

Abstract
Tertiary carbon radicals have notable utility for uniting complex carbon fragments with concomitant formation of new quaternary carbons. This article explores the scope, limitations and certain mechanistic aspects of Okada’s method for forming tertiary carbon radicals from (N-acyloxy)phthalimides by visible-light photocatalysis. Optimized conditions for generating tertiary radicals from (N-acyloxy)phthalimide derivatives of tertiary carboxylic acids by visible-light irradiation in the presence of 1 mol% of commercially available Ru(bpy)3(PF6)2, diethyl 1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (8) and i-Pr2NEt, and their coupling in dichloromethane at room temperature with alkene acceptors were developed. Four representative tertiary (N-acyloxy)phthalimides and 15 alkene radical acceptors were examined. Both reductive couplings with electron-deficient alkenes and radical substitution reactions with allylic and vinylic bromides and chlorides were examined with many such reactions occurring in good yield using only a slight excess (typically 1.5 equiv) of the alkene. In general, the yields of these photocatalytic reactions were higher than the analogous transformations of the corresponding N-phthalimidoyl oxalates. Deuterium labeling and competition experiments reveal that the reductive radical coupling of tertiary (N-acyloxy)phthalimides with electron-deficient alkenes can be terminated by both hydrogen-atom transfer and single-electron reduction followed by protonation, and that this mechanistic duality is controlled by the presence or absence of i-Pr2NEt.
INTRODUCTION
In one of the earliest applications of visible-light photocatalysis to organic synthesis, Okada and co-workers reported in 1991 the coupling with electron-deficient alkenes of primary, secondary and tertiary carbon radicals generated from (N-acyloxy)phthalimides upon visible-light irradiation in the presence of catalytic Ru(bpy)3Cl2 and 1-benzyl-1,4-dihydronicotinamide (BNAH).1,2 The utility of this general method for combining complex carbon fragments has recently been highlighted in several total synthesis investigations in our laboratories.3,4
In our studies, the conditions of Okada were modified to allow the photocatalytic coupling of tertiary carbon radicals to be carried out in non-aqueous solvents. In this article, we provided details of our modification of the Okada method, and a broader survey of the notable utility of this visible-light photocatalytic method for C–C bond formation and the construction of quaternary carbon centers. In particular, we examine a selection of coupling reactions to allow a direct comparison of (N-acyloxy)phthalimides and N-phthalimidoyl oxalates5,6 as precursors of tertiary carbon radicals for C–C bond-forming reductive coupling and allylic and vinylic substitution reactions. We also report investigations that identify both hydrogen-atom transfer and single-electron reduction followed by protonation as viable termination steps of these C–C bond-forming coupling reactions.
RESULTS
Synthesis of (N-acyloxy)phthalimides
We were initially attracted to the use of (N-acyloxy)phthalimide substrates as radical precursors because of their ease of preparation and stability.1,3 These intermediates are reliably formed by carbodiimide-mediated coupling of carboxylic acids with N-hydroxyphthalimide (Figure 1A). To prepare more sterically demanding substrates, coupling of acid chlorides with the potassium salt of N-hydroxyphthalimide in the presence of a crown ether is most effective (Figure 1B).4 As summarized in Figure 1, (N-acyloxy)phthalimides of variable structural complexity were prepared in high yield by these methods. These radical precursors are typically crystalline solids, which are stable to benchtop storage, ambient light, biphasic aqueous purification conditions, and chromatography on silica gel. In preliminary studies comparing (N-acyloxy)phthalimides to the more conventionally-employed Barton esters,7 we observed that the former exhibit superior stability and are more easily synthesized and handled.
Figure 1.

Synthesis of (N-acyloxy)phthalimides by (A) carbodiimide-assisted coupling, and (B) reaction of an acid chloride intermediate with potassium phthalimide N-oxide.
Coupling of (N-acyloxy)phthalimides with alkene acceptors
We next evaluated conditions for generating nucleophilic tertiary radicals from these substrates and their coupling with electron-deficient olefins. In the original report, Okada disclosed the coupling of (N-acyloxy)phthalimides with Michael acceptors such as methyl vinyl ketone (MVK) in the presence of 1 equiv of 1-benzyl-1,4-dihydronicotinamide and the visible light photocatalyst Ru(bpy)3Cl2 in aqueous THF.1 Although various alkyl radicals were generated and coupled with electron-deficient alkene under these conditions, the only tertiary radical generated in this way was the atypical 1-adamantyl radical (eq 1). As we anticipated that an aqueous reaction medium might be problematic with highly lipophilic substrates, we chose to explore related non-aqueous reaction conditions initially disclosed by Gagné for the photocatalytic generation of glycosyl radicals from glucosyl halides (eq 2).8 These conditions employ the Hantzsch ester, diethyl 1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (8), the photocatalyst Ru(bpy)3(BF4)2, and i-Pr2NEt in dichloromethane as solvent.
 |
(1) |
 |
(2) |
Our efforts to optimize the coupling of 1-methyl-1-cyclohexyl (N-acyloxy)phthalimide (4) with methyl vinyl ketone (MVK) quickly identified useful conditions employing 1 mol% of commercially available Ru(bpy)3(PF6)2, 1.5 equiv of Hantzsch ester 8, and 2.2 equiv of i-Pr2NEt in dichloromethane with irradiation at room temperature with blue LEDs. Using these conditions, the coupled product 9 was formed in 85–86% yield (Table 1, entries 1 and 2). Although the coupling of 4 with MVK was complete after stirring for only 1.5 h (entry 2), longer reaction times were not detrimental (entry 1). The inclusion of THF as a solvent, which was found to be beneficial in the visible light photocoupling of related tert-alkyl N-phthalimidoyl oxalates5 did not improve the yield of 9 (entries 3 and 4). Decreasing the excess of Hantzsch ester 8, i-Pr2NEt or MVK proved detrimental (entries 5–9), although the reduction in yield using 1 equiv of the Hantzsch ester was minimal (entry 8). The yield of the coupled product 9 was only slightly diminished when equal amounts of the coupling partners were used (entry 9), suggesting that this photocatalytic reaction would be appropriate for uniting structurally complex fragments. In the absence of light, no conversion was observed (entry 10). Omission of either the Hantzsch ester (entry 11) or i-Pr2NEt (entry 12) resulted in a decreased yield of 9, suggesting that the combination of these reductants was most effective. Significant, albeit slower reactivity was observed in the absence of the photocatalyst, with a 61% yield of 9 being obtained after 18 h (entries 13 and 14). A related observation was noted by Okada and co-workers in their initial report.1
Table 1.
Optimization of the Visible-Light Photoredox Coupling of (N-Acyloxy)phthalimide 4 with MVK
 | ||
|---|---|---|
| Entry | Modification | Yield of 9 (%)a,b |
| 1 | - | 86 |
| 2 | 1.5 h | 85 |
| 3 | CH2Cl2/THF (1:1) | 74 |
| 4 | THF | 68 |
| 5 | i-Pr2NEt (0.2 equiv) | 67 |
| 6 | i-Pr2NEt (1 equiv) | 71 |
| 7 | Hantzsch ester 8 (0.5 equiv) | 65 |
| 8 | Hantzsch ester 8 (1 equiv) | 81 |
| 9 | MVK (1 equiv) | 78 |
| 10 | no light | ND |
| 11 | no Hantzsch ester 8 | 41 |
| 12 | no i-Pr2NEt | 49 |
| 13 | no photocatalyst (18 h) | 61 |
| 14 | no photocatalyst (2 h) | 15 |
4 [0.15 M].
