

In this study, Absmeier et al. used a combination of X-ray crystallography, cross-linking/mass spectrometry, and in vivo and in vitro biochemical functional investigations to investigate the structural organization, functions, and molecular mechanisms of the NTR of the Brr2 helicase. The findings reveal molecular mechanisms that prevent premature and unproductive tri-snRNP disruption and suggest novel regulation of Brr2-dependent splicing.
Keywords: pre-mRNA splicing, RNA helicase structure and function, remodeling of RNA–protein complexes, spliceosome catalytic activation, X-ray crystallography
Abstract
The Brr2 helicase provides the key remodeling activity for spliceosome catalytic activation, during which it disrupts the U4/U6 di-snRNP (small nuclear RNA protein), and its activity has to be tightly regulated. Brr2 exhibits an unusual architecture, including an ∼500-residue N-terminal region, whose functions and molecular mechanisms are presently unknown, followed by a tandem array of structurally similar helicase units (cassettes), only the first of which is catalytically active. Here, we show by crystal structure analysis of full-length Brr2 in complex with a regulatory Jab1/MPN domain of the Prp8 protein and by cross-linking/mass spectrometry of isolated Brr2 that the Brr2 N-terminal region encompasses two folded domains and adjacent linear elements that clamp and interconnect the helicase cassettes. Stepwise N-terminal truncations led to yeast growth and splicing defects, reduced Brr2 association with U4/U6•U5 tri-snRNPs, and increased ATP-dependent disruption of the tri-snRNP, yielding U4/U6 di-snRNP and U5 snRNP. Trends in the RNA-binding, ATPase, and helicase activities of the Brr2 truncation variants are fully rationalized by the crystal structure, demonstrating that the N-terminal region autoinhibits Brr2 via substrate competition and conformational clamping. Our results reveal molecular mechanisms that prevent premature and unproductive tri-snRNP disruption and suggest novel principles of Brr2-dependent splicing regulation.
Splicing entails the removal of noncoding sequences (introns) from primary transcripts and the concomitant ligation of neighboring coding regions (exons). It is mediated by a highly dynamic, multimegadalton RNA protein (RNP) molecular machine, the spliceosome, which consists of five small nuclear RNPs (snRNPs; U1, U2, U4, U5, and U6 in the case of the major spliceosome) and numerous non-snRNPs (Wahl et al. 2009). For each round of splicing, a spliceosome is assembled de novo on a substrate by the stepwise recruitment of snRNPs and non-snRNPs. After assembly of a precatalytic complex, the spliceosome is catalytically activated and carries out the two consecutive steps of a splicing reaction before it is disassembled and its subunits are recycled. Each assembly, activation, catalysis, and disassembly step involves profound rearrangements of the spliceosomal RNP interaction networks, mediated predominantly by eight conserved superfamily 2 (SF2) NTPases/RNA helicases (Staley and Guthrie 1998). The most extensive rearrangements occur during spliceosome activation. In the precatalytic spliceosome, U4 and U6 snRNPs form a di-snRNP by base-pairing of their snRNAs and are associated with U5 snRNP via protein–protein interactions. During spliceosome activation, the U5 snRNP-specific Brr2 helicase unwinds the U4/U6 di-snRNAs (Noble and Guthrie 1996; Laggerbauer et al. 1998; Raghunathan and Guthrie 1998; Kim and Rossi 1999), leading to displacement of U4 and U4/U6-associated proteins and allowing U6 to engage in alternative interactions with the substrate and U2 snRNA as well as form an internal stem–loop that is an essential element in the spliceosome's active site.
Structurally, Brr2 differs decisively from other spliceosomal helicases. It belongs to the Ski2-like subfamily of SF2 helicases and consists of an ∼500-residue N-terminal region (NTR) (Supplemental Fig. 1) of unknown fold and function followed by two structurally similar helicase cassettes. Each cassette contains dual RecA-like domains, a winged helix (WH) domain, and a Sec63 homology unit comprising a helical bundle (HB), a helix–loop–helix (HLH), and an immunoglobulin-like (IG) domain (Santos et al. 2012). During spliceosome activation, Brr2 engages a single-stranded region of U4 snRNA at a tunnel formed by its N-terminal RecA, WH, and HB domains and translocates on this RNA strand in an ATP-dependent manner to dissociate U4 from U6 (Hahn et al. 2012; Mozaffari-Jovin et al. 2012; Santos et al. 2012; Nguyen et al. 2015).
Brr2 already encounters its U4/U6 substrate outside the spliceosome in a preformed U4/U6•U5 tri-snRNP and remains associated, presumably in an inactive state (Hahn et al. 2012; Fourmann et al. 2013), with the spliceosome after catalytic activation. In a cryo-electron microscopic (cryo-EM) structure of a yeast tri-snRNP, Brr2 was modeled in a conformation ready for U4/U6 dissociation, and, indeed, the particles disintegrated upon treatment with 1 mM ATP (Nguyen et al. 2015) as also previously seen with partially purified yeast tri-snRNP (Cheng and Abelson 1987; Raghunathan and Guthrie 1998). Therefore, molecular mechanisms must exist to prevent Brr2 from prematurely unwinding the U4/U6 duplex in the tri-snRNP and during initial stages of spliceosome assembly. Furthermore, in a recent cryo-EM structure of a spliceosome that underwent the first step of splicing, Brr2 could not be modeled due to its flexible anchoring (Yan et al. 2015). Again, it is unclear how Brr2 at this later stage of splicing is prevented from binding and remodeling nontarget RNAs/RNPs.
Here, we investigated the idea that the Brr2 NTR might be crucial for regulating the enzyme's helicase activity at different stages of spliceosome assembly and splicing. Helicases often harbor accessory domains that can modulate their functions in diverse ways (Johnson and Jackson 2013), but only a few helicase structures are known that encompass the helicase core and an accessory domain. The Brr2 NTR is significantly longer than corresponding elements in most other helicases, is evolutionarily conserved, and is predicted to contain large intrinsically unstructured regions (Supplemental Fig. 1A). Using systematic structural and functional analyses, we elucidated the molecular organization of the NTR in the context of the helicase cassettes and showed that the NTR is required for efficient Brr2 binding to the U4/U6•U5 tri-snRNP, for tri-snRNP homeostasis, and for splicing. We also demonstrate that the NTR autoinhibits Brr2 on multiple levels and by multiple mechanisms, fully in line with its structural organization. Our results uncover unique molecular mechanisms of how Brr2 is regulated during splicing.
Results
The Brr2 NTR interlocks the two helicase cassettes
To elucidate how the NTR is positioned with respect to the helicase cassettes of Brr2, we determined a crystal structure of full-length (FL) yeast Brr2 in complex with a Jab1/MPN-like (Jab1) domain of the Prp8 protein (lacking the last 15 C-terminal residues) (Fig. 1A–C; Table 1), a known regulator of Brr2 (Maeder et al. 2009; Mozaffari-Jovin et al. 2013, 2014; Nguyen et al. 2013). During refinement, we used all diffraction data collected up to 2.8 Å resolution (Table 1). The helicase region and Jab1 domain of the present complex resemble the corresponding elements in previously solved structures of truncated yeast (Nguyen et al. 2013) and human (Mozaffari-Jovin et al. 2013) Brr2–Jab1 complexes, with the Jab1 domain resting on top of the N-terminal Sec63 unit (Fig. 1C). Additionally, the present structure reveals that the NTR runs along one entire flank of the helicase region to an extent that is unprecedented in previously analyzed helicase structures (Fig. 1C). Based on the portions visible in the electron density (Supplemental Fig. 1B), the NTR can be divided into four sections (Fig. 1B,C). Residues 113–192 form a domain comprised of two roughly perpendicular helical hairpins that is wedged into a gap between the RecA2 and HB domains of the N-terminal cassette and that we refer to as the “plug.” Residues 258–273, which we term the “intercassette (IC) clamp,” adopt an extended conformation with a central 90° bend and laterally tape the helicase cassettes together. Residues 284–393 form a PWI domain (Absmeier et al. 2015) that is connected to neighboring elements by flexible linkers and is held above the IC clamp by crystal packing contacts. Finally, residues 420–474 embrace the N-terminal cassette, resembling an “N-terminal cassette (NC) clamp.” The C-terminal two-thirds of the NC clamp have been seen before in structures of N-terminally truncated yeast (Nguyen et al. 2013) and human (Santos et al. 2012) Brr2.
