

Fei et al. show that the prenucleosome is a stable alternate conformational isomer of the nucleosome. Prenucleosomes assembled in vitro exhibit properties that are strikingly similar to those of nonnucleosomal histone–DNA particles in the upstream region of active promoters in vivo.
Keywords: nucleosome, prenucleosome, active chromatin, histones, gene expression
Abstract
Chromatin comprises nucleosomes as well as nonnucleosomal histone–DNA particles. Prenucleosomes are rapidly formed histone–DNA particles that can be converted into canonical nucleosomes by a motor protein such as ACF. Here we show that the prenucleosome is a stable conformational isomer of the nucleosome. It consists of a histone octamer associated with ∼80 base pair (bp) of DNA, which is located at a position that corresponds to the central 80 bp of a nucleosome core particle. Monomeric prenucleosomes with free flanking DNA do not spontaneously fold into nucleosomes but can be converted into canonical nucleosomes by an ATP-driven motor protein such as ACF or Chd1. In addition, histone H3K56, which is located at the DNA entry and exit points of a canonical nucleosome, is specifically acetylated by p300 in prenucleosomes relative to nucleosomes. Prenucleosomes assembled in vitro exhibit properties that are strikingly similar to those of nonnucleosomal histone–DNA particles in the upstream region of active promoters in vivo. These findings suggest that the prenucleosome, the only known stable conformational isomer of the nucleosome, is related to nonnucleosomal histone–DNA species in the cell.
Chromatin in the eukaryotic nucleus consists of nucleosomes and nonnucleosomal particles. Nucleosomes have been extensively studied and are well understood. In contrast, little is known about nonnucleosomal chromatin particles. These species are likely to be biologically important because they are found in regions of active chromatin such as promoters. One might imagine canonical nucleosomes as the static component of chromatin and nonnucleosomal particles as the dynamic component of chromatin.
We recently discovered a nonnucleosomal chromatin particle termed the prenucleosome, which is a precursor to the nucleosome in the assembly of chromatin (Torigoe et al. 2011, 2013; Bouazoune and Kingston 2013). Prenucleosomes contain all four core histones, do not supercoil DNA like a canonical nucleosome, and can be converted into periodic nucleosome arrays by an ATP-dependent chromatin assembly motor protein such as ACF or Chd1. By atomic force microscopy, prenucleosomes are indistinguishable from canonical nucleosomes. In addition, prenucleosomes are rapidly formed within seconds, are stable and resistant to challenge by free DNA for at least 2 h, and are more sensitive to digestion by micrococcal nuclease (MNase) than canonical nucleosomes.
The discovery of prenucleosomes provided a resolution to a paradox from the 1970s. At that time, it was observed that nucleosome-like structures (containing at least histones H3 and H2B) form within seconds upon passage of DNA replication forks (for example, see McKnight and Miller 1977; McKnight et al. 1978; Sogo et al. 1986), whereas canonical “mature” nucleosomes, as characterized by nuclease digestion and sedimentation properties, are more slowly generated after ∼10–20 min (for example, see Seale 1975, 1976; Levy and Jakob 1978; Worcel et al. 1978; Schlaeger and Knippers 1979; Klempnauer et al. 1980; Jackson and Chalkley 1981). Thus, the question was how nucleosome-like particles could be formed within seconds when it was also known that it takes at least 10 min to form a canonical nucleosome. It now appears that this paradox can be explained by the rapid formation of prenucleosomes at DNA replication forks.
Because prenucleosomes are formed rapidly, they can provide immediate protection of naked DNA that might be generated during processes such as replication and transcription. In addition, prenucleosomes or prenucleosome-like particles, such as at promoters and enhancers, might function as a more dynamic (less static and repressive) form of histone-bound DNA than canonical nucleosomes.
It was therefore important to gain a better understanding of the prenucleosome. Does it contain a histone octamer or some other combination of the histones? How much DNA is associated with a prenucleosome, and where is the DNA located relative to the histone octamer? Why do prenucleosomes not supercoil DNA like nucleosomes? In previous work, we assembled prenucleosomes onto relaxed plasmid DNA (Torigoe et al. 2011). While this approach has been useful for the characterization of the function of prenucleosomes in the chromatin assembly process, the resulting templates were heterogeneous in terms of the number and locations of the prenucleosomes. Hence, with that system, it was not possible to determine the composition and structure of the prenucleosome.
In this study, we took a new experimental approach to the analysis of prenucleosomes and determined that the prenucleosome is a stable conformational isomer of the nucleosome. We further found that the properties of prenucleosomes assembled in vitro are remarkably similar to those of nonnucleosomal histone–DNA particles at active promoters in vivo. The prenucleosome is the only known stable conformer of the nucleosome. Because there are probably only a limited number of alternate conformations of the nucleosome, prenucleosomes may share a common fundamental structure with native nonnucleosomal particles, such as those present at active chromatin throughout the genome.
Results
Electron microscopy reveals that prenucleosomes associate with ∼70- to 80-base-pair (bp) DNA—the same as that seen with chromatin particles at active promoters in vivo
To investigate the properties of prenucleosomes, we employed psoralen cross-linking and electron microscopy analysis. This method was originally developed for the study of nucleosomes and involves psoralen cross-linking of the linker DNA between nucleosomes followed by electron microscopy under denaturing conditions. The segments of DNA that are bound by histones appear as bubbles of ssDNA (Hanson et al. 1976; Sogo et al. 1986; Brown et al. 2013, 2015).
In our studies of prenucleosomes, we carried out chromatin assembly reactions with relaxed plasmid DNA in the absence or presence of ACF to give prenucleosomes or canonical nucleosomes, and the resulting samples were subjected to psoralen cross-linking, linearization, and electron microscopy. Representative images of the bubbles observed with prenucleosomes and nucleosomes are shown in Figure 1A. Quantitation of the number of bubbles per plasmid DNA revealed approximately the same number of prenucleosomes and nucleosomes per template (Supplemental Fig. S1). This finding is consistent with our previous analysis of prenucleosomes and nucleosomes by atomic force microscopy (Torigoe et al. 2011). The measurement of the bubble sizes revealed a peak at 140–150 nucleotides (nt) with canonical nucleosomes, as expected. With prenucleosomes, however, we observed a peak at ∼70–80 nt (Fig. 1B) as well as larger bubbles that may have been due to the presence of some canonical nucleosomes in the predominantly prenucleosomal samples (as in Torigoe et al. 2011). These findings suggest that prenucleosomes associate with ∼70–80 nt of DNA, which corresponds to less than one turn of the DNA around the histone octamer in a nucleosome. This partial wrapping of DNA around the histones in prenucleosomes (relative to that in nucleosomes) might explain the lack of DNA supercoiling that is observed during the formation of prenucleosomes (Torigoe et al. 2011).
Figure 1.

Psoralen cross-linking and electron microscopy analysis suggests that prenucleosomes associate with ∼70–80 bp of DNA. (A) Analysis of prenucleosomes and nucleosomes by psoralen cross-linking followed by denaturing electron microscopy. Representative images are shown. The histone-free DNA is cross-linked by psoralen, and the resulting bubbles represent the locations of prenucleosomes and nucleosomes. (B) ACF-mediated conversion of NAP1-assembled prenucleosomes to canonical nucleosomes increases the size of psoralen bubbles from ∼70–80 nt to ∼140–150 nt. Prenucleosomes (−ACF) and nucleosomes (+ACF) were subjected to psoralen cross-linking and denaturing electron microscopy. Bubble sizes were measured in ImageJ and converted from nanometers to nucleotides. A total of 4623 prenucleosome bubbles and 5013 nucleosome bubbles was measured in four independent experiments. The plot displays the distribution of bubble sizes as the average ± standard deviation (n = 4) of 10-nt bins. The individual data points are placed at the center of the 10-nt bins. (C) Comparison of the psoralen bubble sizes observed in vitro and in vivo. The data from prenucleosomes and nucleosomes in vitro (shown in B) and at the active versus repressed PHO5 promoters in vivo in Saccharomyces cerevisiae (Brown et al. 2013). The plot shows the distribution of bubble sizes as the average of 10-nt bins.
