

Yoon et al. show that Rev1 is indispensable for translesion synthesis (TLS) mediated by Polη, Polι, and Polκ but is not required for TLS by Polζ. This work implicates a crucial role for Rev1 in the maintenance of genome stability in humans.
Keywords: DNA repair, UV damage, cII mutation analyses, DNA lesion bypass
Abstract
Translesion synthesis (TLS) DNA polymerases (Pols) promote replication through DNA lesions; however, little is known about the protein factors that affect their function in human cells. In yeast, Rev1 plays a noncatalytic role as an indispensable component of Polζ, and Polζ together with Rev1 mediates a highly mutagenic mode of TLS. However, how Rev1 functions in TLS and mutagenesis in human cells has remained unclear. Here we determined the role of Rev1 in TLS opposite UV lesions in human and mouse fibroblasts and showed that Rev1 is indispensable for TLS mediated by Polη, Polι, and Polκ but is not required for TLS by Polζ. In contrast to its role in mutagenic TLS in yeast, Rev1 promotes predominantly error-free TLS opposite UV lesions in humans. The identification of Rev1 as an indispensable scaffolding component for Polη, Polι, and Polκ, which function in TLS in highly specialized ways opposite a diverse array of DNA lesions and act in a predominantly error-free manner, implicates a crucial role for Rev1 in the maintenance of genome stability in humans.
By promoting replication through DNA lesions, translesion synthesis (TLS) DNA polymerases (Pols) ensure the continued progression of the replication fork. In human cells, DNA Polη, Polι, Polκ, and Rev1, which belong to the Y family, and Polζ, which is a member of the B family, play an important role in TLS (Prakash et al. 2005). Biochemical and structural studies with the Y family Pols have indicated that they function in TLS in highly specialized ways. For example, Polη can accommodate the two pyrimidine residues of UV-induced cyclobutane pyrimidine dimer (CPD) in its active site (Biertumpfel et al. 2010; Silverstein et al. 2010), and that enables it to replicate through CPDs efficiently and in a predominantly error-free manner (Johnson et al. 1999b, 2000b; Washington et al. 2000); consequently, inactivation of Polη in humans causes the cancer-prone syndrome, the variant form of xeroderma pigmentosum (XPV) (Johnson et al. 1999a; Masutani et al. 1999). Rev1 is the most intriguing among the Y family Pols; it is highly specialized for inserting a C opposite template G and uses a protein template-directed mechanism for C incorporation (Nair et al. 2005; Swan et al. 2009). Genetic studies in yeast have indicated a noncatalytic role for Rev1 in which it functions as an indispensable component of Polζ (Lawrence and Christensen 1978; Lawrence et al. 1984; Johnson et al. 1998), comprised of the Rev3 catalytic subunit and Rev7, Pol31, and Pol32 accessory subunits (Johnson et al. 2012). Biochemical studies with yeast Polζ have shown that it is highly specialized for extending from the nucleotides inserted opposite DNA lesions by other Pols (Johnson et al. 2000a, 2001, 2003; Haracska et al. 2001; Nair et al. 2006, 2008). Since Polζ can efficiently extend from the correct as well as the incorrect nucleotides and Rev1 further enhances the proficiency of Polζ for extending from the wrong nucleotides opposite DNA lesions (Acharya et al. 2006), Rev1 and Polζ play an important role in damage-induced mutagenesis in yeast.
Human and mouse Polη, Polκ, and Polι as well as Rev7, the accessory subunit of Polζ, have been shown to physically interact with the ∼100-amino-acid C-terminal region of Rev1 (Guo et al. 2003; Ohashi et al. 2004; Tissier et al. 2004). The ability of Rev1 to bind to Y family Pols and to Rev7 has led to the suggestion that Rev1 acts as a scaffold for recruiting the Y family Pols and Polζ to the lesion site. This idea has received further support from nuclear magnetic resonance (NMR) structural studies indicating that the human and mouse Rev1 C-terminal peptide uses independent binding surfaces to simultaneously bind to an interacting peptide of Polη and to the Rev7 protein or to an interacting peptide of Polκ and to Rev7 (Pozhidaeva et al. 2012; Pustovalova et al. 2012; Wojtaszek et al. 2012a,b). Based on these structural observations, it has been proposed that, by simultaneously binding to Y family Pols and the Rev7 subunit of Polζ, Rev1 brings about the assembly of Y family Pols, which generally act as inserters, with Polζ, which acts at the subsequent step of extension in TLS.
Rev1 has also been suggested to function in lesion bypass in other ways. From alkaline sucrose gradient analyses of the size of nascent DNA fragments in UV-damaged Rev1−/− mouse embryonic fibroblasts (MEFs), it has been inferred that Rev1 is not required for the replicative bypass of most CPDs in vivo but is required for the replicative bypass of (6-4) photoproducts (Jansen et al. 2009). From other studies, it has been suggested that, in human or mouse cells, efficient and accurate replication past UV lesions is predominantly performed by Polη and does not require Rev1 (Ito et al. 2012). The requirement for Rev1 arises only when Polη becomes stalled at a lesion site, whereupon Rev1 facilitates the exchange of catalytically inactive and stalled Polη with another TLS Pol (Ito et al. 2012). Accordingly, Rev1 would play a minor and subsidiary role in lesion bypass, required only under very specific conditions.
Overall, the many studies that have been carried out thus far have yielded conflicting results for the role of Rev1 in lesion bypass, and there is no consensus on how Rev1 functions in human cells. Thus, it is not known whether, in human cells derived from normal (noncancerous) tissue, Rev1 functions in TLS during replication in conjunction with Polζ, as it does in yeast; is required for coordinating TLS mediated by the sequential action of Y family Pols and Polζ, as has been suggested from structural studies; or functions in TLS in any of the other ways that have been suggested before. Alternatively, Rev1 may function in lesion bypass in human cells in an entirely different manner, and our evidence shows that to be the case.
To reliably assess the genetic control of TLS in human cells, we devised duplex plasmid systems in which TLS through a DNA lesion can be examined during bidirectional replication initiating from an origin of replication. From analyses of TLS opposite a cis-syn TT dimer carried on the leading or lagging strand DNA template of SV40 origin-based plasmid in human fibroblasts, we determined that Polη plays a prominent role and promotes highly error-free TLS, whereas Polκ and Polζ function in the alternative error-prone TLS pathways (Yoon et al. 2009). We also examined the genetic control of error-free and mutagenic TLS opposite UV-induced CPDs formed at the TT, TC, and CC dipyrimidine sequences in the cII gene carried in the genome in big blue MEFs (BBMEF) and verified that Polη functions in error-free TLS and that Polκ and Polζ promote mutagenic TLS (Yoon et al. 2009). Additionally, from analyses of TLS opposite a (6-4) TT photoproduct carried on the SV40-based plasmid and analyses of error-free and mutagenic TLS opposite (6-4) photoproducts formed in the cII gene in mouse cells, we determined that Polη and Polι function in alternative mutagenic pathways, whereas Polζ controls an error-free mode of TLS opposite (6-4) photoproducts (Yoon et al. 2010b).
To provide further evidence that the conclusions for the genetic control of TLS as derived from studies with the SV40-based plasmid in human cells and from cII mutational analyses in mouse cells are reflective of TLS processes that occur during cellular replication, we carried out TLS studies with a duplex plasmid in which bidirectional replication initiates from the Epstein-Barr virus (EBV) origin of replication (Yoon et al. 2012a). Unlike the SV40-encoded T antigen (which, in addition to origin-binding activity, has a DNA helicase activity), the EBV-encoded binding EBNA1 protein has no DNA helicase activity. The replication of EBV plasmid in human cells has been thoroughly studied and shown to be controlled by the same cellular processes that govern chromosomal replication. For example, the origin recognition complex (ORC1 to ORC6), the regulatory protein Cdc6, the MCM2–MCM7 helicase complex, and the licensing protein Cdt1 are all required for replication of the EBV plasmid (Yates and Guan 1991; Dhar et al. 2001; Schepers et al. 2001; Ritzi et al. 2003; Wang et al. 2006; Lindner and Sugden 2007). Our observations that the genetic control of TLS in EBV plasmid carried in human fibroblasts is identical to that determined from studies with SV40 plasmid in human fibroblasts or from studies with the chromosomal cII gene in mouse fibroblasts have added further evidence that TLS and mutational studies using these different approaches provide a consistent means of unraveling the genetic control of TLS and for elucidating the TLS mechanisms that operate during cellular replication (Yoon et al. 2009, 2010b, 2012a).
