

Abstract
Purpose
Photoreceptor neuronal degenerations are common, incurable causes of human blindness affecting 1 in 2000 patients worldwide. Only half of all patients are associated with known mutations in over 250 disease genes, prompting our research program to identify the remaining new genes. Most retinal degenerations are restricted to the retina, but photoreceptor degenerations can also be found in a wide variety of systemic diseases. We identified an X-linked family from Sri Lanka with a severe choroidal degeneration and postulated a new disease entity. Because of phenotypic overlaps with Bietti's crystalline dystrophy, which was recently found to have systemic features, we hypothesized that a systemic disease may be present in this new disease as well.
Methods
For phenotyping, we performed detailed eye exams with in vivo retinal imaging by optical coherence tomography. For genotyping, we performed whole exome sequencing, followed by Sanger sequencing confirmations and cosegregation. Systemic investigations included electron microscopy studies of peripheral blood cells in patients and in normal controls and detailed fatty acid profiles (both plasma and red blood cell [RBC] membranes). Fatty acid levels were compared to normal controls, and only values two standard deviations above or below normal controls were further evaluated.
Results
The family segregated a REP1 mutation, suggesting choroideremia (CHM). We then found crystals in peripheral blood lymphocytes and discovered significant plasma fatty acid abnormalities and RBC membrane abnormalities (i.e., elevated plasmalogens). To replicate our discoveries, we expanded the cohort to nine CHM patients, genotyped them for REP1 mutations, and found the same abnormalities (crystals and fatty acid abnormalities) in all patients.
Conclusions
Previously, CHM was thought to be restricted to the retina. We show, to our knowledge for the first time, that CHM is a systemic condition with prominent crystals in lymphocytes and significant fatty acid abnormalities.
Keywords: retinal degeneration, retinal tubulations, lymphocyte crystals, fatty acid metabolism, plasmalogens, choroideremia, Bietti's crystalline dystrophy
Choroideremia (CHM) (OMIM 303100) is a relatively common X-linked retinal degeneration first described by Mauthner in 18721 and was incorrectly thought to be a stationary and developmental disorder.2 Currently, it is known as a progressive X-linked retinal dystrophy that causes human blindness by affecting the choriocapillaris, retinal pigment epithelium, and photoreceptors, with an incidence of 1:50,000 and 1:100,000. Choroideremia is thus confined to the retina and choroid. Choroideremia is caused by mutations in the Rab escort protein-1 (REP1), which encodes a chaperone protein for the prenylation of Rab GTPases, which regulates intracellular vesicular trafficking.3 Mutations in REP1 cause a truncation or absence of the normal protein product in affected males, currently with 129 mutations reported.4 Although the CHM protein is required systemically, an autosomal CHM-like gene encodes Rab escort protein-2, which maintains the normal body prenylation of Rab proteins.5–7
Affected males with choroideremia usually begin to experience nyctalopia in the first or second decade of life and demonstrate increasing retinal signs of chorioretinal atrophy. This is followed by visual field constriction. By the fifth to sixth decades of life, the visual field is usually constricted to the central 5° to 10°, which eventually leads to complete blindness. Recently, human experimental clinical trials have started with the goal of testing safety and efficacy of REP1 gene replacement by retinal surgery. It is therefore imperative to know as much about CHM as possible.
Bietti's crystalline retinal dystrophy (BCD) is an autosomal recessive retinal degeneration with very significant clinical overlap with CHM. Bietti's crystalline retinal dystrophy represents a severe progressive retinal degeneration with retinal crystals due to mutations in CYP4V2, which encodes a microsomal fatty acid omega-hydroxylase. Bietti's crystalline retinal dystrophy is a rare disease described by Bietti in 1937 and characterized by multiple small crystalline intraretinal deposits (crystals), with RPE atrophy, pigment clumping, and choroidal sclerosis. The crystals are visible upon clinical funduscopy, but eventually, as the disease worsens and progresses, the crystals become impossible to see and disappear. Initially thought to be restricted to the retina, BCD crystalline inclusions were surprisingly also found throughout the body, including in lymphocytes and skin fibroblasts, with significant elevations of specific fatty acid levels in the serum.8,9 These discoveries led to the insight that BCD is actually a systemic disease.