Isolated yield after silica gel chromatography.
ND = not detected.
We next examined the reaction of (N-acyloxy)phthalimide 4 with a selection of conjugate acceptors to allow (N-acyloxy)phthalimides and the analogous tert-alkyl N-phthalimidoyl oxalates6 to be compared as radical precursors under their respective optimized visible-light photoredox coupling conditions. Reaction of (N-acyloxy)phthalimide 4 with MVK provided product 9 in 85% yield (Table 2), essentially the same efficiency as realized in the reaction of MVK with 1-methyl-1-cyclohexyl N-phthalimidoyl oxalate.9 However, in the coupling with 2-cyclopentenone, (N-acyloxy)phthalimide 4 gave product 10 in 80% yield, 25% higher than the yield obtained from the corresponding N-phthalimidoyl oxalate. The reaction of (N-acyloxy)phthalimide 4 with benzyl methacrylate provided product 11 in 59% yield, again the yield being considerably higher than that achieved from the corresponding N-phthalimidoyl oxalate (41%).9 The greatest difference in reactivity between these two classes of tertiary radical precursors was observed in couplings with methacrylonitrile. (N-acyloxy)phthalimide 4 coupled with this acceptor to give coupled product 12 in 83% yield, while the similar coupling with 1-methyl-1-cyclohexyl N-phthalimidoyl oxalate yielded only trace amounts of 12.9,10,11
Table 2.
Visible-Light Photocatalytic Reductive Coupling of (N-Acyloxy)phthalimide 4 with Various Electron-Deficient Alkenes.
 |
Isolated yield after silica gel chromatography.
Yield measured by NMR relative to an internal standard (1,4-dimethoxybenzene).
We also examined the reaction of (N-acyloxy)phthalimide 4 with styrene (eq 3). In this case, the product of reductive coupling was not obtained, but rather product 13 (83% as a 1:1 mixture of stereoisomers) resulting from the recombination of benzylic radical intermediates.12 Identical products were formed from the corresponding N-phthalimidoyl oxalate precursor.6 As found in that case, efforts to trap the coupled benzylic radical by modifying the reaction conditions, or by adding common hydrogen atom transfer reagents such as Bu3SnH, Et3SiH, Ph3SiH, and PhSH were unsuccessful.13
 |
(3) |
(N-acyloxy)phthalimides were also tested as precursors of tertiary radicals in allylation and vinylation reactions.14 We initiated these studies in a similar fashion by performing control and optimization experiments of the reaction of (N-acyloxy)phthalimide 4 with α-(bromomethyl)styrene (14) (Table 3). Using the conditions optimized for reductive coupling with electron-deficient alkenes, (N-acyloxy)phthalimide 4 reacted with α-(bromomethyl)styrene (14) to give substitution product 15 in 74% yield (entry 1). No synthetically useful product formation was observed in the absence of light or the Hantzsch ester (entries 2 and 3). In contrast to the related reaction with the tert-alkyl N-phthalimidoyl oxalate precursor,6 the absence of the Ru(bpy)3(PF6)2, did not diminish the yield of allylation product 15 after 18 hours (entry 4). However, entries 5 and 6 show that the reaction without the photocatalyst proceeds considerably slower. The yield of 15 was only slightly lower using 1 equiv of the acceptor (entry 7), again showing that the coupling of valuable fragments likely could be accomplished without the need of an excess of either coupling component. Finally, entries 8 to 10 confirmed that an excess of i-Pr2NEt led to higher yields, but even without this additive, product 15 was isolated in 48% yield.
Table 3.
Visible-Light Photoredox Coupling of (N-Acyloxy)phthalimide 4 with α-(Bromomethyl)styrene (14)
 | ||
|---|---|---|
| Entry | Modification | Yield of 15 (%)a |
| 1 | - | 74 |
| 2 | no light | NDb |
| 3 | no Hantzsch ester 8 | 14 |
| 4 | no photocatalyst (18 h) | 75 |
| 5 | no photocatalyst (2 h) | 11 |
| 6 | 2 h | 76 |
| 7 | acceptor (1 equiv) | 70 |
| 8 | no i-Pr2NEt | 48 |
| 9 | i-Pr2NEt (0.2 equiv) | 55 |
| 10 | i-Pr2NEt (1 equiv) | 62 |
Isolated yield after silica gel chromatography.
Product formation after resubjection to light.
ND = not detected.
With suitable reaction conditions in hand, we explored further the scope of radical substitution reactions of this type (Table 4). α-(Chloromethyl)styrene coupled with (N-acyloxy)phthalimide 4 to give allylation product 15 in 80% yield, which was slightly higher than that realized with the corresponding bromide 14. Methyl 2-(bromomethyl)acrylate and methyl 2-(chloromethyl)acrylate coupled in excellent yield with (N-acyloxy)phthalimide 4 to give product 16. Vinylation reactions with methyl 3-bromoacrylate and β-bromostyrene proceeded in lower yield, but with high (>20:1) E stereoselectivity, to form products 17 and 18, respectively. To realize the moderate yields in these vinylic coupling reactions, 5 equiv of the bromide coupling partner had to be employed. Products resulting from a second addition of the tertiary radical were never isolated, although allylation products 15 and 16 are potential excellent radical acceptors themselves. This selectivity likely results from steric shielding by the quaternary carbon fragment in these products.
Table 4.
Substitution Products Formed From Visible-Light Photocatalytic Coupling of Selected Allylic and Vinylic Bromides and Chlorides with (N-Acyloxy)phthalimide 4.a
 |
Isolated yield after silica gel chromatography (average of two experiments).
5 equiv of the halide was used; in other entries 1.5 equiv was used.
To explore further the scope of allylic coupling reactions of tertiary carbon radicals generated by visible-light photocatalysis, we examined the reaction of a selection of (N-acyloxy)phthalimides with α-(bromomethyl)styrene (14) (Table 5). In all cases, the product of 2-phenylallylation was isolated in high yield (71–91%). The coupling reactions of (N-acyloxy)phthalimides 3 and 5, derived from gemfibrozil and 18-β-glycyrrhetinic acid, illustrate the utility of (N-acyloxy)phthalimide derivatives to efficiently elaborate drug and natural product carboxylic acids to products containing new quaternary carbons (entries 3 and 4). Diastereoselectivity in the coupling of chiral (N-acyloxy)phthalimide 5 with allylic bromide 14 was only 3:1, reflecting the lack of dominant steric influence in the proximity to C19 in the 18-β-glycyrrhetinic acid series.
Table 5.
Visible-Light Photocatalytic Coupling of Various Tertiary (N-Acyloxy)phthalimides with (α-Bromomethyl)styrene (14).a
 |
 |
(N-acyloxy)phthalimide (1 equiv), Ru(bpy)3(PF6)2 (1 mol%), 14 (1.5 equiv), Hantzsch ester 8 (1.5 equiv), i-Pr2NEt (2.2 equiv), blue LEDs, 0.15 M in CH2Cl2, rt, 18 h.
