

Abstract
Background
Cyclooxygenase-2-derived prostaglandin E2 (PGE2), a bioactive eicosanoid, has been implicated in many biological processes including reproduction, inflammation and tumor growth. We previously showed that PGE2 stimulated lung cancer cell growth and progression through PGE2 receptor EP2/EP4-mediated kinase signaling pathways. However, the role of PGE2 in controlling lung airway epithelial cell phenotype remains unknown. We evaluated the effects of c-Jun and 3-phosphoinositede dependent protein kinase-1 (PDK1) in mediating epithelial cell hyperplasia induced by PGE2.
Method
The bronchial epithelial cell lines BEAS-2B and HBEc14-KT were cultured and then treated with PGE2. PDK1 small interfering RNA (siRNA) and a PDK1 inhibitor, an antagonist of the PGE2 receptor subtype EP4 and EP4 siRNA, c-Jun siRNA, and overexpressions of c-Jun and PDK1 have been used to evaluate the effects on cell proliferation.
Results
We demonstrated that PGE2 increased normal bronchial epithelial cell proliferation through induction of PDK1, an ankyrin repeat-containing Ser/Thr kinase implicated in the induction of apoptosis and the suppression of tumor growth. PDK1 siRNA and a PDK1 inhibitor blocked the effects of PGE2 on normal cell growth. The PGE2-induced PDK1 expression was blocked by an antagonist of the PGE2 receptor subtype EP4 and by EP4 siRNA. In addition, we showed that induction of PDK1 by PGE2 was associated with induction of the transcription factor, c-Jun protein. Silencing of c-Jun using siRNA and point mutations of c-Jun sites in the PDK1 gene promoter resulted in blockade of PDK1 expression and promoter activity induced by PGE2. In contrast, overexpression of c-Jun induced PDK1 gene promoter activity and expression followed increased cell proliferation.
Conclusion
PGE2 increases normal bronchial epithelial cell proliferation through increased PDK1 gene expression that is dependent on EP4 and induction of c-Jun. Therewith, our data suggest a new role of c-Jun and PDK1 in mediating epithelial cell hyperplasia induced by PGE2.
Keywords: Prostaglandin E2, PDK1, c-Jun, Human normal bronchial epithelial cells
Background
Cancer claims over half a million lives in the United States annually, and lung cancer is the number one cause of cancer death in both men and women. An estimated 226,160 new cases of lung cancer was diagnosed in 2012 in the United States alone, and 160,340 lung cancer deaths are estimated to occur [1]. Chronic inflammation has been associated with an increased risk of cancer. Protein levels of cyclooxygenase-2 (COX-2), a mediator of inflammation, were reported elevated in several cancer types, including colorectal, prostate, and lung cancers, and suppression of either COX-2 expression or COX-2 activation, is being considered for cancer prevention and therapy [2]. Of the five prostaglandins (PG) produced by COX-2, PGE2 appeared to play essential roles in tumor cell proliferation, invasion, angiogenesis, and immunosuppression [3–5]. Four receptor subtypes are known to bind PGE2, and they are named EP1-4. These receptors have also been implicated in tumor cell growth and progression [6, 7]. PGE2 is increased in patients with chronic obstructive pulmonary disease also showing a higher incidence of lung cancer [8].
Three-phosphoinositede dependent protein kinase-1 (PDK1) is a master kinase, which is crucial for the activation of AKT / protein kinase B (PKB) and many other AGC kinases including protein kinase C (PKC), S6 protein kinase (S6K), and serum-and glucocorticoid-induced protein kinase (SGK) [9]. As an upstream regulator of AKT, PDK1 signaling is thought to play a key role in cancer cell growth, survival and tumor angiogenesis [10, 11]. Studies have shown high levels of activated PDK1 in a large percentage of common tumor types, including melanoma, breast, lung, gastric, prostate, hematological, and ovarian cancers [12]. When activated in tumor cells, Akt also has multiple effects that promote disease progression, including suppression of apoptosis and stimulation of tumor cell proliferation, metabolism, and angiogenesis [13–15]. Thus, the PDK1/Akt signaling pathway represents an attractive target for the development of small molecule inhibitors that may be useful in the treatment of cancer. However, the effects of PGE2 on human bronchial epithelial cells are not clear. Here, we explore the effects of PGE2 on human bronchial epithelial cell proliferation and the intracellular signals involved in this process. Our studies show that PGE2 stimulates human bronchial epithelial cell proliferation through the EP4 receptor and activation of c-Jun, which increases the expression of PDK1.
Methods
Cell culture and chemicals
The human bronchial epithelial cell lines BEAS-2B and HBEc14-KT were obtained from John Minna (University of Texas Southwestern Medical Center, Dallas, TX, USA). They were maintained in KERATINOCYTE-SFM medium (Invitrogen Corporation, Carlsbad, CA, USA) supplemented with human recombinant epidermal growth factor 1–53 (EGF 1–53) and bovine pituitary extract (BPE). Cells were plated into six-well culture plates at an initial seeding density of 5 × 104 cells per well. The plates were incubated in a humidified atmosphere of 5 % CO2 in air at 37 °C. Lipofectamine 2000 reagent was purchased from Invitrogen. The CellTiter-Glo Luminescent Cell Viability Assay kit was purchased from Promega. Polyclonal antibodies against PDK1 and phosphorylated PDK1 (ser 241) were purchased from Cell Signaling. The polyclonal antibody against c-Jun was purchased from Santa Cruz Biotechnology, Inc. Polyclonal antibody against EP4, a 16, 16-Dimethyl-PGE2 (dmPGE2) solution in methyl acetate and AH23848, OSU-03012 were purchased from Cayman Chemical Co.
Reverse transcription and real time PCR
Real time PCR was performed to assess whether PDK1 expression was modulated by PGE2. Total RNA was isolated using the RNA Bee kit (Qiagen, Inc., Valencia, CA) as per manufacturer’s instructions. One μg of total RNA was reverse transcribed as per protocol using TaqMan® RT reagents (Applied Biosystems) at 37 °C for 120 min followed by 25 °C for 10 min.