We identified an unusual Sri Lankan X-linked pedigree with severe choroidal sclerosis with phenotypic overlap of BCD (Fig. 1) and ruled out BCD by CYP4V2 sequencing. We initially postulated a new disease entity but found CHM (REP1) mutations (Table 1). We then hypothesized that CHM, like BCD, is also a systemic condition with systemic fatty acid abnormalities and lymphocyte crystals. In analyzing peripheral blood in the Sri Lankan family, to our surprise we found fatty acid abnormalities and lymphocyte crystals. To replicate these novel discoveries we recruited a replication cohort of additional CHM patients and compared them to a group of normal subjects. This paper explores and discusses these findings.
Figure 1.

Images of in vivo retinal architecture of a CHM patient showing the outer retinal tubulations.
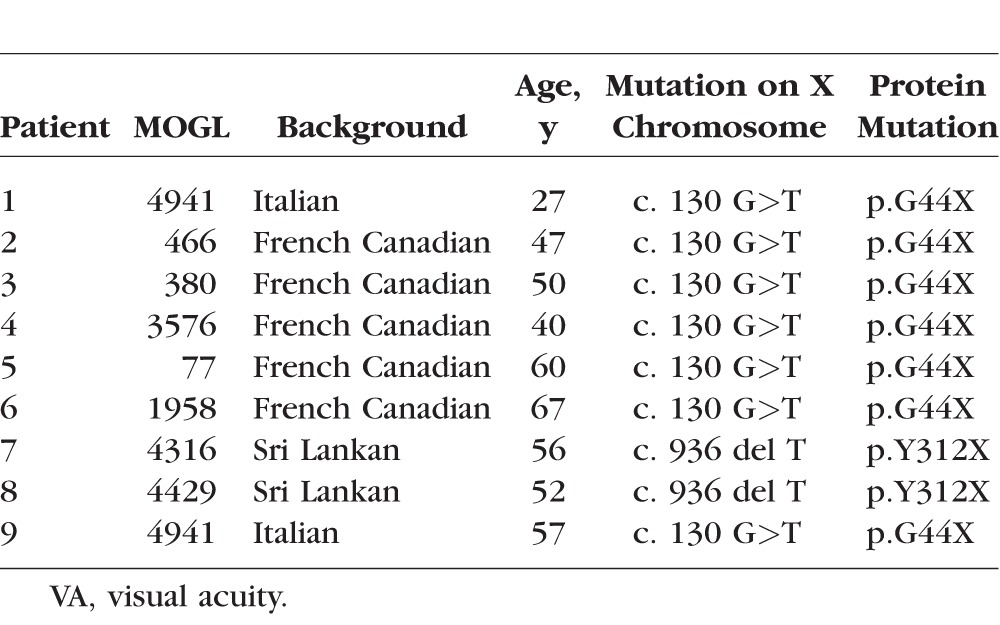
Table 1.
Mutational Analysis
Methods
Patients
The study started when we identified a small Sri Lankan family with severe chorioretinal degeneration and outer retinal tubulations using an optical coherence tomography (OCT). Two males (McGill Ocular Genetics Laboratory [MOGL] 4316 and 4429) were severely affected, while the mother showed retinal mosaicism with mild chorioretinal changes; we thus postulated X-linked inheritance. In the replication cohort, we studied another seven male patients and one female patient of Italian and French Canadian descent. Their ages were varied and ranged from 27 to 60 years old. All patients signed informed consents approved by the Montreal Children's Hospital Research Ethics Board. The protocol and consent forms adhered to the Declaration of Helsinki.
Phenotyping
Characterization of the phenotypes was done through medical history and physical exam. Best-corrected visual acuities were measured using projected Snellen acuities. Kinetic perimetry was measured by Goldmann visual field testing. Fundus dilation and photography were performed, and in vivo retinal microscopy was performed (Spectralis SD-OCT; Heidelberg Engineering, Carlsbad, CA, USA) to confirm disease type, severity, and clinical phenotype. Fundus autofluorescence (FAF) was measured to document abnormalities in lipofuscin metabolism.