Isolated yield after silica gel chromatography (average of two experiments).
Evaluating the generality of the radical coupling in the absence of Ru(bpy)32+
After observing during our optimization studies that the reductive coupling of (N-acyloxy)phthalimides with electron-deficient alkenes occurs in the absence of the Ru(bpy)32+ photocatalyst (Table 1, entries 13 and 14), we investigated further the generality of this reaction. As our preliminary studies had shown that the reductive coupling with MVK was significantly slower in the absence of the photocatalyst, reactions were carried out for 18 h (Table 6). Reductive coupling of (N-acyloxy)phthalimide 4 with MVK or acrylonitrile provided products 9 and 22 in 61% and 57% yield, respectively (entries 1 and 2). Attempted reactions of (N-acyloxy)phthalimide 4 with methacrylonitrile or three cyclopent-1-ene-1-carbonitriles afforded no products of reductive coupling (entries 4–7). In these four cases, both coupling partners were recovered in high yield.
Table 6.
The Reaction of (N-Acyloxy)phthalimide 4 with Various Alkenes in the Absence of a Photocatalyst.
 |
Isolated yield after silica gel chromatography.
50% of 2-cyclopentenone was recovered.
Both 4 and the alkene acceptor were recovered in >90% yield.
Allylic and vinylic substitution reactions were also surveyed in the absence of the Ru(bpy)32+ photocatalyst (Table 7). In contrast to the results of the reductive coupling reactions, allylic substitution reactions of (N-acyloxy)phthalimide 4 with α-(chloromethyl)- and α-(bromomethyl)styrene afforded allylated product 15 in identical high yields to that observed in the presence of Ru(bpy)3(PF6)2. The allylic substitution product 16 and the vinylic substitution products 17 and 18 were also formed in the absence of the photocatalyst, although in these cases yields were approximately 40% lower than those realized in the presence of the photocatalyst (Table 4). Attempted coupling of styrene with (N-acyloxy)phthalimide 4 in the absence of Ru(bpy)3(PF6)2 gave only trace amounts of product 13 (see eq 3) resulting from recombination of benzylic radical intermediates.
Table 7.
Substitution Products Formed from Visible-Light Photocatalytic Coupling of Selected Allylic and Vinylic Bromides and Chlorides With (N-Acyloxy)phthalimide 4 in the Absence of Ru(bpy)3(PF6)2.a
 |
Isolated yield after silica gel chromatography.
5 equiv of the acceptor were used.
Mechanistic investigations
The basic mechanism originally suggested by Okada for the formation of carbon radicals upon visible-light irradiation of (N-acyloxy)phthalimides in the presence of a dihydropyridine reductant and catalytic Ru(bpy)32+ (vide infra)1 is consistent with the results of our investigations. However, our experimental results suggest that the termination of the reductive coupling of (N-acyloxy)phthalimides with electron-deficient alkenes can take place by two pathways (Scheme 1). After addition of a tertiary radical to a C–C π-bond, the product radical can be terminated either by hydrogen atom abstraction (path A) or by a two-step process of single-electron transfer followed by protonation of the resulting anion (path B).
Scheme 1.

Possible Termination Pathways in Reductive Coupling of Tertiary Radicals and Alkenes
To probe the role of the Hantzsch ester in the termination sequence, we examined the coupling of (N-acyloxy)phthalimide 4 with MVK employing 4,4-dideuterio Hantzsch ester 23 (eq 4).15 Coupled product 9 was obtained from this reaction in 47% yield and was determined to have only 33% deuterium incorporation (at C2 of the butanone side chain).16 This result
 |
(4) |
contrasts sharply with the essentially complete deuterium incorporation observed in the related coupling of the corresponding N-phthalimidoyl oxalate.6 As the protic acid generated by oxidation of dideutero Hantzsch ester 23 would be a mixture of protio and deuterio species, the low level of deuterium incorporation in product 9 (eq 4) is consistent with significant termination by the two-step electron transfer/protonation sequence.
To examine in more depth the termination stage of reductive coupling of (N-acyloxy)phthalimides with alkenes, we investigated coupling reactions of (N-acyloxy)phthalimide 4 with various α,β-unsaturated nitriles containing a leaving group at the allylic α' position, a strategy introduced in the preceding article.6 Coupling of 1-methyl-1-cyclohexyl (N-acyloxy)phthalimide (4) with cyanocyclopentene allylic benzoate 24, under our optimized conditions for reductive couplings, gave products 25 and 26 in 28% and 45% yield, respectively (Scheme 2). The product of reductive coupling was formed as a mixture of four stereoisomers, with the major isomer being represented by structure 25.17 As β-scission of intermediate E to eject a high-energy benzoyloxy radical is implausible, the significant formation of allylic substitution product 26 requires that the intermediate radical E first suffers single-electron reduction to α-cyanocarbanion F.18
Scheme 2.
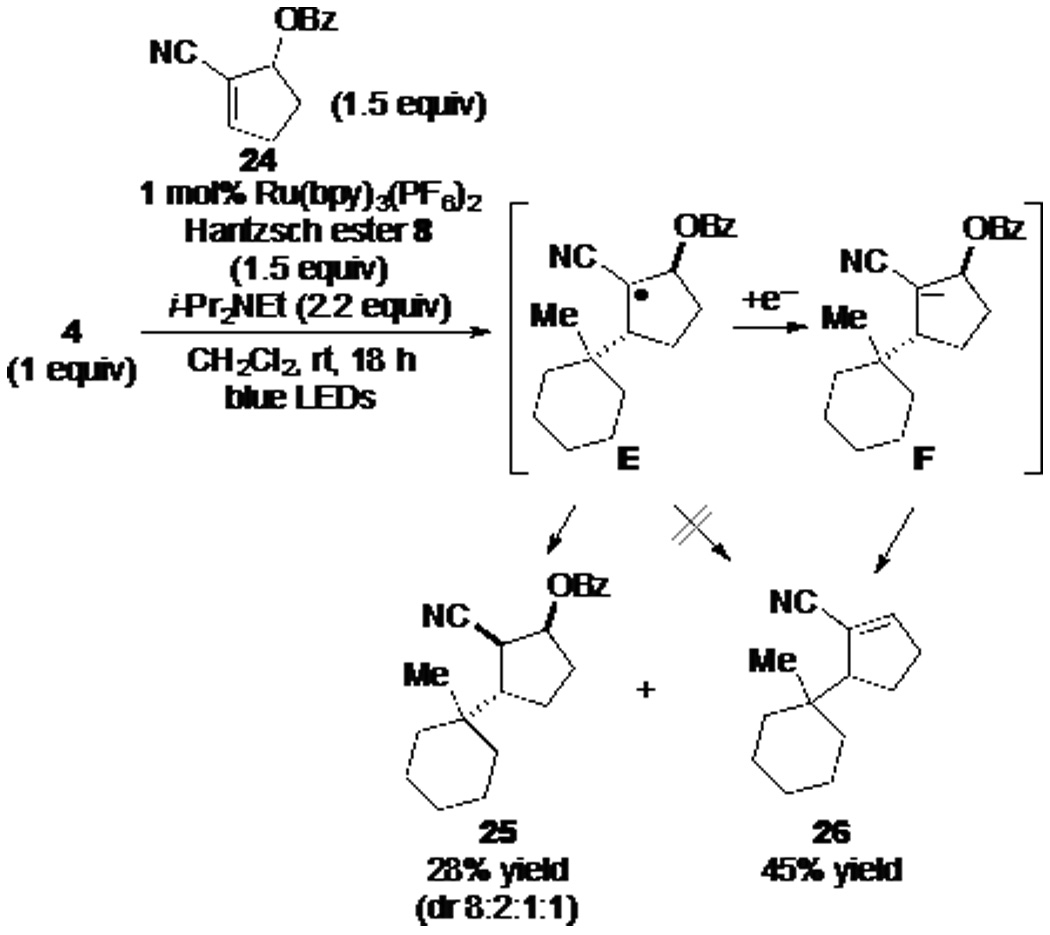
Products Formed Upon Visible-Light Photoredox Coupling of N-(Acyloxy)phthalimide 4 with Cyanocyclopentene Allylic Benzoate 24
In order to examine whether the nature of the radical precursor, or more likely the reaction conditions used for radical generation, has an effect on the termination mechanism, (N-acyloxy)phthalimide 4 was allowed to react with cyclopentene allylic benzoate 24 under the conditions typically used for the reductive coupling of N-phthalimidoyl oxalates (eq 5). In this case, only products of reductive coupling were obtained (67% yield). As the conditions of the reactions reported in Scheme 2 and eq 5 differ most notably in the presence of i-Pr2NEt in the former, the electron-rich trialkylamine appeared to be potentially critical for single-electron reduction of the coupled radical E to generate intermediate carbanion F of Scheme 2.