Here, we carried out a number of studies to analyze the role of Rev1 in TLS in human and mouse cells. We determined the role of Rev1 in mediating replication through a cis-syn TT dimer and a (6-4) TT photoproduct carried on the duplex plasmid in human fibroblasts and showed that Rev1 is indispensable for TLS mediated by Polη, Polκ, and Polι but is not required for TLS mediated by Polζ. To verify these observations in the genomic context, we analyzed the genetic control of error-free and mutagenic TLS opposite CPDs and (6-4) photoproducts induced by UV light in the chromosomal cII gene in mouse fibroblasts and showed that Rev1 functions with the Y family Pols and not Polζ. In addition, we provided evidence that Rev1 is required for the accumulation of Polη, Polι, and Polκ but not of Polζ into replication foci in UV-irradiated human and mouse cells and that, for UV survival, Rev1 interacts epistatically with Polη, Polι, and Polκ and not with Polζ. Altogether, these studies establish a crucial role for human Rev1 in TLS in conjunction with DNA Polη, Polι, and Polκ and show that, in normal human cells, Rev1-dependent TLS operates in a predominantly error-free manner.
Results
The role of Rev1 in TLS opposite the cis-syn TT dimer in human cells
The details of the SV40 plasmid system for TLS studies have been described before (Yoon et al. 2009, 2010b). Briefly, the multiple-cloning site with the lacZ′ gene is replaced with a specific target sequence, one strand of which harbors a DNA lesion in the MfeI restriction site, and, opposite the DNA lesion site, the other strand has an SpeI sequence that contains a +1 frameshift (+1 nucleotide [nt]) (Supplemental Fig. S1). Since the lacZ′ sequence in the lesion-containing strand is in-frame and since functional β-galactosidase (β-gal) is dependent only on the reading frame and not the sequence context of the TLS product, both error-free and error-prone TLS products result in blue colonies. Because the other DNA strand, which has no DNA lesion, contains a +1 frameshift, the lacZ′ gene in this strand is nonfunctional. The two DNA strands are further distinguished in that the plasmid carries the Kan+ (kanamycin resistance) gene in the lesion-containing strand, whereas the other DNA strand has the Kan− gene (Supplemental Fig. S1). TLS frequencies are determined from the number of blue colonies among the total colonies that grow on LB/kan plates. Since a template switch at the lesion site would involve continued synthesis of the Kan+ strand by using the other DNA strand, which harbors a +1 frameshift, the resulting Kan+ colonies will be white. Kan+ white colonies could also result from nucleotide excision repair (NER). However, since blue colonies can only result from TLS through the lesion site, their frequency among the total Kan+ colonies gives a very reliable and reproducible measurement of the TLS frequency.
To determine the role of Rev1 in TLS opposite a cis-syn TT dimer, we examined the effects of siRNA depletions of different TLS Pols individually and in combination with Rev1 depletion. For all of the TLS Pols, including Rev1, we ascertained that the siRNA treatment causes a highly efficient depletion of the protein (Supplemental Fig. S2A) and that the wild-type protein complements the TLS deficiency resulting from the siRNA treatment. To determine that the wild-type protein complements the TLS deficiency engendered by siRNA treatment, we stably expressed the 3xFlag-wild-type protein or its siRNA-resistant (siR) form in normal MRC5 human fibroblasts. As shown in Supplemental Figure S2B, in siRNA-treated cells expressing 3xFlag-tagged wild-type Polη (3xFlag-WT-Polη) or 3xFlag-WT-Rev1, Polη or Rev1 was efficiently depleted by siRNA treatment, indicating that the siRNA treatment conferred efficient knockdown of the proteins expressed from the genome as well as the vector. In contrast, in cells expressing the siR form of Polη or Rev1, the levels of both proteins remained intact in siRNA-treated cells. As shown in Supplemental Table 1, in normal human fibroblasts treated with control siRNA, TLS opposite a cis-syn TT dimer carried on the leading strand template of SV40-based plasmid occurs with a frequency of ∼25%. In human fibroblast cells treated with Polη siRNA and carrying the vector alone or the vector expressing Flag-WT-Polη, TLS frequency declines to ∼11%, whereas in cells expressing the Flag-WT-siR-Polη, TLS is restored to wild-type levels. Similarly, for Rev1, the siRNA depletion of Rev1 in human fibroblast cells expressing the vector or the vector containing the Flag-WT-Rev1 TLS frequency is reduced to ∼7.5%, and TLS is fully restored to wild-type levels in cells expressing the Flag-WT-siR-Rev1 protein. The complementation of the TLS deficiency by the siR Rev1 or Polη shows that the TLS defects engendered by siRNA depletion result from the depletion of that particular protein and not from any off-target effects. Similarly, we verified that the TLS deficiency conferred by the siRNA depletion of other TLS Pols opposite this UV lesion was complemented by the respective siR form of wild-type protein.
In contrast to the TLS frequency of ∼25% in NER-proficient human fibroblasts (Supplemental Table S1), in NER-defective XPA cells treated with control siRNA, TLS opposite a cis-syn TT dimer carried on the leading strand template of a SV40-based plasmid occurs with a frequency of ∼40% (Table 1). Thus, NER removes a considerable proportion of UV lesions from the plasmid before its replication. In Polη-depleted XPA cells, the TLS frequency is reduced to ∼18%, whereas in cells depleted for Polκ or the Rev3 or Rev7 subunit of Polζ, the frequency is reduced to ∼30%. Upon Rev1 depletion, the TLS frequency is reduced to ∼15%, and simultaneous depletion of Rev1 and Polη or Rev1 and Polκ confers the same reduction in TLS frequency as that seen upon Rev1 depletion alone (∼15%). In contrast, simultaneous depletion of Rev1 with Rev3 or Rev7 results in a drastic reduction in TLS frequency (∼5%) compared with that conferred by the depletion of Rev1 or Polζ subunits individually. From the epistasis of Rev1 over Polη and Polκ and the synergistic interaction of Rev1 with Polζ, we deduce that Polη and Polκ function in TLS opposite a cis-syn TT dimer in conjunction with Rev1, whereas Polζ functions independently of Rev1.
Table 1.
The effects of siRNA knockdown of TLS Pols on the replicative bypass of a cis-syn TT dimer carried on the leading or lagging strand DNA template in NER-defective XPA human fibroblasts
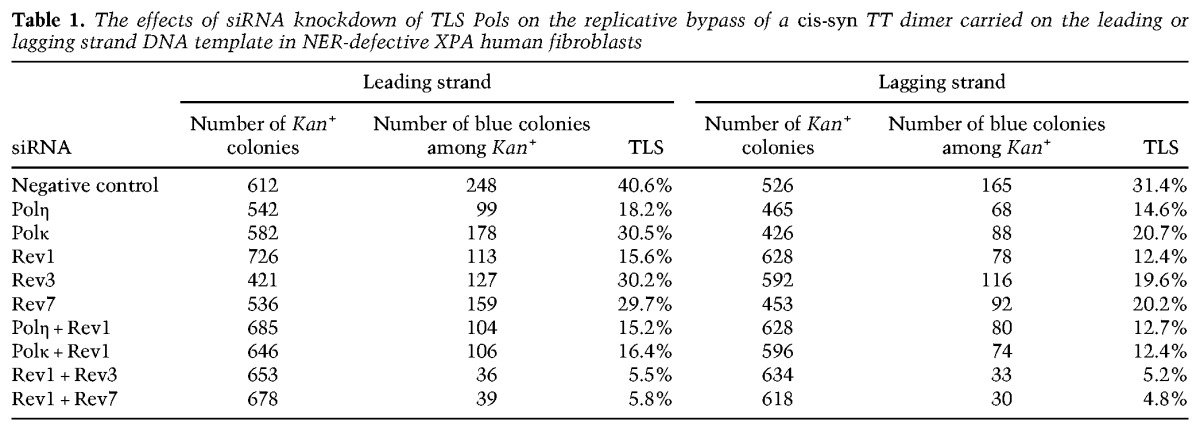
Opposite a cis-syn TT dimer carried on the lagging strand template of SV40-based plasmid, TLS occurs with a frequency of ∼30% in control siRNA-treated XPA cells (Table 1). The TLS frequency is reduced to ∼15% in Polη-depleted cells and ∼20% in Polκ- or Polζ-depleted cells, and Rev1 depletion confers a reduction in TLS frequency to ∼12%. The simultaneous depletion of Rev1 with Polη or Polκ causes no further reduction in TLS frequency than that seen upon Rev1 depletion alone, but the simultaneous depletion of Rev1 with Polζ leads to a larger reduction in TLS frequencies (∼5%) than that conferred by Rev1 depletion (∼12%). Thus, for TLS opposite a cis-syn TT dimer carried on either DNA strand, Rev1 functions together with Polη or Polκ and not with Polζ.
TLS opposite a cis-syn TT dimer occurs predominantly in an error-free manner, as only ∼2% of TLS products harbor mutations. The frequency of mutagenic TLS rises twofold to threefold in Polη-depleted cells, whereas the simultaneous depletion of Polκ and Polζ results in the complete absence of mutagenic TLS. From these and other observations, we concluded previously that Polη functions in an error-free manner, while Polκ and Polζ provide alternative mutagenic pathways of TLS opposite a cis-syn TT dimer (Yoon et al. 2009). As shown in Supplemental Table S2, Rev1 depletion leads to an approximately twofold increase in the frequency of mutagenic TLS, which we expect to have resulted from the combined inactivation of the major Polη-dependent error-free pathway of TLS and the relatively minor Polκ-dependent pathway, which acts in a weakly mutagenic manner.