These studies additionally enabled us to compare the properties of prenucleosomes assembled in vitro and nonnucleosomal chromatin particles observed in vivo. Remarkably, the biochemical data on prenucleosomes versus nucleosomes (Fig. 1B) exhibit a close resemblance to the psoralen bubble size distribution observed in vivo with the active versus repressed PHO5 promoter in yeast (Brown et al. 2013). In the in vivo analysis, a peak bubble size of 70–80 bp of DNA was seen at the activated PHO5 promoter, whereas a peak of 140–150 bp was observed at the repressed PHO5 promoter. To demonstrate the correlation, we directly compared the in vitro data (this study) and in vivo data (Fig. 1C; Brown et al. 2013). These data suggest that the active PHO5 promoter contains both prenucleosomes (or prenucleosome-like particles) and nucleosomes, whereas the repressed PHO5 promoter contains mostly canonical nucleosomes. Thus, although prenucleosomes were initially identified in the analysis of chromatin assembly, these findings indicate that they may also be present at the promoter regions of active genes.
Rapid and efficient assembly of monomeric prenucleosomes (mono-prenucleosome) onto 80-bp DNA fragments
The psoralen cross-linking and electron microscopy experiments additionally led to a new approach to the analysis of prenucleosomes. If prenucleosomes associate with 70–80 bp of DNA, we thought that it might be possible to assemble a mono-prenucleosome with histones and an 80-bp DNA fragment. To test this hypothesis, we deposited core histones onto an 80-bp DNA fragment by using the NAP1 core histone chaperone. We employed the same conditions as those used for the formation of prenucleosomes on plasmid DNA templates (Torigoe et al. 2011), except that a short 80-bp DNA fragment was used instead of relaxed plasmid DNA. These experiments revealed the rapid (complete within 30 sec) formation of a distinct nucleoprotein species with all four core histones (Fig. 2A). There was essentially complete conversion of the free DNA to the nucleoprotein species at a 1:1 histone octamer:DNA ratio. Under these same conditions, we did not see the efficient formation of distinct complexes with equimolar amounts of either H3–H4 or H2A–H2B alone. Based on these data and other results shown in this work, we refer to these nucleoprotein complexes as monomeric prenucleosomes or “mono-prenucleosomes.”
Figure 2.

Rapid and efficient formation of mono-prenucleosomes with 80-bp DNA fragments. (A) The NAP1-mediated formation of mono-prenucleosomes with an 80-bp genomic DNA fragment occurs rapidly and requires all four core histones. Histone–NAP1 complexes were combined with an 80-bp DNA fragment (an 80-bp segment in the coding sequence of the Drosophila melanogaster ISWI gene; henceforth termed the “80-bp genomic DNA”). The samples were incubated at room temperature for 30 sec and then subjected to native (nondenaturing) 5% polyacrylamide gel electrophoresis. The DNA was visualized by staining with ethidium bromide. One octamer equivalent of all four core histones per DNA would be a 1:1 octamer:DNA ratio. Note that one octamer equivalent of H2A+H2B+H3+H4 has the same amount of H3 and H4 as one octamer equivalent of H3+H4. (B) Mono-prenucleosomes can be formed with the central 80 bp of the 601 nucleosome positioning sequence. Mono-prenucleosomes were formed and analyzed as in A, with all four core histones along with either the 80-bp genomic DNA or the central 80 bp of the 601 sequence. (C) Mono-prenucleosomes appear to be the thermodynamically most stable arrangement of the four core histones and 80 bp of DNA at 50 mM NaCl. Mono-prenucleosomes were formed with the dNLP histone chaperone as well as by salt dialysis of the four core histones with the 80-bp genomic DNA fragment. For comparison, H3–H4 monotetrasomes were also generated in parallel by salt dialysis with H3–H4. The histones were used at an octamer equivalent:DNA ratio of 1.0. (D) Mono-tetrasomes can be converted into prenucleosomes by the addition of H2A–H2B. Monotetrasomes were formed by salt dialysis with the 80 bp of genomic DNA as in C. Next, NAP1–H2A–H2B complexes were added as indicated. The samples were incubated for 30 sec at room temperature and then subjected to native 5% polyacrylamide gel electrophoresis. As a reference, a mono-prenucleosome formed by salt dialysis as in C was included (“Mono-prenuc”).
We then tested whether mono-prenucleosomes can be assembled with different DNA segments. In Figure 2A, we used an 80-bp stretch of DNA from the coding sequence of the Drosophila melanogaster ISWI gene (sequence given in Supplemental Table S1); we refer to this fragment as the “80-bp genomic DNA.” We also used the central 80 bp of the 601 nucleosome positioning sequence (for sequence, see Supplemental Table S1; Lowary and Widom 1998); we term this fragment the “central 80 bp of 601 sequence.” As shown in Figure 2B, mono-prenucleosomes can be formed efficiently with either the 80-bp genomic DNA or the central 80 bp of 601 sequence. These two DNA fragments are used throughout this study.
Next, we sought to determine whether mono-prenucleosomes are generally formed under different reaction conditions. To this end, we tested a different core histone chaperone, Drosophila nucleoplasmin-like protein (dNLP) (Ito et al. 1996) as well as salt dialysis techniques (Stein 1989) for the reconstitution of mono-prenucleosomes. These experiments revealed that mono-prenucleosomes are efficiently formed with not only NAP1 but also dNLP and by salt dialysis in the absence of histone chaperones (Fig. 2C). Thus, in a minimal purified reaction consisting of only the four core histones and 80 bp of DNA, mono-prenucleosomes can be formed by salt dialysis under standard conditions used for nucleosome reconstitution, in which the histones and DNA are combined in 1 M NaCl, and the samples are slowly dialyzed in a stepwise manner to 0.05 M NaCl (Stein 1989). These results indicate that the mono-prenucleosome is a thermodynamically stable arrangement of the four core histones and 80 bp of DNA.
It should also be noted that mono-prenucleosome reconstitution requires a histone chaperone, such as NAP1 or dNLP, or initial high-salt conditions that prevent the formation of histone–DNA aggregates. If core histones are added directly to free DNA in low-salt buffer (for example, with ≤100 mM NaCl), insoluble histone–DNA aggregates are formed that do not enter the gel (Fig. 2C).
Comparison of assembly by salt dialysis with all four core histones or equimolar amounts of histones H3–H4 reveals the efficient formation of mono-prenucleosomes with the four core histones and inefficient formation of mono-tetrasomes with histones H3–H4 (Fig. 2C). Moreover, we observed that mono-tetrasomes can be efficiently converted into mono-prenucleosomes by the addition of NAP1–H2A–H2B complexes (Fig. 2D). These findings reveal that mono-tetrasomes are distinct from mono-prenucleosomes.
Analysis of the composition and salt lability of mono-prenucleosomes
We next analyzed the composition of mono-prenucleosomes by sucrose gradient sedimentation analysis. In these experiments, mono-prenucleosomes were reconstituted by either NAP1 deposition or salt dialysis and then subjected to 10%–30% sucrose gradient sedimentation. The presence of mono-prenucleosomes was detected by native gel electrophoresis of the nucleoprotein complexes as in Figure 2, and the histones were detected by SDS–polyacrylamide gel electrophoresis and silver staining. These experiments revealed that mono-prenucleosomes cosediment with all four core histones (Fig. 3A). As a control, histones in mono-prenucleosomes sediment faster than free histones (Supplemental Fig. S2A). In addition, it can be seen that the sedimentation rate of NAP1-assembled mono-prenucleosomes is the same as that of salt dialysis-reconstituted mono-prenucleosomes (Fig. 3A). Throughout this study, we found that mono-prenucleosomes that are prepared by either method have the same biochemical properties.
Figure 3.