 |
(5) |
To further study the role of i-Pr2NEt in the termination process, we subjected (N-acyloxy)phthalimide 4 to photoredox-catalyzed coupling with cyclopentene 24 omitting the Hantzsch ester 8 (eq 6). This experiment resulted in the exclusive formation of allylic substitution product 26 in 36% yield, with no trace of product 25 of reductive coupling being observed.19 These results implicate the combination of i-Pr2NEt and the photocatalyst as the single-electron donors in the reduction of the initially formed product radical,20,21,22 and show that the aminium radical cation generated upon oxidation of the amine during the course of the reaction does not act as a hydrogen-atom donor in the termination step.
 |
(6) |
As expected, the coupling of (N-acyloxy)phthalimide 4 with cyclopentenyl bromide 27 provided only the allylic substitution product 26 (eq 7). As 26 was also formed exclusively in the coupling of 1-methyl-1-cyclohexyl N-phthalimidoyl oxalate with bromoalkene 27–a process that undoubtedly involves homolytic β-scission6–single-electron reduction would not be required for the formation of substitution product 26 in the transformation depicted in eq 7.23
 |
(7) |
We conclude with one additional example of the critical role the stoichiometric reductant can play. As already described, the coupling of (N-acyloxy)phthalimide 4 with α-(bromomethyl)styrene (14) proceeded in the absence of the photocatalyst (Table 7) or i-Pr2NEt, albeit the later in poorer yield (Table 3, entries 1 and 8). In contrast, the reaction of (N-acyloxy)phthalimide 4 with α-(acetoxymethyl)styrene (28) fails in the absence of either i-Pr2NEt or Ru(bpy)3(PF6)2 (eq 8).
 |
(8) |
DISCUSSION
On the basis of our investigation and existing precedent,1,2 we propose the following mechanism for the coupling of tertiary (N-acyloxy)phthalimides with conjugate acceptors (Scheme 3). Irradiation of Ru(bpy)32+ with visible light generates the excited-state catalyst Ru(bpy)32+*, which is reductively quenched by i-Pr2NEt or the Hantzsch ester 8 to provide the strong reductant Ru(bpy)3+. The (N-acyloxy)phthalimide 29 then receives an electron from Ru(bpy)3+ (E1/2 = –1.33 V vs. SCE), or potentially–but less likely–from the Hantzsch ester intermediate H that is produced in the termination step (vide infra), to transiently form radical anion I. Homolytic fragmentation and decarboxylation of I releases phthalimide or phthalimide anion, CO2, and the tertiary radical intermediate A. Addition of this radical to a conjugate acceptor generates stabilized radical J, which can be terminated either by hydrogen-atom abstraction from dihydropyridine 8 or derived intermediate G or by single-electron transfer from Ru(bpy)3+ followed by protonation to provide the product 30.
Scheme 3.

Proposed Mechanism for the Ru(bpy)32+-Catalyzed Coupling of Tertiary (N-Acyloxy)phthalimides with MVK in the Presence of Visible Light
We turn to consider the related transformation in the absence of the photocatalyst, a transformation that is poorly understood at this time. Owing to the observed requirement of the Hantzsch ester 8 for significant reaction progress (Table 1, entry 11), the observed reactivity in the absence of Ru(bpy)32+ most likely is mediated by the Hantzsch ester. This suggestion would be consistent with Okada’s observation of a related transformation in the absence of Ru(bpy)32+ under reaction conditions in which a dihydropyridine was the only reductant present (see eq 1). The non-catalyzed reaction could be initiated by oxidation of the Hantzsch ester 8 by trace amounts of oxygen to form intermediate G (depicted in Scheme 3),24,25 or potentially by electron transfer from photoexcited 8 to the (N-acyloxy)phthalimide. Loss of a proton from radical cation G would form the vinylogous α-amino radical H. Intermediate H is a strong one-electron reductant (E½ = –0.71 V vs SCE);26 however, not sufficiently strong that rapid electron transfer to a (N-acyloxy)phthalimide (E½ = –1.26 to –1.37 V vs. SCE)6,27 would be expected. Presumably single-electron transfer form H occurs in concert with cleavage of the N–O bond of the (N-acyloxy)phthalimide.28–,29,30 The propagation steps of the resulting chain reaction are depicted in Scheme 4.31
Scheme 4.

Potential Chain Mechanism for the Visible-Light Promoted Coupling of Tertiary (N-Acyloxy)phthalimides with MVK in the Absence of Ru(bpy)32+
The observation that the visible-light reductive coupling is slower in the absence of a photocatalyst, and succeeds only with highly reactive coupling partners, would be consistent with a chain mechanism (Scheme 4). However, much additional investigation will be required to elucidate in any detail the mechanism of the reductive coupling in the absence of the photocatalyst.
Conclusion
(N-Acyloxy)phthalimide derivatives of tertiary carboxylic acids are shown to be excellent precursors of tertiary radicals upon visible-light irradiation in the presence of 1 mol% of commercially available Ru(bpy)3(PF6)2, 1.5 equiv of Hantzsch ester 8, and 2.2 equiv of i-Pr2NEt in dichloromethane at room temperature. Tertiary radicals generated in this way reductively couple with a variety of electron-deficient alkenes, and undergo substitution reactions with allylic and vinylic halides, in moderate to excellent yields to form new C–C σ-bonds and new quaternary centers. In nearly all cases examined, the yields of these photocatalytic reactions were higher than the analogous transformations of the corresponding N-phthalimidoyl oxalates.6 In some cases, the coupling of (N-acyloxy)phthalimide derivatives of tertiary carboxylic acids can be accomplished in the absence of the photocatalyst Ru(bpy)3(PF6)2; however, this reaction is of less preparative significance, because it is slower than the photocatalytic reaction and only succeeds with highly reactive radical acceptors.