Figure 1.

Structure of a FL Brr2–Jab1 complex. (A) Simulated annealing composite omit map covering the NTRs (magenta), N-terminal cassette (NC; gray), C-terminal cassette (CC; brown), and Jab1 (gold) in the FL Brr2–Jab1 crystal structure contoured at the 1.0 σ level. Molecular models are shown as ribbons. (B) Scheme of the NTR organization. Numbers above the scheme provide the domain borders, and angled arrows and numbers below the scheme indicate the starting positions of the NTR truncation variants of yeast Brr2. (C) Orthogonal views of the FL Brr2–Jab1 complex structure showing a ribbon of the NTR on the surface of the helicase cassettes and Jab1. The NTR is colored blue to red from the N terminus to the C terminus, the N-terminal cassette is colored dark gray, the C-terminal cassette is beige, and Jab1 is light gray. (D) Close-up views of the plug and IC clamp/PWI region of the NTR (magenta) showing cross-links identified in isolated Brr2. (Solid lines) Zero-length cross-links observed with 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM); (dashed lines) cross-links observed with bis(sulfosuccinimidyl) suberate (BS3). The image is rotated 10° to the left about the vertical axis (top panel) and 45° to the top about the horizontal axis (bottom panel) compared with the top panel in B.
Table 1.
Crystallographic data
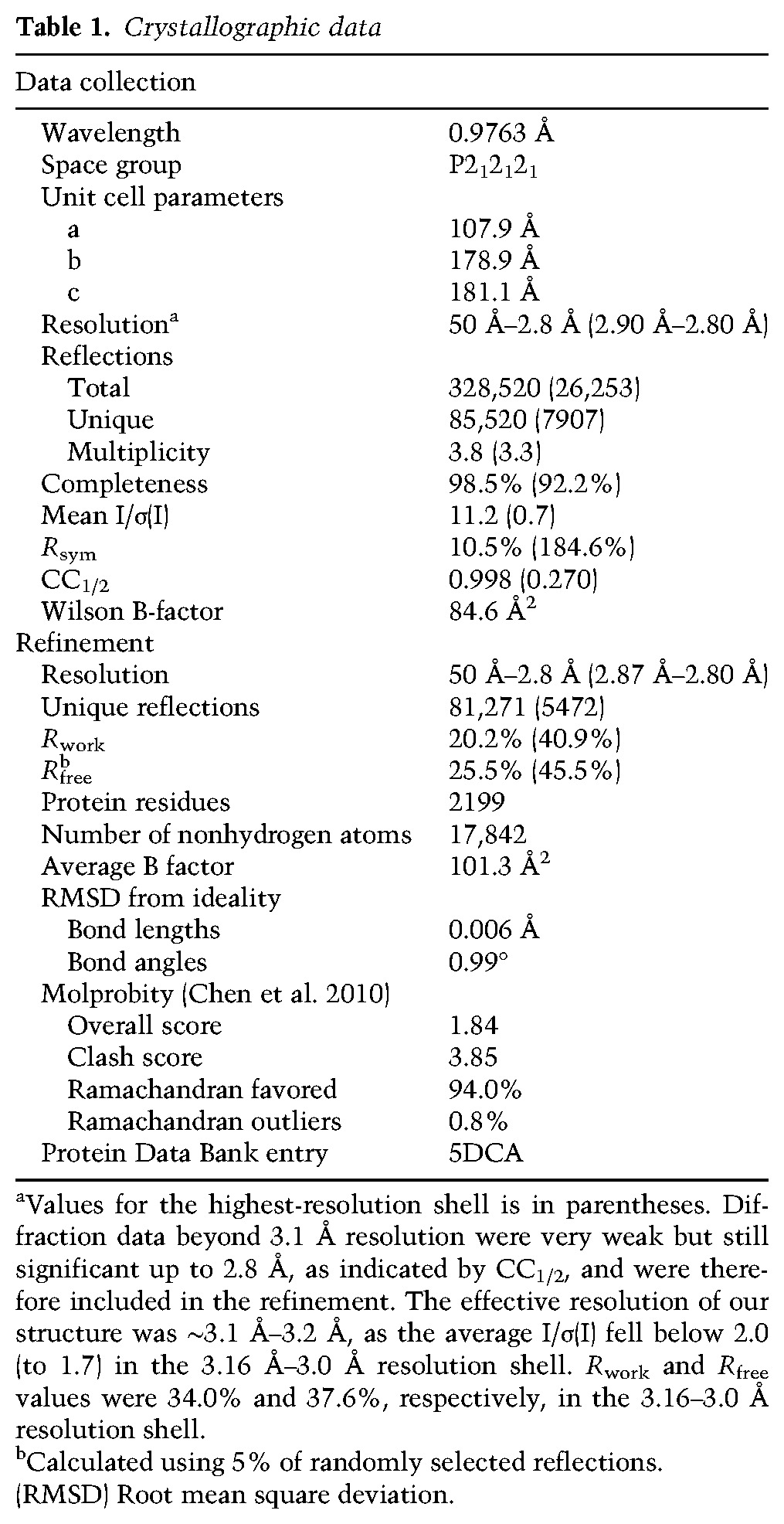
Chemical cross-linking and identification of cross-linked residues by mass spectrometry showed that the helicase region and folded portions of the NTR in isolated Brr2 adopt very similar structures and that the IC clamp of the NTR is positioned in the same fashion along the helicase region as seen in the crystal structure (Fig. 1D; Supplemental Table 1), indicated by the expected distributions of distances between Cα atoms of cross-linked residues [bis(sulfosuccinimidyl) suberate (BS3): 16.4 Å ± 6.1 Å for 52 cross-links seen in the structure; 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM): 13.8 Å ± 7.4 Å for 36 cross-links seen in the structure; excluding interdomain cross-links involving the PWI domain]. However, in isolated Brr2, several cross-links were observed between one edge of the PWI domain and the RecA1 domain of the C-terminal cassette, while, in our crystal structure, this edge of the PWI domain is turned more toward the N-terminal cassette (Fig. 1D; Supplemental Table 1). These results indicate that the PWI domain adopts a different relative position in isolated Brr2 and engages in direct contacts with the C-terminal cassette and the IC clamp. A different positioning of the PWI domain in isolation and in the crystal could be facilitated by the flexible linkers that connect this domain to the remainder of Brr2. As cross-links of the IC clamp are fully consistent with the crystal structure, this repositioning does not seem to change the positions of elements neighboring the PWI domain. Taken together, these results clearly show that the beginning (plug) and end (NC clamp) of the Brr2 NTR interact with the N-terminal cassette and that intervening segments (IC clamp and PWI) traverse to and contact the C-terminal cassette, thereby interlocking both helicase cassettes.