Forty ng of cDNA per reaction were used in the real time PCR using the ABI Prism® 7900HT Sequence Detection System (Foster City, CA). In the presence of AmpliTaq Gold DNA polymerase (ABI Biosystems, Foster City, CA), the reaction was incubated for 2 min at 50 °C followed by 10 min at 95 °C. Then the reaction was run for 40 cycles at 15 s, 95 °C and 1 min, 60 °C per cycle (Table 1). Assays-on-Demand™ primers and probes specific for the oxytocin receptor (Applied Biosystems; ID number Hs00168573-m1) were used in the PCR. GAPDH was measured and used to normalize all samples using the ΔΔCT method (Applied Biosystems). Gene expression of PDK1 is expressed relative to GAPDH and untreated samples in each stimulation study, respectively. At least three replicates were run for each condition.
Table 1.
Reverse transcription and real time PCR primer
| Stage | Temp | Time | |
|---|---|---|---|
| Reverse transcription | hold | 37 °C | 120 min |
| hold | 25 °C | 10 min | |
| PCR | hold | 50 °C | 2 min |
| denature | 95 °C | 15 s | |
| anneal/extend | 60 °C | 1 min | |
| hold cycle (40 cycles) | 95 °C | 10 min | |
| Primer | Sense 5′ AGATGAGTGACCGAGGAG | ||
| Antisense 3′ TATACACTGGGAGTCTTTCT | |||
Western blot analysis
The procedure was performed as previously described [16]. Protein concentrations were determined by the Bio-Rad protein assay. 5 μg protein from whole-cell lysates were solubilized in 5× SDS sample buffer and separated on SDS 10 % polyacrylamide gels. Blots were incubated with antibodies against c-Jun, PDK1, EP4, GAPDH, and phosphorylated PDK1 (c-Jun, PDK1, pPDK1 and EP4 in the concentrations of 1:1,000 for 2 h; GAPDH in the concentration of 1:10000 for 1 h) at room temperature. After washing several times, the blots were followed by incubation with a secondary goat antibody raised against rabbit IgG conjugated to horseradish peroxidase (1:2,000; Santa Cruz). The blots were washed, transferred to fresh chemiluminescence solution (Amersham) for about 1–2 min, and exposed to X-ray film. In controls, the primary antibodies were omitted or replaced with a control rabbit IgG.
Treatments with EP4, PDK1, and c-Jun small interfering RNA and expression vector
The EP4, PDK1, c-Jun siRNA and control nonspecific siRNA oligonucleotides were purchased from Santa Cruz Biotechnology. pWZL-Neo-Myr- Flag PDK1 and pWZL-Neo-Myr-Flag-DEST were purchased from Addgene Inc. The pGME4 c-Jun vector and pGME4 were provided by Dr. Tom Curran (Children’s Hospital of Philadelphia, University of Pennsyvania, USA). For the transfection procedure, cells were grown to 60 % confluence, and EP4, c-Jun and PDK1 siRNAs,control siRNA,and expression vector were transfected using the lipofectamine 2000 reagent according to the manufacturer’s instructions. Briefly, the lipofectamine reagent was incubated with serum-free medium for 5 min. Subsequently, a mixture of respective siRNA was added. After incubation for 20 min at room temperature, the mixture was diluted with medium and added to each well. The final concentration of siRNAs in each well was 100 nmol/L. After culturing for 30 h, cells were washed and resuspended in new culture medium in the presence or absence of dmPGE2 for Western blot and cell growth and gel mobility shift assays.
Transient transfection assay
The human PDK1 promoter ligated to the luciferase reporter gene were a gift from Drs. Michalik and Desvergne (Center for Integrative Genomics, University of Lausanne, Lausanne, Switzerland) and have been reported previously [17]. The PDK1 promoter construct contains ∼ 663 bp of the human PDK1 promoter connected to the pGL2 vector. Briefly, normal bronchial epithelial cells were seeded at a density of 5 × 105 per well in 24-well dishes and grown to 50–60 % confluence. For each well, 0.4 μg of the above PDK1 plasmid DNA constructs, with or without 0.5 μg of the internal control phRL-TK Synthetic Renilla Luciferase Reporter Vector, were co-transfected into the cells using lipofectamine 2000 reagent . After 24 h of incubation, cells were treated with or without dmPGE2 for 4 h. The preparation of cell extracts and measurement of luciferase activities were carried out using the Dual-Luciferase Reporter kit according to recommendations by the manufacturer (Promega). The assays for firefly luciferase activity and Renilla luciferase activity were performed sequentially in a Labsystems Luminoskan Ascent luminometer equipped with dual injectors. Changes in firefly luciferase activity were calculated and plotted after normalization with changes in Renilla luciferase activity within the same sample.
Cell viability assay
Normal bronchial epithelial cells were plated at the indicated densities (2 × 103 cells/well) in 96-well multiwell culture plates (Costar). Cells were treated with inhibitor or antagonist for 2 h before exposure of the cells to PGE2 in the culture medium (containing 10 % FBS). In separate experiments, cells were transfected with control, PDK1, EP4 or c-Jun siRNAs or expression vector for 40 h before exposure to PGE2 for up to 4 days. Cell proliferation was evaluated using the CellTiter-Glo Luminescent Cell Viability Assay, a homogenous method of determining number of viable cells in culture based on quantitation of the ATP present which signals the presence of metabolically active cells.
Statistical analysis
All experiments were repeated a minimum of three times. All data from western blot analysis, real-time PCR, and luciferase assays were expressed as mean ± SD. In cell viability assay, the bar graphs represented the mean ± s.d. of relative cell viability compared to the control group of at least three independent experiments. In western blot analysis, the optical densities (OD) of pPDK1, PDK1, EP4 and C-Jun were normalized to the OD of GAPDH in the same membrane. The data represented the mean ± s.d. of relative OD compared to the control group of at least three independent experiments with three samples in each. In transient transfection assay, the bar graphs represent the mean ± s.d. of relative luciferase activities compared to the control group of at least three independent experiments. One-way anova analyses followed by the Least Significant Difference (LSD) test were performed. Asterisks showed in the figures indicate significant differences of experimental groups in comparison with the corresponding control condition. P-values <0.05 were considered statistically significant.
