

Abstract
Purpose
We critically evaluated the role of the adenosine A1 receptor (A1R) in normal development of retinal vasculature and pathogenesis of retinopathy of prematurity (ROP) by using the A1R knockout (KO) mice and oxygen-induced retinopathy (OIR) model.
Methods
Mice deficient in A1Rs and their wild-type (WT) littermates were examined during normal postnatal development or after being subjected to 75% oxygen from postnatal day (P) 7 to P12 and to room air from P12 to P17 (OIR model of ROP). Retinal vascularization was examined by whole-mount fluorescence and cross-sectional hematoxylin-eosin staining. Cellular proliferation, astrocyte and microglial activation, and tip cell function were determined by isolectin staining and immunohistochemistry. Apoptosis was determined by TUNEL assay.
Results
Genetic deletion of the A1R did not affect normal retinal vascularization during postnatal development with indistinguishable three-layer vascularization patterns in retina between WT and A1R KO mice. In the OIR model, genetic deletion of the A1R resulted in stage-specific effects: reduced hyperoxia-induced retinal vaso-obliteration at P12, but reduced avascular area and attenuated hypoxia-induced intraretinal revascularization without affecting intravitreal neovascularization at P17 and reduced avascular areas in retina at P21. These distinct effects of A1Rs on OIR were associated with A1R control of apoptosis mainly in inner and outer nuclear layers at the vaso-obliterative phase (P12) and the growth of endothelium tip cells at the vasoproliferative phase (P17), without modification of cellular proliferation, astrocytic activation, and tissue inflammation.
Conclusions
Adenosine A1 receptor activity is not required for normal postnatal development of retinal vasculature but selectively controls hyperoxia-induced vaso-obliteration and hypoxia-driven revascularization by distinct cellular mechanisms.
Keywords: adenosine A1 receptor, oxygen-induced retinopathy, proliferative retinopathy, retinal revascularization, neovascularization, endothelial tip cells, vaso-obliteration
Retinopathy of prematurity is the most common cause of blindness in childhood.1 This sight-threatening disease is characterized by two critical phases, hyperoxia- and inflammation-induced damage to retinal vessels and vaso-obliteration followed by hypoxia-driven physiological revascularization and pathologic neovascularization, the processes largely driven by hypoxia-induced factor-1α (HIF-1α) signaling pathway and vascular endothelial growth factor (VEGF) levels in retina.1,2 Conventional therapies for ROP are limited to laser and cryosurgery, which ablate the avascular retina to prevent retinal detachment caused by ROP.3 However, the effects of ablative laser therapy are limited and are associated with destruction to retina, causing clinically significant loss of visual field. Anti-VEGF therapy (e.g., intravitreal injection of anti–VEGF-A antibody bevacizumab) has been proposed3 and has recently been tested in a randomized, controlled, and multicenter trial involving 150 infants, which showed reduction of the recurrence rate,4 but the efficacy of intravitreal bevacizumab remains unclear with reported persistent avascular retina5 and recurrent intravitreal neovascularization.6 Importantly, VEGF acts not only as an angiogenic factor, but also as a neurotrophic factor during retinal development. Thus, there are concerns on the unintended effects of anti-VEGF agents on delayed growth and retinal vasculature development of preterm infants.7,8
Current therapeutic development of ROP focuses on directly targeting VEGF and HIF-1α signaling pathway.1,2,4,9 However, cellular responses to hypoxia are characterized by robust increases in extracellular adenosine production (up to 100-fold) and signaling events through the markedly induced adenosine receptors (up to 50-fold) locally.10 For example, in a canine model of ROP, the expression of 5′ nucleotidase (CD73, an enzyme responsible for generating extracellular adenosine) and adenosine A2A receptor (A2AR) was suppressed during the hyperoxic phase, but markedly increased in hypoxic retina, supporting the possible involvement of adenosine-A2AR signaling in retinal pathologic angiogenesis.11–14 Increased adenosine–adenosine receptor signaling in hypoxic retina not only constitutes a defense mechanism to protect retina by modulating neuroinflammation, cell death, and promoting angiogenesis, but also offers an opportunity of targeting pathologic angiogenesis of ROP with minimal effects on normal retinal vascular development. In support of this view, we have recently demonstrated that genetic inactivation of the A2AR attenuated hypoxia-induced pathologic angiogenesis without affecting normal postnatal retinal vascularization.15 Therapeutic potential of adenosine receptor–based therapy for ROP is supported by the ability of adenosine receptors to modulate inflammation, neuroprotection, and angiogenesis in retina through activation of four G-protein–coupled receptors, namely, A1, A2A, A2B, and A3, all of which have been detected in retina.16,17 Increased adenosine acting at A2ARs suppresses neuroinflammation and protects against diabetic retinopathy (DR)18 and traumatic optic neuropathy,19 through control of retinal microglial function and production of proinflammatory cytokines20,21; studies with genetic inactivation of the A2AR,15 with A2BR antagonists22–24 and with ribozyme approach to inactivating A2BRs, demonstrate that adenosine acting at the A2A and A2B receptors promotes pathologic angiogenesis in retina through modulating VEGF level.25 The translational potential of adenosine receptor–based therapy for controlling proliferative retinopathy is substantiated by a clinical potential, since a recent large clinical trial of 2006 infants has demonstrated that treatment with caffeine, a nonselective adenosine receptor antagonist, reduces ROP-related problems after 2-year follow-up,26 and by genetic identification of the variants of the human A2AR gene that are associated with reduced risk of developing DR in a prospective study.27
Several studies of adenosine receptor control of retinal vascularization have focused on the A2AR effect,11,15,18 with some on the A2Breceptor.25 Adenosine A1 receptor (A1R) mRNA and ligand binding are detected in developing retina.17 However, the A1R effect on retinal vascularization is largely unknown. As an important step in developing adenosine receptor–based treatment for ROP, we critically evaluated the role of the A1R in normal development of retinal vasculature and pathogenesis of proliferative retinopathy by using the A1R knockout (KO) mice and oxygen-induced retinopathy (OIR) model. Our characterization of the A1R KO and wild-type (WT) mice in normal and OIR model demonstrated for the first time that A1R activity did not affect normal retinal vascular development, but distinctly controlled hyperoxia-induced vaso-obliteration at P12 and hypoxia-induced revascularization at P17 by acting at A1Rs in distinct cellular elements.