The role of Rev1 in TLS opposite a (6-4) TT photoproduct in human cells
We showed previously that TLS opposite a (6-4) TT photoproduct is mediated by alternative mutagenic pathways that require Polη or Polι and by another error-free Polζ-dependent pathway (Yoon et al. 2010b). As shown in Table 2, opposite the (6-4) TT lesion carried on the leading strand template in XPA cells, the TLS frequency is not affected by depletion of either Polη or Polι, but the simultaneous depletion of Polη and Polι reduces TLS frequency to ∼27% from ∼37% in control siRNA-treated cells; in contrast, in Rev3-depleted cells, the TLS frequency is reduced to a much greater extent (∼17%). Thus, Polζ plays a major role in TLS opposite the (6-4) TT photoproduct, whereas Polη and Polι provide relatively minor pathways for replicating through this DNA lesion. Depletion of Rev1 in combination with depletion of Polη or Polι conferred no further reduction in TLS frequency than that seen upon Rev1 depletion alone (∼27%), whereas simultaneous depletion of Rev1 and Polζ led to a drastic reduction in TLS frequency (∼6%) from that conferred upon their individual depletions. The epistatic interaction of Rev1 with Polη or Polι and the synergistic interaction of Rev1 with Polζ support the inference that, for TLS opposite the (6-4) TT photoproduct also, Rev1 functions together with Polη and Polι and not with Polζ. Further support for this conclusion was provided by the genetic analysis of TLS opposite the (6-4) TT photoproduct carried on the lagging strand template (Table 2).
Table 2.
The effects of siRNA knockdown of TLS Pols on the replicative bypass of a (6-4) TT photoproduct carried on the leading or lagging strand DNA template in NER-defective XPA human fibroblasts
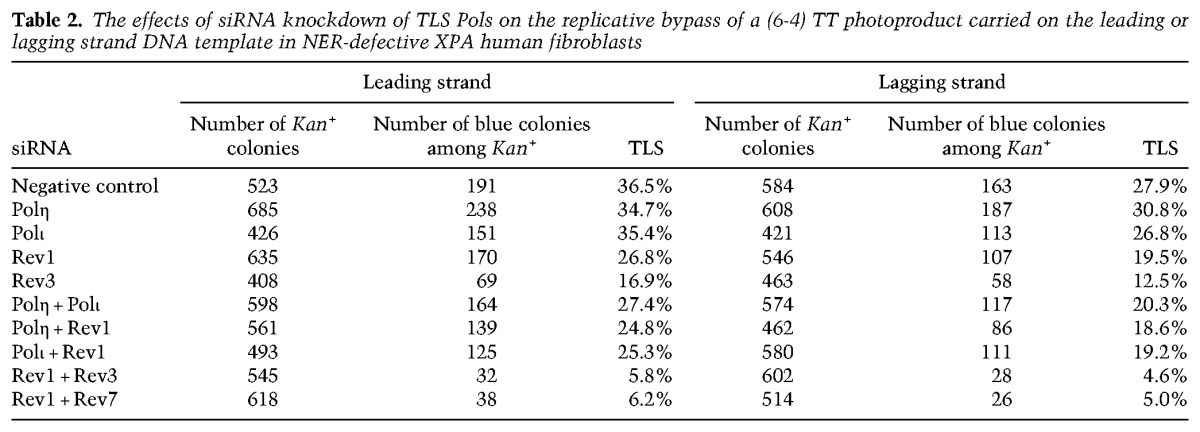
Since Polη and Polι provide alternative mutagenic pathways for TLS opposite a (6-4) TT photoproduct and since mutagenic TLS is abrogated in cells depleted for both Polη and Polι (Yoon et al. 2010b), the requirement of Rev1 for TLS mediated by these Pols would suggest that mutagenic TLS will be similarly absent in Rev1-depleted cells. As shown in Supplemental Table S3, in control siRNA-treated cells, mutagenic TLS opposite a (6-4) TT photoproduct occurs with a frequency of ∼2%, whereas no mutagenic TLS products were recovered from Rev1-depleted cells.
Thus, from the analyses of the role of Rev1 in TLS opposite a cis-syn TT dimer and its contribution to error-free or mutagenic TLS opposite this lesion, we infer a role of Rev1 in Polη- and Polκ-mediated TLS pathways, which act in an error-free or weakly mutagenic manner, respectively. From similar analyses carried out for a (6-4) TT photoproduct, we infer a role of Rev1 in mediating Polη- and Polι-mediated TLS pathways, which act in a weakly mutagenic fashion. Altogether, we conclude that, in human fibroblasts, Rev1 is specifically required for TLS mediated by Polη, Polκ, and Polι.
The effects of Rev1 depletion on UV mutagenesis in the chromosomal cII gene in MEFs
Next, we examined the effects of Rev1 depletion on mutagenesis resulting from TLS opposite CPDs and (6-4) photoproducts formed at TT, TC, and CC dipyrimidine sites in the cII gene that has been integrated into the genome of BBMEF cells (You et al. 2001). This system provides a convenient and reliable way to measure the effects of DNA-damaging agents on mutagenesis and has been shown to exhibit responses similar to those observed with endogenous chromosomal genes (You and Pfeifer 2001; You et al. 2001; Besaratinia and Pfeifer 2006). To examine UV mutagenesis resulting from TLS opposite CPDs, the (6-4) photoproducts were selectively removed from the genome by expressing a (6-4) photoproduct-specific photolyase gene in the BBMEF cell line, and the effects of siRNA depletion of TLS Pols (Supplemental Fig. S2C) were analyzed. As has been reported previously and independently verified by us, this experimental protocol allows for the complete removal of (6-4) photoproducts (You et al. 2001; Yoon et al. 2009).
As shown in Table 3, in unirradiated cells treated with control siRNA, spontaneous mutations in the cII gene in BBMEF cells occur at a frequency of ∼15 × 10−5. In UV-irradiated mouse cells expressing (6-4) photoproduct-specific photolyase and exposed to photoreactivating light to remove (6-4) photoproducts, mutations in the cII gene, which would result from replication through CPDs, are elevated to a frequency of ∼45 × 10−5. In UV-irradiated cells, siRNA depletion of Polη confers an increase in mutation frequency to ∼105 × 10−5, whereas depletion of Polκ or Rev3 results in an about equal reduction in mutation frequencies, to ∼30 × 10−5. As expected from the requirement of Rev1 for error-free TLS mediated by Polη and for Polκ-dependent mutagenic TLS, Rev1 depletion confers a reduction in mutation frequencies to ∼75 × 10−5 compared with that in Polη-depleted cells (∼105 × 10−5). Our observations that simultaneous depletion of Rev1 with Polη or Polκ causes no further reduction in mutation frequencies than that seen upon depletion of Rev1 alone, whereas simultaneous depletion of Rev1 with Rev3 leads to a reduction in mutation frequencies nearly to the level observed in unirradiated cells, lend further support for a role of Rev1 in Polη- and Polκ-mediated TLS opposite CPDs but not in Polζ-dependent TLS.
Table 3.
UV-induced mutation frequencies in the cII gene in BBMEF cells expressing a (6-4)PP photolyase and treated with siRNAs for different TLS Pols

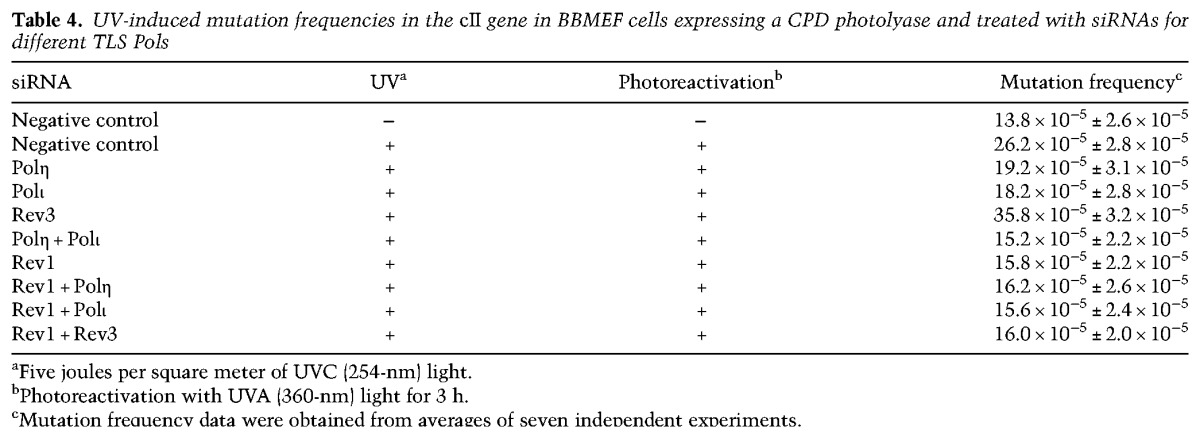
To examine UV mutations that result from TLS opposite (6-4) photoproducts formed at dipyrimidine sites in the cII gene, CPDs were selectively removed from the genome in BBMEF cells by expression of a CPD-specific photolyase gene. As shown in Table 4, spontaneous mutations in this cell line occur at a frequency of ∼14 × 10−5, and in UV-irradiated cells, mutation frequency resulting from TLS opposite (6-4) photoproducts rises to ∼26 × 10−5. Since TLS opposite (6-4) photoproducts occurs by Polη- and Polι-dependent mutagenic pathways and via a Polζ-mediated error-free pathway, UV-induced mutation frequencies resulting from TLS opposite (6-4) photoproducts decline to ∼19 × 10−5 in cells depleted for Polη or Polι, and in cells depleted for both Polη and Polι, mutation frequencies are reduced near to the level that occurs in unirradiated cells. Depletion of Rev1 confers a reduction in UV-induced mutation frequencies to nearly the same level as that in unirradiated cells, and simultaneous depletion of Rev1 with Polη or Polι causes no further reduction in mutation frequencies over that in Rev1-depleted cells. From these observations indicating epistasis of Rev1 over Polη and Polι and from the observation that the elevated mutagenesis in Rev3-depleted cells (∼36 × 10−5) is reduced in cells additionally depleted for Rev1 to nearly spontaneous levels (∼16 × 10−5), we conclude that Rev1 functions in TLS opposite (6-4) photoproducts together with Polη and Polι and not with Polζ.