Blood Sampling
All patients signed consent forms and fasted at least 12 hours before blood taking. Three peripheral blood samples were drawn simultaneously for each patient, from another seven CHM patients, and from three normal adults. One blood sample was used to analyze fasting plasma and red blood cell membrane fatty acid profiles. These blood samples were analyzed at the Kennedy Krieger Institute (Baltimore, MD, USA) for a panel of 57 fatty acids in both plasma and red blood cell membranes. Normal controls are included in the extensive Kennedy Krieger Institute analyses. The second blood sample was used for transmission electron microscopic (EM) analysis of blood cells. The third blood sample was for CHM genotyping and sequencing. For the EM studies, the lavender-top tubes (containing ETDA) were centrifuged for buffy coat isolation and prefixed with 2.5% glutaraldehyde, postfixed with osmium tetraoxide, and sectioned at 80 nm. The samples were seen under magnification from 28,000× to 68,000×. Detailed fatty acid analysis methods and blood sample fixation for EM are found below. The discoveries were initially made in the two Sri Lankan patients by the McGill pathology team who were not familiar with CHM or BCD but who were asked to look for crystals similar to those found in BCD. After the crystalline deposits were found in CHM patients, we recruited seven more CHM patients and a new pathologist (KS), who was completely blinded to the above results. Similar crystals were found in these additional CHM patients and none in our normal controls. For the fatty acid studies, we recruited a world-renowned expert in fatty acid abnormalities (NB).
Fatty Acid Analysis
Blood samples were evaluated at Kennedy Krieger Institute according to established clinical protocols. Briefly, after fasting, patients' plasma from EDTA-treated whole blood was treated to extract free fatty acids and fatty acids from circulating complex lipids. Red blood cells from EDTA-treated whole blood were washed free of plasma and treated to extract fatty acids from membrane lipids. The samples were analyzed by gas chromatography/mass spectrometry. Plasmalogens in the form of dimethylacetals (DMAs) were identified by gas chromatography. The report includes 57 plasma fatty acids and red blood cell (RBC) fatty acids from C10:0 through C30:0, as well as useful ratios and group summaries (e.g., total omega-6 and omega-3 fatty acids, plasmalogen DMA/fatty acid ratios [C16:0DMA/C16:0 and C18:0DMA/C18:0]).10
Methods for EM and Preparation
Blood samples were drawn into lavender tubes. Whole blood was transferred into small Eppendorf tubes using pipettes and were centrifuged for 5 minutes at 2800g. The supernatant was removed, and gluteraldehyde fixative (2.5% glutaraldehyde in 0.1 M phosphate buffer) was added to the centrifuge tube until the tube was full. The sample was centrifuged for 5 minutes at 2800g and fixed in a refrigerator at 4°C for 48 hours. The sample was then centrifuged for 5 minutes at 2800g, and the fixative was removed. After fixation, the top 1 mm of fixed blood was removed using a small wooden applicator stick and placed into a new Eppendorf tube. Isotonic buffer solution was added, and the tube was allowed to remain at room temperature for 10 minutes. The sample was then centrifuged for 5 minutes at 2800g. The supernatant was removed, and care was taken not to disturb the concentrated pellet of cells. Isotonic buffer solution was added, and the solution was centrifuged as previously described two more times in order to rinse off all EM fixative.
Osmium tetraoxide was added and left for 1 hour at room temperature in a fume hood. The tube was centrifuged for 5 minutes. The osmium supernatant was removed, and the solution was rinsed three times using distilled water, then was centrifuged for 5 minutes between each solution change. The supernatant was removed, and Epon was used to infiltrate the solution, centrifuging between steps for 5 minutes. The tube was allowed to polymerize overnight at 60°C. On the next day, the Eppendorf tube was cut and the block of tissue was trimmed for EM analysis.
Whole Exome Sequencing
For the Sri Lankan family only, the DNA library preparation, capturing, sequencing, and bioinformatics analyses were performed at Genome Quebec Innovation Centre (Montreal, Canada) as previously detailed, under the supervision of JM.11 Briefly, genomic DNA (3 μg) of each study participant was captured using a kit (SureSelect Human All Exon Kit, version 4; Agilent Technologies, Inc., Santa Clara, CA, USA) and subsequently a sequencer (Illumina Hiseq2000; Illumina, San Diego, CA, USA) with paired-end 100–base pair reads. The high-quality trimmed paired-end sequences were aligned to the human reference genome (hg19) using BWA software (v. 0.5.9).12 A mean coverage of 93X (MOGL 4316), 90X (MOGL 4429) was obtained for all consensus coding sequence exons. Variant calling and annotation were performed by SAMtools (v. 0.1.17)13 and ANNOVAR,14 respectively. In order to focus on likely disease-causing variants, we filtered out all of the variants with an allele frequency more than 5% in either the 1000-genomes database (available in the public domain at http://www.1000genomes.org) or the NHLBI exomes (http://evs.gs.washington.edu/EVS/, v.0.0.14; provided in the public domain by Exome Variant Server, NHLBI GO Exome Sequencing Project). We also filtered out variants that had previously been seen in more than five individuals in our in-house exome database (containing >460 exomes). Finally, we considered only the most protein-damaging variants (frameshift Indel, nonsense, missense, and canonical splice site) for further analysis. Because of possible consanguinity, we focused on rare homozygous variants as most likely to cause the disease.