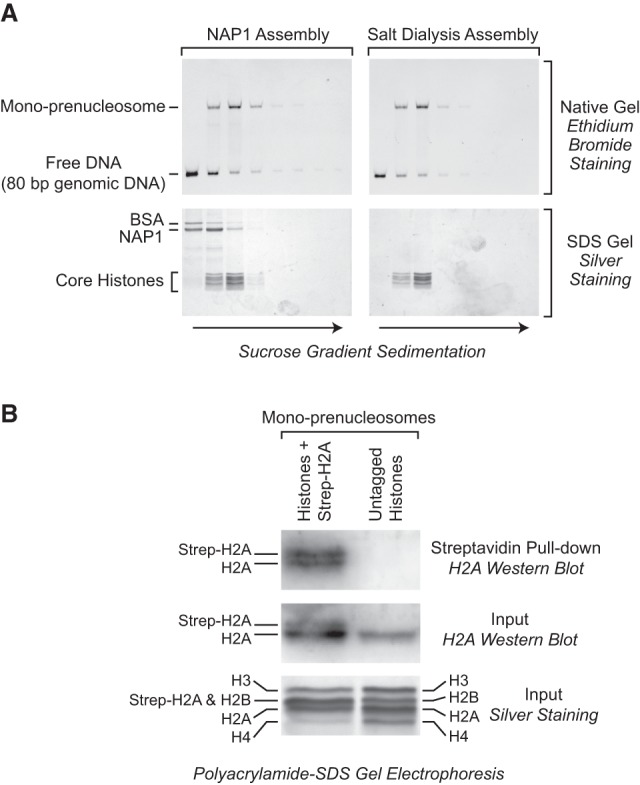
Mono-prenucleosomes contain all four core histones and are distinct from hexasomes. (A) Sucrose gradient sedimentation analysis reveals that mono-prenucleosomes contain all four core histones. Mono-prenucleosomes were prepared by either NAP1-mediated deposition (left panels) or salt dialysis (right panels) and then subjected to 10%–30% (w/v) (left to right) sucrose gradient sedimentation in a Beckman SW41 rotor (32,000 rpm for 18 h at 4°C). The arrows indicate the direction of sedimentation. (Top panels) The presence of mono-prenucleosomes was detected by native polyacrylamide gel electrophoresis and ethidium bromide staining of the DNA. (Bottom panels) The protein composition was analyzed by SDS–polyacrylamide gel electrophoresis and silver staining. The top two fractions and the bottom fraction did not contain histones (for example, see Supplemental Fig. S2A) and are not included. The sedimentation of the free core histones relative to prenucleosomal histones is shown in Supplemental Figure S2A. (B) Mono-prenucleosomes contain two copies of H2A and thus appear to contain a core histone octamer rather than hexamer. Mono-prenucleosomes were reconstituted with recombinant core histones onto the 80-bp genomic DNA by NAP1-mediated histone deposition. The H2A species were a combination of wild-type H2A and Strep-H2A at a 3:1 ratio of H2A:Strep-H2A. Prenucleosomes containing Strep-H2A were pulled down with streptavidin beads and then analyzed by Western blot with antibodies against histone H2A. An H2A Western blot and silver-stained SDS gel are also shown for the input samples. The Western blots were detected and quantitated by using 32P-labeled protein A.
Although the four core histones can be seen in mono-prenucleosomes (Fig. 3A), it was possible that the histones in mono-prenucleosomes exist as a hexamer (with two copies of H3 and H4 and only one copy of H2A and H2B) rather than as an octamer (with two copies each of H2A, H2B, H3, and H4) (for example, see Arimura et al. 2012). To test the hexamer hypothesis, we reconstituted mono-prenucleosomes with a 3:1 molar ratio of wild-type H2A to Strep-tagged H2A along with the other three core histones at a 1:1:1:1 ratio of total H2A (wild-type H2A + Strep-H2A):H2B:H3:H4. We then pulled down the mono-prenucleosomes containing Strep-tagged H2A and examined whether wild-type H2A coprecipitates with Strep-H2A by Western blot with antibodies against H2A (Fig. 3B). If there were a hexamer of histones in a mono-prenucleosome, then wild-type H2A would not coprecipitate with the Strep-H2A because of the single copy of H2A in a hexamer. However, these experiments revealed the presence of two copies of H2A in mono-prenucleosomes, as there was a ratio of approximately 1.0:1.4 of wild-type H2A to Strep-H2A. This ratio is nearly identical to the theoretical expectation of 1.0:1.3 for an octamer (given the 3:1 ratio of wild-type H2A to Strep-tagged H2A prior to precipitation). These results therefore support the conclusion that a mono-prenucleosome comprises 80 bp of DNA and a histone octamer.
We additionally tested the salt lability of mono-prenucleosomes relative to mononucleosomes. In these experiments, we prepared mono-prenucleosomes and mononucleosomes by salt dialysis (with 80-bp and 146-bp DNA fragments, respectively); adjusted the final NaCl concentrations to 0.1 M, 0.3 M, 0.8 M, or 2.0 M; and then subjected the samples to sucrose gradient sedimentation at the same salt concentrations (Supplemental Fig. S2B). As a reference, mononucleosomes partially disassemble at 0.8 M NaCl, as seen with native chromatin (for example, see Germond et al. 1976), and are completely disassembled at 2.0 M NaCl. In contrast, mono-prenucleosomes begin to dissemble at 0.3 M NaCl and are substantially disassembled at 0.8 M NaCl. Thus, consistent with the fewer histone–DNA contacts in prenucleosomes relative to nucleosomes, mono-prenucleosomes are more salt labile than canonical nucleosomes.
Mono-prenucleosomes can be converted into canonical nucleosomes by ACF
A key property of prenucleosomes is their ability to be converted into canonical nucleosomes by ACF (Torigoe et al. 2011). Therefore, we tested whether mono-prenucleosomes can be converted into canonical nucleosomes. To address this question, we formed poly-prenucleosomes by head-to-tail ligation of the 80 bp of DNA in mono-prenucleosomes to free DNA. Next, we incubated the poly-prenucleosomes with ACF and ATP and then digested the reaction products extensively with MNase, which converts nucleosome arrays into core particles that contain ∼147 bp of DNA (Fig. 4A). This experiment revealed that ACF is able to convert poly-prenucleosomes into canonical nucleosomes, as assessed by the generation of the ∼147-bp DNA species that is diagnostic of core particles. In the absence of ACF, we did not detect 147-bp DNA fragments upon digestion of the poly-prenucleosomes with MNase. Thus, the conversion of poly-prenucleosomes to polynucleosomes is dependent on ACF. In addition, the ability of mono-prenucleosomes to be converted into canonical nucleosomes further supports the conclusion that mono-prenucleosomes contain a complete histone octamer.
Figure 4.

Prenucleosomes can be converted into canonical nucleosomes by ACF. (A) ACF-dependent assembly of poly-prenucleosomes to polynucleosomes. Mono-prenucleosomes (prepared by salt dialysis with 80-bp genomic DNA containing two 5-nt overhangs) were ligated in a sequential head-to-tail fashion with free DNA (90 bp with two 5-nt overhangs) to give poly-prenucleosomes, as indicated in the diagram. The resulting poly-prenucleosomes were assembled into polynucleosomes with ACF and ATP. The formation of canonical nucleosomes was verified by MNase digestion of the polynucleosomes into core particles, which contain ∼147 bp of DNA. (B) ACF-dependent assembly of mono-prenucleosomes to canonical nucleosomes. Mono-prenucleosomes (prepared by NAP1-mediated histone deposition with the central 80 bp of 601 DNA containing two 5-nt overhangs) were ligated to two free 80-bp DNA fragments (each containing a single 5-nt overhang) to give mono-prenucleosomes that are flanked by 85-bp DNA extensions, as illustrated in the diagram. The resulting mono-prenucleosomes were assembled into nucleosomes by ACF. The formation of canonical nucleosomes was assessed by MNase digestion into core particles that contain ∼147 bp of DNA. The 80-bp and 165-bp DNA fragments are incomplete ligation products.
In related experiments, we observed the conversion of mono-prenucleosomes to mononucleosomes by ligation of free DNA tails (85 bp) onto mono-prenucleosomes followed by the addition of ACF (Fig. 4B). As seen with the ligated poly-prenucleosomes (Fig. 4A) as well as prenucleosomes formed by deposition of histones onto relaxed plasmid DNA (Torigoe et al. 2011), the conversion of prenucleosomes to canonical nucleosomes is dependent on the ACF motor protein. These experiments, along with the results in Supplemental Figure S3, A and B, further show that prenucleosomes formed with NAP1 or by salt dialysis can be converted into nucleosomes by ACF and that this process can occur with the 80-bp genomic DNA fragment or the central 80 bp of 601 DNA.