Table 4.
UV-induced mutation frequencies in the cII gene in BBMEF cells expressing a CPD photolyase and treated with siRNAs for different TLS Pols
Since CPDs and (6-4) photoproducts account for ∼75% and 25% of UV-induced mutations, respectively, in UV-irradiated cells containing both these lesions, we expect that, because of inactivation of Polκ-dependent mutagenic TLS opposite CPDs and because of inactivation of Polη- and Polι-mediated error-prone TLS opposite (6-4) photoproducts, Rev1 depletion would confer a reduction in UV-induced mutation frequency compared with that observed in Polη-depleted cells. As shown in Supplemental Table S4, UV-induced mutations occur at a frequency of ∼52 × 10−5 in cells treated with control siRNA, and the mutation frequency rises to ∼95 × 10−5 in Polη-depleted cells and is reduced to ∼70 × 10−5 in Rev1-depleted cells. Thus, the overall effects of Rev1 depletion on Polη-dependent error-free TLS and Polκ-dependent mutagenic TLS opposite CPDs and on Polη- and Polι-mediated mutagenic TLS opposite (6-4) photoproducts result in elevated UV mutagenesis, but the level of enhancement of UV mutagenesis is reduced in Rev1-depleted cells compared with that in Polη-depleted cells.
The pattern of UV mutations in the cII gene in Rev1-depleted MEFs
In control siRNA-treated BBMEF cells, UV mutations in the cII gene resulting from TLS opposite CPDs occur primarily at 11 dipyrimidine sites, #1–#11, (Fig. 1A, panel I), and the pattern of UV mutations remains the same in Polη-depleted MEFs (Yoon et al. 2009). However, and interestingly, the pattern of UV mutations differs strikingly in Polκ- versus Polζ-depleted cells, as different hot spots remain in Polκ-depleted cells (#4, #5, #7, and #8) versus Polζ-depleted cells (#1, #2, #3, and #6) (Yoon et al. 2009). As expected from the role of Rev1 in Polη- and Polκ-dependent TLS opposite CPDs, the pattern of hot spots that persists in Rev1-depleted cells (#4, #5, #7, and #8) resembles that in Polκ-depleted cells (Fig. 1A, panel II). Since Polζ function is not affected in Rev1-depleted cells and since hot spots at position #4, #5, #7, and #8 are eliminated upon Polζ depletion, the observed pattern of hot spots is consistent with the involvement of Rev1 in Polκ-mediated mutagenic TLS but not in Polζ-dependent mutagenic TLS.
Figure 1.

UV-induced (5 J/m2) mutational spectra in the cII gene in BBMEF mouse cells. (A) Mutational spectra in the cII gene resulting from TLS opposite CPDs in BBMEFs expressing (6-4) PP photolyase. (Panel I) In control siRNA-treated cells, UV-induced mutational hot spots in the cII gene from nucleotide positions 25–288 are clustered at 11 different positions, #1–#11 (Yoon et al. 2009). A frequently occurring spontaneous mutational hot spot is indicated by “g” at position 223. (Panel II) UV-induced mutations that result from TLS opposite CPDs in Polκ siRNA-treated cells are shown above the sequence, and mutations that occur in Rev1 siRNA-treated cells are shown below the sequence. (B) Mutational spectra in the cII gene resulting from TLS opposite (6-4) photoproducts in BBMEF cells expressing CPD photolyase. UV-induced mutations that result from TLS opposite (6-4) photoproducts in negative control siRNA-treated cells are clustered at four different positions, #1–#4 (Yoon et al. 2010b), and are shown above the sequence, whereas mutations that occur in Rev1 siRNA-treated cells are shown below the sequence. (NC) Negative control.
UV-induced mutations resulting from TLS opposite (6-4) photoproducts in the cII gene occur predominantly at dipyrimidine sites indicated by #1, #2, #3, and #4 (Fig. 1B), and these UV-induced hot spots are absent in cells depleted for Polη and Polι (Yoon et al. 2010b). As would be expected from the requirement of Rev1 for Polη- and Polι-dependent mutagenic TLS opposite (6-4) photoproducts, these hot spots are absent in Rev1-depleted cells also (Fig. 1B).
UV mutagenesis in the cII gene in Rev1−/−, Rev1+/−, and Rev1+/+ primary MEFs
We also generated Rev1+/+, Rev1+/−, and Rev1−/− mice in the C57BL/6 background. Rev1−/− mice suffer from poor viability and growth retardation (Supplemental Fig. S3A). Rev1+/+, Rev1+/−, and Rev1−/− primary embryonic fibroblasts harboring the cII gene were derived as described in the Materials and Methods. As shown in Supplemental Table S5, in unirradiated Rev1+/+, Rev1+/−, and Rev1−/− MEFs, mutations in the cII gene arise at a frequency of ∼5 × 10−5. In Rev1+/+ MEFs, UV irradiation at 5 J/m2 elevates the mutation frequency to ∼32 × 10−5. As expected from the results of siRNA knockdown of Rev1 (Table 3; Supplemental Table S4), the frequency of UV-induced mutations rises to ∼55 × 10−5 in both of the Rev1−/− cell lines that we generated. We also determined whether cII mutation frequencies in UV-irradiated Rev1+/− heterozygous MEFs remain the same as in Rev1+/+ wild-type cells or whether Rev1 protein becomes limiting in Rev1+/− heterozygotes, which then affects UV-induced mutation frequencies. From our observation that, compared with the mutation frequency of ∼32 × 10−5 in Rev1+/+ MEFs, UV-induced mutation frequency in the cII gene rises to ∼41 × 10−5 in Rev1+/− MEFs, we infer that, in the heterozygotes, Rev1 becomes limiting and that the reduced levels of Rev1 adversely affect Polη-mediated error-free TLS opposite CPDs.
The requirement of Rev1 for assembly of Polη, Polκ, and Polι but not Polζ into replication foci in UV-irradiated human and mouse cells
Next, we examined whether Rev1 was required for the accumulation of Polη, Polκ, and Polι into replication foci in UV-irradiated human fibroblasts. For this purpose, XPV cells expressing GFP-Polη, GFP-Polκ, or GFP-Polι were treated with control siRNA or Rev1 siRNA. As shown in Figure 2A, the incidence of Polη foci is greatly enhanced in UV-irradiated XPV cells treated with negative control siRNA, whereas in cells treated with Rev1 siRNA, there is no UV-induced accumulation of Polη into replication foci. We also observed an increase in the frequency of Polκ and Polι foci in UV-irradiated XPV cells, and the incidence of these foci fell dramatically in Rev1-depleted cells, (Fig. 2B,C). In contrast to the large reduction in UV-induced Polη, Polκ, and Polι foci in Rev1-depleted XPV cells, Rev1 depletion had no untoward effect on Rev7 focus formation in UV-irradiated XPV cells (Fig. 2D). Quantification of these data show that, in Rev1-depleted cells, the frequency of cells containing elevated levels of Polη, Polκ, and Polι foci in UV-irradiated cells is reduced to the levels observed in unirradiated cells (Fig. 2 A–C), whereas the frequency of cells containing Rev7 foci remained the same in UV-irradiated cells regardless of whether cells were treated with negative control siRNA or Rev1 siRNA (Fig. 2D). Similarly, Rev1 depletion had no effect on the frequency of cells containing Rev3 foci (data not shown). Thus, Rev1 is indispensable for the accumulation of Polη, Polκ, and Polι into replication foci in UV-irradiated XPV cells but is not required for the accumulation of Polζ into replication foci. We also verified the requirement of Rev1 for the accumulation of Polη, Polκ, and Polι but not Polζ into replication foci in UV-irradiated wild-type human fibroblasts (Fig. 2E).
Figure 2.