Results
PGE2 increases cell proliferation and phosphorylation and expression of PDK1 in HBEc14-KT and BEAS2B cells
PGE2 has been shown to stimulate lung cancer cell proliferation [18]. To examine the effects of PGE2 on human bronchial epithelial cell proliferation, HBEc 14-KT and BEAS2B cells were treated with increased concentrations of dmPGE2 for 72 h. As shown in Fig. 1a, PGE2 stimulates normal bronchial epitheial cell proliferation in a dose-dependent manner with maximal effect at a concentration of 1 μM, compared to the control group (1.604 ± 0.046 vs 1.000 ± 0.046 in BEAS2B, P <0.01; 1.665 ± 0.023 vs 1.000 ± 0.017 in HBEc14-KT, P <0.01). Exposure to PGE2 enhances the phosphorylation and expression of PDK1 in a dose-dependent and time-dependent manner with maximal effect at a concentration of 1 μM at 2–4 h (Fig. 1b). pPDK1 and PDK1 reached their peaks at 4 h with the concentration of 1 μM of dmPGE2, compared to the control group (3.414 ± 0.243 vs 1.000 ± 0.135 and 1.512 ± 0.087 vs 1.001 ± 0.129 in BEAS2B, P <0.01, P <0.05; 1.373 ± 0.092 vs 1.000 ± 0.142 and 1.415 ± 0.726 vs 1.000 ± 0.130 in HBEc14-KT, P <0.01, P < 0.01). pPDK1 and PDK1 reached their peaks with the concentration of 1 μM of dmPGE2 after incubation for 4 h, compared to the control group (2.812 ± 0.317 vs 1.000 ± 0.216 and 5.214 ± 0.478 vs 1.000 ± 0.198 in BEAS2B, P <0.01, P <0.01; 2.754 ± 0.139 vs 1.000 ± 0.141 and 2.351 ± 0.286 vs 1.000 ± 0.127 in HBEc14-KT, P <0.01, P <0.01). To evaluate the role of PDK1 in PGE2-induced cell proliferation, PDK1 was silenced with siRNA or pre-treated with PDK1 inhibitor OSU-03012 (2 μM) in the cell lines (Fig. 1c and d). PDK1 inhibitor (OSU-03012) decreased cell proliferation induced by treatment with PGE2 (1.279 ± 0.030 vs 1.463 ± 0.005 in BEAS2B, P <0.01; 1.211 ± 0.142 vs 1.918 ± 0.038 in HBEc14-KT, P <0.01). PDK1 siRNA decreased cell proliferation induced by treatment with PGE2 (1.177 ± 0.038 vs 1.708 ± 0.127 in BEAS2B, P <0.05; 1.272 ± 0.052 vs 2.428 ± 0.073 in HBEc14-KT, P <0.01). In contrast, transfection of cells with PDK1 plasmid increases cell proliferation induced by PGE2 (1.653 ± 0.042 vs 1.381 ± 0.067 in BEAS2B, P <0.05; 1.681 ± 0.033 vs 1.395 ± 0.057 in HBEc14-KT, P <0.01) (Fig. 1e).
Fig. 1.
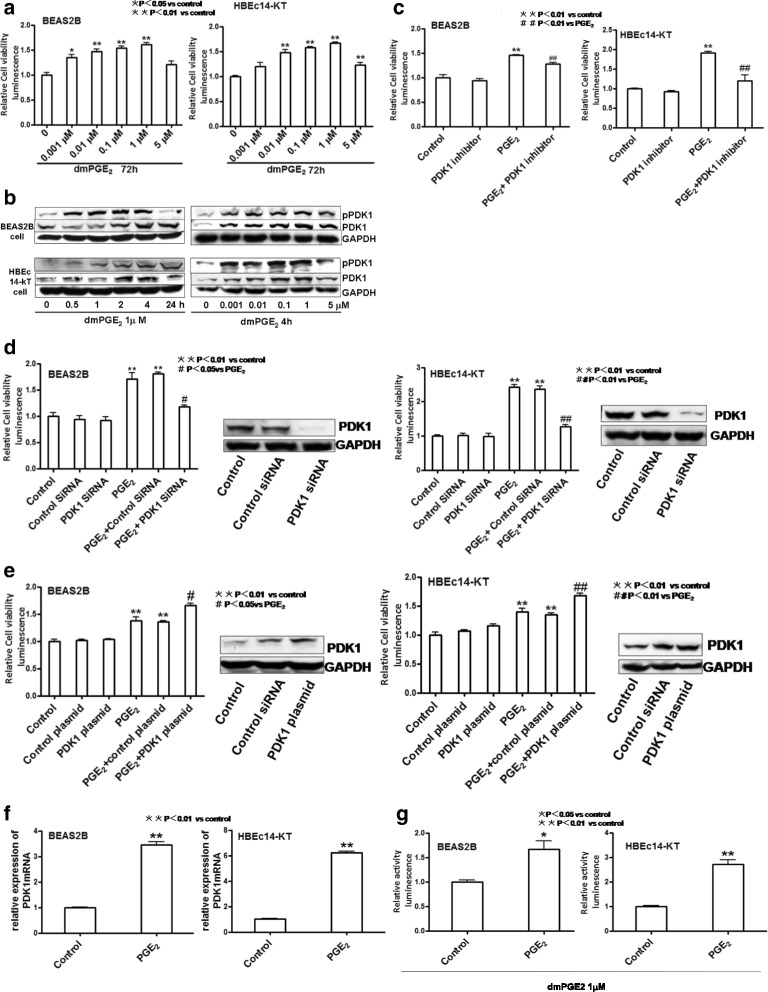
The effect of dmPGE2 on human bronchial epithelial cell proliferation and induction of expression and activation of PDK1. a Effects on cell proliferation. b dmPGE2 increases the phosphorylation and expression of PDK1 in a time- and dose-dependent manner in BEAS2B and HBEc14-KT cells. c PDK1 inhibitor OSU-03012 decreases cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. d PDK1 siRNA decreases the cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. e PDK1 plasmid increases the cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. f Effect on mRNA expression. g Effect on PDK1 promoter activity
Having established a role for PDK1 in PGE2-stimulated normal bronchial epithelial cell proliferation, we examined whether the effects of PGE2 on PDK1 expression occur at the transcriptional level. We found that PGE2 induces the expression of PDK1 mRNA (3.462 ± 0.103 vs 1.000 ± 0.020 in BEAS2B, P <0.01; 6.218 ± 0.138 vs 1.008 ± 0.069 in HBEc14-KT, P <0.01) (Fig. 1f) and activity of PDK1 gene promoter (1.666 ± 0.177 vs 1.000 ± 0.039 in BEAS2B, P <0.05; 2.714 ± 0.187 vs 1.000 ± 0.043 in HBEc14-KT, P <0.01) (Fig. 1g). We therefore conclude that PGE2 increases cell proliferation in normal bronchial epithelial cells by activation of the PDK1.