Materials and Methods
Congenic A1R KO and WT Littermates in C57BL/6 Background
All experimental procedures were conducted in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research and the Animal Experimentation Regulation of Wenzhou Medical University. Generation of congenic A1R KO mice and WT littermates has been described previously.28 Heterozygous A1R KO (A1R−/+) mice were then interbred to generate homozygous A1R KO (A1R−/−), heterozygous A1R KO (A1R−/+), and WT littermates (A1R+/+). The mouse genotype was determined by PCR analysis of genomic DNA extracted from mouse tails. Genetic deletion of the A1R did not affect normal development and most physiological measurements including the body weight, heart rate, blood pressure and body temperature28 and survival rate in this study.
Mouse Model of Oxygen-Induced Retinopathy
The mouse model of OIR was performed according to the protocol as previously described by Smith and colleagues.29 Briefly, pups with their dams were kept in a 75% ± 2% oxygen chamber for 5 days from postnatal day (P) 7 to P12. Nursing mothers were prepared to replace dams for feeding pups every 12 hours during the hyperoxic stage (P7–P12). At P12, the animals were returned to room air for 5 days (from P12 to P17). Age-matched mice raised in room air (P0–P17) were used as the “room air” control group for study of normal postnatal development retinal vascularization.
Fluorescence Immunostaining in Whole-Mount Retinas
Fluorescence staining of whole-mounted retinas was performed as previously described.30 Eyes were fixed in 4% paraformaldehyde and the retinas were dissected and stained with 10 μg/mL isolectin B4 (Molecular Probes, Life Technologies, Carlsbad, CA, USA) after being blocked and permeabilized. The retinas were then incubated with anti–glial fibrillary acidic protein (GFAP) mouse monoclonal antibody (1:500; Sigma-Aldrich Corp., St. Louis, MO, USA) for 12 hours at 4°C, followed by incubation with fluorescence-conjugated second antibody (1:500; Invitrogen, Life Technologies) for 2 hours, and then whole-mounted. Eight nonoverlapping and randomly selected microscopic fields per retina were imaged by confocal scanning laser microscopy (LSM 710; Carl Zeiss, Oberkochen, Germany) to assess the formation of endothelial tip cells.
To assess normal postnatal development of retinal angiogenesis, A1R KO pups and WT littermates (breeding in room air) were killed and the eyes were harvested at P3, P5, P7, P12, and P17. Whole-mount retinas were stained with isolectin B4. The development of superficial vascular layer from P0 to P7 was quantified as the ratio of vascular area to total retinal area. Three nonoverlapping and randomly selected microscopic fields per retina and whole-mounted retina were assessed for morphology and distribution of retinal vessels from P7 to P17.
Neovascular Nuclei Quantification
To quantify neovascular nuclei, eyes were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned (5 μm), and stained with hematoxylin-eosin. The nuclei beyond the internal limiting membrane on the vitreous side were defined as neovascular nuclei and counted by an investigator in a blinded manner.
TUNEL Staining for Detection of Cell Apoptosis
The TUNEL staining was performed by using a commercial in situ cell death detection kit and following the procedure of the manufacturer's manual (Roche Diagnostics, Basel, Switzerland). The TUNEL-positive cells were counted from three sections crossing the optic nerve per retina to quantify the change of TUNEL signal.
Immunofluorescence Analysis
At P17, mouse eyes were dissected and embedded in paraffin. After being deparaffinized and heated in 10 mM sodium citrate for antigen repairing, retinal paraffin sections were blocked and permeabilized and then were incubated with anti–proliferating cell nuclear antigen (PCNA) rabbit polyclonal antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-GFAP mouse monoclonal antibody (1:500; Sigma-Aldrich Corp.), or anti–Iba-1 rabbit monoclonal antibody (Wako, Chuo-ku, Osaka, Japan) overnight at 4°C. Fluorescence-conjugated second antibodies (1:500; Invitrogen, Life Technologies) were applied to detect the positive signals. The GFAP/PCNA-positive cells were counted from three sections crossing the optic nerve per retina.
Quantitative Analysis of A1R mRNA Levels
Adenosine A1 receptor mRNA level in retina harvested at P17 was done by the quantitative real-time polymerase chain reaction (qPCR) procedure as we have described previously15 using the following forward and reverse primers for A1R mRNA: 5′-CATCCTGGCTCTGCTTGCTATT-3′ and 5′-TTGGCTATCCAGGCTTGTTCC-3.′
Statistical Analysis
All data were expressed as mean ± SEM. Statistical analyses were conducted by using Student's t-test, with P < 0.05 being considered statistically significant.
Results
Genetic Deletion of the A1R Did Not Affect the Normal Development of Retinal Vascularization in Mice
To determine the effect of A1R gene deletion on normal development of retinal vascularization, we analyzed development of the retinal vascular networks at P3, P7, P12, and P17 of homozygous A1R KO (A1R−/−) and WT (A1R+/+) littermates by fluorescein staining of whole-mounted retinas (Fig. 1A). At P3, the vessels grew radially from the optic nerve head toward the edge of the retina and became progressively interconnected and formed an initial superficial vascular layer. At P5, the deep vascular layer was apparent under the superficial layer, and the vasculature of both WT and A1R KO mice covered approximately half of the entire retinal area (data not shown). At P7, the average vascular area ratio of the WT and A1R KO group was 78% and 79%, respectively. At P12, the vasculature covered all the retina for WT and A1R KO mice, and an intermediate vascular layer developed between the superficial and deep layer. Quantitative analysis revealed that the vascular area at P3 (n = 3/group) and P7 (n = 3/group) was comparable between WT and A1R KO mice (Fig. 1A). At P12 and P17, retinal vascularization was near complete and the arborous pattern of retinal vasculature formed over the entire retina with very few avascular areas. Thus, we adapted “branch point” analysis31 for quantification of retinal vascularization at P12 and P17. Importantly, branch point analysis showed that retinal vascularization was indistinguishable between WT and A1R KO mice at P12 (n = 6/group) and P17 (n = 8/group), indicating that the superficial retinal vasculature grew with similar rates with indistinguishable patterns of the vessel distribution between WT and A1R KO mice. Moreover, distinct morphologies of three vessel layers were also observed under the confocal scanning laser microscopy at P12 and P17 (Fig. 1B). Arterioles in the superficial layer had numerous dichotomous branches. The vessels in the intermediate layer presented short capillary segments. The vessels in the deep layer formed anastomotic grids. Similarly, the morphology and distribution of retinal blood vessels were indistinguishable between WT and A1R KO mice.
Figure 1.

