169.Chiarugi P, Cirri P, Taddei L, et al. The low M(r) protein-tyro sine phosphatase is involved in Rho-mediated cytoskeleton rearrangement after integrin and platelet-derived growth factor stimulation. J Biol Chem. 2000;275:4640–6. doi: 10.1074/jbc.275.7.4640. [DOI] [PubMed] [Google Scholar]
- 170.Chiarugi P, Taddei ML, Cirri P, et al. Low molecular weight protein-tyrosine phosphatase controls the rate and the strength of NIH-3T3 cells adhesion through its phosphorylation on tyrosine 131 or 132. J Biol Chem. 2000;275:37619–27. doi: 10.1074/jbc.M006375200. [DOI] [PubMed] [Google Scholar]
- 171.Vepa S, Scribner WM, Parinandi NL, English D, Garcia JG, Natarajan V. Hydrogen peroxide stimulates tyrosine phosphorylation of focal adhesion kinase in vascular endothelial cells. Am J Physiol. 1999;277:L150–8. doi: 10.1152/ajplung.1999.277.1.L150. [DOI] [PubMed] [Google Scholar]
- 172.Basuroy S, Dunagan M, Sheth P, Seth A, Rao RK. Hydrogen peroxide activates focal adhesion kinase and c-Src by a phosphatidylinositol 3 kinase-dependent mechanism and promotes cell migration in Caco-2 cell monolayers. Am J Physiol Gastrointest Liver Physiol. 2010;299:G186–95. doi: 10.1152/ajpgi.00368.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Abdelsaid MA, El-Remessy AB. S-glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J Cell Sci. 2012;125:4751–60. doi: 10.1242/jcs.103481. [DOI] [PubMed] [Google Scholar]
- 174.Maraldi T, Prata C, Caliceti C, et al. VEGF-induced ROS generation from NAD(P)H oxidases protects human leukemic cells from apoptosis. Int J Oncol. 2010;36:1581–9. doi: 10.3892/ijo_00000645. [DOI] [PubMed] [Google Scholar]
- 175.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–83. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 176.Lee CF, Ullevig S, Kim HS, Asmis R. Regulation of monocyte adhesion and migration by Nox4. PLoS One. 2013;8:e66964. doi: 10.1371/journal.pone.0066964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fernandez IM-GA, Clempus RE, Seidel-Rogol B, et al. TGF-β mediates focal adhesion maturation by a Smad/Nox4-dependent mechanism that involves regulation of Hsp27 and Hic5. Free Radic Biol Med. 2013;65:S157–8. [Google Scholar]
- 178.Shibanuma M, Mashimo J, Kuroki T, Nose K. Characterization of the TGF beta 1-inducible hic-5 gene that encodes a putative novel zinc finger protein and its possible involvement in cellular senescence. J Biol Chem. 1994;269:26767–74. [PubMed] [Google Scholar]
- 179.Nishiya N, Shirai T, Suzuki W, Nose K. Hic-5 interacts with GIT1 with a different binding mode from paxillin. J Biochem. 2002;132:279–89. doi: 10.1093/oxfordjournals.jbchem.a003222. [DOI] [PubMed] [Google Scholar]
- 180.Shibanuma M, Mori K, Kim-Kaneyama JR, Nose K. Involvement of FAK and PTP-PEST in the regulation of redox-sensitive nuclear-cytoplasmic shuttling of a LIM protein, Hic-5. Antioxid Redox Signal. 2005;7:335–47. doi: 10.1089/ars.2005.7.335. [DOI] [PubMed] [Google Scholar]
- 181.Ishino K, Kaneyama, Shibanuma M, Nose K. Specific decrease in the level of Hic-5, a focal adhesion protein, during immortalization of mouse embryonic fibroblasts, and its association with focal adhesion kinase. J Cell Biochem. 2000;76:411–9. doi: 10.1002/(sici)1097-4644(20000301)76:3<411::aid-jcb9>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 182.Avraamides C, Bromberg ME, Gaughan JP, Thomas SM, Tsygankov AY, Panetti TS. Hic-5 promotes endothelial cell migration to lysophosphatidic acid. Am J Physiol Heart Circ Physiol. 2007;293:H193–203. doi: 10.1152/ajpheart.00728.2006. [DOI] [PubMed] [Google Scholar]