EP4 mediates the cell proliferation and increased expression and activation of PDK1 induced by PGE2 in HBEc14-KT and BEAS2B cells
PGE2 binds to and activates four distinct receptor subtypes named EP1-4. EP4 has been shown to a target molecule in cancer cell proliferation [19]. To investigate the mechanisms involved in PGE2-mediated bronchial epithelial cell proliferation, we transfected cells with EP4 siRNA or pre-treated cells with the EP4 antagonist AH23848 (10 μM). After pre-treatment with EP4 siRNA or antagonist, cell proliferations induced by PGE2 decreased (Fig. 2a and b). EP4 antagonist AH23848 decreased cell proliferation induced by PGE2 (1.456 ± 0.046 vs 2.078 ± 0.299 in BEAS2B, P <0.01; 1.131 ± 0.052 vs 1.838 ± 0.074 in HBEc14-KT, P <0.01). EP4 siRNA decreased cell proliferation induced by treatment with PGE2 (1.243 ± 0.018 vs 1.480 ± 0.029 in BEAS2B, P <0.05; 1.086 ± 0.124 vs 1.866 ± 0.015 in HBEc14-KT, P <0.01). This effect was associated with inhibition of phosphorylation and expression of PDK1 (Fig. 2c and d). EP4 antagonist AH23848 decreased the phosphorylation (1.213 ± 0.013 vs 1.457 ± 0.021 in BEAS2B, P <0.05; 1.198 ± 0.019 vs 1.562 ± 0.029 in HBEc14-KT, P <0.05) and expression of PDK1 (1.012 ± 0.008 vs 1.452 ± 0.025 in BEAS2B, P <0.05; 1.213 ± 0.019 vs 1.483 ± 0.024 in HBEc14-KT, P <0.05) induced by dmPGE2. EP4 siRNA decreased the phosphorylation (0.987 ± 0.019 vs 1.621 ± 0.038 in BEAS2B, P <0.05; 1.242 ± 0.022 vs 1.765 ± 0.031 in HBEc14-KT, P <0.05) and expression of PDK1 (1.056 ± 0.011 vs 1.513 ± 0.018 in BEAS2B, P <0.05; 1.356 ± 0.028 vs 2.107 ± 0.039 in HBEc14-KT, P <0.05) induced by dmPGE2. It also decreased PDK1 gene promoter activity (1.104 ± 0.113 vs 1.716 ± 0.088 in BEAS2B, P <0.05; 1.187 ± 0.111 vs 1.468 ± 0.101 in HBEc14-KT, P <0.01) induced by dmPGE2 (Fig. 2e). So we conclude that EP4 mediates cell proliferation induced by PGE2 in normal bronchial epithelial cells.
Fig. 2.
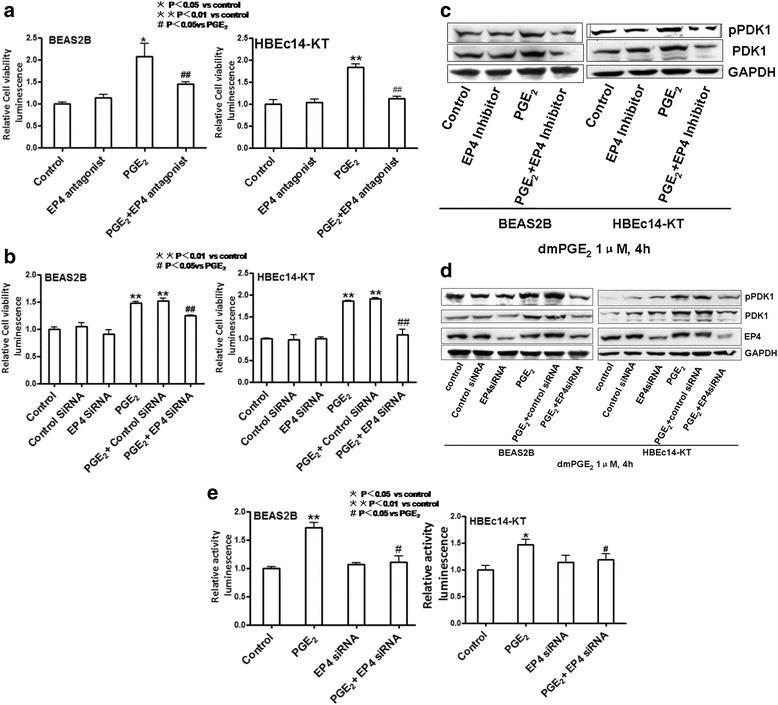
EP4 Mediates the Cell Proliferation and increased expression and activation of PDK1 induced by PGE2 in HBEc14-KT and BEAS2B cells. a EP4 antagonist AH23848 decreased the cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. b EP4 siRNA decreases the cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. c EP4 antagonist AH23848 decreased the phosphorylation and expression of PDK1 induced by dmPGE2 in BEAS2B and HBEc14-KT cells. d EP4 siRNA decreased the phosphorylation and expression of PDK1 induced by dmPGE2 in BEAS2B and HBEc14-KT cells. e Effect on PDK1 promoter activity
The role of transcription factor c-Jun in PGE2 induction of PDK1 and cell growth
The PDK1 gene promoter contains multiple transcription factor-binding sites, including two sites for c-Jun. Then we explored the role of c-Jun in mediating PGE2-induced PDK1 expression in normal bronchial epithelial cells. PGE2 induces the expression of c-Jun protein in a time-dependent and dose-dependent manner (Fig. 3a). C-Jun reached its peaks at 2 h in BEAS2B cells and 4 h in HBEc14-KT cells with the concentration of 1 μM of dmPGE2, compared to the control group (2.561 ± 0.189 vs 1.000 ± 0.063 in BEAS2B, P <0.01; 1.347 ± 0.132 in HBEc14-KT, P <0.01). c-Jun reached its peak with the concentration of 1 μM of dmPGE2 after incubation for 4 h, compared to the control group (1.499 ± 0.108 vs 1.001 ± 0.063 in BEAS2B, P <0.01; 1.536 ± 0.174 vs 1.000 ± 0.024 in HBEc14-KT, P <0.01). Silencing of c-Jun with its siRNA decreases cell proliferation (1.115 ± 0.054 vs 1.386 ± 0.072 in BEAS2B, P <0.05; 1.030 ± 0.104 vs 1.495 ± 0.072 in HBEc14-KT, P <0.01) (Fig. 3b) and the expression of PDK1 induced by PGE2 (1.105 ± 0.011 vs 2.534 ± 0.029 in BEAS2B, P <0.01; 1.157 ± 0.021 vs 1.892 ± 0.027 in HBEc14-KT, P <0.01) induced by dmPGE2 (Fig. 3d). Transfection with c-Jun plasmid increases the cell proliferation (1.741 ± 0.051 vs 1.518 ± 0.059 in BEAS2B, P <0.05; 1.772 ± 0.258 vs 1.521 ± 0.027 in HBEc14-KT, P <0.05) (Fig. 3c) and the expression of PDK1 induced by PGE2 (2.278 ± 0.134 vs 1.196 ± 0.095 in BEAS2B, P <0.05; 2.453 ± 0.141 vs 2.056 ± 0.163 in HBEc14-KT, P <0.05) (Fig. 3e).