It is particularly notable that the ligation of free DNA to mono-prenucleosomes does not result in the wrapping of the DNA around the histones to give a canonical nucleosome. We hypothesize that the histone octamer is slightly unfolded or expanded in prenucleosomes relative to nucleosomes and that this alternate conformation of the histones in the prenucleosome does not enable facile wrapping of the DNA into a nucleosome. This postulated alternate conformation of the octamer in prenucleosomes could be due to charge repulsion between the histones that occurs in the absence of the extra histone–DNA contacts in canonical nucleosomes.
We also found that the Chd1 motor protein can be used in place of ACF for the conversion of ligated mono-prenucleosomes to nucleosomes (Supplemental Fig. S3C). Hence, mono-prenucleosomes can be converted to nucleosomes in a variety of different conditions. Most importantly, these experiments reveal that mono-prenucleosomes are functionally active as precursors to canonical nucleosomes.
MNase analysis indicates that prenucleosomes contain ∼80 bp of DNA
To complement the results from the psoralen cross-linking and electron microscopy experiments (Fig. 1), we used the mono-prenucleosome system to re-examine the length of DNA that is closely associated with prenucleosomes. For these experiments, we employed MNase as a probe of histone–DNA interactions. In our studies of the ACF-mediated conversion of prenucleosomes to nucleosomes (Fig. 4), we observed that MNase digestion of prenucleosomes with ligated free DNA (in the absence of ACF) yields a set of bands of ∼80 bp. We mapped the ends of these MNase digestion products by primer extension analysis and found that the majority of the DNA fragments ranged from ∼78 to 85 bp in length and correlated with the central 80-bp 601 DNA that was used in the assembly of the mono-prenucleosomes (Fig. 5). The boundaries of MNase digestion of prenucleosomes are not as distinct as those seen with MNase digestion of canonical nucleosomes. Nevertheless, these results provide independent confirmation of the estimates of 70–80 bp of DNA per prenucleosome based on electron microscopy of prenucleosomes in relaxed plasmid DNA (Fig. 1). Collectively, these results indicate that prenucleosomes are closely associated with ∼80 bp of DNA.
Figure 5.

MNase digestion analysis reveals that prenucleosomes are associated with ∼80 bp of DNA. Mono-prenucleosomes were reconstituted by NAP1-mediated deposition onto the central 80 bp of the 601 DNA (with two 5-nt overhangs) and ligated to two free 80-bp DNA fragments (each containing a single 5-nt overhang) to give mono-prenucleosomes that are flanked by 85-bp DNA extensions, as indicated in the diagram. The samples were digested with MNase, and the 5′ ends of the resulting DNA fragments were mapped by primer extension analysis. The 5′ ends of the primers corresponded to the ends of the central 80 bp of the 601 sequences; thus, the majority of the MNase-digested fragments ranged in size from ∼78 to 85 bp. The control lanes show the ends of the unligated (single white dots) and ligated (double white dots) DNA fragments prior to MNase digestion.
We also examined whether the DNA in a prenucleosome is accessible to restriction enzymes. To address this question, we assembled mono-prenucleosomes onto a variant of the 80-bp genomic DNA fragment that contains an EcoRV site (the center of the restriction site is 25/55 bp from the DNA ends) and an XhoI site (the center of the site is 46/34 bp from the ends). The addition of EcoRV or XhoI with mono-prenucleosomes versus naked DNA controls revealed that the packaging of DNA into mono-prenucleosomes blocks the access of the DNA to restriction enzymes (Supplemental Fig. S4), as seen with canonical nucleosomes. This restriction enzyme accessibility assay could be useful in the analysis of the properties of prenucleosomes.
The central region of the DNA in a mono-prenucleosome is located at approximately the same position as the analogous stretch of DNA in a core particle
We next sought to determine the location of the 80 bp of DNA in the mono-prenucleosome. To this end, we employed the histone-directed DNA cleavage method of Flaus et al. (1996) with the use of N-(1,10-phenanthrolin-5-yl)iodoacetamide (OP) as the histone-modifying reagent (Brogaard et al. 2012; Henikoff et al. 2014). In these experiments, we used wild-type core histones as well as histone octamers containing the H4S47C or H2BT87C mutant histones. In particular, it should be noted that H4S47C in a nucleosome is located near the dyad (Flaus et al. 1996). Alkylation of the cysteine sulfhydryl group by OP results in a covalent linkage between an o-phenanthroline moiety and the cysteine residue. In the presence of Cu(II) and hydrogen peroxide, this o-phenanthroline group mediates the generation of hydroxyl radicals that cleave the nearby DNA.
We thus alkylated wild-type and mutant (H4S47C or H2BT87C) histones with OP, reconstituted mono-prenucleosomes with the modified histones and the central 80 bp of 601 DNA, and then examined the cleavage of DNA upon addition of Cu(II) and hydrogen peroxide. These experiments revealed distinct cleavage sites near the middle of the 601 DNA sequence with the modified H4S47C histones but not with the wild-type or H2BT87C histones (Fig. 6). The DNA cleavage was also dependent on the addition of Cu(II). Strikingly, with the H4S47C histones, the sites of DNA cleavage of the central 80-bp 601 fragment in mono-prenucleosomes are identical to those seen in canonical nucleosomes with the full 147-bp 601 DNA fragment (Henikoff et al. 2014); that is, the central 80 bp of 601 sequence in the mono-prenucleosome is located at the position analogous to the same DNA segment in a canonical nucleosome with the full 147-bp 601 DNA. We also observed a related DNA cleavage pattern of the 80-bp genomic DNA with the H4S47C histones (Supplemental Fig. S5), but the results were less distinct, possibly due to some heterogeneity in the interaction of the 80-bp genomic DNA to prenucleosomes relative to that of the well-positioned central 80 bp of 601 DNA. Hence, these findings indicate that the central region of the DNA in a mono-prenucleosome is located at approximately the same position as the analogous stretch of DNA in a core particle.
Figure 6.
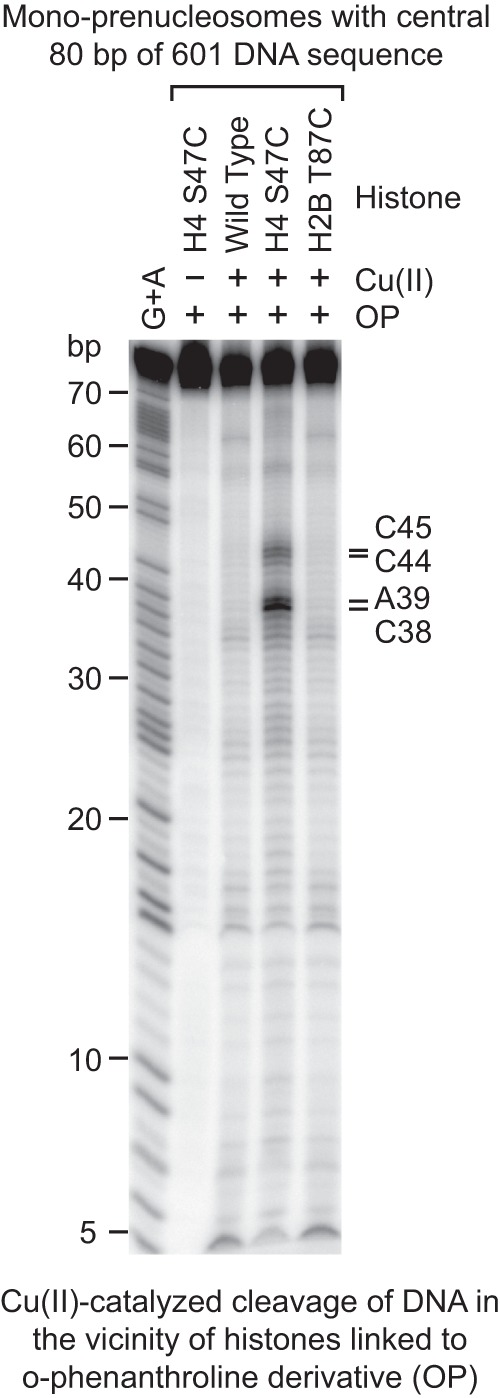
Mapping of the histone–DNA contacts in a mono-prenucleosome. Core histones containing the wild-type or the indicated mutant histones were modified with OP, which links an o-phenanthroline moiety onto the histones via alkylation of the thiol group on cysteine residues. The resulting derivatized histones were reconstituted by salt dialysis into mono-prenucleosomes with the central 80 bp of 601 DNA that is 32P-labeled at the 5′ end. The hydroxyl radical cleavage reactions were initiated by the addition of Cu(II), hydrogen peroxide, and mercaptopropionic acid. The cleavage products were purified and analyzed by electrophoresis on a 10% polyacrylamide–urea gel. The Maxam-Gilbert G+A ladder was used to identify the OP cleavage products, which are indicated at the right side of the autoradiogram. In a canonical nucleosome, H4S47C is located near the dyad.