NTR truncations lead to splicing defects, reduced Brr2 association with the tri-snRNP, and increased tri-snRNP disruption
To investigate functions of the NTR, we tested the physiological consequences of sequential NTR truncations (T1–T4) (Fig. 1B; Supplemental Fig. 1B) in a strain deleted for Brr2. T1 and T2 strains exhibited growth defects at 30°C and 37°C, T3 additionally showed defects at 25°C (Fig. 2A), and strains lacking almost the entire NTR (T4) were not viable anymore. Extracts of viable strains contained comparable levels of FL, T1, T2, or T3 with the expected molecular sizes (Fig. 2B). Coproduction of FL Brr2 with N-terminal deletion variants showed no growth defects at 30°C (Fig. 2C), suggesting that N-terminal deletions do not lead to unspecific off-target effects but rather to splicing-related effects that can be rescued by FL Brr2. Indeed, transcripts originating from the intron-containing ACT1, RPL17B, and TEF4 genes showed increased intron retention in strains expressing NTR deletion variants compared with FL Brr2, and the degree of intron retention for all three genes correlated with the extent of the NTR deletions and thus with the associated growth defects (Fig. 2D).
Figure 2.

Physiological effects of Brr2 NTR truncations. (A) Yeast growth assay comparing strains that produce the indicated Brr2 variants (FL, T1, T2, and T3) as the sole type of Brr2 protein. The T4 strain was not viable. (B) Western blot analysis monitoring the types and amounts of Brr2 variants relative to the U5 protein Snu114 produced in extracts of the strains shown in A. (C) Yeast growth assay comparing strains that produce the indicated Brr2 variants (FL, T1, T2, T3, and T4) in a FL Brr2 background. (D) Accumulation of intron-containing transcripts of the indicated genes (ACT1, RPL17B, and TEF4) relative to the amounts of intronless THD1 transcripts in yeast strains producing the indicated Brr2 variants (FL, T1, T2, and T3) as the sole type of Brr2 protein. Values and error bars represent means ± standard error of the mean (SEM) of two technical duplicates of at least four independent experiments. Significance (Student's unpaired t-test) is indicated by asterisks. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (E) Western blots of odd-numbered glycerol gradient fractions of extracts from yeast strains producing the indicated Brr2 variants (FL, T1, T2, and T3). (Left panels) Without pretreatment (− ATP). (Right panels) After preincubation with ATP (+ ATP). Proteins are identified at the right. (F) Northern blots of even-numbered glycerol gradient fractions. snRNPs contained in the fractions are indicated below the gels, and snRNAs are identified at the right. (G) Quantification of the data in F. The left panel shows the amounts of U4 plus U6 snRNAs in U4/U6 di-snRNP fractions relative to U4 plus U6 snRNAs in combined di-snRNP and tri-snRNP fractions (before and after the addition of ATP). The right panel shows the amounts of U5 snRNA in U5 snRNP fractions relative to U5 snRNA in combined U5 snRNP and tri-snRNP fractions (before and after the addition of ATP). (H) Solution hybridization analysis (probing for U4 snRNA) of RNAs extracted from the fractions in F and separated by nondenaturing PAGE. Migration positions of the U4/U6 duplex (syn. U4/U6; obtained by annealing of in vitro transcribed RNAs) and single-stranded U4 snRNA are shown at the right. Boil time was chosen so that part of U4/U6 remained associated.
To test whether NTR truncations affect the association of Brr2 with U5 snRNP and the tri-snRNP, we analyzed cellular extracts on glycerol gradients to separate snRNPs and probed odd-numbered gradient fractions by Western blotting for Brr2 and another U5 protein, Snu114. Irrespective of preincubation of the extracts with ATP, strains producing NTR truncation variants T1, T2, or T3 contained relatively more free Brr2 (fractions 11–15) and relatively less tri-snRNP-associated Brr2 (fractions 23–25) compared with the FL strain (Fig. 2E). T2 and T3 strains also show reduced Snu114 association with the tri-snRNP, in particular after ATP treatment, suggesting that the tri-snRNP is destabilized when the respective NTRs of Brr2 are lacking. U5 snRNP association of Brr2 and Snu114 (fractions 17–19) was not affected in a similar way. Thus, Brr2 is in part anchored to the tri-snRNP via its NTR. As the shortest truncation of the NTR, T1, already led to a significant loss of Brr2 from the tri-snRNP but not from U5, the very N-terminal Brr2 residues might contact tri-snRNPs that are not contained in U5 snRNP. This idea is consistent with yeast two-hybrid interactions between the 250 N-terminal residues of Brr2 and the U4/U6 proteins Prp31 and Prp3 (Absmeier et al. 2015) and with decreased pull-down of several tri-snRNPs via a Brr2 variant lacking the first 120 residues (Zhang et al. 2015).
To see whether the stability of the remaining tri-snRNPs is affected by Brr2 NTR truncations, we monitored even-numbered gradient fractions by Northern blotting for snRNAs. U5 snRNP migrated in later fractions in truncated Brr2 extracts compared with the FL strain (Fig. 2E,F), possibly as a consequence of different conformations of the U5 snRNPs due to loss of some interactions between the Brr2 NTR and U5 snRNPs or U5 snRNA (Absmeier et al. 2015; Zhang et al. 2015). While ATP treatment did not change the distribution of U4/U6 di-snRNP (fractions 12–14), U5 snRNP (fractions 14–20), and U4/U6•U5 tri-snRNP (fractions 22–24) in the FL and T1 extracts, it led to reduced tri-snRNP levels and increased levels of U4/U6 di-snRNP and U5 snRNP in extracts of the T2 and T3 strains (Fig. 2F, quantification in G).
To test whether the increased disruption of tri-snRNP in the Brr2 truncation strains is associated with unwinding of U4/U6 di-snRNAs by the Brr2 variants, RNAs were extracted from the gradient fractions, annealed to a U4-specific DNA probe, and separated by native PAGE. In untreated extracts containing FL Brr2 or the T2 variant, the majority of U4 snRNA was detected in the tri-snRNP fractions base-paired to U6 (Fig. 2H). Consistent with the previous analysis, ATP treatment did not lead to changes of this profile in the FL Brr2 extract but led to reduced levels of U4/U6 in the tri-snRNP and increased levels of the U4/U6 duplex, but not free U4, in the U4/U6 di-snRNP fractions of the T2 strain (Fig. 2H). These results suggest that, upon truncation of the N terminus to beyond the plug domain, Brr2 has the tendency to disrupt the tri-snRNP into U4/U6 and U5 in an ATP-dependent manner without unwinding U4/U6 di-snRNAs. Alternatively, truncated variants of Brr2 might disrupt tri-snRNPs by increased unwinding of the U4/U6 di-snRNAs followed by fast reannealing of the RNAs and reassembly of U4/U6 di-snRNPs.
The NTR autoinhibits Brr2 by substrate competition and conformational clamping
We next investigated potential modulatory effects of the NTR on Brr2 RNA-binding, ATPase, and U4/U6-unwinding activities. T2 and T3 variants of yeast and human Brr2 (the only truncation variants that we could produce in soluble form for these Brr2 orthologs) exhibited increased RNA affinities (Fig. 3A, top and middle), RNA-stimulated ATPase activities (Fig. 3B, top and middle), and U4/U6-unwinding activities (Fig. 3C, top and middle) compared with the respective FL proteins (Table 2). To investigate these effects in a more systematic manner, we resorted to the Chaetomium thermophilum enzyme, for which we could recombinantly produce the complete truncation series (Table 2; Supplemental Fig. 1B). Stepwise shortening of the NTR of C. thermophilum Brr2 led to a gradual increase in U4/U6 affinity (except for the T4 variant; see below) (Fig. 3A, bottom), RNA-stimulated ATPase activity (Fig. 3B, bottom), and U4/U6-unwinding activity (Fig. 3C, bottom). These results show that the NTR autoinhibits Brr2.
Figure 3.