Fig. 3.
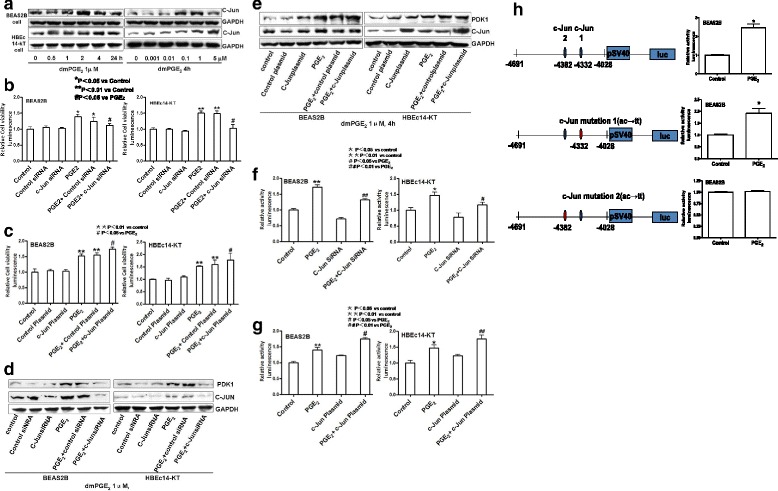
c-Jun Mediates the Cell Proliferation and increased expression and activation of PDK1 induced by PGE2 in HBEc14-KT and BEAS2B cells. a dmPGE2 increases expression of c-Jun in a time- and dose-dependent manner in BEAS2B and HBEc14-KT cells. b c-Jun siRNA decreases the cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. c c-Jun plasmid increases the cell proliferation induced by dmPGE2 in normal bronchial epithelial cells. d c-Jun siRNA decreases expression of PDK1 induced by dmPGE2 in BEAS2B and HBEc14-KT cells. e c-Jun plasmid increases expression of PDK1 induced by dmPGE2 in BEAS2B and HBEc14-KT cells. f Effect of c-Jun siRNA on PDK1 promoter activity. g Effect of c-Jun plasmid on PDK1 promoter activity. h Effects of c-Jun point-moutations on PDK1 promoter activity
Having shown the important role of PDK1 in PGE2-related NSCLC proliferation, we further investigated whether the PGE2-mediated up-regulation of PDK1 reflected transactivation of the gene. To this end, we performed transient transcription experiments using human PDK1 promoter-reporter constructs connected to a luciferase reporter gene and found that PGE2 increased PDK1 promoter activity. The induction of PDK1 promoter activity by PGE2 was inhibited by c-Jun siRNA (1.319 ± 0.028 vs 1.716 ± 0.069 in BEAS2B, P <0.01; 1.171 ± 0.071 vs 1.468 ± 0.101 in HBEc14-KT, P <0.05) (Fig. 3f). Exogenous overexpression of c-Jun enhanced the PDK1 promoter activity (1.753 ± 0.037 vs 1.401 ± 0.075 in BEAS2B, P <0.05; 1.762 ± 0.117 vs 1.468 ± 0.101 in HBEc14-KT, P <0.01) (Fig. 3g). The induction of PDK1 gene promoter activity by PGE2 was abrogated when the one of c-Jun sites at -4382 bp was mutated in the PDK1 gene promoter, suggesting a critical role for c-Jun in mediating the effect of PGE2 on PDK1 gene promoter activity (Fig. 3h). dmPGE2 increased PDK1 promoter activity (2.432 ± 0.358 vs 1.000 ± 0.039 in BEAS2B, P <0.05); point mutation of c-Jun with ac to tt at −4028 site could not derease PDK1 promoter activity (1.9832 ± 0. vs 1.000 ± 0.024 in BEAS2B, P <0.05); point mutation of c-Jun site with ac to tt at -4382 bp dereased PDK1 promoter activity (1.010 ± 0.011 vs 1.000 ± 0.039 in BEAS2B, P <0.05);
Discussion
The transformation is driven by both endogenous and exogenous factors including chemical agents that induce the activation of cancer-promoting genes. Accumulating epidemiologic and clinical data provide a strong link between inflammation and cancer initiation and progression [20, 21], but the molecular inflammatory determinants remain to be established. COX-2 derived PGE2 is a proinflammatory bioactive lipid and is the major prostaglandin produced in many human solid tumors, including cancer of the colon [22], stomach [23], breast [24] and lung [25]. PGE2 and its receptors play an essential role in promoting cancer progression. For example, PGE2 significantly enhanced carcinogen induced colon tumor incidence and multiplicity in rats. PGE2 accelerates intestinal adenoma growth in ApcMin mice [26]. However, the effects of PGE2 on human bronchial epithelial cells are not clear.. Therefore, we will focus on PGE2 and its receptors and its downstream targets in human bronchial epithelial cells, and explore the potential mechanism. This is the first study on the effects of PGE2 in inducing the activation of cancer-promoting genes in normal bronchial epithelial cells. In concordance with studies performed in tumor cells before, we demonstrate PGE2 increases normal bronchial epithelial cell proliferation, through increased PDK1 gene expression that is dependent on EP4 and induction of c-Jun. It unveils a novel role of c-Jun and PDK1 in mediating epithelial cell hyperplasia induced by PGE2.