It is also relevant to note that the H2BT87C residue, which is located at the opposite side of the dyad in a canonical nucleosome (for example, see Ferreira et al. 2007), was included as a probe in case the prenucleosomal DNA were located asymmetrically on the histone octamer. With its symmetric location, the prenucleosomal DNA would be cleaved by the OP-modified H2BT87C residue ∼4–6 nt from the end (if the prenucleosome had the same histone–DNA contacts as a nucleosome) and yield small fragments that could not be clearly resolved. Thus, the absence of distinct cleavage sites with H2BT87C is not definitive but is consistent with the proposed central location of the DNA in the prenucleosome.
p300 specifically acetylates H3K56 in prenucleosomes relative to nucleosomes
We further investigated the properties of prenucleosomes by subjecting prenucleosomes and nucleosomes to acetylation by purified p300 and then analyzing the resulting histones by mass spectrometry. Among the possible candidates for prenucleosome-specific acetylation by p300, H3K56 is the only amino acid residue that was found to exhibit this property (Fig. 7A). Acetylation of H3K56 by p300 occurs with prenucleosomes but not with NAP1–histone complexes or nucleosomes. In addition, the acetylation at H3K56 was confirmed by the parallel analysis of H3K56A, with which acetylation was not detected.
Figure 7.
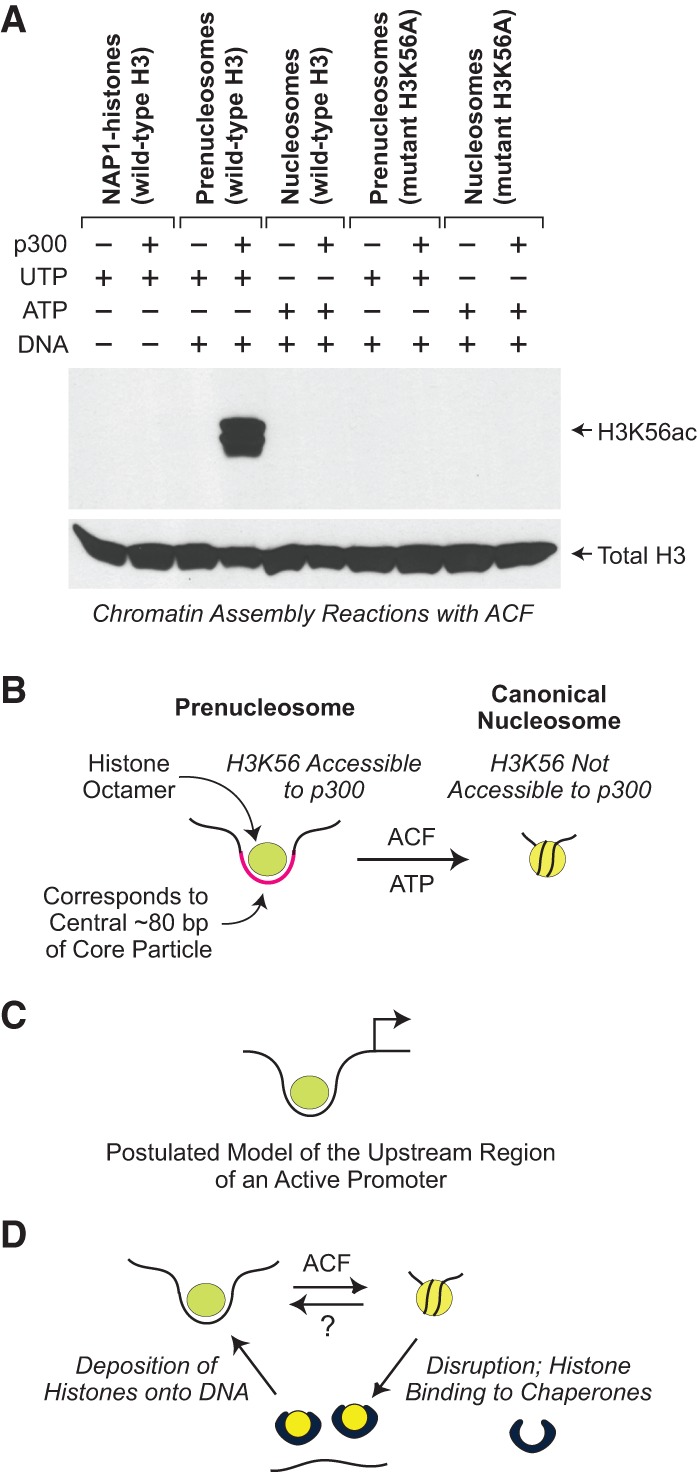
The prenucleosome, a conformational isomer of the nucleosome. (A) p300 specifically acetylates histone H3K56 in prenucleosomes relative to nucleosomes. Chromatin assembly reactions with ACF (Fyodorov and Kadonaga 2003; Torigoe et al. 2011) were performed with relaxed circular plasmid DNA in the presence of acetyl-CoA. ATP (or UTP as the −ATP control), DNA, and p300 were included as indicated. In addition, as a test for acetylation at H3K56, we performed parallel reactions with the mutant histone H3K56A, which cannot be acetylated at H3 residue 56. The resulting samples were then subjected to Western blot analysis with H3K56ac-specific antibodies (Millipore, catalog no. 07-677). As a reference, the blot was stripped and reprobed with anti-total H3 antibodies (Abcam, catalog no. AB1791). (B) Prenucleosomes comprise a core histone octamer and 80 bp of DNA at a location that is analogous to that of the central 80 bp of the core particle. H3K56 is accessible to p300 in a prenucleosome but not a nucleosome. Prenucleosomes can be converted into canonical nucleosomes by a motor protein such as ACF or Chd1. (C) Prenucleosomes or prenucleosome-related particles may be present in the upstream region of active promoters. (D) Model for the productive dynamic interconversion between prenucleosomes and nucleosomes. Prenucleosomes can be formed by the deposition of histones onto DNA and converted into nucleosomes by an ATP-driven motor protein such as ACF or Chd1. Nucleosomes can be disrupted by the action of enzymes such as polymerases as well as some ATP-driven chromatin remodeling factors. The resulting free histones are bound by the chaperones and then reassembled into prenucleosomes. It is not known whether a canonical nucleosome can be directly converted into a prenucleosome.
In metazoans, H3K56 is acetylated by CBP/p300 proteins (Das et al. 2009). H3K56 is located at the DNA entry and exit points of the nucleosome (for example, see Masumoto et al. 2005; Xu et al. 2005). Hence, the greater accessibility of p300 to H3K56 in prenucleosomes relative to nucleosomes is consistent with the location of prenucleosomal DNA in the region that corresponds to the central 80 bp of the nucleosome (Fig. 6).
H3K56ac has been found to be involved in chromatin assembly during DNA replication and repair (for example, see Masumoto et al. 2005; Han et al. 2007; Chen et al. 2008; Li et al. 2008). In addition, genome-wide chromatin immunoprecipitation (ChIP) experiments in yeast, Drosophila, and human cells have shown that H3K56ac is highly enriched at active promoters as well as enhancers (for example, see Lo et al. 2011; Venkatesh et al. 2012; Skalska et al. 2015). Moreover, in Drosophila, the increase in H3K56 acetylation at promoters and enhancers by Notch activation was found to occur rapidly by a mechanism that requires CBP acetyltransferase activity but not transcriptional elongation (Skalska et al. 2015). These findings suggest that pre-existing H3K56 can be acetylated by CBP.
Thus, the specific acetylation of H3K56 in prenucleosomes relative to nucleosomes provides another link between the properties of prenucleosomes and dynamic chromatin in cells. It is also possible that the presence of H3K56ac at promoters and enhancers may reflect the occurrence of prenucleosomes or prenucleosome-like particles. In the future, it should be interesting and informative to examine the relation between H3K56 acetylation and prenucleosome function in greater detail.