Effects of NTR truncations on yeast, human, and C. thermophilum Brr2 activities. (A) Apparent Kd values of the indicated Brr2 variants binding to U4/U6 di-snRNAs (organisms are indicated at the left, and protein variants are indicated below the graphs). Values represent means ± SEM of at least two independent experiments. Apparent Kd values were obtained by fitting quantified data from electrophoretic gel mobility shift assays (EMSA) titrations to a single exponential Hill function {fraction bound = A[protein]n/([protein]n + Kd n), where A is the fitted maximum of RNA bound, and n is the Hill coefficient} (Ryder et al. 2008). (B) ATPase activities of the indicated Brr2 variants determined by thin-layer chromatography (organisms are indicated at the left, and protein variants are indicated below the graphs). (−) Intrinsic ATPase activities; (+) U4/U6-stimulated ATPase activities. Values represent means ± SEM of at least three independent experiments. (C) Quantification of U4/U6-unwinding time courses using the indicated Brr2 variants (organisms are indicated at the left, and protein variants are indicated below the graphs). Radioactive bands on gels monitoring the unwinding reactions were quantified by densitometry and fit to a first-order reaction [fraction unwound = A{1 − exp(−ku t)}, where A is the amplitude of the reaction, ku is the apparent first-order rate constant of unwinding, and t is time]. Data points and error bars represent means ± SEM of at least three independent experiments. (D–F) Close-up views of the structure of the FL Brr2–Jab1 complex illustrating the effects of NTR elements removed from the various truncations. (D) Orthogonal views illustrating steric hindrance of U4/U6 di-snRNA binding by the plug. (E) Conformational clamping of N-terminal and C-terminal cassettes by the IC clamp and the PWI domain. (F) Conformational clamping of the N-terminal cassette by the NC clamp. Views in the left panel of D and in E and F are the same as in the top of Figure 1B.
Table 2.
RNA affinities, ATPase activities, and U4/U6-unwinding activitiesa
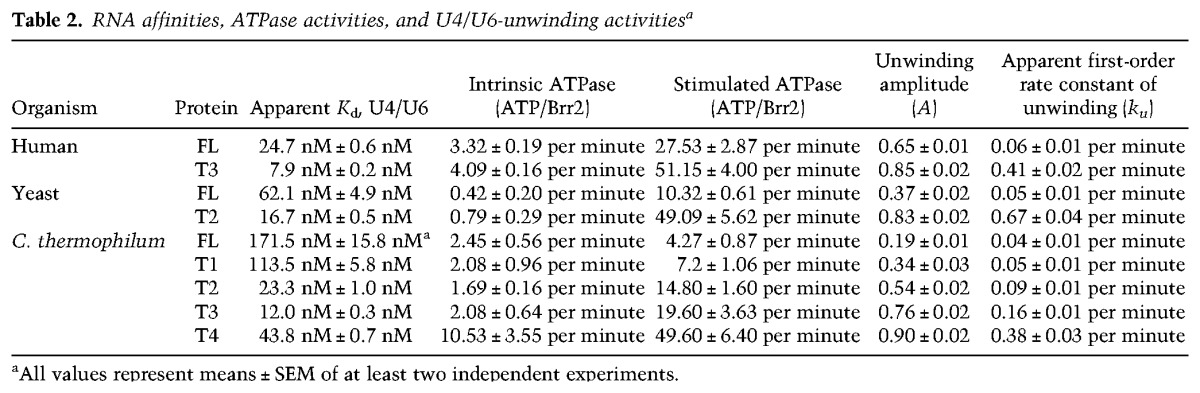
Our structural analyses provide mechanistic explanations for the detailed trends in activities upon stepwise shortening of the NTR. We observed cross-links among the N-terminal 111 residues, which are removed in T1, to domains of the N-terminal cassette (Supplemental Table 1). Therefore, the very N terminus can fold back onto the N-terminal cassette and thereby reinforce positioning of the neighboring plug (Fig. 1C). Truncation of T2 removes N-terminal portions, including the plug, and both T1 and T2 are associated with strong increases in RNA affinity (Fig. 3A). Modeling of how Brr2 engages U4/U6 di-snRNAs based on the crystal structure of the related Hel308 helicase in complex with DNA (Büttner et al. 2007) shows that the plug resides in a position where it would obstruct access of the RNAs to the enzyme (Fig. 3D). The additional observed increase in stimulated ATPase activity of T1 and T2 thus most likely reflects the stronger RNA binding of these fragments upon weakening of the inhibitory position of the plug (T1) or removal of it entirely (T2). These effects are likely enhanced by concomitant removal of the IC clamp in T2, which cross-struts the helicase cassettes and whose removal will thus increase conformational flexibility in the active N-terminal cassette (Fig. 3E). We showed previously that the N-terminal cassette has to open between the HB and RecA2 domains to engage U4 snRNA and that ATP initially docks to a nonhydrolytic conformation of the RecA domains (Santos et al. 2012). Thus, the IC clamp likely hinders conformational changes in the N-terminal cassette required for productive RNA and nucleotide binding by fastening it to the C-terminal cassette. T3 additionally lacks the PWI domain that, based on our cross-linking analysis, acts like the IC clamp to interconnect the helicase cassettes (Fig. 3E). Consistently, T3 shows a further increase in RNA affinity and stimulated ATPase activity compared with T2. Finally, truncated T4 also lacks about a third of the NC clamp, which interconnects the N-terminal RecA and WH domains (Fig. 3F). The NC clamp thus reinforces the nonhydrolytic conformation of the two RecA domains, explaining the strong up-regulation of both intrinsic and stimulated ATPase activities when truncated (Fig. 3B). While this manipulation could even further facilitate conformational changes required for RNA binding, loss of NC clamping might also prevent the helicase from efficiently holding on to the RNA, likely explaining the somewhat reduced RNA affinity of T4 compared with T3 (Fig. 3A). Taken together, the modulated helicase activities associated with NTR truncations (Fig. 3C) can be understood from the stepwise removal of substrate competitive elements (plug) and conformational clamps (IC clamp/PWI and NC clamp).
Discussion
Many SF2 helicases contain accessory regions appended at either end or inserted into the helicase cores (Johnson and Jackson 2013). The lengths, sequences, and domain compositions of these accessory regions are highly variable, and thus their functions typically cannot be predicted based on sequence information. As such accessory regions frequently include intrinsically unstructured segments, they are often excluded from structural and mechanistic studies. Here, we conducted a systematic structural and functional analysis of the long NTR of the spliceosomal Brr2 helicase, showing that (1) the NTR contains two folded domains (plug and PWI) and neighboring extended elements, which run along and interlock the two helicase cassettes; (2) the complete NTR is required for full cell viability and efficient splicing; (3) the NTR is required for stable association of Brr2 with the tri-snRNP, most likely involving its N-terminal portions contacting other tri-snRNPs; (4) NTR truncations lead to increased, ATP-dependent disruption of the tri-snRNP; and (5) regions of the NTR autoinhibit Brr2 by competing with U4/U6 di-snRNA binding (plug) and restricting the conformational flexibility of the helicase region (IC clamp/PWI and NC clamp). While, in principle, the snRNP- and splicing-related effects may be indirect consequences of the Brr2 truncations, the known crucial involvement of Brr2 in splicing and our observation that growth defects are suppressed by coproduction of FL Brr2 suggest that the Brr2 variants directly affect snRNP distributions and splicing.
The Sad1 protein promotes tri-snRNP formation (Huang et al. 2014) and is required for splicing of almost all intron-containing genes in yeast (Hadjivassiliou et al. 2014). Noncanonical, Brr2-dependent, and ATP-dependent disruption of the tri-snRNP into U4/U6 di-snRNP and U5 snRNP occurs upon depletion of Sad1 from splicing extracts (Huang et al. 2014). In that study, a yeast strain that lacks significant amounts of the U4/U6 reassembly factor Prp24 was used, ruling out U4/U6 reannealing and U4/U6 di-snRNP reassembly as possible explanations for the observed effect. We likewise observed increases in U4/U6 di-snRNP and U5 snRNP at the expense of tri-snRNP upon truncating the Brr2 NTR to beyond the plug domain and addition of ATP. As in our studies, Prp24 is present in the extracts; this result could indicate that, in the presence of ATP, truncated Brr2 variants lead to increased canonical disruption of the tri-snRNP via complete U4/U6 unwinding followed by fast reannealing of U4/U6 di-snRNAs and fast U4/U6 di-snRNP reassembly. This interpretation would be consistent with the increased helicase activity that we observed in truncated Brr2 variants in vitro.