The ability to include or exclude the electron-rich trialkylamine i-Pr2NEt in photocatalytic reactions of (N-acyloxy)phthalimides allows one to dictate whether the coupling reaction is terminated by hydrogen-atom transfer or single-electron reduction followed by protonation. With some alkene radical acceptors, this choice can dictate the reaction outcome.
The ease of synthesis and purification and high crystallinity of (N-acyloxy)phthalimides, together with their efficient photocatalytic generation of tertiary radicals at room temperature upon irradiation with visible light, combine to make these carboxylic acid derivatives highly attractive precursors of tertiary carbon radicals for use in C–C bond formation.
EXPERIMENTAL SECTION
Materials and Methods
Unless stated otherwise, reactions were conducted in oven-dried glassware under an atmosphere of nitrogen or argon using anhydrous solvents (either freshly distilled or passed through activated alumina columns). For all radical coupling reactions, CH2Cl2 was sparged with argon for 5 min prior to use. Commercially obtained reagents were used as received. Ru(bpy)3(PF6)2 was obtained from Sigma Aldrich. Methyl vinyl ketone (MVK), acrylonitrile, benzyl methacrylate and methacrylonitrile were distilled from neat solutions prior to use. Hantzsch ester 8 is commercially available; however, we prepared it by a straightforward literature procedure.31 The 4,4-d2-Hantzsch ester 23,14a i-Pr2NEt•HBF4,8 methyl 2-(bromomethyl)acrylate,32 methyl 2-(chloromethyl)acrylate,33 (E)-methyl 3-bromoacrylate,34 α-(chloromethyl)styrene,35 α-(bromomethyl)styrene (14),35 α-(acetoxymethyl)styrene (28)36 were prepared according to literature procedures. The syntheses of 2,37,4 24,6 and 276 have been reported previously. Usually one representative coupling reaction and yield of the product is described in detail; isolated yields reported in the Results section are the average yields obtained from duplicate experiments. Reaction temperatures were controlled using a temperature modulator, and unless stated otherwise, reactions were performed at room temperature (rt, approximately 23 °C). Thin-layer chromatography (TLC) was conducted with E. Merck silica gel 60 F254 pre-coated plates (0.25 mm), and was visualized by exposure to UV light (254 nm) or anisaldehyde, ceric ammonium molybdate, iodine, or potassium permanganate. EMD silica gel 60 (particle size 0.040–0.063 mm) was used for flash column chromatography. 1H NMR spectra were recorded at 500 or 600 MHz and are reported relative to deuterated solvent signals. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz) and integration. 13C NMR spectra were recorded at 125 or 150 MHz. Data for 13C NMR spectra are reported in terms of chemical shift. IR spectra were recorded on an FT-IR spectrometer and are reported in terms of frequency of absorption (cm−1). Blue LEDs (30 cm, 1 watt) were purchased from Creative Lighting (http://www.creativelightings.com, product code CL-FRS5050-12WP-12V) and powered by 8 AA batteries.
(N-Acyloxy)phthalimide 1
A round-bottom flask was charged with adamantane-1-carboxylic acid (1.08 g, 6.00 mmol, 1 equiv) and THF (28 mL) under argon. After sequential addition of N-hydroxyphthalimide (1.64 g, 10.0 mmol, 1.66 equiv), DMAP (35 mg, 0.29 mmol, 0.05 equiv) and N,N’-diisopropylcarbodiimide (1.4 mL, 8.93 mmol, 1.5 equiv), the reaction mixture was maintained at rt with stirring overnight. After this time, the heterogeneous mixture was filtered and the filtrate concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (10% EtOAc/hexanes). Subsequent recrystallization from hot hexanes gave 1 (1.75 g, 5.38 mmol, 90%) as a colorless solid.1 Rf 0.53 (20% EtOAc/hexanes); mp: 143–144 °C; 1H NMR (500 MHz, CDCl3): δ 7.88–7.86 (m, 2H), 7.78–7.76 (m, 2H), 2.14 (s, 6H), 2.10 (s, 3H), 1.78 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 173.4, 162.3, 134.8, 129.2, 124.0, 40.7, 38.6, 36.3, 27.8; IR (thin film): 2907, 2852, 1776, 1741, 1466, 1356 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C19H19NO4NH4 343.1658, found 343.1643.
(N-Acyloxy)phthalimide 3
Following the procedure described for the preparation of 1, gemfibrozil (1.50 g, 6.00 mmol, 1 equiv), N-hydroxyphthalimide (1.64 g, 10.0 mmol, 1.66 equiv), DMAP (35 mg, 0.29 mmol, 0.05 equiv), N,N’-diisopropylcarbodiimide (1.4 mL, 8.90 mmol, 1.5 equiv) in THF (28 mL) gave, after purification of the crude product by silica gel chromatography (10% EtOAc/hexanes), 3 (2.12 g, 5.37 mmol, 90%) as a colorless solid. Rf 0.52 (20% EtOAc/hexanes); mp: 79–80 °C; 1H NMR (500 MHz, CDCl3): δ 7.89–7.88 (m, 2H), 7.80–7.78 (m, 2H), 7.00 (d, J = 7.2, 1H), 6.67-6.65 (m, 2H), 4.03-4.00 (m, 2H), 2.32 (s, 3H), 2.19 (s, 3H), 1.97-1.92 (m, 4H), 1.45 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 173.9, 162.2, 157.1, 136.6, 134.8, 130.4, 129.2, 124.0, 123.7, 120.8, 112.1, 67.8, 42.1, 37.5, 25.3, 25.1, 21.6, 15.9; IR (thin film): 2923, 2870, 1782, 1744, 1509, 1468, 1370, 1264, 1130, 1043 cm−1; HRMS-ESI (m/z) [M + H]+ calculated for C23H25NO5H 396.1811, found 396.1823.
(N-Acyloxy)phthalimide 4
A round bottom flask was charged with 1-methyl-1-cyclohexane carboxylic acid (5.00 g, 35.2 mmol, 1 equiv), N-hydroxyphthalimide (8.61 g, 52.8 mmol, 1.5 equiv) and N,N’-dicyclohexylcarbodiimide (10.90 g, 52.8 mmol, 1.5 equiv) under argon. After sequential addition of THF (350 mL) and DMAP (430 mg, 3.52 mmol, 0.1 equiv), the reaction mixture was maintained at rt with stirring overnight. After this time, the heterogeneous mixture was concentrated under reduced pressure and the resulting residue suspended in Et2O (400 mL). The mixture was filtered through cotton, transferred to a separatory funnel, and washed with saturated aqueous NH4Cl (3×200 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (7% EtOAc/hexanes) to give 4 (9.05 g, 31.5 mmol, 90%) as a colorless solid. Rf 0.26 (10% EtOAc/hexanes); mp: 52–54 °C; 1H NMR (600 MHz, CDCl3): δ 7.90–7.84 (m, 2H), 7.79–7.75 (m, 2H), 2.26–2.20 (m, 2H), 1.69–1.51 (m, 5H), 1.42 (s, 3H), 1.40–1.34 (m, 2H), 1.33–1.23 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 173.7, 162.3, 134.7, 129.2, 123.9, 43.2, 35.8, 26.8, 25.5, 23.1; IR (thin film): 2934, 2860, 1807, 1782, 1743 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C16H17NO4Na 310.1055, found 310.1051.