Genetic inactivation of A1Rs does not affect the normal development of retinal vessels in mice. (A) The development of retina vasculature of WT and A1R KO mice at P3, P7, P12, and P17 in room air was visualized by isolectin B4 staining in whole-mount retinas. Vascularized areas and whole retinal surface are shown by yellow dotted line and white dotted line, respectively. The superficial vascularized areas of retinas at P3 (n = 3/group) and P7 (n = 3/group) were quantified as a percentage of the whole retinal area. At P12 (n = 6/group) and P17 (n = 8/group), “branch point” analysis was used for quantification of retinal vascularization under room air. Data are presented as mean ± SEM. Scale bar: 500 μm. (B) The distributions of three retinal vascular layers were displayed in distinct confocal planes. The retinal images from P12 and P17 of WT and A1R KO in room air were examined by isolectin B4 staining of whole-mount retinas. The development of morphology and distribution of superficial, intermediate, and deep plexuses were indistinguishable between WT and A1R KO mice. Scale bar: 50 μm.
Collectively, quantitative analysis of retinal vascularization under room air with total 20 WT and 20 A1R KO mice at different stages (P3, P7, P12, and P17) demonstrates that A1R activity is not required for normal postnatal development of retinal vascularization in mice.
Genetic Deletion of A1Rs Reduced Hyperoxia-Induced Retinal Vascular Regression and Cellular Apoptosis at P12
As the first evidence for the involvement of A1R in OIR, we found that A1R mRNA in retina of OIR harvested at P17 was markedly reduced compared to the room air condition (n = 8, P < 0.01) (Fig. 2). This reduction of A1R mRNA may reflect the reduced protective effect by adenosine acting at A1Rs in retina.
Figure 2.
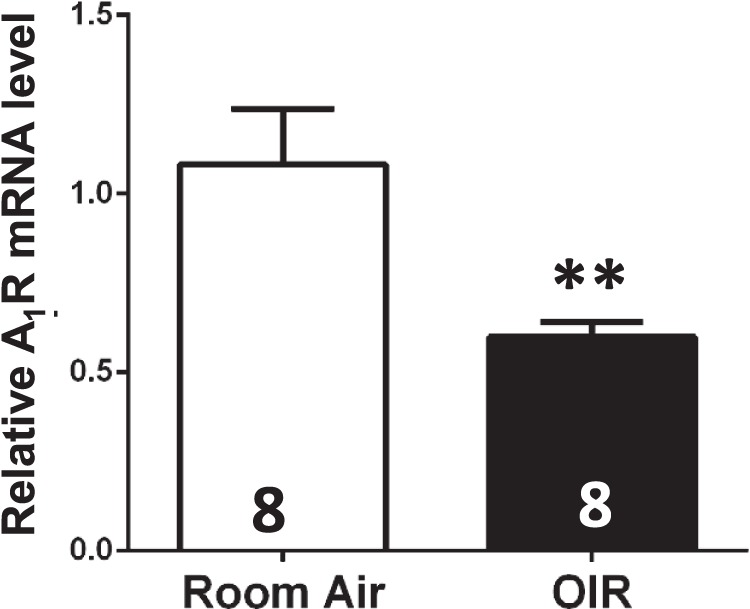
Adenosine A1 receptor mRNA level was reduced in retina of OIR compared to that of room air at P17. Retina of OIR and room air was dissected and harvested at P17, and A1R mRNA expression in retina was determined by qPCR analysis. Compared to the room air group, A1R mRNA in retina of OIR was markedly reduced (Fig. 2, n = 8/group). Data are presented as the mean ± SEM. *P < 0.05, Student's t-test.
To assess the effect of A1R gene deletion at the hyperoxic stage of OIR, we analyzed the vascular regression of WT and A1R KO mice in whole-mounted retinas with isolectin B4 staining. Both WT and KO groups clearly revealed large areas of vaso-obliteration in the central retina (Fig. 3A). Quantitative analysis of retinal vaso-obliteration showed that the percentage of avascular areas of total retina in the A1R KO group was significantly smaller than that of the WT group by ∼20% (6.64% absolute difference over 33.23% of total avascular area = ∼20%) (Fig. 3B), indicating that A1R activation increased hyperoxia-induced retinal vaso-obliteration.
Figure 3.

Genetic deletion of A1Rs reduced hypoxia-induced retinal vascular regression and cellular apoptosis at P12. (A) Retinal blood vessels of both WT and KO groups were visualized by isolectin B4 staining of whole-mount retinas at P12 of OIR. The whole retinal surface is shown by white dotted line. Avascular area is indicated as yellow. Scale bar: 500 μm. (B) Avascular area (%) was quantified as a percentage of the whole retinal surface (n = 7 retinas from seven WT mice and n = 9 retinas from nine A1R KO mice). Data are presented as the mean ± SEM. *P < 0.05, Student's t-test. (C, D) The apoptotic cells in retinal cross-sections of WT and A1R KO mice were detected by TUNEL staining at P12 of OIR. TUNEL-positive cells are indicated by yellow arrows. TUNEL signals were detected mainly in INL and ONL layers of retina in both WT and A1R KO mice. Coimmunostaining of TUNEL and CD31 (a marker for endothelial cell) revealed that CD31+ cells (white arrowhead) were largely segregated from the TUNEL signal (yellow arrowhead) with little coimmunostaining of TUNEL/CD31 (yellow arrow). Scale bar: 20 μm. (E) The TUNEL-positive cells of WT and A1R KO groups were quantified. Data are presented as mean ± SEM. *P < 0.05 comparing A1R KO group with WT group (n = 6 retinas from six WT mice and n = 6 retinas from six A1R KO mice).
Vaso-obliteration in OIR is a result of neuronal and endothelial apoptosis induced by a combination of direct toxicity of reactive oxygen species to neurons and a downregulation of HIF-1α–dependent VEGF expression to endothelial cells.32 We performed TUNEL analysis to determine whether the larger avascular region in WT mice at P12 was attributed to neuronal and endothelial apoptosis. Consistent with other studies,33–35 TUNEL-positive signals were detected within the inner nuclear layer (INL; mainly neurons such as amacrine cells and bipolar cells) and outer nuclear layer (ONL) of the avascular area, as well as the ganglion cell layer (GCL) of both WT and A1R KO groups. Furthermore, TUNEL signal was detected in both INL and ONL layers but largely segregated from the CD31+ cells (a marker for endothelium cells) with only very few TUNEL/CD31 costaining signals, indicating that apoptosis was mainly attributed to neuronal apoptosis in retina (Figs. 3C, 3D). Quantitative analysis showed that TUNEL-positive cells were significantly higher in WT mice than A1R KO mice at P12 (Fig. 3E). Thus, A1R gene deletion reduces avascular areas at least partly by reducing hyperoxia-induced neuronal apoptosis in retina.