However, given the complete lack of any increase in free U4 upon the above manipulation, we favor the alternative explanation that truncated Brr2 variants promote increased noncanonical disruption of the tri-snRNP into U4/U6 di-snRNP and U5 snRNP, as seen upon depletion of Sad1. Therefore, data presented here and previously (Huang et al. 2014) might indicate that Sad1 is a direct or indirect regulator of the Brr2 helicase in the tri-snRNP and that it cooperates with the Brr2 NTR to stabilize the tri-snRNP against nonproductive Brr2-mediated disruption before tri-snRNP incorporation into the spliceosome. As the NTR in the conformation seen in isolated Brr2 autoinhibits the enzyme via multiple mechanisms, Sad1 might stabilize this autoinhibited conformation upon tri-snRNP formation.
In a recent cryo-EM structure of a yeast tri-snRNP, Brr2 was modeled bound to U4 snRNA and ready to dissociate the U4/U6 di-snRNAs (Nguyen et al. 2015). Thus, Brr2 as modeled in the cryo-EM structure could not adopt the autoinhibited state described here, as its loading on U4 snRNA would be in conflict with the autoinhibitory position of the plug. The complex used for cryo-EM analysis disintegrated in an uncharacterized fashion upon addition of 1 mM ATP (Nguyen et al. 2015), showing that additional mechanisms must exist to prevent wasteful tri-snRNP disruption in vivo, where ATP levels range between 1 and 2.8 mM (Ozalp et al. 2010). Consistent with this proposal, we observed that yeast tri-snRNPs in extracts remain stable in the presence of ATP. Notably, Sad1 was lost during the tri-snRNP preparation for EM analysis (Nguyen et al. 2015), suggesting that the EM preparation in the presence of ATP also falls apart into U4/U6 and U5. In the absence of Sad1, tri-snRNP stability may be compromised due to the lack of interactions involving Sad1 and the inability to reinforce the autoinhibitory conformation of Brr2. Conformational changes associated with the onset of Brr2-mediated U4/U6 unwinding could thus lead to the observed noncanonical mode of dissociation. These comparisons support our notion of Sad1–Brr2 NTR cooperation in the tri-snRNP and suggest that, in the presence of Sad1, Brr2 is positioned differently in the tri-snRNP than modeled in the cryo-EM structure.
Brr2 in an alternative, autoinhibited conformation and position in the tri-snRNP would also be prevented from binding and remodeling noncognate RNAs upon assembly of a precatalytic spliceosome. The helicase could then be set into action specifically when required for splicesome catalytic activation; e.g., via the Snu114 G protein (Bartels et al. 2002, 2003; Small et al. 2006). After spliceosome catalytic activation, Brr2 remains associated with the spliceosome, and regulatory mechanisms may be required to either shut off its helicase activity in later stages of the splicing cycle (Hahn et al. 2012; Fourmann et al. 2013) or specifically put it to work during additional Brr2-dependent remodeling events (Small et al. 2006). In a recent, near-atomic resolution EM structure of a spliceosome that underwent the first step of splicing (Yan et al. 2015), no portion of Brr2 could be modeled, although the helicase was present, showing that it is flexibly anchored at this stage. Adoption of its autoinhibited state during this stage of splicing catalysis would again be a facile mechanism to prevent the enzyme from reaching out to and remodeling noncognate RNA duplexes.
Recently, we reported another reversible inhibitory mechanism of Brr2 based on the intermittent insertion of the C-terminal tail of the Prp8 Jab1 domain into the RNA-binding tunnel of the helicase (Mozaffari-Jovin et al. 2013). As the plug domain of the NTR blocks the entrance to the RNA-binding tunnel of Brr2 but leaves the tunnel itself unobstructed, both inhibitory mechanisms could be at work at the same time during certain stages of splicing (the tail could not have been seen in the present structure, as we used a tail-deleted variant of the Jab1 domain for crystallization). Alternatively, the two inhibitory mechanisms could act sequentially. As, also after removal of the plug domain, Brr2 remains bound to the Jab1 domain (Mozaffari-Jovin et al. 2013; Nguyen et al. 2013, 2015), the Jab1 C-terminal tail could still inhibit Brr2 after separation of the helicase core from the plug domain and thereby counteract noncognate RNA binding and unwinding; e.g., after step 1 of splicing.
Recently, it was reported that removal of the N-terminal 120 residues of Brr2 leads to increased loss of U5 and U6 snRNAs during spliceosome activation (Zhang et al. 2015), but the molecular mechanism underlying this effect remained entirely unclear. In light of our finding that NTR deletions correlate with nonproductive disruption of the tri-snRNP into U4/U6 and U5, the above effect suggests that the NTR also guides productive tri-snRNP disruption during splicing. One possibility is that, during the rearrangements required to initiate Brr2-mediated spliceosome activation, parts of the NTR remain in places where they fasten tri-snRNP components that have to be kept together during U4/U6 unwinding (Fig. 4). This model is attractive because separation of the helicase region from the NTR would render Brr2 fully active, as required for efficient U4/U6 unwinding. Obviously, upon artificial deletion of the NTR, Brr2 might engage U4/U6 already in the tri-snRNP even when Sad1 is present, and initial ATP-driven U4/U6 unwinding without tri-snRNP fastening by the NTR could lead to the observed noncanonical disruption of the tri-snRNP before complete U4/U6 separation.
Figure 4.

Model for the function of the Brr2 NTR. The NTR (magenta) serves to stably anchor Brr2 to the tri-snRNP and autoinhibits Brr2 in isolation and in the tri-snRNP, possibly avoiding engagement of off-target RNAs. Upon formation of a precatalytic spliceosome by association of the tri-snRNP with the A complex, conformational changes are required for Brr2 to engage U4 snRNA. To achieve productive Brr2-mediated tri-snRNP disruption (separation of U4/U6 and release of U4), parts of the Brr2 NTR need to fasten portions of the tri-snRNP that have to remain associated. After U4/U6 unwinding, the NTR might rebind the helicase region of Brr2 and thus shut off the enzyme again.
All spliceosomal helicases, except Brr2, have been shown to remodel suboptimal substrates in a nonproductive manner, thereby channeling them into discard pathways to enhance splicing fidelity or regulate alternative splicing (Wahl and Lührmann 2015). Although speculative, it is interesting to note that our findings of possible noncanonical Brr2-dependent rearrangements show that, in principle, Brr2 also has the potential to regulate splicing by discarding suboptimal intermediates through the off-pathway separation of U4/U6 from U5.
Materials and methods
Cloning and expression
Codon-optimized DNA fragments encoding regions of human Brr2 (FL [residues 1–2136] and T3 [395–2129]), C. thermophilum Brr2 (FL [1–2205], T1 [119–2193], T2 [287–2193], T3 [426–2193], and T4 [473–2193]) and yeast Brr2 (FL [1–2163] and T2 [271–2163]) were cloned into a modified pFL vector (EMBL, Grenoble) to produce proteins with a TEV-cleavable N-terminal His10 tag. Virus production and expression were performed as described (Santos et al. 2012). A codon-optimized DNA fragment encoding yeast Jab1 (residues 2147–2398 of yeast Prp8, lacking the C-terminal 15 residues) was cloned into the pETM-11 vector (EMBL, Heidelberg) under the control of a T7 promotor for production of the recombinant protein bearing a TEV-cleavable N-terminal His6 tag. Escherichia coli Rosetta2 DE3 cells were transformed with the vector and cultivated in autoinducing medium (Studier 2005).