(N-Acyloxy)phthalimide 5
A round bottom flask was charged with 18β-glycyrrhetinic acid (1.00 g, 2.12 mmol, 1 equiv) and THF (21 mL) under argon. After sequential addition of N-hydroxyphthalimide (520 mg, 3.19 mmol, 1.5 equiv), DMAP (52 mg, 0.43 mmol, 0.2 equiv) and N,N’-diisopropylcarbodiimide (0.4 mL, 2.6 mmol, 1.2 equiv), the reaction mixture was maintained at rt while stirring overnight. After this time, the heterogeneous mixture was filtered and the filtrate quenched with saturated aqueous NaHCO3 (20 mL). The aqueous phase was extracted with Et2O (3×50 mL). The combined organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (20–40% EtOAc/hexanes) to give 5 (1.02 g, 1.66 mmol, 78%) as a colorless solid. Rf 0.27 (40% EtOAc/hexanes); mp 279–283 °C (dec); [α]D24 +179, [α]57724 +188, [α]54624 +216, [α]43524 +384, [α]40524 +486 (c 0.78 (CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.89–7.87 (m, 2H), 7.80–7.78 (m, 2H), 5.76 (s, 1H), 3.22 (dd, J = 11.0, J = 5.2, 1H), 2.78 (td, J = 6.6, 3.1, 1H), 2.54 (dd, J = 13.4, J = 3.3, 1H), 2.33 (s, 1H), 2.13 (d, J= 13.5, 1H), 2.09-2.01 (m, 2H), 1.86 (dt, J = 13.6, 4.5, 1H), 1.78 (t, J= 13.8, 1H), 1.69-1.58 (m, 5H), 1.51-1.40 (m, 7H), 1.38 (s, 3H), 1.34-1.24 (m, 1H), 1.21 (d, J= 13.4, 1H), 1.14 (s, 3H), 1.12 (s, 3H), 1.09-1.04 (m, 1H), 1.00 (s, 3H), 0.98-0.93 (m, 1H), 0.91 (s, 3H), 0.80 (s, 3H), 0.70 (d, J= 11.0, 1H); 13C NMR (125 MHz, CDCl3): δ 200.2, 172.7, 168.5, 162.2, 134.9, 129.2, 129.0, 124.1, 78.9, 61.9, 55.1, 47.8, 45.5, 44.0, 43.2, 41.3, 39.3, 39.2, 37.4, 37.2, 32.9, 32.0, 31.6, 28.5, 28.2, 28.1, 27.4, 26.6, 26.5, 23.5, 18.8, 17.6, 16.5, 15.7; IR (thin film): 3519, 2968, 2931, 2868, 1806, 1782, 1744, 1655, 1039 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C38H49NO6Na 638.3458, found 638.3438.
4-(1-Methylcyclohexyl)butan-2-one (9)
(Table 1, entry 1 and general procedure for optimization experiments). A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (2 mg, 2.6 µmol, 0.01 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1.7 mL, sparged with Ar for 5 min), methyl vinyl ketone (33 µL, 0.39 mmol, 1.5 equiv) and i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm-loop of blue LEDs (see the Supporting Information for a picture of the reaction setup). The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (2.5% EtOAc/hexanes) to provide 9 (40 mg, 0.24 mmol, 91%) as a colorless oil. Characterization data obtained for 9 matched those previously reported.5
3-(1-Methylcyclohexyl)cyclopentan-1-one (10)
Following the general procedure for optimization experiments, (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (2 mg, 2 µmol, 0.01 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv), cyclopentenone (33 µL, 0.39 mmol, 1.5 equiv), i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv) in CH2Cl2 (1.7 mL, sparged with Ar for 5 min) gave 10 (37 mg, 0.21 mmol, 80%) as a colorless oil. Characterization data obtained for 10 matched those previously reported.5
(±)-Benzyl 2-methyl-3-(1-methylcyclohexyl)propanoate (11)
Following the general procedure for optimization experiments, (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (2 mg, 2.6 µmol, 0.01 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv), benzyl methacrylate (66 µL, 0.39 mmol, 1.5 equiv), i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv) in CH2Cl2 (1.7 mL, sparged with Ar for 5 min) gave a crude product. The yield of 11 (59%) was determined by examining the relative integration of NMR signals in this crude mixture using an internal standard (1,4-dimethoxybenzene). An analytically pure sample of 11 was obtained by silica gel chromatography (2.5% EtOAc/hexanes) to provide 11 as a colorless oil. Characterization data for 11 are included in the preceding article.6
(±)-2-Methyl-3-(1-methylcyclohexyl)propanenitrile (12)
Following the general procedure for optimization experiments, (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (2 mg, 2.6 µmol, 0.01 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv), methacrylonitrile (33 µL, 0.39 mmol, 1.5 equiv), i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv) in CH2Cl2 (1.7 mL, sparged with Ar for 5 min) gave 12 (36 mg, 0.22 mmol, 83%) as a colorless oil. Rf 0.47 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3): δ 2.64–2.57 (m, 1H), 1.77 (dd, J = 14.1, 13.4, 1H), 1.58–1.4 (m, 5H), 1.39–1.22 (m, 8H), 0.97 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 124.5, 38.0, 37.6, 33.2, 26.3, 22.0, 21.9, 20.8, 20.2; IR (thin film): 2927, 2853, 2237, 1455, 1382 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C11H19NNa 188.1415, found 188.1408.
(±)-(1,4-bis(1-methylcyclohexyl)butane-2,3-diyl)dibenzene (13) (eq 3)
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv), Ru(bpy)3(PF6)2 (1 mg, 1.5 µmol, 0.01 equiv), Hantzsch ester 8 (57 mg, 0.23 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (0.5 mL, sparged with Ar for 5 min), styrene (26 µL, 0.23 mmol, 1.5 equiv) and i-Pr2NEt (55 µL, 0.33 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (100% pentane) to provide 13 (25 mg, 0.063 mmol, 83%), a 1:1 mixture of stereoisomers, as a colorless solid. Characterization data for 13 are included in the preceding article.6
(3-(1-Methylcyclohexyl)prop-1-en-2-yl)benzene (15)
(Table 3, entry 1, coupling with α-(bromomethyl)styrene and general procedure for allylic and vinylic substitution). A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv), Ru(bpy)3(PF6)2 (1 mg, 1.5 µmol, 0.01 equiv), Hantzsch ester 8 (57 mg, 0.23 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1 mL, sparged with Ar for 5 min), α-(bromomethyl)styrene (14) (33 µL, 0.23 mmol, 1.5 equiv) and i-Pr2NEt (55 µL, 0.33 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (100% pentane) to provide 15 (24 mg, 0.11 mmol, 74%) as a colorless oil. Characterization data for 15 are included in the preceding article.6
Preparation of 15 from α-(chloromethyl)styrene
In an identical fashion, (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv) was coupled with α-(chloromethyl)styrene (32 µL, 0.23 mmol, 1.5 equiv) to provide 15 (26 mg, 0.12 mmol, 82%).