DNA analyses by Sanger sequencing of the REP1 gene was performed on the third blood sample for each patient. A FlexiGene kit (Qiagen, Germantown, MD, USA) and the QIAamp DNA blood kit (Qiagen) were used to extract the DNA from whole blood according to the manufacturer's protocol. NanoDrop (ThermoFisher Scientific, Burlington, ON, Canada) was used to verify the DNA quantity and quality. REP1 primers were designed by Primer3 software (http://frodo.wi.mit.edu/primer3/ [in the public domain]). A PCR kit (HotStarTaq Master Mix Kit; Qiagen) was used to perform the PCR following the manufacturer's protocol. Gel electrophoresis was used to verify the purity of the fragment and then sequenced by Sanger sequencing. The results were analyzed using the Sequencer software program to identify REP1 mutations, which were analyzed in silico by Ensembl software, Blocks Substitution Matrix (BLOSUM) analyses, the SIFT algorithm, and the (PolyPhen) tool to determine their significance.
Results
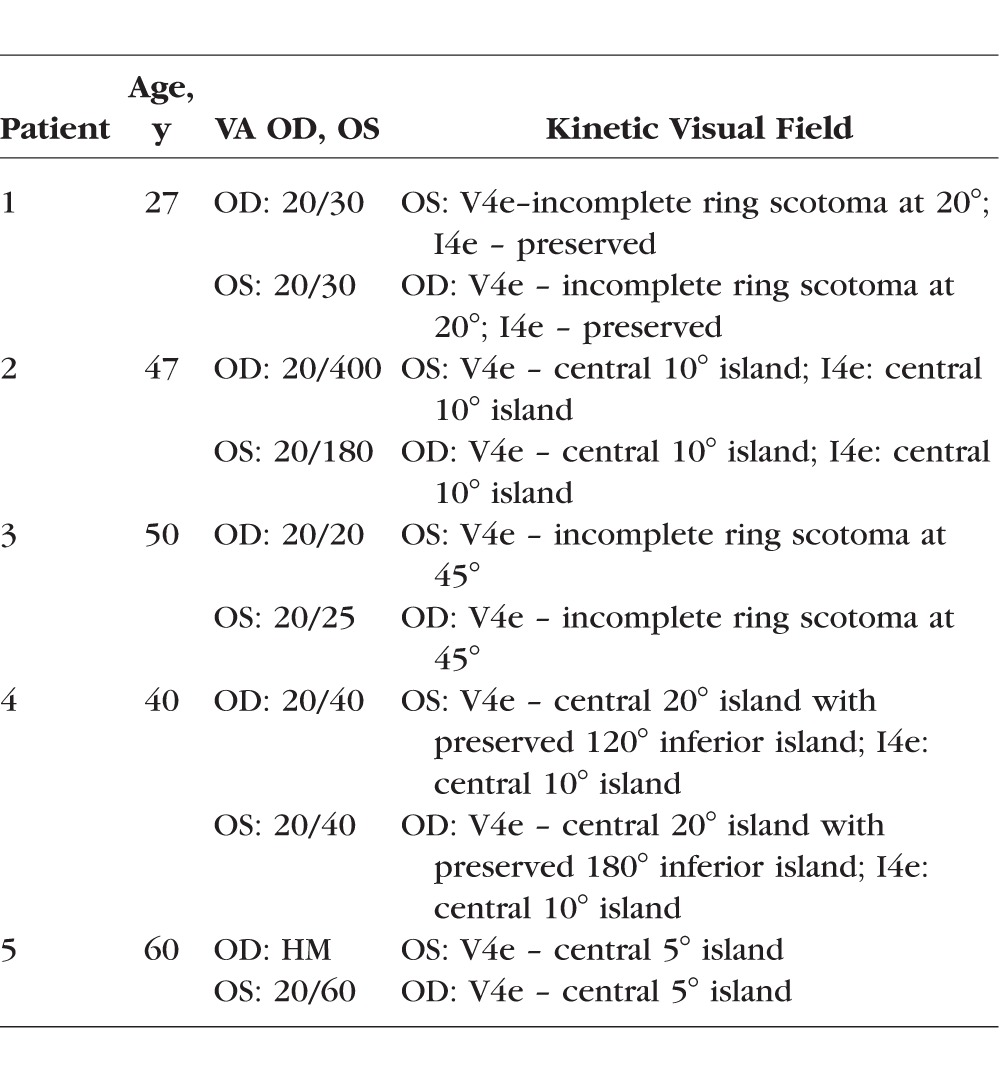
We identified a Sri Lankan X-linked retinal dystrophy family with two affected males and one carrier female. We found severe retinal degeneration with outer retinal tubulations with significant overlap with BCD, which is an autosomal recessive entity (Figs. 1, 2). We then postulated a new retinal disease with mutations in a new retinal gene. Bietti's crystalline dystrophy has subretinal crystals, lymphocyte crystals, and extensive fatty acid abnormalities, and it is autosomal recessive. Because of this, we excluded CYP4V2 mutations by Sanger sequencing. To identify the possible novel gene, we performed whole exome sequencing of the two brothers from Sri Lanka and reasoned that they must share the same mutation in the new X-linked retinal crystalline dystrophy gene. To our surprise the only mutation they shared was a p.Tyr312* (Y312X) mutation in REP1 (Table 1), suggesting that they have CHM but with a novel phenotype. To test the hypothesis that CHM, like BCD, has systemic features, we performed EM studies on the proband's (patient 7) peripheral blood cells and found significant intracellular rod-like crystals (Figs. 3E, 3F) in lymphocytes. To replicate this discovery, we recruited another seven CHM patients and performed EM studies on the peripheral blood cells in four of these patients plus three normal controls. All patients and controls in this replication study underwent Sanger sequencing for REP1 mutations. All patients had known and confirmed CHM mutations (Table 1); the controls did not. All our CHM patients had outer retinal tubulations and a wide range of severity in retinal appearance and FAF phenotypes (Figs. 4, 5). These patients' visual acuities and descriptions of their kinetic visual fields are found in the Appendix (Table A1).