Protein purification
For all preparations, cell pellets were resuspended, supplemented with protease inhibitors (Roche), and lysed by sonication using a Sonoplus ultrasonic homogenizer HD 3100 (Bandelin), and the lysates were cleared by centrifugation. The yeast FL Brr2 and T2 fragment were purified as previously described for human Brr2395–2129 (Santos et al. 2012) except that a HiPrep Heparin FF 16/10 (GE Healthcare) column was used instead of a MonoQ column.
For yeast Jab1, lysate in 100 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10 mM imidazole, and 2 mM DTT was loaded on a 5-mL HisTrap FF column (GE Healthcare) equilibrated with 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10 mM imidazole, and 2 mM DTT, and eluted in a linear gradient to 500 mM imidazole. Peak fractions were mixed with TEV protease; dialyzed overnight against 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10 mM imidazole, and 2 mM DTT; and again passed through a HisTrap column. The flowthrough was further purified by Superdex 75 (GE Healthcare) size exclusion chromatography in 10 mM Tris-HCl (pH 7.5), 200 mM NaCl, and 2 mM DTT.
All other proteins (in 40 mM HEPES-NaOH at pH 8.0, 600 mM NaCl, 10 mM imidazole, 20% [v/v] glycerol, 2 mM DTT) were captured on a 5-mL HisTrap FF column and eluted with a linear gradient to 250 mM imidazole. Peak fractions were diluted to 40 mM HEPES-NaOH (pH 8.0), 50 mM NaCl, 5% (v/v) glycerol, and 2mM DTT; loaded on a HiPrep Heparin FF 16/10 column; and eluted with a linear gradient to 1.5 M NaCl. The proteins were further purified using Superdex 200 size exclusion chromatography in 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 20% (v/v) glycerol, and 2 mM DTT.
Crystallographic procedures
FL yeast Brr2 and Jab1 were mixed in a 1:5 molar ratio in 10 mM Tris-HCl (pH 7.5), 200 mM NaCl, and 2 mM DTT and separated by Superdex 200 (GE Healthcare) size exclusion chromatography. Fractions containing the target complex were pooled, concentrated to 2.5 mg/mL, and used for crystallization. Crystals were grown in 48-well plates using the sitting-drop vapor diffusion technique at 18°C with drops containing 1.3 µL of protein complex solution and 1 µL of reservoir solution (0.1 M MES-NaOH at pH 6.5, 9% [w/v] PEG 3350, 0.2 M MgCl2). Crystals were cryoprotected by transfer into mother liquor containing 22.5% (v/v) ethylene glycol and flash-cooled in liquid nitrogen.
Diffraction data were collected at 100 K on beamline 14.1 of the BESSY II storage ring (Berlin, Germany) (Mueller et al. 2015) and on EMBL beamline P14 of the Petra III synchrotron (Hamburg, Germany) using a monochromated X-ray beam (λ = 0.9184 Å) and processed with XDS (Table 1; Kabsch 2010). The structure was solved by molecular replacement with Molrep (Vagin and Teplyakov 2010) using a truncated yeast Brr2–Jab1 structure as the search model (Protein Data Bank [PDB] ID 4BGD) (Nguyen et al. 2013) and completed by manual model building in Coot (Emsley and Cowtan 2004), guided in part by the crystal structure of a C. thermophilum Brr2 PWI domain (PDB ID 4RVQ) (Absmeier et al. 2015). The model was refined by alternating rounds of manual model building with Coot and automated refinement with RefMac5 (Murshudov et al. 2011). Coordinates and structure factors have been deposited in the Research Collaboratory for Structural Bioinformatics PDB (http://www.pdb.org) with accession code 5DCA.
Cross-linking analysis
For cross-linking, we used the homobifunctional, primary amino group-reactive BS3, which can bridge N termini or lysine side chains when their Cα atoms are <35 Å apart, or the hetero-bifunctional “zero-length” DMTMM, which mediates formation of amide bonds between proximal carboxylate (C termini or Asp or Glu side chains) and primary amine (N termini or Lys side chains) moieties. For cross-linking by BS3, 25 pmol of yeast Brr2 was cross-linked by the addition of 6 nmol of BS3 (Pierce, Thermo Scientific) in 20 mM HEPES-NaOH (pH 7.3), 200 mM NaCl, 2 mM DTT, and 5% (v/v) glycerol (final volume of 17.5 µL) and incubated for 30 min at room temperature. The reaction was quenched by the addition of 1 µL of 1 M Tris-HCl. Cross-linking efficiency was analyzed by SDS-PAGE on a 4%–12% Bis-Tris gel (Invitrogen). The band corresponding to cross-linked and monomeric Brr2 was excised, and the polypeptide was digested in the gel with trypsin as previously described (Schmidt and Urlaub 2009). The proteolytic peptides were dissolved in 20 µL of 5% (v/v) acetonitrile (ACN) and 0.1% (v/v) formic acid (FA) and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Six microliters of the cross-linking sample was injected into a nano-LC system (UltiMate 3000 RSLCnano system) equipped with a 3 cm × 150 µm (inner diameter) C18 trapping column and in line with a 30 cm × 75 µm (inner diameter) C18 analytical column (both packed in-house with 1.9 µm of C18 resin; Dr. Maisch GmbH). Peptides were loaded on the trapping column and desalted for 3 min at a flow rate of 10 µL/min in 95% mobile phase A (0.1% [v/v] FA in H2O) and 5% mobile phase B (80% [v/v] ACN and 0.05% [v/v] FA in H2O). After desalting, peptides were eluted and separated on the analytical column using a 43-min linear gradient of 15%–46% mobile phase B at a flow rate of 300 nL/min. Separated peptides were analyzed online using an Orbitrap fusion mass spectrometer (Thermo Scientfic). The 20 most intense precursor ions with charge states 3–8 in the survey scan (380–1580 m/z scan range) were isolated in the quadrupole mass filter (isolation window 1.6 m/z) and fragmented in the higher-energy collisional dissociation (HCD) cell with 30% normalized energy. A dynamic exclusion of 20 sec was used. Both the survey scan (MS1) and the product ion scan (MS2) were performed in the Orbitrap at 120,000 and 30,000 resolution at 200 m/z, respectively. Spray voltage was set at 2.3 kV, and 60% of S-lens RF level was used. Automatic gain control (AGC) targets were set at 5 × 105 (MS1) and 5 × 104 (MS2). The raw data were converted to MGF files by Proteome Discoverer 2.0.0.802 software (Thermo Scientific). The MGF files were searched against a FASTA database containing the sequence of yeast Brr2 by pLink 1.22 software (Yang et al. 2012) using a target decoy strategy. Database search parameters included mass accuracies of MS1 <10 parts per million (ppm) and MS2 <20 ppm, carbamidomethylation on cysteine as a fixed modification, and oxidation on methionine as a variable modification. The number of residues of each peptide on a cross-link pair was set between 4 and 40. A maximum of two missed trypsin cleavage sites was allowed. The results were obtained with a 3% false discovery rate.
For zero-length cross-linking, all of the conditions were identical to BS3 cross-linking except 30 mM DMTMM (Sigma-Aldrich) was used instead of BS3 for incubation with Brr2, and the quenching step was omitted.