- 183.Tumbarello DA, Brown MC, Hetey SE, Turner CE. Regulation of paxillin family members during epithelial-mesenchymal transformation: a putative role for paxillin delta. J Cell Sci. 2005;118:4849–63. doi: 10.1242/jcs.02615. [DOI] [PubMed] [Google Scholar]
- 184.Srinivasan R, Forman S, Quinlan RA, Ohanian J, Ohanian V. Regulation of contractility by Hsp27 and Hic-5 in rat mesenteric small arteries. Am J Physiol Heart Circ Physiol. 2008;294:H961–9. doi: 10.1152/ajpheart.00939.2007. [DOI] [PubMed] [Google Scholar]
- 185.Hetey SE, Lalonde DP, Turner CE. Tyrosine-phosphorylated Hic-5 inhibits epidermal growth factor-induced lamellipodia formation. Exp Cell Res. 2005;311:147–56. doi: 10.1016/j.yexcr.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 186.Tumbarello DA, Turner CE. Hic-5 contributes to epithelial-mesenchymal transformation through a RhoA/ROCK-dependent pathway. J Cell Physiol. 2007;211:736–47. doi: 10.1002/jcp.20991. [DOI] [PubMed] [Google Scholar]
- 187.Miron T, Vancompernolle K, Vandekerckhove J, Wilchek M, Geiger B. A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. J Cell Biol. 1991;114:255–61. doi: 10.1083/jcb.114.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Patil SB, Tsunoda Y, Pawar MD, Bitar KN. Translocation and association of ROCK-II with RhoA and HSP27 during contraction of rabbit colon smooth muscle cells. Biochem Biophys Res Commun. 2004;319:95–102. doi: 10.1016/j.bbrc.2004.04.159. [DOI] [PubMed] [Google Scholar]
- 189.Patil SB, Pawar MD, Bitar KN. Phosphorylated HSP27 essential for acetylcholine-induced association of RhoA with PKCalpha. Am J Physiol Gastrointest Liver Physiol. 2004;286:G635–44. doi: 10.1152/ajpgi.00261.2003. [DOI] [PubMed] [Google Scholar]
- 190.Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res. 1997;80:383–92. doi: 10.1161/01.res.80.3.383. [DOI] [PubMed] [Google Scholar]
- 191.Piotrowicz RS, Hickey E, Levin EG. Heat shock protein 27 kDa expression and phosphorylation regulates endothelial cell migration. FASEB J. 1998;12:1481–90. doi: 10.1096/fasebj.12.14.1481. [DOI] [PubMed] [Google Scholar]
- 192.Tangkijvanich P, Melton AC, Chitapanarux T, Han J, Yee HF. Platelet-derived growth factor-BB and lysophosphatidic acid distinctly regulate hepatic myofibroblast migration through focal adhesion kinase. Exp Cell Res. 2002;281:140–7. doi: 10.1006/excr.2002.5657. [DOI] [PubMed] [Google Scholar]
- 193.Hedges JC, Dechert MA, Yamboliev IA, et al. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J Biol Chem. 1999;274:24211–9. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- 194.Thiele JJ, Hsieh SN, Briviba K, Sies H. Protein oxidation in human stratum corneum: susceptibility of keratins to oxidation in vitro and presence of a keratin oxidation gradient in vivo. J Invest Dermatol. 1999;113:335–9. doi: 10.1046/j.1523-1747.1999.00693.x. [DOI] [PubMed] [Google Scholar]