Genetic Deletion of A1Rs Attenuated Hypoxia-Induced Intraretinal Revascularization but Not Intravitreal Neovascularization at P17
In contrast to the reduced avascular area of A1R KO mice at P12, quantitative analysis revealed that the A1R KO mice in fact had increased avascular area, compared to WT group, at P17 (Figs. 4A, 4B). This increased avascular area in A1R KO mice at P17 could likely be attributed to a perturbation of revascularization in A1R KO mice at the hypoxic phase. We also investigated the effect of A1R KO on pathologic angiogenesis and showed that there was no significant difference in neovascularization tufts toward the vitreous and neovascular nuclei that formed at the hypoxia phase between A1R KO and WT groups (Figs. 4A–D). Furthermore, we also analyzed the expression of PCNA, an indicator of cell proliferation, in retina at P17. Consistent with earlier studies,15,36 we detected PCNA-positive signals mainly in the superficial vasculature and neovascular tufts surrounding the lumen in the GCL of WT and A1R KO mice (Fig. 4E). However, the numbers of PCNA-positive cells were indistinguishable between A1R KO and WT mice at P17, consistent with the lack of A1R KO on neovascularization tufts (Fig. 4F). Additional analysis showed that scatter TUNEL-positive cells were detected in retina (mainly in the inner and outer nuclear layer) and largely segregated from CD31+ cells (Figs. 4G, 4H). There was no difference in retinal TUNEL-positive cells between WT and A1R KO groups, indicating that the reduced revascularization was not due to increased apoptosis (Fig. 4I). These results showed that at the hypoxic phase, A1R activation promoted intraretinal revascularization without affecting intravitreal neovascularization, an effect not being attributed to cellular apoptosis or endothelial cellular proliferation in retina.
Figure 4.

Genetic deletion of A1Rs reduced hypoxia-induced physiological intraretinal revascularization without affecting intravitreal neovascularization, hypoxia-induced cellular apoptosis, and proliferation at P17. (A) Retinal vasculature was stained by isolectin B4 of whole-mount retinas at P17 of OIR. Whole retinal area was circumscribed by white dotted line (n = 13 retinas from 13 WT mice and n = 12 retinas from 12 A1R KO mice). Avascular area and the areas of neovascularization tufts were highlighted in yellow and green, respectively. Scale bar: 500 μm. (B) The avascular area (%) was quantified as the ratio of central avascular area to whole retinal area. The neovascularization tufts area (%) was quantified as a percentage of whole retinal area. (C) Retinal pathologic angiogenesis at P17 was observed by using hematoxylin and eosin staining (n = 7 retinas from 7 WT mice and n = 7 retinas from 12 A1R KO mice). Nuclei on the vitreal side of the inner limiting membrane are indicated by black arrows. Scale bar: 50 μm. (D) The number of neovascular nuclei was quantified. (E) Hypoxia-induced retinal cellular proliferation of WT and A1R KO mice at P17 of OIR was detected by PCNA immunohistochemistry (n = 7 retinas from 7 WT mice and n = 7 retinas from 12 A1R KO mice). The PCNA-positive cells are indicated by yellow arrows. Scale bar: 20 μm. (F) The quantification of PCNA-positive cells of WT and A1R KO mice is shown. (G, H) Hypoxia-induced apoptotic cells of WT and A1R KO retinas at P17 of OIR were assayed by TUNEL staining and costaining with CD31. TUNEL-positive cells are indicated by yellow arrows (n = 14 retinas from 14 WT mice and n = 15 retinas from 15 A1R KO mice). Coimmunostaining of TUNEL and CD31+ cells revealed that CD31+ cells (white arrowhead) were largely segregated from the TUNEL signal (yellow arrowhead) with little coimmunostaining of TUNEL/CD31 signal (yellow arrow). Scale bar: 20 μm. (I) The quantification of TUNEL-positive cells of WT and A1R KO mice is shown. Data in (B, D, F, I) are presented as mean ± SEM. ***P < 0.001 (Student's t-test), comparing A1R KO group with WT group.
Genetic Deletion of the A1R Did Not Affect the Number of Microglial Cells and Astrocytes but Reduced the Formation of Endothelial Tip Cells to Attenuate Intraretinal Revascularization in Hypoxia Phase
We further sought to identify the cellular mechanisms underlying the effects of A1R activation on promoting revascularization by analyzing function of microglial cells, astrocytes, and endothelial tip cells in retina of WT and A1R KO mice at P17 of OIR.
Immunohistochemistry analysis of Iba-1 expression in retina showed that microglial activation in retina (mainly in the GCL and INL layers) was indistinguishable between A1R KO and WT littermates (Figs. 5A, 5B). Thus, inflammatory response (as indirectly indicated by microglial activation) is unlikely a major contributing factor in A1R control of retinal vascularization and retinopathy. Furthermore, retinal astrocytes participate in angiogenesis in response to hypoxia through their high expression of VEGF37 and their support for the growth of the leading edge of the developing superficial vascular network.38,39 Consistent with previous studies,40 GFAP-positive cells were mainly detected in the GCL of retina by immunofluorescence staining. However, GFAP-positive astrocytes in retina were not significantly affected by the genetic inactivation of A1R (Figs. 5C, 5D). Endothelial tip cells play an important role during angiogenesis by sensing the VEGF gradient produced by astrocytes and by leading the extension of typical filopodia in retinal sprout tips. We therefore further evaluated the formation of endothelial tip cells in retina of WT and A1R KO groups at P17 of OIR. As seen in Figures 5E and 5F, tip cell filopodia adhered to GFAP-positive astrocytes. Quantitative analysis revealed that the number of endothelial tip cells in retina was significantly reduced in A1R KO group compared with WT group (Fig. 5G). This finding suggests that A1R activation enhanced function of endothelial tip cells to promote hypoxia-induced revascularization in retina at P17 of OIR.
Figure 5.

Genetic deletion of A1Rs did not affect microglial and astrocytic activation but reduced formation of endothelial tip cells at P17. (A) Microglial activation in retina was assessed by immunohistochemistry of Iba-1 expression during OIR. The expression of Iba-1 was detected by immunohistochemistry in mainly the GCL and INL layers of retina. Iba-1–positive cells are indicated by yellow arrows. Scale bar: 20 μm. (B) The quantification of Iba-1–positive cells in retina of WT (n = 7 retinas from seven mice) and KO (n = 7 retinas from seven mice) is shown. Data are presented as the mean ± SEM. (C) The number of astrocytes of WT and A1R KO retinas at P17 of OIR was determined by GFAP immunofluorescence staining. The GFAP-positive cells are indicated by yellow arrows. Scale bar: 20 μm. (D) The GFAP-positive cells of retinas from WT (n = 7 retinas from seven mice) and KO (n = 7 retinas from seven mice) were quantified and presented as mean ± SEM. (E) Hypoxia-induced growth of endothelium tip cells and astrocytes in retina of WT and A1R KO mice at P17 of OIR was stained with isolectin B4 and anti-GFAP, respectively. Representative retinal endothelial tip cells are indicated by yellow arrows. Scale bar: 20 μm. (F) Higher magnifications of the regions from WT retina (the yellow pane) are shown. The interactions between endothelial tip cells and GFAP-positive astrocytes were indicated by their close contact between these cells. Scale bar: 10 μm. (G) The number of endothelial tip cells in WT and A1R KO groups was quantified. Data are presented as mean ± SEM. **P < 0.01 for comparing A1R KO group with WT group (n = 7 retinas from seven mice for each group).