Requirement of Rev1 for accumulation of Polη, Polι, and Polκ into replication foci in UV-damaged human fibroblasts. XPV (XP30R0) cells were transfected with GFP-Polη (A), GFP-Polκ (B), GFP-Polι (C), or GFP-Rev7 (D), and, after 16 h, cells were treated with control siRNA or Rev1 siRNA followed by treatment with 20 J/m2 UVC. Representative images of GPF-Polη (A), GFP-Polκ (B), GFP-Polι (C) , and GFP-Rev7 (D) foci are shown at the left, and quantification of cells containing these foci is shown at the right. Error bars represent the standard deviation of three independent experiments. (E) Wild-type human fibroblasts were transfected with GFP-Polη, GFP-Polκ, GFP-Polι, or GFP-Rev7 and exposed to control siRNA or Rev1 siRNA, and the percentage of cells containing foci of these TLS Pols in UVC-treated or untreated cells was quantified. (NC) Negative control.
To determine the requirement of Rev1 for recruitment of Y family Pols into replication foci in mouse cells, we examined the assembly of GFP-Polη and GFP-Polκ into foci in UV-irradiated Rev1+/+ and Rev1−/− MEFs. The incidence of Polη and Polκ foci is greatly enhanced in UV-irradiated Rev1+/+ MEFs (Supplemental Fig. S4A) whereas in UV-irradiated Rev1−/− MEFs, the incidence of Polη and Polκ foci remained the same as in unirradiated MEFs (Supplemental Fig. S4B). The incidence of UV-induced Rev7 foci, however, was not affected in the absence of Rev1, as it remained the same in Rev1−/− and Rev1+/+ primary MEFs (Supplemental Fig. S4C). Thus, in mouse cells as well, Rev1 is indispensable for the UV-induced assembly of Y family Pols into replication foci but is not required for Polζ assembly into foci.
Requirement of Polη, Polκ, and Polι for accumulation of Rev1 into replication foci in UV-irradiated human cells
To determine whether the accumulation of Rev1 into foci in UV-damaged cells occurs independently of Polη, Polκ, and Polι or whether these Pols are required for the assembly of Rev1 into replication foci, we examined whether the incidence of Rev1 foci was reduced in UV-irradiated cells depleted of Polη, Polκ, and Polι (Supplemental Fig. S5). Approximately 15% of unirradiated wild-type cells contain Rev1 foci, whereas the frequency of cells containing Rev1 foci rises to ∼50% in UV-irradiated cells (Supplemental Fig. S5A,B). Interestingly, in UV-irradiated cells, Polη depletion led to an almost 50% reduction in the frequency of cells containing Rev1 foci (Supplemental Fig. S5A,B); this large effect of Polη depletion on the proficiency of Rev1 to assemble into foci closely parallels the major contribution of Polη to TLS opposite CPDs (Table 1). A similar reduction in Rev1 foci is observed in UV-irradiated XPV fibroblasts; moreover, the simultaneous depletion of Polκ and Polι in XPV cells abrogated all of the UV-induced Rev1 foci in XPV cells (Supplemental Fig. S5C). In contrast to the requirement of Polη, Polκ, and Polι for enabling the assembly of Rev1 into foci, depletion of Rev7 had no inhibitory effect on Rev1 focus formation (Supplemental Fig. S5B).
Requirement of Rad18 for the assembly of TLS Pols into replication foci in UV-irradiated human fibroblasts
Previously, we showed that Rad18 is indispensable for promoting TLS through UV lesions in human and mouse cells (Yoon et al. 2012b). This requirement of Rad18 derives from its roles as the E3 component of the Rad6-Rad18 ubiquitin-conjugating enzyme complex, which ubiquitylates PCNA at the Lys164 residue, which is an essential prerequisite for TLS to occur. Since the UV sensitivity of cells simultaneously depleted for Rad18 and a TLS Pol such as Polη, Polκ, or Polζ remains the same as that of cells depleted for Rad18 alone, Rad18 exhibits an epistatic relationship with the various TLS Pols (Yoon et al. 2012b).
To determine whether Rad6–Rad18-mediated PCNA ubiquitylation was required for the accumulation of TLS Pols into replication foci, we examined the effects of Rad18 depletion (Supplemental Fig. S6A) on the assembly of TLS Pols into replication foci in UV-irradiated human fibroblasts. Our observations that the UV-induced assembly of Polη, Polκ, Rev1, or Rev7 does not occur in Rad18-depleted cells (Supplemental Fig. S6B,C) show that Rad6–Rad18-mediated PCNA ubiquitylation is a necessary prerequisite for initiating the assembly of TLS Pols at DNA lesion sites.
Epistasis of Rev1 over Polη, Polκ, and Polι for UV survival in human and mouse fibroblasts
The requirement of Rev1 for TLS mediated by Polη and Polκ opposite CPDs and for TLS by Polη and Polι opposite (6-4) photoproducts predicts that Rev1 depletion would confer a greater increase in UV sensitivity than that imparted by depletion of Polη, Polκ, or Polι alone, and, importantly, the UV sensitivity of Rev1-depleted cells would not increase upon depletion of either Polη, Polκ, or Polι. To determine whether such an epistatic relationship exists between Rev1 and these other Y family Pols, we examined the UV sensitivity of wild-type human fibroblasts treated with control siRNA, Rev1 siRNA, or siRNAs for other TLS Pols. As shown in Figure 3A, UV survival was reduced by ∼55% in Rev1-depleted cells, whereas among the other Y family Pols, UV survival was affected the most upon Polη depletion, no reduction in UV survival occurred in cells depleted for Polι, and a modest reduction in UV survival was noted upon Polκ depletion. These effects of TLS Pols on UV survival approximate their relative contributions to the replication of UV-damaged DNA (Tables 1, 2). Our observations that the UV sensitivity of cells depleted for Rev1 in combination with depletion of Polη, Polκ, or Polι remains the same as that of cells depleted for Rev1 alone are in accord with an epistatic interaction of Rev1 with Polη, Polκ, and Polι (Fig. 3A).
Figure 3.

UV sensitivity of human and mouse fibroblasts depleted for Rev1 and other TLS Pols. Human and mouse cells were treated with the siRNA for 48 h and then irradiated with 10 J/m2 of UVC light. Cells were incubated for an additional 48 h after UV irradiation, and UV sensitivity was determined by the MTT assay. The data represent the mean and standard deviation of results from five independent experiments. (A) UV survival of human fibroblast (HF) cells. (B) UV survival of BBMEF cells.
Since Polζ functions in TLS opposite UV lesions in a Rev1-independent manner, we expect that simultaneous depletion of Rev1 with Polζ will confer an increase in UV sensitivity compared with that in Rev1-depleted cells. Accordingly, we found that depletion of Rev1 in conjunction with depletion of Rev3 or Rev7 results in a greater reduction in UV survival than that observed in Rev1-depleted cells (Fig. 3A). The increased UV sensitivity of cells simultaneously depleted for Rev1 and Polζ adds further support for a Polζ-independent role of Rev1 in lesion bypass. As expected, the UV sensitivity of human fibroblasts simultaneously depleted for Rad18 and Rev1 remains the same as that of Rad18-depleted cells, indicating epistasis (Fig. 3A).
We also verified that the UV sensitivity of MEFs depleted for Rev1 in combination with depletion of Polη, Polκ, or Polι remains the same as that of cells depleted for Rev1 alone (Fig. 3B). Furthermore, the simultaneous depletion of Rev1 and Polζ resulted in a greater reduction in UV survival than that in Rev1-depleted cells, and the UV survival of Rad18-depleted cells is not affected upon Rev1 depletion (Fig. 3B). Thus, in MEFs also, Rev1 exhibits epistatic interactions with Polη, Polκ, and Polι and not Polζ, and Rev1 functions in TLS in a Rad18-dependent manner.
UV survival of Rev1−/−, Rev1+/−, and Rev1+/+ primary MEFs
Our observations that the frequency of UV-induced mutations is elevated in Rev1+/− primary MEFs suggested that the levels of Rev1 needed for its role in TLS become limiting in these cells (Supplemental Table S5). To determine whether such a semidominant effect of Rev1 knockout mutation extends to other phenotypes, we examined the UV survival of Rev1+/+, Rev1+/−, and Rev1−/− primary MEFs. As shown in Supplemental Figure S3C, compared with the UV survival of Rev1+/+ MEFs, the UV survival of Rev1+/− cells was reduced by ∼30%, whereas the survival of Rev1−/− cells was reduced by ∼70%. Thus, the complete absence of Rev1 has a very drastic effect on UV survival, and a single copy of Rev1 does not suffice for wild-type levels of UV survival.