Discussion
The prenucleosome is a stable conformational isomer of the nucleosome
The prenucleosome was initially identified as a nonnucleosomal histone–DNA complex that is a precursor to the nucleosome in the assembly of chromatin in vitro (Torigoe et al. 2011). However, the ability to study prenucleosomes was limited by the heterogeneity of the prenucleosome samples that were assembled onto plasmid DNA templates. Hence, we lacked a fundamental understanding of the composition, structure, and organization of prenucleosomes.
In this study, we observed by psoralen cross-linking and electron microscopy that prenucleosomes appear to be associated with ∼70–80 bp of DNA (Fig. 1). This finding led to the development of the mono-prenucleosome system that involves the assembly of mono-prenucleosomes from the four core histones and an 80-bp DNA fragment (Fig. 2). Prenucleosomes contain a histone octamer and are distinct from species such as tetrasomes or hexasomes that contain less than a complete octamer (Figs. 2, 3). Moreover, mono-tetrasomes can be converted into mono-prenucleosomes with no apparent accumulation of stable hexasome species (Fig. 2D). It is particularly notable that mono-prenucleosomes can be formed by the deposition of histones by NAP1 or dNLP as well as salt dialysis (Fig. 2C). Importantly, mono-prenucleosomes are functionally active, as they can be ligated to free (naked) DNA and then converted into canonical nucleosomes by a motor protein such as ACF or Chd1 (Fig. 4; Supplemental Fig. S3).
By using histone-directed DNA cleavage methodology (Flaus et al. 1996; Brogaard et al. 2012; Henikoff et al. 2014), we mapped the location of the 80 bp of DNA relative to the histone octamer in mono-prenucleosomes (Fig. 6; Supplemental Fig. S5). These experiments revealed that the central region of the 80 bp of DNA in a mono-prenucleosome is at the location analogous to the central region of the DNA near the dyad in a canonical nucleosome. We additionally mapped the ends of the DNA fragments generated by MNase digestion of prenucleosomes and found that the amount of DNA that is closely associated with prenucleosomes is ∼80 bp (Fig. 5). This is similar to the prenucleosomal DNA length that was independently estimated by psoralen cross-linking and electron microscopy (Fig. 1). The partial wrapping of DNA relative to that in a nucleosome is likely to be responsible for the lack of DNA supercoiling by prenucleosomes as well as the ability of p300 to acetylate H3K56 in prenucleosomes but not in canonical nucleosomes (Fig. 7A).
These findings indicate that the prenucleosome is a stable conformational isomer of the nucleosome (Fig. 7B). Moreover, no other histone–DNA particle was observed to be formed as efficiently and rapidly as prenucleosomes in the presence of the four core histones. Because there are probably only a small number of stable alternate conformations of the nucleosome, prenucleosomes may share a common fundamental structure with native nonnucleosomal particles such as those present at active chromatin throughout the genome.
As briefly discussed above, it is interesting to note that mono-prenucleosomes that are ligated to free DNA do not fold into canonical nucleosomes. The prenucleosomal histones appear to be in a different conformation than the histone octamer in a canonical nucleosome, possibly due to charge repulsion between the histones because of the reduced histone–DNA contacts in prenucleosomes relative to nucleosomes.
Based on the ability of prenucleosomes to be formed rapidly and then converted into canonical nucleosomes by a motor protein such as ACF or Chd1, we imagine that prenucleosomes are generated and assembled into nucleosomes during processes in which nucleosomes are disrupted, such as DNA replication, transcription, and repair. For instance, prenucleosomes have the same properties as histone–DNA complexes (in which H2B and H3 can be detected) at DNA replication forks that resemble nucleosomes but are formed much more rapidly than canonical nucleosomes (for example, see McKnight and Miller 1977; McKnight et al. 1978; Worcel et al. 1978). In addition, some factors might be able to convert canonical nucleosomes directly into prenucleosomes, but such activities have not yet been identified.
Prenucleosomes appear to be related to nonnucleosomal histone–DNA complexes at active promoters
In our psoralen cross-linking and electron microscopy analysis of prenucleosomes versus nucleosomes, we observed a striking similarity between the distributions of psoralen bubble sizes with prenucleosomes versus nucleosomes compared with those obtained with active versus repressed promoters in vivo in yeast (Fig. 1C). These findings suggest that the “nucleosome-depleted regions” (NDRs; also termed “nucleosome-free regions” [NFRs]) that are located immediately upstream of the transcription start site of active genes contain prenucleosomes or prenucleosome-like particles (Fig. 7C).
In further support of this hypothesis, methidiumpropyl-EDTA sequencing (MPE-seq) analyses in mouse embryonic stem cells revealed that subnucleosome-sized chromatin fragments (including those containing 50–100 bp of DNA) are located specifically in the upstream promoter region of active genes (Ishii et al. 2015). Notably, these noncanonical chromatin particles contain histones H2A and H3 by ChIP-seq (ChIP combined with deep sequencing) analysis. Moreover, the degree of enrichment of the subnucleosome-sized, histone-containing particles in the upstream promoter region correlates with the level of gene transcription, as assessed by RNA sequencing (RNA-seq) (Ishii et al. 2015). Hence, these 50- to 100-bp-sized histone-containing particles that are present at active promoters have features that are similar to those of prenucleosomes.
In addition, the ability of prenucleosomes, but not nucleosomes, to be acetylated at H3K56 by p300 (Fig. 7A) may be responsible, at least in part, for the observed enrichment of H3K56ac at active promoters and enhancers (for example, see Lo et al. 2011; Venkatesh et al. 2012; Skalska et al. 2015). p300 is associated with transcriptional enhancers (for example, see Heintzman et al. 2007; Visel et al. 2009), and it is possible that the enrichment of H3K56ac at enhancers is due to p300-mediated acetylation of prenucleosomes. In promoter regions, the averaged peak of H3K56 acetylation has been observed to flank the NDR at about −250 bp or about +250 bp relative to the +1 transcription start site. The absence of an H3K56ac peak precisely at the NDR could be due to the increased fragmentation of the sensitive DNA at the NDR (relative to the DNA in bulk chromatin) during the sonication of the chromatin. To clarify this issue, the generation of chromatin fragments for H3K56ac ChIP might optimally be performed with mild DNA cleaving reagents such as methidiumpropyl-EDTA-Fe(II) [MPE-Fe(II)] or low concentrations of MNase as in Ishii et al. (2015).
Prenucleosomes may also be related to “fragile” nucleosomes, which are MNase-sensitive nucleosomes that have been seen in yeast promoters (for example, see Weiner et al. 2010; Xi et al. 2011; Knight et al. 2014; Kubik et al. 2015). In addition, in HeLa (human) cells, salt-labile nucleosomes containing histones H2A.Z and H3.3 have been found at active promoters (Jin et al. 2009). Thus, the NDRs of active promoters appear to contain prenucleosomes or prenucleosome-related species (Fig. 7C).
Why might prenucleosomes or prenucleosome-like particles be present in the NDRs of active promoters? Because prenucleosomes interact with only ∼80 bp of DNA, they would be more easily altered or disrupted than canonical nucleosomes. Also, if disrupted or displaced, prenucleosomes could be rapidly reassembled. In these respects, prenucleosomes appear to be compatible with the function of transcription factors. An alternate but related viewpoint is that prenucleosomes are intermediates in the dynamic process of nucleosome disassembly and reassembly at active promoters (Fig. 7D; see also Brown et al. 2013). It is even possible that prenucleosomes enhance transcription, such as in the establishment of the optimal structure of the active promoter. This notion is supported by the observation that the intensities of the H2B and H3 ChIP signals associated with the subnucleosome-sized DNA fragments correlate with transcriptional activity as measured by RNA-seq (Ishii et al. 2015). In the future, it will be interesting and important to investigate the potential role of prenucleosomes or prenucleosome-like structures at active promoters.