Lastly, we analyzed avascular area of A1R KO and WT littermates of OIR at P21. In contrast to the A1R KO effect at P17, the avascular area in retina was in fact smaller in A1R KO than in their WT littermates (Figs. 6A, 6B; n = 7 per group), indicating that A1R activation at the P17 to P21 period apparently slows down normal vascularization of retina in OIR model. Thus, A1R inactivation exerted distinct effects on retinal vascularization at P12, P17, and P21, likely depending on the different levels of adenosine and different cellular populations involved in these different stages of OIR.
Figure 6.

Genetic deletion of A1Rs reduced retinal normal vascularization at P21 of OIR. (A) Retinal blood vessels of both WT and KO groups were visualized by isolectin B4 staining of whole-mount retinas at P21 of OIR. The whole retinal surface is shown by white dotted line. Avascular area is indicated as yellow. Scale bar: 500 μm. (B) Avascular area (%) was quantified as a percentage of the whole retinal surface (n = 7 retinas from seven mice for each group). Data are presented as the mean ± SEM. *P < 0.05, Student's t-test.
Discussion
Adenosine A1 Receptor Activity Selectively Modulates Oxygen-Induced Retinopathy Without Affecting Normal Retinal Vascular Development
One of the critical concerns in developing therapeutic strategy for treating ROP is to achieve maximal therapeutic effect on proliferative retinopathy with minimal unintended side effects on neurovascular development in retina. In agreement with our previous finding that A2AR inactivation selectively modulates pathologic angiogenesis without affecting normal retinal vasculature development,15 genetic deletion of A1R activity preferentially affected retinal vascularization under pathologic condition of OIR, but postnatal retinal vascularization developed normally with typical morphology, density, and distribution of retinal vessels from P3 to P17 in the absence of the A1R (Fig. 1). This finding reinforces our contention that interruption of adenosine signaling through A1R and A2AR has no effect on normal retinal vascular development. This selectivity is further supported by the finding that A1R mRNA was reduced in retina of OIR model compared to that of room air at P17. The finding confers a critical advantage for the proposed adenosine receptor–based therapeutic strategy over other treatment strategies (such as anti-VEGF antibody), since activity of these molecular targets may be necessary not only for pathologic angiogenesis, but also for normal retinal vascularization during development.41
This A1R and A2AR selective control of pathologic retinal vasculature growth in OIR may be in part attributed to the surge of extracellular adenosine and the marked induction of adenosine receptors in response to hypoxia.10 In models of ROP, hypoxia triggers the surge in extracellular adenosine as a result of transcriptional induction of CD73 and equivalent nucleotide transporter 1 as well as suppression of adenosine kinase, thereby elevating the capacity of local tissues for extracellular adenosine production.11,42 Adenosine accumulating locally during hypoxia permits the local control of retinal vessel growth.11 Furthermore, pathologic conditions are accompanied by the increases of local inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor–α, which lead to a delayed (∼24 hours), marked, and sustained increase in adenosine receptor (particularly A2AR and the A2BR) expression in tissues and inflammatory cells.43–45 Locally increased adenosine levels and adenosine receptor signaling might represent a local “find-me” signal and service a unique “purinergic chemotaxis” for a local resolution to pathologic conditions (as revealed by genetic KO studies).10 Thus, the surge of adenosine level and the induction of adenosine receptors in the hypoxic phase of OIR11 may constitute a negative feedback and defense mechanism countering such proangiogenic states triggered by hypoxia and HIF-1α–mediated expression of VEGF in retina. Further dissection of the A1R and A2AR signaling interacting with the molecular and cellular pathways, leading to distinct physiological development and pathologic angiogenesis, is needed to fully understand this selectivity of A1R and A2AR control of OIR.
Adenosine A1 Receptor Inactivation Reduces Hyperoxia-Induced Vaso-obliteration by Attenuating Neural Apoptosis in Retina at P12
A hallmark of ROP is abnormal and excessive blood vessel growth, but paradoxically it is early vaso-obliteration that initiates proliferative retinopathy. Thus, preventing early retinal vessel loss at the hyperoxic phase can prevent the devastating latter stage (the hypoxic phase) of the disease. In the vaso-obliteration phase, hyperoxia induces apoptosis of ganglia and developing endothelial cells and inhibits endothelial cell proliferation and migration, resulting in vaso-obliteration.32,46 Notably, despite clear vaso-obliteration at the retina center in the hyperoxic phase, there is no “hypoxia” in retina as shown by in vivo detection with nitroimidazole EF5.47 Since the adenosine concentration and the expression of ecto-5′ nucleotidase (CD73) are low during the hyperoxic phase, we presumed that A1Rs play a limited role at this stage. Surprisingly, we found that the avascular area and TUNEL-positive cells in the INL of retina in A1R KO mice were reduced as compared to WT littermates, suggesting that A1R activation probably aggravated hyperoxia-induced damage to developing retinal vessels by affecting neuronal apoptosis. Activation of retinal A1Rs has been shown to inhibit Ca2+ channels in retinal ganglion cells of mini-slices,48,49 to protect NMDA-induced cell death in cultured retinal neurons,50 and to mediate the IL-6 effect on the survival of cultured retinal ganglion cells.51 Consistent with the A1R-mediated neuroprotective effect, early studies28,52,53 indicate that cytotoxicity and cell death are generally more pronounced in neurons and astrocytes derived from A1R KO mice. Thus, additional studies are clearly warranted to clarify how A1R KO may confer cellular protection at the hyperoxic phase of OIR. Nonetheless, our findings highlight for the first time the important function of adenosine-A1Rs in modulating retinal vascular function even under hyperoxic environments with low extracellular adenosine level.