Discussion
Here we determined the role of Rev1 in mediating replication through the UV lesions in human and mouse fibroblasts and provided several lines of evidence that show that Rev1 functions with Polη, Polι, and Polκ and not Polζ. Briefly, we found that (1) for TLS opposite a cis-syn TT dimer carried on the DNA template for leading or lagging strand replication in SV40-based duplex plasmid in human cells, Rev1 interacts epistatically with Polη and Polκ but not Polζ; (2) for TLS opposite a (6-4) TT photoproduct carried on the SV40-based duplex plasmid in human cells, Rev1 interacts epistatically with Polη and Polι but not Polζ; (3) for UV-induced mutations resulting from replication through CPDs or (6-4) photoproducts in the genomic cII gene in mouse cells, Rev1 displays epistasis with Polη, Polι, and Polκ but not Polζ; (4) the accumulation of Polη, Polι, and Polκ into replication foci in UV-damaged human and mouse cells requires Rev1, but the accumulation of Polζ into replication foci is not affected in Rev1-depleted cells; (5) in both human and mouse fibroblasts, the UV sensitivity of Rev1-depleted cells is not affected upon the additional depletion of Polη, Polι, or Polκ; however, the UV sensitivity of Rev1-depleted cells is enhanced upon the additional depletion of Polζ; and (6) since the above noted studies were carried out with human and mouse fibroblasts that have been immortalized, to confirm that the genetic mechanisms of TLS in these cells do not differ from those in normal cells, we derived primary embryonic fibroblasts from Rev1+/+, Rev1+/−, and Rev1−/− mice generated in the C57BL/6 genetic background. As expected from the predominant role of Rev1 in the replication of UV-damaged DNA in an error-free manner, the frequency of UV-induced mutations rises in Rev1−/− cells, and the lack of Rev1 confers a large reduction in UV survival.
Since the Y family Pols Polη, Polι, Polκ, and Rev1 function in highly specialized ways in promoting replication through DNA lesions that include UV lesions (Johnson et al. 1999b; Biertumpfel et al. 2010; Silverstein et al. 2010), DNA cross-links (Ummat et al. 2012), lesions that disrupt Watson-Crick pairing (Nair et al. 2006), and lesions that protrude into the DNA minor groove (Nair et al. 2008), these Pols would play a major role in promoting replication through a large variety of DNA lesions. The dual requirement of Rev1 as a DNA Pol that can specifically act in TLS opposite bulky N2-dG adducts (Nair et al. 2005; Swan et al. 2009) and its more significant scaffolding role as an indispensable component of TLS mediated by Polη, Polι, and Polκ would suggest that Rev1 deficiency will have a more pronounced effect on genomic stability than the deficiency of Polη, Polι, or Polκ. In accord with this, Rev1-null mice in the C57Bl/6 background display poor viability, a greatly reduced body size, and developmental defects (Supplemental Fig. S3A; Jansen et al. 2006), whereas null mutants of Polη, Polκ, or Polι are viable and exhibit no growth or developmental defects. Since the mutational inactivation of the DNA Pol function of Rev1 has no obvious debilitating effects on viability and development (Kano et al. 2012), presumably these severe effects of the Rev1-null mutation derive at least in part from the defects engendered in lesion bypass by Rev1 deficiency impacting on the ability of Polη, Polι, and Polκ to function in TLS and, to a much lesser extent, from the lack of its Pol function.
The requirement of Rev1 for TLS mediated by Polη, Polι, and Polκ opposite UV lesions observed in our studies differs from the role that Rev1 plays in TLS that occurs during gap-filling reactions in human or mouse cells. The observations that TLS opposite a (6-4) TT photoproduct carried on a gapped plasmid is greatly reduced in cells lacking Rev1 or Rev3 have suggested that Rev1 and Polζ function together in mediating TLS opposite this lesion in the gapped plasmid (Jansen et al. 2009; Shachar et al. 2009). However, since no epistasis analyses had been carried out in those previous studies, to ascertain that Rev1 interacts epistatically with Polζ, we examined the effects of depletions of Rev1 and Rev3 individually and simultaneously on TLS opposite a (6-4) TT photoproduct carried on the gapped plasmid in human fibroblasts (Supplemental Fig. S7). From our observation that depletion of Rev1, Rev3, or both Rev1 and Rev3 generates the same level of reduction in gap filling (Supplemental Table S6), we conclude that, for gap filling opposite a (6-4) TT photoproduct, Rev1 functions together with Polζ. Polη plays a major role in TLS opposite a cis-syn TT dimer carried on the gapped plasmid in human cells (Supplemental Table S7). Our observations that depletion of Rev1 or Rev3 has no effect on the frequency of gap filling opposite this lesion have indicated that Rev1 is not required for Polη-mediated TLS in gap-filling reactions (Supplemental Table S7). Thus, for TLS in a gapped plasmid, Rev1 functions together with Polζ and not Polη.
The requirement of Rev1/Polζ for TLS opposite a (6-4) TT photoproduct in a gapped plasmid suggested that, similar to that in yeast, in human cells also, this complex may function in a highly error-prone manner. To examine this, we analyzed the mutagenicity of Rev1/Polζ opposite (6-4) TT photoproduct in the gapped plasmid. Sequence analyses of 288 TLS products obtained from negative control siRNA-treated cells showed that ∼12% of them harbor mutational changes that include incorporation of G or T opposite the 3′ or 5′ site of the photoproduct, whereas ∼2% of mutations occurred in the flanking 3′ or 5′ base and not opposite the two Ts of the photoproduct (Supplemental Table S8). In contrast, the (6-4) TT photoproduct carried in the same sequence context in the duplex plasmid as in the gapped plasmid generated only ∼2% mutational products (Supplemental Table S3; Yoon et al. 2010b). Thus, Rev1/Polζ-dependent TLS opposite a (6-4) TT photoproduct in the gapped plasmid incurs approximately sixfold more mutational events than the mutagenic TLS that occurs opposite this lesion during replication and is mediated by the Rev1-dependent Polη and Polι pathways.
In the lack of requirement of Rev1 for Polη function but in its requirement for Polζ function, the role of Rev1 in TLS in the gapped plasmid in human cells resembles that in yeast, where Rev1 acts as an indispensable component for TLS mediated by Polζ but not for Polη-dependent TLS. Moreover, since TLS in yeast occurs post-replicatively in gaps (Daigaku et al. 2010; Karras and Jentsch 2010) and since a concordance exists in the role of Rev1 for TLS in yeast and in the gapped plasmid in human cells, we surmise that the requirement of Rev1 for Polζ-mediated TLS in normal mammalian cells may be indicative of TLS that occurs post-replicatively rather than in coordination with the replication fork.
Because of the highly specialized roles of Y family Pols in inserting nucleotides opposite DNA lesions and because their active sites are adapted to incorporate a correct nucleotide opposite the DNA lesions, Rev1, via its role as an indispensable component of Y family Pols, would function in predominantly error-free lesion bypass in human cells. Hence, Rev1 would contribute to genome stability in normal human cells. In cancer cells, however, Rev1 may function together with Polζ, and the Rev1/Polζ complex may mediate a highly mutagenic mode of TLS (Doles et al. 2010; Xie et al. 2010). The adoption of error-prone TLS processes by cancer cells may contribute to their high mutagenicity and their ability to acquire resistance to chemotherapeutic drugs (Doles et al. 2010; Xie et al. 2010). Such a role of Rev1 in conjunction with Polζ in mediating highly error-prone TLS in cancer cells may be similar to the role that the Rev1/Polζ complex plays in yeast or in gapped plasmid in human cells.
Intriguingly, in chicken DT40 cells, Rev1 functions in a Rad18-, PCNA ubiquitylation-, and Polη-independent manner (Edmunds et al. 2008). In its lack of requirement for Rad6–Rad18-mediated PCNA ubiquitylation, the role of Rev1 in DT40 cells differs vastly from that in human or mouse fibroblast cells, as Rad18 is indispensable for TLS in these cells (Yoon et al. 2012b). Furthermore, we show here that Rad18 is indispensable for the accumulation of TLS Pols into replication foci in UV-irradiated human fibroblasts; thus, Rad6–Rad18-mediated PCNA ubiquitylation is essential for the targeting of TLS Pols to lesion sites in normal human cells. Although the evolutionary divergence of TLS processes between aves and mammals may account for the strikingly different roles of Rev1 in chicken DT40 cells versus that in normal human fibroblasts, a more likely explanation is that the very different role of Rev1 in TLS in chicken DT40 cells results from the myriad genetic changes that would have occurred during the transition of normal chicken cells to tumor cells. The DT40 cell line originates from avian leucosis virus (ALV)-induced lymphomas; these cells contain proviral DNA sequences integrated upstream of the c-myc proto-oncogene and express elevated levels of c-myc mRNA (Baba et al. 1985). Since the regulatory processes in DT40 cells differ from that in normal cells, the genetic control of lesion bypass processes would have diverged in DT40 cells from that in normal chicken cells. Furthermore, our observations imply that the genetic control of TLS processes as inferred from studies in DT40 cells has no bearing on the understanding of TLS processes that occur in normal human cells.