Conclusion and perspective
Chromatin in the eukaryotic nucleus is multidimensional. There are covalent modifications of the histones, histone variants, ATP-driven chromatin remodeling factors, nonhistone chromosomal proteins, and nonnucleosomal chromatin particles. Notably, each of these dimensions of chromatin affects gene expression. The prenucleosome is the only known stable conformer of the nucleosome and the only distinct nonnucleosomal histone–DNA particle that has been observed to be rapidly and efficiently formed on DNA in the presence of the four core histones, as in the nucleus. (For instance, as seen in Figure 2, C and D, H3–H4 tetrasomes are inefficiently formed and rapidly converted into prenucleosomes in the presence of H2A–H2B.) It thus seems likely that many nonnucleosomal particles in the cell are prenucleosomes or prenucleosome-related particles. An additional attractive feature of this hypothesis is the ability of prenucleosomes to be converted into nucleosomes by ATP-dependent motor proteins such as ACF or Chd1. Hence, in this model, there is a productive dynamic interconversion between prenucleosomes and nucleosomes (Fig. 7D). It is our hope that, in the future, the new knowledge of nonnucleosomal components of chromatin will contribute to an integrated understanding of the dynamic structure and function of our genome.
Materials and methods
Reagents and methodology
DNA oligonucleotides were synthesized by Integrated DNA Technologies. The sequences of the oligonucleotides are in Supplemental Table S1. D. melanogaster NAP1 and ACF complexes were synthesized in Sf9 cells by using baculovirus expression vectors and purified as described previously (Fyodorov and Kadonaga 2003). D. melanogaster dNLP was synthesized in Escherichia coli and purified by the method of Ito et al. (1996). D. melanogaster Chd1 was synthesized in E. coli and purified as described (Torigoe et al. 2013). Native core histones were purified from D. melanogaster embryos that were collected from 0 to 12 h after egg deposition (Fyodorov and Levenstein 2002). Recombinant D. melanogaster histones were synthesized in E. coli and purified by the method of Luger et al. (1999). Human p300 protein was purified and used as described by Kraus and Kadonaga (1998). All experiments were performed independently at least twice to ensure the reproducibility of the data.
Reconstitution of mono-prenucleosomes
Mono-prenucleosomes were prepared by either histone chaperone-mediated deposition or salt dialysis. In chaperone-mediated reconstitution reactions, the core histones were incubated with either NAP1 (at a 5:1 mass ratio of NAP1:core histones) or dNLP (at a 10:1 mass ratio of dNLP:core histones) for 20 min on ice in a volume of 10 µL in the following buffer: 10 mM K-HEPES (pH 7.6), 0.1 M KCl, 0.1 mM EDTA, 1 mM DTT, and 70 µg/mL bovine serum albumin. Eight microliters of HEG buffer (25 mM K-HEPES at pH 7.6, 0.1 M KCl, 0.1 mM EDTA, 10% [v/v] glycerol) was then added. Next, 2 µL of the preannealed DNA oligonucleotides (in 10 mM Tris-HCl at pH 7.5, 0.1 mM EDTA, 0.05 M NaCl) was added to the histone chaperone mixture to give a final volume of 20 µL. The samples were mixed immediately by gentle vortexing, incubated for 30 sec, and analyzed by native 5% polyacrylamide gel electrophoresis.
Salt dialysis reconstitution of mononucleosomes, mono-prenucleosomes, and mono-tetrasomes was performed by the method of Stein (1989). In a standard reaction, the DNA fragment (as indicated for each experiment) (50 pmol; 6 µL in TE buffer: 10 mM Tris-HCl at pH 7.5, 1 mM EDTA) was combined with 140 µL of TE buffer containing 1.07 M NaCl and 0.011% (v/v) NP-40, and core histones (50 pmol; 4 µL in 10 mM K-HEPES at pH 7.6, 0.1 M KCl, 1 mM DTT, 10% [v/v] glycerol) were added to give final concentrations of 1.0 M NaCl and 0.01% (v/v) NP-40 in a total volume of 150 µL. The resulting histone–DNA mixture was subjected to dialysis at room temperature in a ThermoFisher Slide-A-Lyzer dialysis cassette (molecular weight cutoff of 3500 Da) for 2 h against TE containing 0.8 M NaCl for 1.5 h against TE containing 0.6 M NaCl and 2 h against TE containing 0.05 M NaCl. The resulting products were analyzed by native 5% polyacrylamide gel electrophoresis. Mono-prenucleosomes that were reconstituted by salt dialysis were stable for at least a few weeks at 4°C. For storage, prenucleosomes were dialyzed overnight at 4°C against histone storage buffer (10 mM K-HEPES at pH 7.6, 0.1 mM EDTA, 0.1 M KCl, 10% [v/v] glycerol, 1 mM DTT).
Ligation of mono-prenucleosomes to free DNA and assembly into canonical nucleosomes by ACF
Mono-prenucleosomes were ligated to free (naked) DNA by using methods similar to those employed for the ligation of nucleosomes to free DNA (Clark and Felsenfeld 1992; Stein et al. 2002). Typically, mono-prenucleosomes (85 pmol of DNA with sticky ends) and the flanking DNA strands (85 pmol each, and each with a single sticky end that is complementary to one end of the prenucleosomal DNA) were combined in 10 mM K-HEPES (pH 8.0), 1 mM EGTA, 1 mM DTT, 0.5 mM ATP, an ATP regeneration system (3 mM phosphoenolpyruvate, 20 U/µL pyruvate kinase), 2.5 mM MgCl2, 12.5 µg of bovine serum albumin, and 3000 U of T4 DNA ligase (New England Biolabs) in a volume of 138 µL. HEG (82 µL) was added to give a final volume of 220 µL, and the ligation was carried out overnight at 16°C. In the assembly of prenucleosomes into nucleosomes, the ligated prenucleosomes (20 pmol; 52 µL directly from the ligation reaction) were combined with a solution of 20 mM ATP and 5 µL of 33 mM MgCl2, 1.0 µL of 700 nM ACF (to a final ACF concentration of 6 nM), and 17 µL of HEG buffer to a final volume of 70 µL. The reaction was carried out for 1.5 h at 27°C. The samples were analyzed by MNase digestion as described by Torigoe et al. (2011).
Trimethylpsoralen cross-linking
Cross-linking was performed essentially as described (Sogo and Thoma 1989; Brown et al. 2015). The specific conditions were as follows: A 175-µL sample of chromatin assembly reaction (Fyodorov and Kadonaga 2003; Torigoe et al. 2011) was diluted with 125 µL of dilution buffer (15 mM K-HEPES at pH 7.6, 100 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 6.6% [v/v] glycerol, 1% [w/v] polyvinyl alcohol [average molecular weight 10,000], 1% [w/v] polyethylene glycol 8000, 20 µg/mL bovine serum albumin) and transferred to a single well of a 24-well plate. The 24-well plate containing all samples was placed on an ice-water slurry and positioned 5 cm away from five 366-nm 15 W UV bulbs in a Stratalinker 2400 (Stratagene). Seven rounds of the following steps were performed: (1) addition of 15 µL of 400 μg/mL 4,5′,8-trimethylpsoralen (Sigma) in 100% ethanol, (2) incubation in the dark for 5 min on ice, and (3) irradiation by UV for 5 min. Following cross-linking, 5 µL of 20 mg/mL proteinase K and 20 µL of 10% (w/v) SDS were added, and the samples were incubated for 1 h at 55°C. DNA was extracted with phenol–chloroform and precipitated. DNA was resuspended, digested with EcoRI, purified by using a DNA clean and concentrator kit (Zymo Research), and eluted from the column with 8 µL of TEN (30 mM tetraethylammonium chloride, 20 mM EDTA, 10 mM NaCl). Maximal cross-linking was verified with naked, relaxed DNA.
Sample preparation for electron microscopy
DNA (1–3 µL in TEN) was denatured in 70% (v/v) deionized formamide (Sigma) and 0.5 M glyoxal (Sigma) in a total volume of 13 µL for 30 min at 37°C. Samples were immediately placed on ice and diluted with 5 µL of TEN. Benzalkonium chloride (Sigma) was added to 0.001% (w/v) to facilitate spreading of the DNA. A 10-cm petri dish with a mica ramp resting on the rim was filled with distilled water. Graphite powder was lightly dusted on the surface of the water. A portion (5 μL) of the denatured DNA sample was run down the mica ramp and spread on top of the water, which pushed back the graphite dusting. DNA near the graphite border or mica ramp was picked up by using carbon-coated copper grids (Electron Microscopy Sciences) pretreated with 30 µg/mL ethidium bromide. Grids were stained with 0.5 mM uranyl acetate, washed with 100% ethanol, and air-dried. Following staining, they were secured to a rotary tilt stage in a 208C high-vacuum turbo carbon coater equipped with a metal evaporation accessory (Cressington) and shadowed at an angle of 3° with platinum:palladium (80:20; Electron Microscopy Sciences) until a thickness monitor (Cressington, MTM-10) reported 100 nm of metal deposition on the sensor.