Three-phosphoinositide-dependent protein kinase-1 (PDK1) is a major mediator of cellular signaling between phosphoinositide-3 kinase and various intracellular serine/threonine kinases, including PKB, p70 ribosomal S6 kinase, serum and glucocorticoid-inducible kinase, and protein kinase. PDK1 is able to phosphorylate Thr-308 on PKBα [27, 28], which has been shown to play a crucial role in normal and pathophysical conditions. Activation of PDK1 has been shown to regulate cell survival and growth, cell cycle progression, gene expression and differention [9, 29]. Autophosphorylation and growth-factor-induced phosphorylation in its activation loop is required for PDK1 activity [30]. S241A mutation abolished PDK1 catalytic activity completely, which suggests phosphorylation at Ser-241 is important for PDK1 signaling [31]. There is increasing evidence that PDK1 is involved in cancer progression and invasion. Tissue microarray analysis of human invasive breast cancer has revealed that phosphorylation of PDK1 on Ser-241 was strongly enhanced in 90 % of the samples tested [11]. Immunohistochemical analysis using anti-phospho-Tyr-9 antibodies demonstrated that the level of Tyr-9 phosphorylation is increased markly in diseased lung, liver, colon, and breast tissue compared to normal tissue [12]. Studies have shown that angiotension-II-induced focal adhesion formation is inhibited by infection with Adeno-PDK1-Y9F via paxillin [10]. This regulation of focal adhesion suggests that PDK1 participates in integrating signals that control cell growth, apoptosis and migration. PDK1 gene has been assoiated with poor differentiation of late stage lung cancer [32]. PDK1 was shown capable of augmenting tumorigenesis in tissues harboring ERBB2 amplifications [33], PTEN deletions [34], and mutations in the catalytic subunit of phosphoinositide 3-kinase (PIK3CA) [35]. Inhibition of PDK1 is therefore expected to attenuate tumors associated with deregulated PIK3CA/PTEN signaling. Indeed, hypomorphic mutation of PDK1 in Pten +/− mice delays the onset of tumorigenesis, and small molecule inhibitors of PDK1 inhibit tumor xenografts and lung colonization [36]. Further, PDK1 inactivation effectively attenuated the development of Kras oncogene-driven pancreatic cancer, but not NSCLC, further supporting the importance of PDK1 in tumor development, albeit, in select cancer types [37]. Here, we found that PGE2 increased the expression of PDK1 in bronchial epithelial cells. Overexpression of PDK1 enhanced cell proliferation, while cells stimulated with PDK1 inhibitor or siRNA reduced cell growth. These findings provides the first genetic evidence that PDK1 may also play an essential role in abnormally enhanced cell proliferation induced by PGE2 in bronchial epithelial cells, which was convinced in tumor cells in previous studies. Consistent with former studies, PDK1 inactivations by small molecule inhibitor of PDK1 or siRNA effectively attenuated cell proliferation in bronchial epithelial cells, supporting the potential importance of PDK1 in NSCLC development. PDK1 also plays a crucial role in metastasis. This kinase mediates mammary epithelial cell growth and invasion in the transformed phenotype, in part, by membrane type 1 matrix metalloproteinase (MMP) induction, which in turn activates MMP-2 and modulates the extracellular matrix proteins decorin and collagen [11]. Knockdown of PDK1 inhibits spontaneous migration and epidermal-growth-factor-induced chemotaxis in breast cancer cells. In severe combined immunodeficiency mice, PDK1-depleted human breast cancer cells form tumors more slowly and are defective in extravasation to the lungs after intravenous injection [38]. These results indicate that PDK1 plays an important role in regulating malignancy in breast cancer cells. Moreover, reducing PDK1 expression in PTEN +/− mice protects these animals from developing a wide range of tumors [36], thereby providing genetic evidence that PDK1 is a key effector in mediating neoplasia that result from loss of PTEN. In addition, PDK1 is an upstream kinase of AKT, which regulates activities of AKT, in turn, mediates the regulation of tumor cell growth, metastasis, and angiogenesis [39]. All these results validate PDK1 is a promising anticancer target for the prevention of tumors.
PGE2 exerts its cellular effects by binding to its cognate receptors (EP1-4) that belong to the family of seven transmembrane G protein coupled rhodopsin-type receptors [40]. The central role of PGE2 in tumor-genesis has been further confirmed through homozygous deletion of its receptors. Mice with homozygous deletion in EP1 and EP4 receptors, but not EP3, were partially resistant to colon carcinogen mediated induction of aberrant crypt foci [41, 42]. EP2 disruption decreases the number and size of intestinal polyps in APC△716 knockout mice [43]. In addition to colorectal cancer, it has been shown that EP1, 2, and 4 receptors were elevated whereas EP3 receptor levels were decreased in mammary tumors in COX-2-MMTV mice [44]. It was showed that PGE2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src pathway [45]. In this study, blockade of EP4 by antagonists or by EP4 siRNA resulted in reduced PDK1 protein expression and activity of PDK1 promoter, suggesting that EP4 mediates the effect of PGE2 on PDK1 expression in normal bronchial epithelial cells. The EP4 receptor can activate several pathways that involve cell proliferation, adhesion, invasion and migration. These main molecules include G protein, adenylyl cyclase, protein kinase A, exchange protein directly activated by cAMP, phosphodiesterase, phosphatidylinositol 3-Kinase, extracellular signal-regulated kinase. Most recently, we found that the EP4 receptor could increase the expression of JNK [16], which has been found to play a pivotal role in activating transcription factors (including c-Jun) that increase cellular growth and tumor formation in NSCLC cells. We also showed that activation of PI3-K, PKA, and JNK induced by EP4 was involved in the effect of PGE2 on c-jun in NSCLC [46].
Specific mitogen-activated protein kinase (MAPK) cascades control the activation of fos and jun family protooncogenes and their protein products (c-Jun and c-Fos) are known as the AP-1 family members. These “early response protooncogene” products dimerize to form the AP-1 transcription factor, a converging point that regulates the expression of genes involved in cell proliferation, differentiation, transformation, inflammation, pulmonary defense, and autoregulation of AP-1 gene transcription [46]. Early studies [47, 48] suggested that c-Jun had a role in early events in the pathogenesis of lung cancers because it was highly expressed in 31–50 % of patients with non- small cell cancers, and it was also upregulated in atypical bronchial epithelium. One study showed a transgenic mouse model directing conditional expression of the dominant-negative c-Jun mutant TAM67 in lung epithelial cells decreased tumor number and overall lung tumor burden in chemically induced mouse lung tumor models [49]. c-Jun is an important transcriptional activator of PDK1. Notably, expression of PDK1 is sufficient to restore tumor growth after c-Jun knockdown in melanoma cells, suggesting that PDK1 is an important mediator of c-Jun oncogenic activities [50]. Consistent with this, we found that PGE2 increased the expression of c-Jun and exogenous overexpression of c-Jun enhanced bronchial epithelial cells proliferation and expression of PDK1, whereas inhibition by PDK1 inhibitor or siRNA caused inhibitions of cell growth and expression of PDK1. This suggests that c-Jun is an upstream regulator of PDK1 signaling. Induction of c-Jun by PGE2 regulates the expression and activation of PDK1 and cell growth. It indicated c-Jun might play an important role in the potential mechanism of abnormal cell proliferation in brochial epithelial cells.