Figure 2.

Spectral-domain OCT for the nine CHM patients (one carrier female, eight affected males). (A) Patient 1 OD, (B) patient 2 OS, (C) patient 3 OS, (D) patient 4 OD, (E) patient 5 OS, (F) patient 6 (carrier) OS, (G) patient 7 OD, (H) patient 8 OS, and (l) patient 9 OS.
Figure 3.

Electron microscopic studies of four CHM patients' peripheral lymphocytes. Photos illustrate striking intracytoplasmic crystalline structures with their magnifications. (A) Patient 2 with 23,000× magnification, (B) patient 3 with 49,000×, (C) patient 3 with 68,000×, (D) patient 4 with 49,000×, (E) patient 7 with 8900×, and (F) patient 7 with 22,000×.
Figure 4.

Fundus photos of the posterior pole of our nine CHM patients (one carrier female, eight affected males). (A) Patient 1 OS, (B) patient 2 OD, (C) patient 3 OD, (D) patient 4 OD, (E) patient 5 OS, (F) carrier patient 6 OS, (G) patient 7 OS, (H) patient 8 OS, and (I) patient 9 OS.
Figure 5.

Fundus autofluorescence of nine CHM patients show the diversity of autofluorescent retinal deposits and patterns (A–I).
In addition to patient 7, the EM studies on the other CHM patients and normal controls confirmed that all CHM patients in our replication cohort had peripheral blood cells with crystals, but the controls did not (Fig. 3). At this point, to confirm our discoveries, we recruited a new pathologist (KS) who was not familiar with CHM or BCD and was completely blinded to our previous discoveries, data, and discussions. We found two types of lymphocyte crystals. In Figures 3E and 3F (Sri Lankan CHM patients), we documented rod-like crystals, while in the other three CHM patients we found needle-like intracellular lymphocyte crystals (Figs. 3A–D). Furthermore, EM studies on blood samples from three normal controls confirm a lack of crystals in blood cells (Figs. 6A–C). Blood cell crystals in CHM patients are a novel discovery and suggest that CHM is a systemic condition. Since lipid abnormalities were identified in BCD patients, and to further investigate the content of the blood cell crystals in the CHM patients, we performed extensive serum lipid (plasma lipid and RBC membrane content) studies to explore the systemic disease hypothesis. We also recruited a fatty acid expert (NB).
Figure 6.

Electron microscope images of normal controls, illustrating the lack of crystals in the lymphocytes (A–C). Magnification 30,000×.