- 195.Lee SH, Miyamoto K, Goto T, Oe T. Non-invasive proteomic analysis of human skin keratins: screening of methionine oxidation in keratins by mass spectrometry. J Proteomics. 2011;75:435–49. doi: 10.1016/j.jprot.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 196.Sattayakhom A, Ittiwat W, Stremmel W, Chamulitrat W. Redox regulation of cytokeratin 18 protein by NADPH oxidase 1 in preneoplastic human epithelial cells. J Cancer Res Clin Oncol. 2011;137:1669–78. doi: 10.1007/s00432-011-1041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rogers KR, Morris CJ, Blake DR. Oxidation of thiol in the vimentin cytoskeleton. Biochem J. 1991;275(Pt 3):789–91. doi: 10.1042/bj2750789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lii CK, Lin AH, Lee SL, Chen HW, Wang TS. Oxidative modifications of proteins by sodium arsenite in human umbilical vein endothelial cells. Environ Toxicol. 2011;26:459–71. doi: 10.1002/tox.20572. [DOI] [PubMed] [Google Scholar]
- 199.Pekovic V, Gibbs-Seymour I, Markiewicz E, et al. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell. 2011;10:1067–79. doi: 10.1111/j.1474-9726.2011.00750.x. [DOI] [PubMed] [Google Scholar]
- 200.Janue A, Odena MA, Oliveira E, Olive M, Ferrer I. Desmin is oxidized and nitrated in affected muscles in myotilinopathies and desminopathies. J Neuropathol Exp Neurol. 2007;66:711–23. doi: 10.1097/nen.0b013e3181256b4c. [DOI] [PubMed] [Google Scholar]
- 201.Janue A, Olive M, Ferrer I. Oxidative stress in desminopathies and myotilinopathies: a link between oxidative damage and abnormal protein aggregation. Brain Pathol. 2007;17:377–88. doi: 10.1111/j.1750-3639.2007.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Canton M, Neverova I, Menabo R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol. 2004;286:H870–7. doi: 10.1152/ajpheart.00714.2003. [DOI] [PubMed] [Google Scholar]
- 203.Kaneko K, Nakamura A, Yoshida K, Kametani F, Higuchi K, Ikeda S. Glial fibrillary acidic protein is greatly modified by oxidative stress in aceruloplasminemia brain. Free Radic Res. 2002;36:303–6. doi: 10.1080/10715760290019327. [DOI] [PubMed] [Google Scholar]
- 204.Santpere G, Ferrer I. Delineation of early changes in cases with progressive supranuclear palsy-like pathology. Astrocytes in striatum are primary targets of tau phosphorylation and GFAP oxidation. Brain Pathol. 2009;19:177–87. doi: 10.1111/j.1750-3639.2008.00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Smerjac SM, Bizzozero OA. Cytoskeletal protein carbonylation and degradation in experimental autoimmune encephalomyelitis. J Neurochem. 2008;105:763–72. doi: 10.1111/j.1471-4159.2007.05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Choi J, Forster MJ, McDonald SR, Weintraub ST, Carroll CA, Gracy RW. Proteomic identification of specific oxidized proteins in ApoE-knockout mice: relevance to Alzheimer’s disease. Free Radic Biol Med. 2004;36:1155–62. doi: 10.1016/j.freeradbiomed.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 207.Kato Y, Ono S, Kitamoto N, Kettle AJ. Covalent modification of cytoskeletal proteins in neuronal cells by tryptamine-4,5-dione. Redox Biol. 2014;2C:983–990. doi: 10.1016/j.redox.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kim NH, Kang JH. Protective effects of histidine dipeptides on the modification of neurofilament-L by the cytochrome c/hydrogen peroxide system. J Biochem Mol Biol. 2007;40:125–9. doi: 10.5483/bmbrep.2007.40.1.125. [DOI] [PubMed] [Google Scholar]