Conclusion
In summary, our findings demonstrate that PGE2 stimulates human bronchial epithelial cell proliferation through activations of EP4-related signal and c-Jun, which, in turn, increases activation and expression of PDK1. This suggests that PDK1 and c-Jun might play an important role in human bronchial epithelial cells thereby influencing their growth.
Acknowledgement
We are grateful to Dr. John Minna (the Hamon Center for Therapeutic Oncology Research, Simmons Cancer Center, University of Texas Southwestern Medical Center, TX) for providing the normal bronchial epithelial cell lines, and Dr. Tom Curran (Children’s Hospital of Philadelphia, University of Pennsylvania) for providing c-Jun expression vector. This work was supported by Sichuan Province Science and Technology Support Program (No. 2013SZ0001).
Footnotes
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
YF and KW carried out the molecular genetic studies and drafted the manuscript. YW participated in the design of the study and performed the statistical analysis. KW conceived of the study. All authors read and approved the final manuscript.
Contributor Information
Yu Fan, Email: 350177646@qq.com.
Ye Wang, Email: doctordream@163.com.
Ke Wang, Phone: (86)28-85423660, Email: wang2ke@126.com.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Baratelli F, Krysan K, Heuze-Vourc’h N, Zhu L, Escuadro B, Sharma S, et al. PGE2 confers survivin-dependent apoptosis resistance in human monocyte-derived dendritic cells. J Leukoc Biol. 2005;78:555–564. doi: 10.1189/jlb.1004569. [DOI] [PubMed] [Google Scholar]
- 3.Hawcroft G, Volpato M, Marston G, Ingram N, Perry SL, Cockbain AJ, et al. The omega-3 polyunsaturated fatty acid eicosapentaenoic acid inhibits mouse MC-26 colorectal cancer cell liver metastasis via inhibition of PGE2-dependent cell motility. Br J Pharmacol. 2012;166:1724–1737. doi: 10.1111/j.1476-5381.2012.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Liu Q, Zhang M, Yu Y, Liu X, Cao X. Fas signal promotes lung cancer growth by recruiting myeloid-derived suppressor cells via cancer cell-derived PGE2. J Immunol. 2009;182:3801–3808. doi: 10.4049/jimmunol.0801548. [DOI] [PubMed] [Google Scholar]
- 6.Rundhaug JE, Simper MS, Surh I, Fischer SM. The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev. 2011;30:465–480. doi: 10.1007/s10555-011-9317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reader J, Holt D, Fulton A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011;30:449–463. doi: 10.1007/s10555-011-9303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martey CA, Pollock SJ, Turner CK, O’Reilly KM, Baglole CJ, Phipps RP, et al. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287:L981–991. doi: 10.1152/ajplung.00239.2003. [DOI] [PubMed] [Google Scholar]
- 9.Fyffe C, Falasca M. 3-Phosphoinositide-dependent protein kinase-1 as an emerging target in the management of breast cancer. Cancer Manag Res. 2013;5:271–280. doi: 10.2147/CMAR.S35026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taniyama Y, Weber DS, Rocic P, Hilenski L, Akers ML, Park J, et al. Pyk2- and Src-dependent tyrosine phosphorylation of PDK1 regulates focal adhesions. Mol Cell Biol. 2003;23:8019–8029. doi: 10.1128/MCB.23.22.8019-8029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Z, Yuan H, Yin Y, Zeng X, Bai R, Glazer RI. 3-phosphoinositide-dependent protein kinase-1 (PDK1) promotes invasion and activation of matrix metalloproteinases. BMC Cancer. 2006;6:77. doi: 10.1186/1471-2407-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang KJ, Shin S, Piao L, Shin E, Li Y, Park KA, et al. Regulation of 3-phosphoinositide-dependent protein kinase-1 (PDK1) by Src involves tyrosine phosphorylation of PDK1 and Src homology 2 domain binding. J Biol Chem. 2008;283:1480–1491. doi: 10.1074/jbc.M706361200. [DOI] [PubMed] [Google Scholar]
- 13.Kang X, Kong F, Wu X, Ren Y, Wu S, Wu K, et al. High glucose promotes tumor invasion and increases metastasis-associated protein expression in human lung epithelial cells by upregulating heme oxygenase-1 via reactive oxygen species or the TGF-beta1/PI3K/Akt signaling pathway. Cell Physiol Biochem. 2015;35:1008–1022. doi: 10.1159/000373928. [DOI] [PubMed] [Google Scholar]
- 14.Im YK, La Selva R, Gandin V, Ha JR, Sabourin V, Sonenberg N, et al. The ShcA adaptor activates AKT signaling to potentiate breast tumor angiogenesis by stimulating VEGF mRNA translation in a 4E-BP-dependent manner. Oncogene. 2015;34:1729–1735. doi: 10.1038/onc.2014.110. [DOI] [PubMed] [Google Scholar]
- 15.Wu DM, Zhang P, Liu RY, Sang YX, Zhou C, Xu GC, et al. Phosphorylation and changes in the distribution of nucleolin promote tumor metastasis via the PI3K/Akt pathway in colorectal carcinoma. FEBS Lett. 2014;588:1921–1929. doi: 10.1016/j.febslet.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 16.Zhong X, Fan Y, Ritzenthaler JD, Zhang W, Wang K, Zhou Q, et al. Novel link between prostaglandin E2 (PGE2) and cholinergic signaling in lung cancer: the role of c-Jun in PGE2-induced alpha7 nicotinic acetylcholine receptor expression and tumor cell proliferation. Thorac Cancer. 2015;6:488–500. doi: 10.1111/1759-7714.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di-Poï N, Tan NS, Michalik L, Wahli W, Desvergne B. (2002). Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell 10: 721–733. [DOI] [PubMed]
- 18.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xin X, Majumder M, Girish GV, Mohindra V, Maruyama T, Lala PK. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab Invest. 2012;92:1115–1128. doi: 10.1038/labinvest.2012.90. [DOI] [PubMed] [Google Scholar]