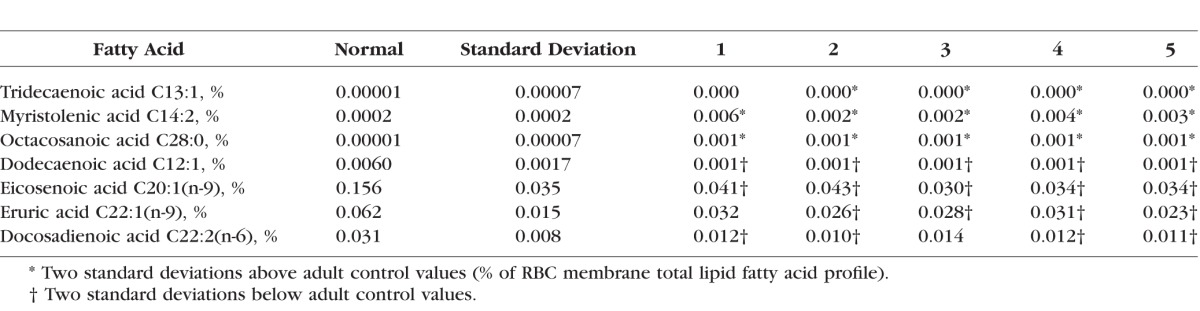
We were able to obtain fasting blood for fatty acid testing on five of the cohort of nine CHM patients. From these patients, compared to the hundreds of controls in the KKI Laboratory, we found significant abnormalities (P < 0.01; 2 standard deviations above or below normal levels) for at least 5 RBC fatty acids and 13 plasma fatty acids of the total of 57 fatty acids tested in all five patients. The 4 out of the 5 RBC fatty acids and 7 out of the 13 plasma fatty acids presented in Tables 2 and 3 were selected because at least 4 out of our 5 CHM patients tested demonstrated these significant abnormalities. From the RBC fatty acids, we identified significant elevations in capric acid (C10:0), nervonic acid (C24:1[n-9]), and the plasmalogen derivative, dimethylacetal acid (16:0), as well as a significant decrease in eicosenoic acid (C20:1[n-9]) (Table 2). The seven abnormal FA profiles are noted below. Plasma fatty acid profiles revealed elevations of tridecaenoic acid (C13:1), myristolenic acid (C14:2), and octacosanoic acid (C28:0) (Table 3). Levels of other plasma fatty acids were found to be significantly lower than normal: dodecaenoic acid (C12:1), eicosenoic acid (C20:1[n-9]), erucic acid (C22:1[n-9]), and docosadienoic acid (C22:2[n-6]).
Table 2.
Red Blood Cell Membrane Total Lipid Fatty Acid Profile
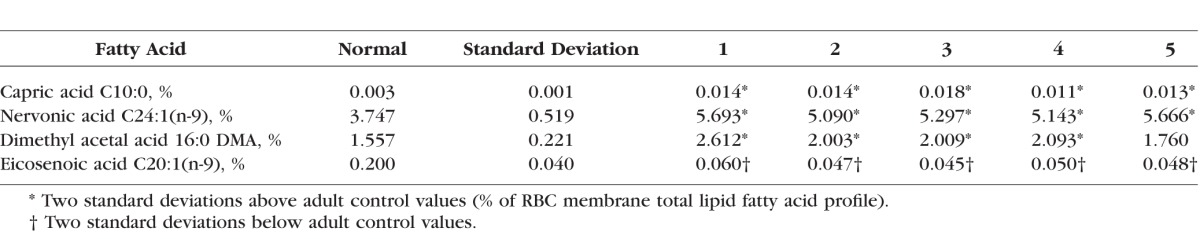
Table 3.
Plasma Total Lipid Fatty Acid Profile
Discussion
Choroideremia is a well-known retinal degeneration that has been known for over 140 years. Choroideremia is thought to be an isolated ocular disorder, confined to the retina and choroid, and it has never been considered a systemic disease. In 1990, the gene for CHD was localized to Xq133 and found to encode the Rab escort protein-1 (REP1). The Rab protein family is involved in intracellular vesicular trafficking, motility, and fusion of organelles in cells. While complete loss of REP function would be lethal,15 the partially redundant protein REP2 prevents this and is encoded by a CHD-like gene on 1q42. However, in the eye, REP2 does not compensate fully for the deficiency of REP1. Although initial clinical reports suggest that the loss of the RPE is the primary event,16 the exact pathogenesis and retinal layer (i.e., photoreceptors, RPE, choroid) initially affected is still unclear. Rodrigues et al.17 reported on the eye pathology of a deceased CHM patient and identified retinal rosette-like structures with pigment-filled macrophages. The authors suggested that there is impairment in the phagocytosis of outer segments. Macdonald et al.4 also found occasional rosettes and T-cell infiltration of choroidal vessels and gliosis of the retina of an autopsy specimen with CHM. Systemic features, to our knowledge, have never been investigated. Similar to patients with CHM, BCD patients exhibit severe progressive chorioretinal atrophy and degeneration.