Electron microscopy
Images were taken on a JEOL 1230 electron microscope at 120 keV at 20,000× magnification and were processed and analyzed in ImageJ. Chromatin assembly reactions were performed four separate times, and totals of 380 and 376 molecules were analyzed from chromatin assembly reactions without and with the addition of ACF, respectively.
Additional Materials and Methods are included in the Supplemental Material.
Supplementary Material
Acknowledgments
This work is dedicated to Professor E. Peter Geiduschek in recognition of his many fundamental and important contributions to molecular biology. We thank E. Peter Geiduschek, Russell F. Doolittle, Long Vo Ngoc, and Yuan Wang for critical reading of the manuscript. We are grateful to Kristin Brogaard and Ji-Ping Wang for their advice on the use of OP. We also thank Arnold Stein for his advice on the ligation of mono-prenucleosomes to free DNA. J.T.K. is the Amylin Chair in the Life Sciences. This work was supported by grants from the National Science Foundation (1243957) to H.B. and from the National Institute of General Medical Sciences/National Institutes of Health (R01 GM058272) to J.T.K.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.272633.115.
References
- Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H. 2012. Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry 51: 3302–3309. [DOI] [PubMed] [Google Scholar]
- Bouazoune K, Kingston RE. 2013. Assembly, remodelled. eLife 2: e01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang JP, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Mao C, Falkovskaia E, Jurica MS, Boeger H. 2013. Linking stochastic fluctuations in chromatin structure and gene expression. PLoS Biol 11: e1001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Eskin JA, Hamperl S, Griesenbeck J, Jurica MS, Boeger H. 2015. Chromatin structure analysis of single gene molecules by psoralen cross-linking and electron microscopy. Methods Mol Biol 1228: 93–121. [DOI] [PubMed] [Google Scholar]
- Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. 2008. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134: 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DJ, Felsenfeld G. 1992. A nucleosome core is transferred out of the path of a transcribing polymerase. Cell 71: 11–22. [DOI] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, Tyler JK. 2009. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira H, Somers J, Webster R, Flaus A, Owen-Hughes T. 2007. Histone tails and the H3 αN helix regulate nucleosome mobility and stability. Mol Cell Biol 27: 4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus A, Luger K, Tan S, Richmond TJ. 1996. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci 93: 1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyodorov DV, Kadonaga JT. 2003. Chromatin assembly in vitro with purified recombinant ACF and NAP-1. Methods Enzymol 371: 499–515. [DOI] [PubMed] [Google Scholar]
- Fyodorov DV, Levenstein ME. 2002. Chromatin assembly using Drosophila systems. Curr Protoc Mol Biol 21: 21.7.1–21.7.27. [DOI] [PubMed] [Google Scholar]
- Germond JE, Bellard M, Oudet P, Chambon P. 1976. Stability of nucleosomes in native and reconstituted chromatins. Nucleic Acids Res 3: 3173–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. 2007. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315: 653–655. [DOI] [PubMed] [Google Scholar]
- Hanson CV, Shen CK, Hearst JE. 1976. Cross-linking of DNA in situ as a probe for chromatin structure. Science 193: 62–64. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ramachandran S, Krassovsky K, Bryson TD, Codomo CA, Brogaard K, Widom J, Wang JP, Henikoff JG. 2014. The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. eLife 3: e01861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Kadonaga JT, Ren B. 2015. MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc Natl Acad Sci 112: E3457–E3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Tyler JK, Bulger M, Kobayashi R, Kadonaga JT. 1996. ATP-facilitated chromatin assembly with a nucleoplasmin-like protein from Drosophila melanogaster. J Biol Chem 271: 25041–25048. [DOI] [PubMed] [Google Scholar]
- Jackson V, Chalkley R. 1981. A new method for the isolation of replicative chromatin: selective deposition of histone on both new and old DNA. Cell 23: 121–134. [DOI] [PubMed] [Google Scholar]
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. 2009. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet 41: 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempnauer KH, Fanning E, Otto B, Knippers R. 1980. Maturation of newly replicated chromatin of simian virus 40 and its host cell. J Mol Biol 136: 359–374. [DOI] [PubMed] [Google Scholar]
- Knight B, Kubik S, Ghosh B, Bruzzone MJ, Geertz M, Martin V, Dénervaud N, Jacquet P, Ozkan B, Rougemont J, et al. 2014. Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev 28: 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, Kadonaga JT. 1998. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev 12: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone JL, Rougemont J, Shore D. 2015. Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol Cell 60: 422–434. [DOI] [PubMed] [Google Scholar]
- Levy A, Jakob KM. 1978. Nascent DNA in nucleosome-like structures from chromatin. Cell 14: 259–267. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. 2008. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 134: 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KA, Bauchmann MK, Baumann AP, Donahue CJ, Thiede MA, Hayes LS, des Etages SA, Fraenkel E. 2011. Genome-wide profiling of H3K56 acetylation and transcription factor binding sites in human adipocytes. PLoS One 6: e19778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276: 19–42. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ. 1999. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol 119: 1–16. [DOI] [PubMed] [Google Scholar]
- Masumoto H, Hawke D, Kobayashi R, Verreault A. 2005. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436: 294–298. [DOI] [PubMed] [Google Scholar]
- McKnight SL, Miller OL Jr. 1977. Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell 12: 795–804. [DOI] [PubMed] [Google Scholar]
- McKnight SL, Bustin M, Miller OL Jr. 1978. Electron microscopic analysis of chromosome metabolism in the Drosophila melanogaster embryo. Cold Spring Harbor Symp Quant Biol 42: 741–754. [DOI] [PubMed] [Google Scholar]
- Schlaeger EJ, Knippers R. 1979. DNA-histone interaction in the vicinity of replication points. Nucleic Acids Res 6: 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale RL. 1975. Assembly of DNA and protein during replication in HeLa cells. Nature 255: 247–249. [DOI] [PubMed] [Google Scholar]
- Seale RL. 1976. Studies on the mode of segregation of histone nu bodies during replication in HeLa cells. Cell 9: 423–429. [DOI] [PubMed] [Google Scholar]
- Skalska L, Stojnic R, Li J, Fischer B, Cerda-Moya G, Sakai H, Tajbakhsh S, Russell S, Adryan B, Bray SJ. 2015. Chromatin signatures at Notch-regulated enhancers reveal large-scale changes in H3K56ac upon activation. EMBO J 34: 1889–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Thoma F. 1989. Electron microscopy of chromatin. Methods Enzymol 170: 142–165. [DOI] [PubMed] [Google Scholar]
- Sogo JM, Stahl H, Koller T, Knippers R. 1986. Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J Mol Biol 189: 189–204. [DOI] [PubMed] [Google Scholar]
- Stein A. 1989. Reconstitution of chromatin from purified components. Methods Enzymol 170: 585–603. [DOI] [PubMed] [Google Scholar]
- Stein A, Dalal Y, Fleury TJ. 2002. Circle ligation of in vitro assembled chromatin indicates a highly flexible structure. Nucleic Acids Res 20: 5103–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torigoe SE, Urwin DL, Ishii H, Smith DE, Kadonaga JT. 2011. Identification of a rapidly formed nonnucleosomal histone-DNA intermediate that is converted into chromatin by ACF. Mol Cell 43: 638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torigoe SE, Patel A, Khuong MT, Bowman GD, Kadonaga JT. 2013. ATP-dependent chromatin assembly is functionally distinct from chromatin remodeling. eLife 2: e00863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. 2012. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489: 452–455. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. 2010. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20: 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worcel A, Han S, Wong ML. 1978. Assembly of newly replication chromatin. Cell 15: 969–977. [DOI] [PubMed] [Google Scholar]
- Xi Y, Yao J, Chen R, Li W, He X. 2011. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res 21: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang K, Grunstein M. 2005. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121: 375–385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.