- 20.van Verschuer VM, Hooning MJ, van Baare-Georgieva RD, Hollestelle A, Timmermans AM, Koppert LB, et al. Tumor-associated inflammation as a potential prognostic tool in BRCA1/2-associated breast cancer. Hum Pathol. 2015;46:182–190. doi: 10.1016/j.humpath.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 21.Sridharan DM, Asaithamby A, Bailey SM, Costes SV, Doetsch PW, Dynan WS, et al. Understanding cancer development processes after HZE-particle exposure: roles of ROS, DNA damage repair and inflammation. Radiat Res. 2015;183:1–26. doi: 10.1667/RR13804.1. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, DuBois RN. An inflammatory mediator, prostaglandin E2, in colorectal cancer. Cancer J. 2013;19:502–510. doi: 10.1097/PPO.0000000000000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piazuelo MB, Epplein M, Correa P. Gastric cancer: an infectious disease. Infect Dis Clin North Am. 2010;24:853–869. doi: 10.1016/j.idc.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau GT, Huang H, Lin SM, Leung LK. Butein downregulates phorbol 12-myristate 13-acetate-induced COX-2 transcriptional activity in cancerous and non-cancerous breast cells. Eur J Pharmacol. 2010;648:24–30. doi: 10.1016/j.ejphar.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 25.Maeng HJ, Lee WJ, Jin QR, Chang JE, Shim WS. Upregulation of COX-2 in the lung cancer promotes overexpression of multidrug resistance protein 4 (MRP4) via PGE2-dependent pathway. Eur J Pharm Sci. 2014;62:189–196. doi: 10.1016/j.ejps.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 26.Hull MA, Faluyi OO, Ko CW, Holwell S, Scott DJ, Cuthbert RJ, et al. Regulation of stromal cell cyclooxygenase-2 in the ApcMin/+ mouse model of intestinal tumorigenesis. Carcinogenesis. 2006;27:382–391. doi: 10.1093/carcin/bgi236. [DOI] [PubMed] [Google Scholar]
- 27.Tsoi H, Li L, Chen ZS, Lau KF, Tsui SK, Chan HY. The SARS-coronavirus membrane protein induces apoptosis via interfering with PDK1-PKB/Akt signalling. Biochem J. 2014;464:439–447. doi: 10.1042/BJ20131461. [DOI] [PubMed] [Google Scholar]
- 28.Voordeckers K, Kimpe M, Haesendonckx S, Louwet W, Versele M, Thevelein JM. Yeast 3-phosphoinositide-dependent protein kinase-1 (PDK1) orthologs Pkh1-3 differentially regulate phosphorylation of protein kinase A (PKA) and the protein kinase B (PKB)/S6K ortholog Sch9. J Biol Chem. 2011;286:22017–22027. doi: 10.1074/jbc.M110.200071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barile E, De SK, Pellecchia M. PDK1 inhibitors. Pharm Pat Anal. 2012;1:145–163. doi: 10.4155/ppa.12.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calleja V, Laguerre M, de Las H-MG, Parker PJ, Requejo-Isidro J, Larijani B. Acute regulation of PDK1 by a complex interplay of molecular switches. Biochem Soc Trans. 2014;42:1435–1440. doi: 10.1042/BST20140222. [DOI] [PubMed] [Google Scholar]
- 31.Sato S, Fujita N, Tsuruo T. Regulation of kinase activity of 3-phosphoinositide-dependent protein kinase-1 by binding to 14-3-3. J Biol Chem. 2002;277:39360–39367. doi: 10.1074/jbc.M205141200. [DOI] [PubMed] [Google Scholar]
- 32.Shen H, Zhu Y, Wu YJ, Qiu HR, Shu YQ. Genomic alterations in lung adenocarcinomas detected by multicolor fluorescence in situ hybridization and comparative genomic hybridization. Cancer Genet Cytogenet. 2008;181:100–107. doi: 10.1016/j.cancergencyto.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69:6299–6306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finlay DK, Sinclair LV, Feijoo C, Waugh CM, Hagenbeek TJ, Spits H, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med. 2009;206:2441–2454. doi: 10.1084/jem.20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr Biol. 2005;15:1839–1846. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 37.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 38.Sephton CF, Zhang D, Lehmann TM, Pennington PR, Scheid MP, Mousseau DD. The nuclear localization of 3′-phosphoinositide-dependent kinase-1 is dependent on its association with the protein tyrosine phosphatase SHP-1. Cell Signal. 2009;21:1634–1644. doi: 10.1016/j.cellsig.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Chan CH, Jo U, Kohrman A, Rezaeian AH, Chou PC, Logothetis C, et al. Posttranslational regulation of Akt in human cancer. Cell Biosci. 2014;4:59. doi: 10.1186/2045-3701-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta. 1851;2015:414–421. doi: 10.1016/j.bbalip.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, et al. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 1999;59:5093–5096. [PubMed] [Google Scholar]
- 42.Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- 43.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 44.Chang SH, Ai Y, Breyer RM, Lane TF, Hla T. The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res. 2005;65:4496–4499. doi: 10.1158/0008-5472.CAN-05-0129. [DOI] [PubMed] [Google Scholar]
- 45.Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res. 2010;8:569–577. doi: 10.1158/1541-7786.MCR-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaulian E. AP-1--The Jun proteins: oncogenes or tumor suppressors in disguise? Cell Signal. 2010;22:894–899. doi: 10.1016/j.cellsig.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Volm M, Drings P, Wodrich W. Prognostic significance of the expression of c-fos, c-jun and c-erbB-1 oncogene products in human squamous cell lung carcinomas. J Cancer Res Clin Oncol. 1993;119:507–510. doi: 10.1007/BF01686458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szabo E, Riffe ME, Steinberg SM, Birrer MJ, Linnoila RI. Altered cJUN expression: an early event in human lung carcinogenesis. Cancer Res. 1996;56:305–315. [PubMed] [Google Scholar]
- 49.Tichelaar JW, Yan Y, Tan Q, Wang Y, Estensen RD, Young MR, et al. A dominant-negative c-jun mutant inhibits lung carcinogenesis in mice. Cancer Prev Res (Phila) 2010;3:1148–1156. doi: 10.1158/1940-6207.CAPR-10-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopez-Bergami P, Kim H, Dewing A, Goydos J, Aaronson S, Ronai Z. c-Jun regulates phosphoinositide-dependent kinase 1 transcription: implication for Akt and protein kinase C activities and melanoma tumorigenesis. J Biol Chem. 2010;285:903–913. doi: 10.1074/jbc.M109.075630. [DOI] [PMC free article] [PubMed] [Google Scholar]