Adenosine A1 Receptor Activation Is Required for Hypoxia-Driven Physiological Revascularization, but Not Neovascularization in Retina at P17
In the vaso-proliferation phase of OIR, both regrowth of normal (intraretinal) revascularization and pathologic (intravitreal) angiogenesis occur during P12 to P17, the former promoting retinal vascular recovery but the latter resulting in pathologic neovascularization tufts. Current research on ROP has been largely focused on the mechanism that drives pathologic (intravitreal) neovascularization in the retina and treatment strategies to stop it.1,2,4,9 On the other hand, intraretinal revascularization has received much less attention.54 An early study55 has reported that the A1R agonist CPA increases membrane vessel in the chick chorioallantoic membrane model. This study provides the first in vivo demonstration that A1R activation is critical to beneficial intraretinal revascularization at the hypoxic phase by showing that A1R inactivation increases the avascular area without changes in neovascularization.
Control of retinal vascularization during development and OIR likely involves close interactions among neurons, astrocytes, microglial cells, and endothelial cells. Impairment in beneficial intraretinal revascularization by genetic deletion of the A1R can be attributed to several factors such as reduced endothelial cell proliferation, increased intravitreal neovascularization, and increased cell apoptosis, leading to reduced astrocytic functions or reduced tip cell formation. Despite the fact that ROP is fundamentally a vascular proliferating disorder, our findings suggest that cell proliferation and vitreal neovascularization tufts are apparently not important factors for A1R control of intraretinal revascularization. Moreover, control of avascular area in A1R KO mice is also not associated with cellular apoptosis, since the numbers of TUNEL-positive cells at P17 were comparable between WT and A1R KO groups. Similarly, retinal inflammatory response is unlikely a major contributing factor in A1R control of retinal vascularization and retinopathy, since microglial activation was not affected by A1R inactivation. Astrocytes play a significant role in angiogenesis in response to hypoxia through their high expression of VEGF.37 Indeed, studies in OIR models have shown that the density of astrocytes in the retina decreases during hyperoxia and then increases following hypoxia,56,57 and that restoring retinal astrocytes reduces vascular pathology associated with OIR.40,58 However, our analysis revealed that the GFAP-positive cells (astrocytes) in retina at P17 were indistinguishable between WT and A1R KO mice. Thus, under our experimental condition, cell types other than astrocytes are likely responsible for the A1R-mediated modulation of hypoxia-induced intraretinal vascularization.
At the tips of vascular sprouts with long filopodia are endothelial tip cells that play an important role in developmental blood vessel formation and physiological revascularization.59 In retina, endothelial tip cells are mainly located at the leading edge of vascular plexus and the fusion sites of the remodeling area.60 In sprouting angiogenesis, though, the filopodia of endothelial tip cells participate in intercellular communication, cell migration, and cell adhesion to lead the outgrowth of blood vessels toward the gradients of VEGF-A.61 Consistent with this functional view of tip cells, we detected filopodia of endothelial tip cells that were closely attached to the astrocytes in retina. Importantly, our analysis revealed that the quantity of endothelial tip cells is reduced by genetic deletion of the A1R at P17. Thus, activation of A1Rs promotes revascularization by increasing the number of endothelial tip cells, with normal morphology of filopodia to ensure vascular extension in the right direction along the astrocyte template. Whether the A1R control of tip cells is achieved by a direct effect of A1Rs in tip cells or an indirect effect of A1Rs in neurons (by stimulating platelet derived growth factor) or astrocytes (by releasing VEGF) on tip cells needs to be clarified in future studies. Moreover, future studies of A1R control of expression of the tip cell–specific candidate genes that are significantly upregulated in the tip cell fraction of sprouting vessels in a mouse model of OIR62 would shed light on the transcriptional mechanism underlying A1R control of tip cells in promoting retinal vascularization.
Adenosine A1 Receptor Inactivation Promotes Retinal Normal Vascularization at P21 of OIR
As further demonstration of the stage-specific effect of A1R inactivation, we uncovered that A1R activation at the P17 to P21 period apparently slows down retinal vascularization in OIR model. This new level of complexity of A1R activity reinforces the notion that adenosine acting at A1Rs exerts distinct functions at different courses of the disease (P12, P17, and P21). Distinct effects of A1R KO at P12, P17, and P21 may be due to the different levels of adenosine and different cellular populations involved in these different processes. Consequently, adenosine-based therapeutic strategy should be disease-stage–specific.
In summary, the findings that A1Rs (this study) and A2ARs15 selectively modulate OIR without affecting normal retinal vascular development confer a critical advantage for the proposed adenosine receptor–based therapeutic strategy over other treatment strategies (such as anti-VEGF antibody) that also compromise normal retinal vasculature during development. We uncovered distinct A1R control of hyperoxia-induced vaso-obliteration at P12 and hypoxia-induced revascularization at P17 by acting on neurons and tip cells. These findings advance the prospective of adenosine receptor–based therapy for ROP with two novel strategies: (1) to reduce hyperoxia-induced retinal vessel loss (with A1R antagonists) to effectively control OIR, rather than direct inhibition of pathologic (intravitreal) neovascularization; and (2) to promote intraretinal revascularization (with A1R agonists) at the hypoxic phase, thus shifting from blocking pathologic intravitreal neovascularization toward beneficial intraretinal revascularization. From the translational perspective, it would be interesting to study the genetic association between the single nucleotide polymorphism of the human genes encoding the A1 receptor (AdoraA1) and ROP, as revealed in other pathologic conditions such as apnea of prematurity,63 ischemic cardiomyopathy,64 and development of posttraumatic seizures.65 Lastly, the demonstration of the distinct and coordinated control of OIR by retinal A1Rs and A2ARs also provides biological basis for the clinical finding that the use of caffeine (the nonselective A1R and A2AR antagonist) in treatment of apnea in premature infants is associated with reduced ROP in a 2-year follow-up study.
Acknowledgments
The authors thank Jing Lin (Mount Sinai School of Medicine, New York, NY, USA) for his critical reading of the manuscript.
Supported by the Start-up Fund from Wenzhou Medical University (No. 89211010; No. 89212012), the Zhejiang Provincial Special Funds (No. 604161241), Key Laboratory of Vision Science, Ministry of Health, China (No. 601041241), the National Key Basic Research Program of China (2012CB910402), the Central Government Special Fund for Local Universities' Development (No. 474091314), National Natural Science Foundation of China (No. 81100672), and Zhejiang Provincial Natural Science Foundation Grant (LY12H12007) and by US National Institutes of Health Grants (DK095862, HL095556, and HL108922) and Boston University School of Medicine Special Research Fund DTD 4-30-14.