Plasmid shuffling and yeast growth assays
Saccharomyces cerevisiae strain PRY118 (MATa, brr2::LEU2, ade2, lys2, his3, ura3, leu2 [pSE360-BRR2]) (Raghunathan and Guthrie 1998) was transformed with plasmid pPR150 (ARS, CEN, HIS3, and BRR2-PYA; PYA [polyomavirus medium T-antigen]) or pPR150-derived plasmids encoding N-terminally truncated yeast Brr2 (T1 [112–2163], T2 [271–2163], T3 [391–2163], and T4 [442–2163]) with a C-terminal polyoma tag. Positive transformants carrying both FL BRR2 and truncated brr2 genes were selected on complete minimal (CM) agar plates lacking histidine, leucine, and uracil. Replicas were plated onto CM agar plates lacking histidine and leucine and supplemented with 5-fluoroorotic acid (5-FOA). The 5-FOA plates were again replica-plated on CM agar lacking histidine, leucine, and uracil. Strains producing constructs FL, T1, T2, and T3, which grew on 5-FOA plates but did not grow on the replica plates lacking uracil (indicating loss of the pSE360-BRR2 plasmid and retention of a pPR150 plasmid bearing a FL BRR2 or truncated brr2 copy) were used for further experiments. Three clones were picked for each construct. Additionally, for constructs FL, T1, T2, T3, and T4, four clones that grew on CM agar lacking histidine and uracil but failed to grow on 5-FOA plates were picked. These clones carried both the pSE360-borne wild-type BRR2 gene and the pPR150-borne BRR2 or brr2 gene. Clones carrying only pPR150-derived plasmids were diluted to an OD600 of 1 and then diluted 1:100 into yeast extract, peptone, and dextrose (YPD) medium. YPD cultures were grown for 2 d at 30°C. Cultures were diluted in YPD to an OD600 of 1, and 10-fold serial dilutions were spotted onto YPD agar plates. Plates were incubated for 2 d at 25°C, 30°C, and 37°C. Clones carrying both pSE360-BRR2-derived and pPR150-derived plasmids were diluted to an OD600 of 1 and then diluted 1:100 into CM lacking leucine, histidine, and uracil (CM −LHU) and grown for 2 d at 30°C. Cultures were diluted in CM −LHU to an OD600 of 1, and 10-fold serial dilutions were spotted onto CM −LHU agar plates. Plates were incubated for 4 d at 30°C.
Quantitative RT–PCR (qRT–PCR)
Yeast cells were grown at 30°C in CM medium lacking leucine and histidine. Total RNA was prepared from yeast strains producing pPR150-derived FL, T1, T2, and T3 instead of the endogenous Brr2 as described (Chomczynski and Sacchi 2006). For qRT–PCRs, the following primers were used: Act1_Int_fwd (5′-CGCTTGCACCATCCCATTTA-3′), Act1_Int_rev (5′-AGGAGGTTATGGGAGAGTGAA-3′), Tef4_E1_fwd (5′-TGGGTCCAAAGGGCTTAAAG-3′), Tef4_I1_rev (5′-TGCCAAATAAACGAACGGGA-3′), RPL17B_E1_fwd (5′-GTTCGTTCAAGGTTTGTTGCA-3′), RPL17B_I1_rev (5′-TCATCTCCCGTCAACACATCA-3′), Thd1_fwd (5′-AGACCCAGCTAACTTGCCAT-3′), and THD1_rev (5′-ACCAGCGTCAATGTGCTTTT-3′). One microgram of total RNA was reverse-transcribed using specific primers for ACT1, TEF4, RPL17B, and THD1 transcripts in one RT reaction. qPCR was then performed in a 96-well format using the ABsolute qPCR SYBR Green mix (Thermo Fisher) on a Mx3000P thermocycler (Stratagene). Analyses were carried out in technical duplicates on at least four independent preparations of total RNA for each strain. Mean values were used to normalize intron-containing products to intronless THD1 transcript (DCT), and D(DCT)s were calculated for different conditions. Fold changes were calculated assuming that a D(DCT) of 1 corresponds to a twofold change in mRNA. P-values were calculated using Student's unpaired t-test. All PCR products were confirmed by sequencing.
Glycerol gradient analysis
Yeast cells producing FL or truncated variants of Brr2 were grown in YPD medium at 30°C, and whole-cell extracts were prepared as described (Umen and Guthrie 1995). Cell extracts were incubated in the absence or presence of 2 mM ATP/MgCl2 for 30 min at 23°C, diluted with an equal volume of the G100 buffer (20 mM HEPES-KOH at pH 7.0, 100 mM KCl, 0.2 mM EDTA), and sedimented on 10%–30% (v/v) glycerol gradients in G100 buffer. The gradients were ultracentrifuged in a Sorvall TST41.14 rotor at 37,000 rpm for 15 h and separated into 27 fractions.
The distributions of Brr2 variants and Snu114 across the gradient fractions were monitored by Western blotting of gradient fractions and immunostaining using antibodies against Brr2 and Snu114 and the Amersham ECL detection kit (GE Healthcare) as previously described (Liu et al. 2015). To analyze relative levels of snRNPs in cell extracts, proteins in the gradient fractions were digested by proteinase K for 45 min at 37°C in the gradient buffer supplemented with 150 mM NaCl, 10 mM EDTA, and 1% (w/v) SDS. The RNAs were extracted by phenol/chloroform/isoamylalcohol, precipitated by ethanol, and separated by denaturing 8% PAGE followed by Northern blotting using 5′ end radiolabeled DNA probes against U4, U6, and U5 snRNAs (Mozaffari-Jovin et al. 2013).
Analysis of U4/U6 dissociation by solution hybridization
To monitor the dissociation of U4/U6 di-snRNAs in the presence of Brr2 variants before and after ATP treatment of cell extracts, gradient fractions were supplemented with 5 mM EDTA, and proteins were digested using proteinase K for 30 min at 37°C. The RNAs in each fraction were extracted at 4°C, precipitated by ethanol, and dissolved in 10 µL of the solution hybridization buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM EDTA). The U4 snRNA was probed using 2 pmol of 5′ end fluorescently labeled DNA probe 5′-ATTO 550-AGGTATTCCAAAAATTCCCTAC-3′ for 15 min at 37°C (Li and Brow 1993). The probed RNAs were separated by nondenaturing 8% PAGE in 0.5× TBE at 4°C and visualized by a Typhoon Trio+ imager (GE Healthcare).
Electrophoretic mobility shift assays
Yeast U4 and U6 snRNAs were produced by T7 RNA polymerase-based in vitro transcription and 5′ end-labeled with [γ-32P]ATP using T4 polynucleotide kinase. U4/U6 complex (1 nM) was titrated with increasing amounts of proteins in 40 mM HEPES-NaOH (pH 7.9), 15 mM NaCl, 2.5 mM MgOAc, 1 mM DTT, and 0.1 mg/mL acetylated BSA. Samples were separated using 4% (75:1) nondenaturing PAGE. Gels were scanned on a Storm PhosphorImager (GE Healthcare), bands were quantified by densitometry, and apparent Kd values were obtained by fitting the resulting data points to a single exponential Hill function {fraction bound = A[protein]n/([protein]n + Kdn), where A is the fitted maximum of RNA bound, and n is the Hill coefficient} (Ryder et al. 2008) using GraphPad Prism (GraphPad Software, Inc.).
ATPase assay
Brr2 constructs were diluted to 100 nM in 40 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1.5 mM DTT, and 0.1 mg/mL acetylated BSA without or with 1 µM U4/U6 di-snRNAs. The reactions were started by the addition of 1 mM ATP/MgCl2 (supplemented with 0.0125 mCi/mL [α-32P]ATP), and samples were incubated for 25 min at 30°C. The reactions were stopped by the addition of 1 vol of 100 mM EDTA (pH 8.0) and separated by thin-layer chromatography in 20% (v/v) ethanol, 6% (v/v) acetic acid, and 0.5 M LiCl2. Chromatograms were scanned on a PhosphorImager and quantified by densitometry, and ATPase activities were calculated as the number of ATP molecules hydrolyzed per Brr2 molecule per minute.