How does Rev1 affect the TLS function of Polη, Polι, and Polκ in human cells? Since we found no effect of Rev1 on DNA synthesis by Polη or other Y family Pols opposite DNA lesions, Rev1 must affect the function of these TLS Pols in other ways. The requirement of Rev1 for the assembly of Polη, Polι, and Polκ into replication foci in UV-damaged cells suggests a role of Rev1 in the placement of these Pols at the damage site; however, our observations that Polη, Polι, and Polκ also affect the UV-induced assembly of Rev1 into replication foci would suggest that, in the binary complex of Rev1 with Polη or with other TLS Pols, both the proteins are involved in modulating their accumulation at DNA lesion sites. To account for such a role, we suggest that both of the proteins in the binary complex are involved in physical and functional interactions with other proteins and that they all together form a multiprotein assembly and are required for the formation of stable and functional assemblages at DNA damage sites. This proposal raises many interesting questions, such as the identity of the proteins that are components of such multiprotein assemblies and how they affect the proficiency and fidelity of lesion bypass. The identification of Rev1 as an indispensable component of Polη, Polι, and Polκ lays the groundwork for deciphering the various components of the TLS machinery and analyzing their respective roles in lesion bypass in normal human cells.
Concluding remarks
The major findings of this study and their possible implications are as follows: (1) In TLS that occurs during replication of UV-damaged DNA in human cells, Rev1 functions as an indispensable component of Y family Pols but not of Polζ and promotes predominantly error-free lesion bypass. (2) Rev1 plays an essential role in the assembly of Y family Pols at UV lesion sites but is not required for Polζ assembly. (3) A role of Rev1 in the assembly of multiprotein complexes of Y family Pols at DNA lesion sites, as proposed here, raises the interesting possibility that all of the Y family Polη, Polι, and Polκ and Rev1 share identical multiprotein assemblies. This suggests that the mechanism of TLS by Y family Pols would differ from that of Polζ or other TLS Pols. (4) The genetic mechanisms and mutagenicity of TLS in a gapped plasmid differ vastly from TLS that occurs during replication in human cells.
Materials and methods
Construction of plasmid vectors containing a cis-syn TT dimer or a (6-4) TT photoproduct
The heteroduplex vectors containing a cis-syn TT dimer or a (6-4) TT photoproduct on the leading or lagging strand template were constructed as described previously (Yoon et al. 2009, 2010a).
In vivo TLS assays in human cells
For human Rev1 siRNA knockdown, high-performance liquid chromatography (HPLC)-purified duplex siRNA for human Rev1 was purchased from Ambion. The sense sequence of human Rev1 siRNA was 5′-GCAUCAAAGCUGGACGACU-3′, and the efficiency of its knockdown was verified by Western blot analysis (Supplemental Fig. S2). Anti-Rev1 antibody and anti-Rev7 antibody were purchased from Santa Cruz Biotechnology and used for Western blot analysis. The antibodies used for determining the siRNA knockdown efficiency of other TLS Pols as well as the detailed methods for TLS assays have been described previously (Yoon et al. 2009, 2010a,). For complementation assay, normal human fibroblast (MRC5) cells were transfected with pCMV7.1-3xFlag vectors (Sigma) containing the wild-type or siR form of Polη or Rev1. Cells stably expressing these recombinant proteins were selected by zeocin (Invitrogen), and protein expression and siRNA knockdown efficiency were analyzed by Western blot analysis (Supplemental Fig. S2B). Stably transfected cells were used for TLS assay as described previously (Yoon et al. 2014).
UV survival assays
Normal human fibroblasts or BBMEFs were transfected with siRNAs, and, 48 h after siRNA transfection, cells were treated with UV. For the UV irradiation, cells were washed with PBS buffer and irradiated at 10 J/m2 of UVC light in PBS buffer. After irradiation, fresh growth medium without caffeine was added, and cells were incubated for an additional 48 h. UV survival was determined by the MTT assay (Promega) as described in the manufacturers’ manuals. Briefly, 200 µL of MTT assay solution was added to each well and incubated for 30 min. Cell viability was determined by the measurement of OD at 490 nm.
Big blue transgenic mouse cell line and siRNA knockdown
The transgenic BBMEF cells expressing either the (6-4) PP photolyase or the CPD photolyase were grown in Dulbecco modified Eagle's medium (DMEM) containing 10% FBS (GenDEPOT) and antibiotics. HPLC-purified duplex siRNA for mouse Rev1 was purchased from Ambion. The sense sequence of mouse Rev1 siRNA was 5′-GGCACUAUGUCAGUGUUGA-3′, and the efficiency of its knockdown was confirmed by Western blotting with mouse Rev1 antibodies that we raised in rabbits. The siRNA knockdown efficiency of other mouse TLS genes has been described previously (Yoon et al. 2009; Yoon et al. 2010a). For the cII mutation assay, cells were plated on 100-mm plates at 50% confluence (∼5 × 106 cells), and 500 pmol of synthetic duplex siRNAs was transfected using 50 μL of Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's instructions.
UV irradiation, photoreactivation, and cII mutational assays in siRNA-treated BBMEFs
The methods for cII mutational analyses opposite CPDs and (6-4) photoproducts have been described previously (Yoon et al. 2009, 2010a).
Fluorescence microscopy in human cells
Full-length human Polη, Polι, Polκ, Rev1, and Rev7 were subcloned into the pEGFP N1 vector (Clonetech) and transiently transfected into SV40 transformed XPV (XP30R0) or normal human fibroblasts. After 16 h of incubation, cells were suspended and treated with Rev1 siRNA and cultured on a coverslip in six-well plates with 50% confluence. After 48 h, cells were treated with 20 J/m2 of UVC. For the UV irradiation, cells were washed with PBS buffer and irradiated with UVC light in the presence of PBS buffer. After irradiation, cells were incubated in fresh growth medium for 6 h. After washing with PBS buffer, cells were fixed with 4% paraformaldehyde for 30 min. Fixed cells were permeablized with 0.2% Triton X-100 in PBS buffer. Nuclear staining was performed with DAPI (Molecular Probe) in PBS buffer for 20 min. The fluorescent images were visualized and captured by fluorescence microscope (Nikon fluorescence microscope).
Generation of Rev1−/− mice and Rev1−/− transgenic BBMEF cell lines
Rev1+/− mice (TIGM, IST10468C11) generated by the gene trap method (Supplemental Fig. S3B) were purchased from TIGM. Rev1+/− mice were crossed with C57BL/6 to generate male and female Rev1+/− mice. Rev1+/− mice were then intercrossed to generate Rev1−/− mice. All animal studies were approved by the University of Texas Medical Branch Institutional Animal Care and Use Committee. To identify the Rev1 knockout, tail DNA genotyping was performed using the primers IST10468C11F (5′-CATGTGAAGTGGAGAGATCAAAGC-3′) and IST10468C11R (5′-AGTACACAGCTACAAGAGTATGC-3′) for the wild-type allele and primers IST10468C11F (5′-CATGTGAAGTGGAGAGATCAAAGC-3′) and LTR-rev (5′-ATAAACCCTCTTGCAGTTGCATC-3′) for the Rev1-null allele. All primers were purchased from Sigma-Aldrich. To produce Rev1−/− transgenic primary BBMEFs, big blue transgenic male mice (C57BL/6 background) were purchased from Agilent-Stratagene and crossed with Rev1+/− females. The big blue transgene was identified by tail DNA genotyping with primers cII forward primer and cII reverse primer, as described previously (You et al. 1998), and Rev1 genotype was determined by tail DNA genotyping as described above. Primary MEFs were isolated from embryos derived from intercrossing of Rev1+/− big blue transgenic parents according to published procedures (Tommasi et al. 2005). In brief, mouse embryos harvested in utero at 13.5 d of gestation were roughly minced and incubated with trypsin for 20 min at room temperature. Homogenous cell suspensions were then added to 25 mL of DMEM (GeneDepot) supplemented with 10% fetal calf serum. Early-passage (P < 5) MEFs were used for all experiments. To check mouse Rev1 expression, cell extracts were prepared from Rev1+/+ and Rev1−/− MEFs, and Western blot analysis was done with rabbit anti-mouse Rev1 antibody against mouse Rev1 protein.
Fluorescence microscopy in Rev1+/+ and Rev1−/− MEFs
To examine UV-induced Polη and Polκ focus formation in MEFs, Rev1+/+ and Rev1−/− MEFs were immortalized by lentiviral expression of SV40-T antigen (GeneCopoeia). GFP-Polη or GFP-Polκ was transiently transfected into transformed MEFs. After 16 h of incubation, cells were suspended and cultured on a coverslip in a six-well plate with 50% confluence. After 48 h, cells were treated with 20 J/m2 of UVC. After 6 h of incubation, cells were fixed with 4% paraformaldehyde for 30 min. Nuclear staining was performed with DAPI (Molecular Probe) in PBS buffer for 20 min. To examine Rev7 foci in MEFs, primary Rev1+/+ and Rev1−/− MEFs were cultured on a coverslip and incubated for 20 h. Cells were treated with 20 J/m2 of UVC, and, after 6 h incubation, cells were fixed with 4% paraformaldehyde for 30 min. Fixed cells were permeablized with 0.2% Triton X-100 in PBS buffer. Cells were immunostained with Rev7 antibody (BD Bioscience) and then incubated with Alexa fluor 488 secondary antibody (Molecular Probe). Nuclear DNA was stained with DAPI (Molecular Probe) for 20 min. The fluorescent images were visualized and captured by fluorescence microscope (Nikon fluorescence microscope).