Disclosure: S. Zhang, None; H. Li, None; B. Li, None; D. Zhong, None; X. Gu, None; L. Tang, None; Y. Wang, None; C. Wang, None; R. Zhou, None; Y. Li, None; Y. He, None; M. Chen, None; Y. Huo, None; X.-L. Liu, None; J.-F. Chen, None
References
- 1. Penn JS,, Madan A,, Caldwell RB,, Bartoli M,, Caldwell RW,, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008; 27: 331–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cavallaro G,, Filippi L,, Bagnoli P,, et al. The pathophysiology of retinopathy of prematurity: an update of previous and recent knowledge. Acta Ophthalmol. 2014; 92: 2–20. [DOI] [PubMed] [Google Scholar]
- 3. Clark D,, Mandal K. Treatment of retinopathy of prematurity. Early Hum Dev. 2008; 84: 95–99. [DOI] [PubMed] [Google Scholar]
- 4. Mintz-Hittner HA,, Kennedy KA,, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. New Engl J Med. 2011; 364: 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tokunaga CC,, Mitton KP,, Dailey W,, et al. Effects of anti-VEGF treatment on the recovery of the developing retina following oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2014; 55: 1884–1892. [DOI] [PubMed] [Google Scholar]
- 6. Hu J,, Blair MP,, Shapiro MJ,, Lichtenstein SJ,, Galasso JM,, Kapur R. Reactivation of retinopathy of prematurity after bevacizumab injection. Arch Ophthalmol. 2012; 130: 1000–1006. [DOI] [PubMed] [Google Scholar]
- 7. Nishijima K,, Ng YS,, Zhong L,, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007; 171: 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saint-Geniez M,, Maharaj AS,, Walshe TE,, et al. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS One. 2008; 3: e3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hartnett ME,, Penn JS. Mechanisms and management of retinopathy of prematurity. New Engl J Med. 2012; 367: 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen JF,, Eltzschig HK,, Fredholm BB. Adenosine receptors as drug targets—what are the challenges? Nat Rev Drug Discov. 2013; 12: 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lutty GA,, McLeod DS. Retinal vascular development and oxygen-induced retinopathy: a role for adenosine. Prog Retin Eye Res. 2003; 22: 95–111. [DOI] [PubMed] [Google Scholar]
- 12. Takagi H,, King GL,, Robinson GS,, Ferrara N,, Aiello LP. Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells. Invest Ophthalmol Vis Sci. 1996; 37: 2165–2176. [PubMed] [Google Scholar]
- 13. Taomoto M,, McLeod DS,, Merges C,, Lutty GA. Localization of adenosine A2a receptor in retinal development and oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2000; 41: 230–243. [PubMed] [Google Scholar]
- 14. Lutty GA,, Merges C,, McLeod DS. 5′ nucleotidase and adenosine during retinal vasculogenesis and oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2000; 41: 218–229. [PubMed] [Google Scholar]
- 15. Liu XL,, Zhou R,, Pan QQ,, et al. Genetic inactivation of the adenosine A2A receptor attenuates pathologic but not developmental angiogenesis in the mouse retina. Invest Ophthalmol Vis Sci. 2010; 51: 6625–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui D,, Trier K,, Zeng J,, Wu K,, Yu M,, Ge J. Adenosine receptor protein changes in guinea pigs with form deprivation myopia. Acta Ophthalmol. 2010; 88: 759–765. [DOI] [PubMed] [Google Scholar]
- 17. Brito R,, Pereira MR,, Paes-de-Carvalho R, Calaza Kda C. Expression of A1 adenosine receptors in the developing avian retina: in vivo modulation by A(2A) receptors and endogenous adenosine. J Neurochem. 2012; 123: 239–249. [DOI] [PubMed] [Google Scholar]
- 18. Ibrahim AS,, El-Shishtawy MM,, Zhang W,, Caldwell RB,, Liou GI. A((2)A) adenosine receptor (A((2)A)AR) as a therapeutic target in diabetic retinopathy. Am J Pathol. 2011; 178: 2136–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ahmad S,, Fatteh N,, El-Sherbiny NM,, et al. Potential role of A2A adenosine receptor in traumatic optic neuropathy. J Neuroimmunol. 2013; 264: 54–64. [DOI] [PubMed] [Google Scholar]
- 20. Liou GI,, Auchampach JA,, Hillard CJ,, et al. Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Invest Ophthalmol Vis Sci. 2008; 49: 5526–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Santiago AR,, Baptista FI,, Santos PF,, et al. Role of microglia adenosine A(2A) receptors in retinal and brain neurodegenerative diseases. Mediators Inflamm. 2014; 2014: 465694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mino RP,, Spoerri PE,, Caballero S,, et al. Adenosine receptor antagonists and retinal neovascularization in vivo. Invest Ophthalmol Vis Sci. 2001; 42: 3320–3324. [PubMed] [Google Scholar]
- 23. Grant MB,, Davis MI,, Caballero S,, Feoktistov I,, Biaggioni I,, Belardinelli L. Proliferation, migration, and ERK activation in human retinal endothelial cells through A(2B) adenosine receptor stimulation. Invest Ophthalmol Vis Sci. 2001; 42: 2068–2073. [PubMed] [Google Scholar]
- 24. Grant MB,, Tarnuzzer RW,, Caballero S,, et al. Adenosine receptor activation induces vascular endothelial growth factor in human retinal endothelial cells. Circ Res. 1999; 85: 699–706. [DOI] [PubMed] [Google Scholar]
- 25. Afzal A,, Shaw LC,, Caballero S,, et al. Reduction in preretinal neovascularization by ribozymes that cleave the A2B adenosine receptor mRNA. Circ Res. 2003; 93: 500–506. [DOI] [PubMed] [Google Scholar]
- 26. Schmidt B,, Roberts RS,, Davis P,, et al. Long-term effects of caffeine therapy for apnea of prematurity. New Engl J Med. 2007; 357: 1893–1902. [DOI] [PubMed] [Google Scholar]
- 27. Charles BA,, Conley YP,, Chen G,, et al. Variants of the adenosine A(2A) receptor gene are protective against proliferative diabetic retinopathy in patients with type 1 diabetes. Ophthalmic Res. 2011; 46: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johansson B,, Halldner L,, Dunwiddie TV,, et al. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001; 98: 9407–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith LE,, Wesolowski E,, McLellan A,, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994; 35: 101–111. [PubMed] [Google Scholar]
- 30. Connor KM,, Krah NM,, Dennison RJ,, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009; 4: 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suchting S,, Freitas C,, le Noble F,, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A. 2007; 104: 3225–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alon T,, Hemo I,, Itin A,, Pe'er J,, Stone J,, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995; 1: 1024–1028. [DOI] [PubMed] [Google Scholar]