Unwinding assays
Unwinding assays were conducted and evaluated as described (Santos et al. 2012; Mozaffari-Jovin et al. 2013). Briefly, 2 nM U4/U6 complex and 100 nM Brr2 constructs were mixed in 40 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1.5 mM DTT, and 0.1 mg/mL acetylated BSA. After incubation for 3 min at 30°C, reactions were started by the addition of 1 mM ATP/MgCl2. Ten-microliter samples were withdrawn at the respective time points; mixed with 10 µL of 40 mM Tris-HCl (pH 7.4), 50 mM NaCl, 25 mM EDTA, 1% (w/v) SDS, 10% (v/v) glycerol, 0.05% (w/v) xylene cyanol, and 0.05% (w/v) bromophenol blue; and separated using 6% native PAGE (19:1). Gels were scanned on a PhosphorImager, bands were quantified by densitometry, and data were fit to a first-order reaction {fraction unwound = A[1 − exp(−ku t)], where A is the amplitude of the reaction, ku is the apparent first-order rate constant of unwinding, and t is time}.
Data deposition
Coordinates and diffraction data for the FL Brr2–Jab1 complex from yeast have been deposited with the Protein Data Bank (http://www.pdb.org) under accession code 5DCA and will be released upon publication.
Supplementary Material
Acknowledgments
We acknowledge access to beamline BL14.1 of the BESSY II storage ring (Berlin, Germany) via the Joint Berlin MX Laboratory sponsored by the Helmholtz Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Universität zu Berlin, the Max-Delbrück Centrum, and the Leibniz-Institut für Molekulare Pharmakologie. Final data were collected at EMBL beamline P14 of the Petra III synchrotron (Hamburg, Germany). This work was funded by the Deutsche Forschungsgemeinschaft (SFB 740 to M.C.W., and SFB 860 to R.L. and H.U.), the Bundesministerium für Bildung und Forschung (grant 05K10KEC to M.C.W.) and the Einstein Foundation Berlin (grant A-2012-140 to M.C.W.). K.F.S. was supported by a Dahlem International Network PostDoc Fellowship from Freie Universität Berlin. E.A., J.W., S.M.-J., C.B., C.-T.L., M.P., and K.F.S. performed experiments. F.H., H.U., R.L., K.F.S., and M.C.W. coordinated and supervised the studies. All authors planned experiments and analyzed and interpreted the data. E.A., R.L., and M.C.W. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.271528.115.
References
- Absmeier E, Rosenberger L, Apelt L, Becke C, Santos KF, Stelzl U, Wahl MC. 2015. A noncanonical PWI domain in the N-terminal helicase-associated region of the spliceosomal Brr2 protein. Acta Crystallogr D 71: 762–771. [DOI] [PubMed] [Google Scholar]
- Bartels C, Klatt C, Lührmann R, Fabrizio P. 2002. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep 3: 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Urlaub H, Lührmann R, Fabrizio P. 2003. Mutagenesis suggests several roles of Snu114p in pre-mRNA splicing. J Biol Chem 278: 28324–28334. [DOI] [PubMed] [Google Scholar]
- Büttner K, Nehring S, Hopfner KP. 2007. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol 14: 647–652. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Abelson J. 1987. Spliceosome assembly in yeast. Genes Dev 1: 1014–1027. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc 1: 581–585. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fourmann JB, Schmitzova J, Christian H, Urlaub H, Ficner R, Boon KL, Fabrizio P, Lührmann R. 2013. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev 27: 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjivassiliou H, Rosenberg OS, Guthrie C. 2014. The crystal structure of S. cerevisiae Sad1, a catalytically inactive deubiquitinase that is broadly required for pre-mRNA splicing. RNA 20: 656–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn D, Kudla G, Tollervey D, Beggs JD. 2012. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev 26: 2408–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Chung CS, Kao DI, Kao TC, Cheng SC. 2014. Sad1 counteracts Brr2-mediated dissociation of U4/U6.U5 in tri-snRNP homeostasis. Mol Cell Biol 34: 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SJ, Jackson RN. 2013. Ski2-like RNA helicase structures: common themes and complex assemblies. RNA Biol 10: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Rossi JJ. 1999. The first ATPase domain of the yeast 246-kDa protein is required for in vivo unwinding of the U4/U6 duplex. RNA 5: 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B, Achsel T, Lührmann R. 1998. The human U5–200kD DEXH-box protein unwinds U4/U6 RNA duplices in vitro. Proc Natl Acad Sci 95: 4188–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Brow DA. 1993. A rapid assay for quantitative detection of specific RNAs. Nucleic Acids Res 21: 4645–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Mozaffari-Jovin S, Wollenhaupt J, Santos KF, Theuser M, Dunin-Horkawicz S, Fabrizio P, Bujnicki JM, Lührmann R, Wahl MC. 2015. A composite double-/single-stranded RNA-binding region in protein Prp3 supports tri-snRNP stability and splicing. Elife 4: e07320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder C, Kutach AK, Guthrie C. 2009. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat Struct Mol Biol 16: 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffari-Jovin S, Santos KF, Hsiao HH, Will CL, Urlaub H, Wahl MC, Lührmann R. 2012. The Prp8 RNase H-like domain inhibits Brr2-mediated U4/U6 snRNA unwinding by blocking Brr2 loading onto the U4 snRNA. Genes Dev 26: 2422–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Lührmann R, Wahl MC. 2013. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science 341: 80–84. [DOI] [PubMed] [Google Scholar]
- Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Lührmann R, Wahl MC. 2014. Novel regulatory principles of the spliceosomal Brr2 RNA helicase and links to retinal disease in humans. RNA Biol 11: 298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller U, Forster R, Hellmig M, Huschmann FU, Kastner A, Malecki P, Puhringer S, Rower M, Sparta K, Steffien M, et al. 2015. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: current status and perspectives. Eur Phys J Plus 130: 141. [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. Refmac5 for the refinement of macromolecular crystal structures. Acta Crystallogr D 67: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Li J, Galej WP, Oshikane H, Newman AJ, Nagai K. 2013. Structural basis of Brr2–Prp8 interactions and implications for U5 snRNP biogenesis and the spliceosome active site. Structure 21: 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Galej WP, Bai XC, Savva CG, Newman AJ, Scheres SH, Nagai K. 2015. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 523: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Guthrie C. 1996. Identification of novel genes required for yeast pre-mRNA splicing by means of cold-sensitive mutations. Genetics 143: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozalp VC, Pedersen TR, Nielsen LJ, Olsen LF. 2010. Time-resolved measurements of intracellular ATP in the yeast Saccharomyces cerevisiae using a new type of nanobiosensor. J Biol Chem 285: 37579–37588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C. 1998. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol 8: 847–855. [DOI] [PubMed] [Google Scholar]
- Ryder SP, Recht MI, Williamson JR. 2008. Quantitative analysis of protein–RNA interactions by gel mobility shift. Methods Mol Biol 488: 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos KF, Mozaffari-Jovin S, Weber G, Pena V, Lührmann R, Wahl MC. 2012. Structural basis for functional cooperation between tandem helicase cassettes in Brr2-mediated remodeling of the spliceosome. Proc Natl Acad Sci 109: 17418–17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Urlaub H. 2009. iTRAQ-labeling of in-gel digested proteins for relative quantification. Methods Mol Biol 564: 207–226. [DOI] [PubMed] [Google Scholar]
- Small EC, Leggett SR, Winans AA, Staley JP. 2006. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol Cell 23: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326. [DOI] [PubMed] [Google Scholar]
- Studier FW. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expres Purif 41: 207–234. [DOI] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. 1995. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev 9: 855–868. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr D 66: 22–25. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Lührmann R. 2015. SnapShot: spliceosome dynamics II. Cell 162: 456–456.e1. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Yan C, Hang J, Wan R, Huang M, Wong CCL, Shi Y. 2015. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 349: 1182–1191. [DOI] [PubMed] [Google Scholar]
- Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, et al. 2012. Identification of cross-linked peptides from complex samples. Nat Methods 9: 904–906. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li X, Hill RC, Qiu Y, Zhang W, Hansen KC, Zhao R. 2015. Brr2 plays a role in spliceosomal activation in addition to U4/U6 unwinding. Nucleic Acids Res 43: 3286–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.