Supplementary Material
Acknowledgments
The Rev1 knockout mouse was obtained from the Texas A&M Institute for Genomic Medicine in College Station, TX. This study was supported by National Institutes of Health grants ES020833 and ES022948.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.272229.115.
References
- Acharya N, Johnson RE, Prakash S, Prakash L. 2006. Complex formation with Rev1 enhances the proficiency of yeast DNA polymerase ζ for mismatch extension and for extension opposite from DNA lesions. Mol Cell Biol 26: 9555–9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba TW, Giroir BP, Humphries EH. 1985. Cell lines derived from avian lymphomas exhibit two distinct phenotypes. Virology 144: 139–151. [DOI] [PubMed] [Google Scholar]
- Besaratinia A, Pfeifer GP. 2006. Investigating human cancer etiology by DNA lesion footprinting and mutagenicity analysis. Carcinogenesis 27: 1526–1537. [DOI] [PubMed] [Google Scholar]
- Biertumpfel C, Zhao Y, Kondo Y, Ramon-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F, Yang W. 2010. Structure and mechanism of human DNA polymerase η. Nature 465: 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD. 2010. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465: 951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SK, Yoshida K, Machida Y, Khaira P, Chaudhuri B, Wohlschlegel JA, Leffak M, Yates J, Dutta A. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106: 287–296. [DOI] [PubMed] [Google Scholar]
- Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT. 2010. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci 107: 20786–20791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds CE, Simpson LJ, Sale JE. 2008. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30: 519–529. [DOI] [PubMed] [Google Scholar]
- Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. 2003. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J 22: 6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L, Unk I, Johnson RE, Johansson E, Burgers PMJ, Prakash S, Prakash L. 2001. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev 15: 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito W, Yokoi M, Sakayoshi N, Sakurai Y, Akagi J, Mitani H, Hanaoka F. 2012. Stalled Polη at its cognate substrate initiates an alternative translesion synthesis pathway via interaction with REV1. Genes Cells 17: 98–108. [DOI] [PubMed] [Google Scholar]
- Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. 2006. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J Exp Med 203: 319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders LHF, Haracksa L, et al. 2009. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol 29: 3113–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Torres-Ramos CA, Izumi T, Mitra S, Prakash S, Prakash L. 1998. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes Dev 12: 3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Kondratick CM, Prakash S, Prakash L. 1999a. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285: 263–265. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash S, Prakash L. 1999b. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 283: 1001–1004. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. 2000a. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406: 1015–1019. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Prakash S, Prakash L. 2000b. Fidelity of human DNA polymerase η. J Biol Chem 275: 7447–7450. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Haracska L, Prakash S, Prakash L. 2001. Role of DNA polymerase η in the bypass of a (6-4) TT photoproduct. Mol Cell Biol 21: 3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Yu S-L, Prakash S, Prakash L. 2003. Yeast DNA polymerase zeta (ζ) is essential for error-free replication past thymine glycol. Genes Dev 17: 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Prakash L, Prakash S. 2012. Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc Natl Acad Sci 109: 12455–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano C, Hanaoka F, Wang JY. 2012. Analysis of mice deficient in both REV1 catalytic activity and POLH reveals an unexpected role for POLH in the generation of C to G and G to C transversions during Ig gene hypermutation. Int Immunol 24: 169–174. [DOI] [PubMed] [Google Scholar]
- Karras GI, Jentsch S. 2010. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141: 255–267. [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Christensen RB. 1978. Ultraviolet-induced reversion of cyc1 alleles in radiation-sensitive strains of yeast. I. rev1 mutant strains. J Mol Biol 122: 1–21. [DOI] [PubMed] [Google Scholar]
- Lawrence CW, O'Brien T, Bond J. 1984. UV-induced reversion of his4 frameshift mutations in rad6, rev1, and rev3 mutants of yeast. Mol Gen Genet 195: 487–490. [DOI] [PubMed] [Google Scholar]
- Lindner SE, Sugden B. 2007. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid 58: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. 1999. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399: 700–704. [DOI] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2005. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 309: 2219–2222. [DOI] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2006. Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase iota. Nat Struct Mol Biol 13: 619–625. [DOI] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2008. Protein-template directed synthesis across an acrolein-derived DNA adduct by yeast Rev1 DNA polymerase. Structure 16: 239–245. [DOI] [PubMed] [Google Scholar]
- Ohashi E, Murakumo Y, Kanjo N, Akagi J-i, Masutani C, Hanaoka F, Ohmori H. 2004. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 9: 523–531. [DOI] [PubMed] [Google Scholar]
- Pozhidaeva A, Pustovalova Y, D'Souza S, Bezsonova I, Walker GC, Korzhnev DM. 2012. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase η. Biochemistry (Mosc) 51: 5506–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L. 2005. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74: 3173–3153. [DOI] [PubMed] [Google Scholar]
- Pustovalova Y, Bezsonova I, Korzhnev DM. 2012. The C-terminal domain of human Rev1 contains independent binding sites for DNA polymerase η and Rev7 subunit of polymerase ζ. FEBS Lett 586: 3051–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, Schepers A. 2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J Cell Sci 116: 3971–3984. [DOI] [PubMed] [Google Scholar]
- Schepers A, Ritzi M, Bousset K, Kremmer E, Yates JL, Harwood J, Diffley JF, Hammerschmidt W. 2001. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J 20: 4588–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reiβner T, Chanev S, Friedberg EC, Wang Z, Carell T, et al. 2009. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J 28: 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, Aggarwal AK. 2010. Structural basis for the suppression of skin cancers by DNA polymerase η. Nature 465: 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2009. Structure of the human REV1–DNA–dNTP ternary complex. J Mol Biol 390: 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier A, Kannouche P, Reck M-P, Lehmann AR, Fuchs RPP, Cordonnier A. 2004. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase polη and REV1 protein. DNA Repair 3: 1503–1514. [DOI] [PubMed] [Google Scholar]
- Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. 2005. Tumor susceptibility of Rassf1a knockout mice. Cancer Res 65: 92–98. [PubMed] [Google Scholar]
- Ummat A, Rechkoblit O, Jain R, Roy Choudhury J, Johnson RE, Silverstein TD, Buku A, Lone S, Prakash L, Prakash S, et al. 2012. Structural basis for cisplatin DNA damage tolerance by human polymerase η during cancer chemotherapy. Nat Struct Mol Biol 19: 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lindner SE, Leight ER, Sugden B. 2006. Essential elements of a licensed, mammalian plasmid origin of DNA synthesis. Mol Cell Biol 26: 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington MT, Johnson RE, Prakash S, Prakash L. 2000. Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase η. Proc Natl Acad Sci 97: 3094–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek J, Lee CJ, D'Souza S, Minesinger B, Kim H, D'Andrea AD, Walker GC, Zhou P. 2012a. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) ζ, and Polκ. J Biol Chem 287: 33836–33846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek J, Liu J, D'Souza S, Wang S, Xue Y, Walker GC, Zhou P. 2012b. Multifaceted recognition of vertebrate Rev1 by translesion polymerases ζ and κ. J Biol Chem 287: 26400–26408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Doles J, Hemann MT, Walker GC. 2010. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci 107: 20792–20797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Guan N. 1991. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J Virol 65: 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Prakash L, Prakash S. 2009. Highly error-free role of DNA polymerase η in the replicative bypass of UV induced pyrimidine dimers in mouse and human cells. Proc Natl Acad Sci 106: 18219–18224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Bhatia G, Prakash S, Prakash L. 2010a. Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases κ and ζ in human cells. Proc Natl Acad Sci 107: 14116–14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Prakash L, Prakash S. 2010b. Error-free replicative bypass of (6-4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev 24: 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Prakash S, Prakash L. 2012a. Genetic control of translesion synthesis on leading and lagging DNA strands in plasmids derived from Epstein-Barr virus in human cells. MBio 3: 00271–00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Prakash S, Prakash L. 2012b. Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc Natl Acad Sci 109: 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Acharya N, Park J, Basu D, Prakash S, Prakash L. 2014. Identification of two functional PCNA-binding domains in human DNA polymerase κ. Genes Cells 19: 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y-H, Pfeifer GP. 2001. Similarities in sunlight-induced mutational spectra of CpG-methylated transgenes and the p53 gene in skin cancer point to an important role of 5-methylcytosine rsidues in solar UV mutagenesis. J Mol Biol 305: 389–399. [DOI] [PubMed] [Google Scholar]
- You YH, Halangoda A, Buettner V, Hill K, Sommer S, Pfeifer G. 1998. Methylation of CpG dinucleotides in the lacI gene of the Big Blue transgenic mouse. Mutat Res 420: 55–65. [DOI] [PubMed] [Google Scholar]
- You Y-H, Lee D-H, Yoon J-H, Nakajima S, Yasui A, Pfeifer GP. 2001. Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J Biol Chem 276: 44688–44694. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.