- 33. Duan LJ,, Takeda K,, Fong GH. Prolyl hydroxylase domain protein 2 (PHD2) mediates oxygen-induced retinopathy in neonatal mice. Am J Pathol. 2011; 178: 1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ludewig P,, Flachsbarth K,, Wegscheid C,, et al. CEACAM1 confers resistance toward oxygen-induced vessel damage in a mouse model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2014; 55: 7950–7960. [DOI] [PubMed] [Google Scholar]
- 35. Narayanan SP,, Xu Z,, Putluri N,, et al. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis. 2014; 5: e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gendron RL,, Good WV,, Miskiewicz E,, Tucker S,, Phelps DL,, Paradis H. Tubedown-1 (Tbdn-1) suppression in oxygen-induced retinopathy and in retinopathy of prematurity. Mol Vis. 2006; 12: 108–116. [PubMed] [Google Scholar]
- 37. Dorrell MI,, Friedlander M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog Retin Eye Res. 2006; 25: 277–295. [DOI] [PubMed] [Google Scholar]
- 38. Dorrell MI,, Aguilar E,, Friedlander M. Retinal vascular development is mediated by endothelial filopodia a preexisting astrocytic template and specific R-cadherin adhesion. Invest Ophthalmol Vis Sci. 2002; 43: 3500–3510. [PubMed] [Google Scholar]
- 39. Scott A,, Powner MB,, Gandhi P,, et al. Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PLoS One. 2010; 5: e11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weidemann A,, Krohne TU,, Aguilar E,, et al. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia. 2010; 58: 1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lutty GA,, McLeod DS,, Bhutto I,, Wiegand SJ. Effect of VEGF trap on normal retinal vascular development and oxygen-induced retinopathy in the dog. Invest Ophthalmol Vis Sci. 2011; 52: 4039–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elsherbiny NM,, Naime M,, Ahmad S,, et al. Potential roles of adenosine deaminase-2 in diabetic retinopathy. Biochem Biophys Res Commun. 2013; 436: 355–361. [DOI] [PubMed] [Google Scholar]
- 43. Frick JS,, MacManus CF,, Scully M,, Glover LE,, Eltzschig HK,, Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009; 182: 4957–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schingnitz U,, Hartmann K,, Macmanus CF,, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol. 2010; 184: 5271–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Linden J. Regulation of leukocyte function by adenosine receptors. Adv Pharmacol. 2011; 61: 95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aiello LP,, Avery RL,, Arrigg PG,, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994; 331: 1480–1487. [DOI] [PubMed] [Google Scholar]
- 47. Scott A,, Fruttiger M. Oxygen-induced retinopathy: a model for vascular pathology in the retina. Eye (Lond). 2010; 24: 416–421. [DOI] [PubMed] [Google Scholar]
- 48. Sun X,, Barnes S,, Baldridge WH. Adenosine inhibits calcium channel currents via A1 receptors on salamander retinal ganglion cells in a mini-slice preparation. J Neurochem. 2002; 81: 550–556. [DOI] [PubMed] [Google Scholar]
- 49. Santos PF,, Caramelo OL,, Carvalho AP,, Duarte CB. Adenosine A1 receptors inhibit Ca2+ channels coupled to the release of ACh, but not of GABA, in cultured retina cells. Brain Res. 2000; 852: 10–15. [DOI] [PubMed] [Google Scholar]
- 50. Oku H,, Goto W,, Kobayashi T,, et al. Adenosine protects cultured retinal neurons against NMDA-induced cell death through A1 receptors. Curr Eye Res. 2004; 29: 449–455. [DOI] [PubMed] [Google Scholar]
- 51. Perigolo-Vicente R,, Ritt K,, Pereira MR,, Torres PM,, Paes-de-Carvalho R,, Giestal-de-Araujo E. IL-6 treatment increases the survival of retinal ganglion cells in vitro: the role of adenosine A1 receptor. Biochem Biophys Res Commun. 2013; 430: 512–518. [DOI] [PubMed] [Google Scholar]
- 52. Bjorklund O,, Shang M,, Tonazzini I,, Dare E,, Fredholm BB. Adenosine A1 and A3 receptors protect astrocytes from hypoxic damage. Eur J Pharmacol. 2008; 596: 6–13. [DOI] [PubMed] [Google Scholar]
- 53. Dunwiddie TV,, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001; 24: 31–55. [DOI] [PubMed] [Google Scholar]
- 54. Wan T,, Xu Z,, Zhou HJ,, et al. Functional analyses of TNFR2 in physiological and pathological retina angiogenesis. Invest Ophthalmol Vis Sci. 2013; 54: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Clark AN,, Youkey R,, Liu X,, et al. A1 adenosine receptor activation promotes angiogenesis and release of VEGF from monocytes. Circ Res. 2007; 101: 1130–1138. [DOI] [PubMed] [Google Scholar]
- 56. Chan-Ling T,, Tout S,, Hollander H,, Stone J. Vascular changes and their mechanisms in the feline model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1992; 33: 2128–2147. [PubMed] [Google Scholar]
- 57. Downie LE,, Pianta MJ,, Vingrys AJ,, Wilkinson-Berka JL,, Fletcher EL. AT1 receptor inhibition prevents astrocyte degeneration and restores vascular growth in oxygen-induced retinopathy. Glia. 2008; 56: 1076–1090. [DOI] [PubMed] [Google Scholar]
- 58. Dorrell MI,, Aguilar E,, Jacobson R,, et al. Maintaining retinal astrocytes normalizes revascularization and prevents vascular pathology associated with oxygen-induced retinopathy. Glia. 2010; 58: 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu Z,, Gong J,, Maiti D,, et al. MEF2C ablation in endothelial cells reduces retinal vessel loss and suppresses pathologic retinal neovascularization in oxygen-induced retinopathy. Am J Pathol. 2012; 180: 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gerhardt H,, Golding M,, Fruttiger M,, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003; 161: 1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wood W,, Martin P. Structures in focus—filopodia. Int J Biochem Cell Biol. 2002; 34: 726–730. [DOI] [PubMed] [Google Scholar]
- 62. del Toro R,, Prahst C,, Mathivet T,, et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010; 116: 4025–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kumral A,, Tuzun F,, Yesilirmak DC,, Duman N,, Ozkan H. Genetic basis of apnoea of prematurity and caffeine treatment response: role of adenosine receptor polymorphisms: genetic basis of apnoea of prematurity. Acta Paediatr. 2012; 101: e299–e303. [DOI] [PubMed] [Google Scholar]
- 64. Tang Z,, Diamond MA,, Chen JM,, et al. Polymorphisms in adenosine receptor genes are associated with infarct size in patients with ischemic cardiomyopathy. Clin Pharmacol Ther. 2007; 82: 435–440. [DOI] [PubMed] [Google Scholar]
- 65. Wagner AK,, Miller MA,, Scanlon J,, Ren D,, Kochanek PM,, Conley YP. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res. 2010; 90: 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]