Because of the significant clinical overlap between CHM and BCD, we were prompted to investigate our Sri Lankan patients systemically. Initially thought to be restricted to the retina, BCD crystalline inclusions were surprisingly also found throughout the body, including in lymphocytes and skin fibroblasts, with elevations of specific fatty acid levels in the serum.8 Patients with BCD were found to have significantly higher concentrations of octadecanoic acid (18:0) but lower concentration of octadecadienoic acid (18:1[n-9]), while the total monounsaturated fatty acid concentration was significantly lower in BCD as well as lower activity of the delta-9-desaturase enzyme present.9
Our current EM studies of the Sri Lankan CHM patient plus the replication cohort of additional CHM patients demonstrate intracellular crystals in RBCs, in lymphocytes, and in platelets. We found two types of systemic (lymphocyte) crystals: rod-like crystals and needle-like crystals. To explore these crystals further and to compare CHM to BCD at the biochemical level, we then performed an extended fatty acid analysis to ascertain if there are metabolic derangements. We also found significant fatty acid alterations in the CHM patients not found in controls. We postulate that these fatty acid alterations are the result of the same pathologic process resulting in the deposition of crystals in the blood cells and in other, not yet identified tissues of CHM patients. We postulate that the crystal deposition is likely due to dysregulation of fatty acid synthesis and/or degradation due to deficiencies in the vesicular trafficking system caused by the REP1 mutations. Interestingly, delta-9 desaturase was shown to be downregulated in BCD patients, suggesting a defect in the delta-9 desaturase enzyme. However, in our patients we found elevated plasma levels of myristolenic acid, indicating that delta-9 desaturase enzyme was intact, since the formation of myristolenic acid requires the delta-9 desaturase enzyme (Tables 2, 3).
This suggests that the overlapping clinical phenotypes in CHM and BCD (choroidal sclerosis, retinal and lymphocyte crystals, and fatty acid abnormalities) may be the result of different disease mechanisms that result in abnormal fatty acid metabolism, which in the end causes deposits. Mutations in REP1 result in defective vesicular transport, and although effects on different Rab proteins are now being investigated, the vesicle cargo affected is unknown.18 In fibroblasts and monocytes from CHM patients, reduced rates of lysosomal protein degradation were reported.7
In red blood cell membranes of our CHM patients, we found significant elevations in plasmalogens, measured by dimethylacetal acids (Table 2). Plasmalogens are a group of membrane glycerophospholipids thought to play a role in membrane structural properties, including signaling, fission, and fusion, as well as reducing oxidative stress on cell membranes.19 Although reduced plasmalogens are more commonly associated with disease, significant increases in plasmalogens have also been observed in autosomal dominant and recessive RP patients.20 In addition, nervonic acid levels were significantly increased in our CHM cohort (Table 2). Nervonic acid is a monounsaturated omega-9 acid, important in the biosynthesis of neuronal myelin, and it is also found in sphingolipids in the white matter of the human brain.21 The effects of increased levels of nervonic acid in the peripheral blood plasma of our CHM patients are currently not known.
In summary, we found consistent fatty acid changes in our CHM patients that may occur secondary to a deficiency in REP1 and can be further validated by other investigators in the future. These lipids could reflect a yet unknown trafficking function of REP1, which ultimately results in crystalline inclusion deposition.
Choroideremia was first discovered, described, and initially thought to be a primary retinal degeneration, without systemic disease. To the best of our knowledge, our study is the first in the literature showing crystals in blood cells and elevated plasma levels of fatty acids in CHM, illustrating that CHM is a systemic condition. Future work must include elucidating the content of these crystals, determining the exact mechanism for the increases in fatty acid and their consequences to the systemic health of CHM patients. Choroideremia thus joins BCD and a growing list of photoreceptor degenerations that are part of a multisystem human disease.
Acknowledgments
We gratefully thank all the patients for their participation.
Supported by funding (to RKK) from the following: Foundation Fighting Blindness Canada, Canadian Institutes of Health Research, National Institutes of Health, National Eye Institute, Fonds de la Recherche en Santé du Québec, Foundation for Retinal Research, and Réseau de Recherche en Santé de la Vision.