Methyl 2-((1-methylcyclohexyl)methyl)acrylate (16)
In an identical fashion, (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv) was coupled with methyl 2-(bromomethyl)acrylate (27 µL, 0.23 mmol, 1.5 equiv) to give a crude residue, which was purified by silica gel chromatography (3% diethyl ether/pentane) to provide 16 (25 mg, 0.13 mmol, 83%) as a colorless oil. Characterization data for 16 are included in the preceding article.6
Preparation of 16 from methyl 2-(chloromethyl)acrylate
In an identical fashion, (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv) was coupled with methyl 2-(chloromethyl)acrylate (26 µL, 0.23 mmol, 1.5 equiv) to provide 16 (25 mg, 0.13 mmol, 83%) as a colorless oil.6
Methyl (E)-3-(1-methylcyclohexyl)acrylate (17)
In an identical fashion, (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv) was coupled with (E)-methyl 3-bromoacrylate (72 µL, 0.75 mmol, 5 equiv) to provide a crude residue, which was purified by silica gel chromatography (5% diethyl ether/pentane) to give 17 (16 mg, 0.088 mmol, 59%) as a colorless oil. Characterization data for 17 are included in the preceding article.6
(E)-(2-(1-methylcyclohexyl)vinyl)benzene (18)
In an identical fashion, (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv) was coupled with β-bromostyrene (97 µL, 0.75 mmol, 5 equiv) to give a crude residue, which was purified by silica gel chromatography (100% pentane) to provide 18 (18 mg, 0.089 mmol, 59%) as a colorless oil. Characterization data for 18 are included in the preceding article.6
1-(2-Phenylallyl)adamantane (19)
In an identical fashion, (N-acyloxy)phthalimide 1 (49 mg, 0.15 mmol, 1 equiv) was coupled with α-(bromomethyl)styrene (14) (33 µL, 0.23 mmol, 1.5 equiv) to give a crude residue, which was purified by silica gel chromatography (100% pentane) to provide 19 (27 mg, 0.11 mmol, 72%) as a colorless solid. Characterization data for 19 are included in the preceding article.6
2-((4,4-Dimethyl-6-phenylhept-6-en-1-yl)oxy)-1,4-dimethylbenzene (20)
In an identical fashion, (N-acyloxy)phthalimide 3 (59 mg, 0.15 mmol, 1 equiv) was coupled with α-(bromomethyl)styrene (14) (33 µL, 0.23 mmol, 1.5 equiv) to give a crude residue, which was purified by silica gel chromatography (0–2% diethyl ether/pentane) to provide 20 (44 mg, 0.14 mmol, 91%) as a colorless oil. Rf 0.16 (100% hexanes); 1H NMR (500 MHz, CDCl3): δ 7.41 (d, J = 7.4, 2H), 7.33 (t, J = 7.5, 2H), 7.28–7.25 (m, 1H), 7.04 (d, J = 7.5, 1H), 6.69 (d, J = 7.5, 1H), 6.61 (s, 1H), 5.28 (d, J = 1.9, 1H), 5.07 (s, 1H), 3.81 (t, J = 6.6, 2H), 2.54 (s, 2H), 2.34 (s, 3H), 2.21 (s, 3H), 1.79-1.72 (m, 2H), 1.37-1.31 (m, 2H), 0.82 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 157.2, 147.4, 144.0, 136.5, 130.4, 128.3, 127.1, 126.7, 123.7, 120.7, 116.9, 112.1, 68.7, 47.0, 38.6, 34.2, 27.8, 24.5, 21.6, 16.0; IR (thin film): 2953, 2925, 2867, 1616, 1585, 1509, 1469, 1265, 1157, 1130, 1044 cm−1; HRMS-CI (m/z) [M + H]+ calculated for C23H30OH 323.2375, found 323.2386.
2-Phenylallylation of the (N-acyloxy)phthalimide derivative 5 of 18β-glycyrrhetinic acid to form 21
In an identical fashion, (N-acyloxy)phthalimide 5 (92 mg, 0.15 mmol, 1 equiv) was coupled with α-(bromomethyl)styrene (14) (33 µL, 0.23 mmol, 1.5 equiv). After 18 h, the reaction mixture was diluted with CH2Cl2, the resulting organic phase was washed with 4 M HCl (3×10 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel chromatography (20–30% acetone/hexanes) to provide an inseparable 3:1 mixture of C19 epimers of 21 (61 mg, 0.11 mmol, 75%) as a colorless solid. Mixture of two epimers: Rf 0.53 (30% acetone/hexanes); mp 72–74 °C; 1H NMR (500 MHz, CDCl3): δ 7.37-7.27 (m, 5.7H), 7.25-7.21 (m, 1H), 5.49 (s, 1H), 5.25 (d, J = 1.8, 1H), 5.21 (d, J = 1.7, 0.3H), 5.10 (s, 0.3H), 5.04 (s, 0.3H), 5.00 (s, 1H), 3.24-3.19 (m, 1.3H), 2.77 (d, J = 13.4, 1.3H), 2.72 (d, J = 13.3, 0.3H), 2.51 (d, J = 13.3, 1H), 2.43-2.36 (m, 1.3H), 2.25 (d, J = 14.5, 1.3H), 2.08-1.99 (m, 1.7H), 1.91-1.80 (m, 1.3H), 1.80-1.69 (m, 1.7H), 1.67-1.54 (m, 7.7), 1.45-1.31 (m, 6.7H), 1.29 (s, 1.3H), 1.26-1.19 (m, 2.3H), 1.18-1.02 (m, 14.7), 1.02-0.91 (m, 8H), 0.90-0.77 (m, 15H), 0.70-0.64 (m, 2.7H); 13C NMR (125 MHz, CDCl3): δ 200.5, 200.4, 170.7, 170.1, 147.3, 146.5, 144.1, 143.8, 128.4, 128.3, 128.1, 127.4, 127.2, 126.7, 126.5, 117.3, 117.2, 78.9, 61.8, 55.0, 55.0, 50.7, 47.4, 46.9, 45.5, 43.4, 43.3, 43.2, 42.4, 40.4, 39.3, 37.2, 36.2, 36.1, 35.2, 34.8, 34.0, 32.9, 32.8, 32.5, 32.3, 32.2, 30.8, 29.0, 28.7, 28.2, 27.4, 26.8, 26.5, 26.4, 23.5, 23.2, 22.7, 18.8, 17.6, 16.5, 15.7; IR (thin film): 3435, 2925, 2863, 1654, 1461, 1386, 1208, 1044 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C38H54O2Na 565.4022, found 565.3996.