- 209.Valencia A, Sapp E, Kimm JS, et al. Striatal synaptosomes from Hdh140Q/140Q knock-in mice have altered protein levels, novel sites of methionine oxidation, and excess glutamate release after stimulation. J Hunting Dis. 2013;2:459–75. doi: 10.3233/JHD-130080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kang JH. Salsolinol, a tetrahydroisoquinoline-derived neurotoxin, induces oxidative modification of neurofilament-L protection by histidyl dipeptides. BMB Rep. 2012;45:114–9. doi: 10.5483/BMBRep.2012.45.2.114. [DOI] [PubMed] [Google Scholar]
- 211.Kim NH, Jeong MS, Choi SY, Hoon Kang J. Oxidative modification of neurofilament-L by the Cu,Zn-superoxide dismutase and hydrogen peroxide system. Biochimie. 2004;86:553–9. doi: 10.1016/j.biochi.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 212.Furukawa A, Kawamoto Y, Chiba Y, et al. Proteomic identification of hippocampal proteins vulnerable to oxidative stress in excito-toxin-induced acute neuronal injury. Neurobiol Dis. 2011;43:706–14. doi: 10.1016/j.nbd.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 213.Gianni D, Taulet N, Zhang H, et al. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol. 2010;5:981–93. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shi J, Ross CR, Leto TL, Blecha F. PR-39, a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase activity by binding to Src homology 3 domains of p47 phox. Proc Natl Acad Sci USA. 1996;93:6014–8. doi: 10.1073/pnas.93.12.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ohtake T, Fujimoto Y, Ikuta K, et al. Proline-rich antimicrobial peptide, PR-39 gene transduction altered invasive activity and actin structure in human hepatocellular carcinoma cells. Br J Cancer. 1999;81:393–403. doi: 10.1038/sj.bjc.6690707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kadi A, de Isla N, Moby V, et al. The Nebivolol action on vascular tone is dependent on actin cytoskeleton polymerization and Rho-A activity into ECs and SMCs. Clin Hemorheol Microcirc. 2014;56:231–46. doi: 10.3233/CH-131718. [DOI] [PubMed] [Google Scholar]
- 217.Yoshida LS, Abe S, Tsunawaki S. Fungal gliotoxin targets the onset of superoxide-generating NADPH oxidase of human neutrophils. Biochem Biophys Res Commun. 2000;268:716–23. doi: 10.1006/bbrc.2000.2192. [DOI] [PubMed] [Google Scholar]
- 218.Hagens WI, Beljaars L, Mann DA, et al. Cellular targeting of the apoptosis-inducing compound gliotoxin to fibrotic rat livers. J Pharmacol Exp Ther. 2008;324:902–10. doi: 10.1124/jpet.107.132290. [DOI] [PubMed] [Google Scholar]
- 219.Pozo M, Izquierdo MC, de Nicolas R, Egido J, Ortiz A, Gonzalez-Cabrero J. Gliotoxin inhibits neointimal hyperplasia after vascular injury in rats. J Vasc Res. 2009;46:278–89. doi: 10.1159/000176043. [DOI] [PubMed] [Google Scholar]
- 220.Toblli JE, Cao G, Giani JF, Munoz MC, Angerosa M, Dominici FP. Long-term treatment with nebivolol attenuates renal damage in Zucker diabetic fatty rats. J Hypertens. 2011;29:1613–23. doi: 10.1097/HJH.0b013e328349064c. [DOI] [PubMed] [Google Scholar]
- 221.Munnamalai V, Weaver CJ, Weisheit CE, et al. Bidirectional interactions between NOX2-type NADPH oxidase and the F-actin cytoskeleton in neuronal growth cones. J Neurochem. 2014;130:526–40. doi: 10.1111/jnc.12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rehman AU, Dugic E, Benham C, Lione L, Mackenzie LS. Selective inhibition of NADPH oxidase reverses the over contraction of diabetic rat aorta. Redox Biol. 2013;2C:61–64. doi: 10.1016/j.redox.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]