Disclosure: A.Y. Zhang, None; N. Mysore, None; H. Vali, None; J. Koenekoop, None; S.N. Cao, None; S. Li, None; H. Ren, None; V. Keser, None; I. Lopez-Solache, None; S.N. Siddiqui, None; A. Khan, None; J. Mui, None; K. Sears, None; J. Dixon, None; J. Schwartzentruber, None; J. Majewski, None; N. Braverman, None; R.K. Koenekoop, None
Table A1.
Visual Acuity and Visual Field Sizes for the CHM Patients
References
- 1. Mauthner, H. Ein fall von choroideremia. Ber Naturw Med Ver Innsbruck. 1872; 2: 191. [Google Scholar]
- 2. Wolff S. Choroideremia. Arch Ophthalmol. 1930; 3: 80–87. [Google Scholar]
- 3. Cremers FP,, van de Pol DJ,, van Kerkhoff LP,, Wieringa B,, Ropers HH. Cloning of a gene that is rearranged in patients with choroideraemia. Nature. 1990; 347: 674–677. [DOI] [PubMed] [Google Scholar]
- 4. MacDonald IM,, Russell L,, Chan CC. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol. 2009; 54: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cremers FP,, Armstrong SA,, Seabra MC,, Brown MS,, Goldstein JL. REP-2 a Rab escort protein encoded by the choroideremia-like gene. J Biol Chem. 1994; 269: 2111–117. [PubMed] [Google Scholar]
- 6. Wavre-Shapton ST,, Tolmachova T,, Lopes da Silva M,, Futter CE,, Seabra MC. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS One. 2013; 8: e57769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strunnikova NV Barb J, Sergeev YV,, et al. Loss-of-function mutations in Rab escort protein 1 (REP-1) affect intracellular transport in fibroblasts and monocytes of choroideremia patients. PLoS One. 2009; 4: e8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee J, Jiao X, Hejtmancik F,, et al. The metabolism of fatty acids in human Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2001; 42: 1707–1714. [PubMed] [Google Scholar]
- 9. Lai TY Chu KO, Chan KP,, et al. Alterations in serum fatty acid concentrations and desaturase activities in Bietti crystalline dystrophy unaffected by CYP4V2 genotypes. Invest Ophthalmol Vis Sci. 2010; 51: 1092–1097. [DOI] [PubMed] [Google Scholar]
- 10. Steinberg S,, Jones R,, Tiffany C,, Moser A. Investigational methods for peroxisomal disorders. Curr Protoc Hum Genet. 2008; Chapter 17:Unit 17.6.1. [DOI] [PubMed]
- 11. Fahiminiya S,, Majewski J,, Roughley P,, et al. Whole-exome sequencing reveals a heterozygous LRP5 mutation in a 6-year-old boy with vertebral compression fractures and low trabecular bone density. Bone. 2013; 57: 41–46. [DOI] [PubMed] [Google Scholar]
- 12. Li H,, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li H Handsaker B, Wysoker A,, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang K,, Li M,, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van den Hurk JA Schwarz M, van Bokhoven H,, et al. Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat. 1997; 9: 110–117. [DOI] [PubMed] [Google Scholar]
- 16. Flannery JG,, Bird AC,, Farber DB,, Weleber RG,, Bok D. A histopathologic study of a choroideremia carrier. Invest Ophthalmol Vis Sci. 1990; 31: 229–236. [PubMed] [Google Scholar]
- 17. Rodrigues MM,, Ballintine EJ,, Wiggert BN,, et al. Choroideremia: a clinical, electron microscopic, and biochemical report. Ophthalmology. 1984; 91: 873–883. [PubMed] [Google Scholar]
- 18. Kohnke M,, Delon C,, Hastie ML,, et al. Rab GTPase prenylation hierarchy and its potential role in choroideremia disease. PLoS One. 2013; 8: e81758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braverman NE,, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta. 2012; 1822: 1442–1452. [DOI] [PubMed]
- 20. Schaefer EJ Robins SJ, Patton GM,, et al. Red blood cell membrane phosphatidylethanolamine fatty acid content in various forms of retinitis pigmentosa. J Lipid Res. 1995; 36: 1427–1433. [PubMed] [Google Scholar]
- 21. Mohanty BP,, Bhattacharjee S,, Paria P,, Mahanty A,, Sharma AP. Lipid biomarkers of lens aging. Appl Biochem Biotechnol. 2013; 169: 192–200. [DOI] [PubMed] [Google Scholar]