General procedure for coupling reactions in the absence of a photocatalyst (Tables 6 and 7)
Preparation of 9. A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1.7 mL, sparged with Ar for 5 min), methyl vinyl ketone (33 µL, 0.39 mmol, 1.5 equiv) and i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (2.5% EtOAc/hexanes) to provide 9 (27 mg, 0.16 mmol, 61%) as a colorless oil. Characterization data obtained for 9 matched those previously reported.5
Deuterium incorporation in product 9 using 4,4-d2-Hantzsch ester 23
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (3 mg, 2.6 µmol, 0.01 equiv), 4,4-d2-Hantzsch ester 23 (100 mg, 0.39 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1.7 mL, sparged with Ar for 5 min), methyl vinyl ketone (32 µL, 0.39 mmol, 1.5 equiv) and i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. Purification by silica gel chromatography (2.5% EtOAc/hexanes) provided ketone 9 (21 mg, 0.13 mmol, 48%) as a colorless oil. Deuterium incorporation was determined by comparing the relative 1H NMR integrations of the α-keto methyl singlet resonance with the multiplet signal corresponding to protons at C2 (see reference 6 for an 1H NMR spectrum of this product with high deuterium incorporation). 1H NMR analysis determined the deuterium incorporation to be 33%.6
Preparation of the product of reductive coupling 25 and allylation 26 (Scheme 2)
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (3 mg, 2.6 µmol, 0.01 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1.7 mL, sparged with Ar for 5 min), acceptor6 24 (84 mg, 0.39 mmol, 1.5 equiv) and i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm-loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was diluted with Et2O (30 mL) and transferred to a separatory funnel. The ether layer was washed with aqueous 4 N HCl (4 × 20 mL) and aqueous 2 N NaOH (3 × 20 mL) and dried over MgSO4. The organic layer was filtered and concentrated under reduced pressure. The crude residue was subjected to silica gel chromatography (4% acetone/hexanes) to provide 25 (23 mg, 0.07 mmol, 29%, dr 8:2:1:1) and 26 (25 mg, 0.13 mmol, 51%) as colorless oils. Characterization data for 25 and 26 are included in the preceding article.6
Preparation of reductive-coupling product 25 (eq 5)
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (100 mg, 0.35 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (3 mg, 3.5 µmol, 0.015 equiv), Hantzsch ester 8 (88 mg, 0.35 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (50 mg, 0.23 mmol, 1 equiv) and a magnetic stir bar under argon. After sequential addition of THF (1.1 mL, sparged with Ar for 5 min), CH2Cl2 (1.1 mL, sparged with Ar for 5 min), and acceptor6 24 (49 mg, 0.23 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was diluted with Et2O (30 mL) and transferred to a separatory funnel. The ether layer was washed with aqueous 4 N HCl (4 × 20 mL) and aqueous 2 N NaOH (3 × 20 mL) and dried over MgSO4. The organic layer was filtered and concentrated under reduced pressure. The crude residue was subjected to silica gel chromatography (4% acetone/hexanes) to provide 25 (52 mg, 0.17 mmol, 72%, dr 8:2:1:1) as a colorless oil.6
Preparation of allylated product 26 (eq 6)
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (3 mg, 2.6 µmol, 0.01 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1.7 mL, sparged with Ar for 5 min), acceptor6 24 (84 mg, 0.39 mmol, 1.5 equiv) and i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was diluted with Et2O (30 mL) and transferred to a separatory funnel. The ether layer was washed with aqueous 4 N HCl (4×20 mL) and aqueous 2 N NaOH (3 × 20 mL) and dried over MgSO4. The organic layer was filtered and concentrated under reduced pressure. The crude residue was subjected to silica gel chromatography (4% acetone/hexanes) to provide 26 (19 mg, 0.10 mmol, 39%) as colorless oils.6
Preparation of allylated product 26 (eq 7)
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (75 mg, 0.26 mmol, 1 equiv), Ru(bpy)3(PF6)2 (3 mg, 2.6 µmol, 0.01 equiv), Hantzsch ester 8 (100 mg, 0.39 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1.7 mL, sparged with Ar for 5 min), acceptor6 27 (67 mg, 0.39 mmol, 1.5 equiv) and i-Pr2NEt (100 µL, 0.57 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. The crude residue was subjected to silica gel chromatography (2% EtOAc/hexanes) to provide 26 (30 mg, 0.16 mmol, 60%) as a colorless oil.6
Preparation of allylated product 15 (eq 8)
A 1-dram vial was charged with (N-acyloxy)phthalimide 4 (43 mg, 0.15 mmol, 1 equiv), Ru(bpy)3(PF6)2 (1 mg, 1.5 µmol, 0.01 equiv), Hantzsch ester 8 (57 mg, 0.23 mmol, 1.5 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1 mL, sparged with Ar for 5 min), α-(acetoxymethyl)styrene (28) (38 µL, 0.23 mmol, 1.5 equiv) and i-Pr2NEt (55 µL, 0.33 mmol, 2.2 equiv), the vial was capped and placed in the center of a 30-cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (100% pentane) to provide 15 (19 mg, 0.090 mmol, 60%) as a colorless oil.6
Supplementary Material
Acknowledgments
Financial support was provided by the National Science Foundation (CHE1265964) and the National Institute of General Medical Sciences (R01-GM098601). We thank the Alexander von Humboldt Foundation for the support of G.P. by a Feodor Lynen Postdoctoral Research Fellowship and the ACS Organic Chemistry Division for partial support of G.L.L. by a Graduate Fellowship. NMR and mass spectra were determined at UC Irvine using instruments purchased with the assistance of NSF and NIH shared instrumentation grants. We are grateful to Dr. Martin Schnermann for early optimization of the generation of carbon radicals from (N-acyloxy)phthalimides in aprotic solvents, and Kyle B. Schenthal for assistance in synthesizing (N-acyloxy)phthalimide starting materials.
Footnotes
Supporting Information. Copies of 1H and 13C NMR spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J. Am. Chem. Soc. 1991;113:9401–9402. [Google Scholar]
- 2.For a comprehensive review of visible-light photocatalysis which discusses the reactivity and electrochemical potentials of common photoredox catalysts see: Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. For a complementary method for generating carbon radicals from carboxylic acids using visible-light photocatalysis see: Chu L, Ohta C, Zuo Z, MacMillan DWC. J. Am. Chem. Soc. 2014;136:10886–10889. doi: 10.1021/ja505964r.
- 3.Schnermann MJ, Overman LE. Angew. Chem. Int. Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller DS, Untiedt NL, Dieskau AP, Lackner GL, Overman LE. J. Am. Chem. Soc. 2015;137:660–663. doi: 10.1021/ja512527s. [DOI] [PubMed] [Google Scholar]
- 5.Lackner GL, Quasdorf KW, Overman LE. J. Am. Chem. Soc. 2013;135:15342–15345. doi: 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lackner GL, Quasdorf KW, Pratsch G, Overman LE. preceding article. [Google Scholar]
- 7. Barton DHR, Serebryakov EPA. Proc. Chem. Soc. 1962:309. Barton DHR, Dowlatshahi HA, Motherwell WB, Villemin D. J. Chem. Soc. Chem. Commun. 1980:732–733. Barton DHR, Crich D, Motherwell WB. Tetrahedron Lett. 1983;24:4979–4982. Barton D, Chern C-Y, Jaszberenyi J. Tetrahedron. 1995;51:1867–1886. For a brief review see: Saraiva MF, Couri MRC, Hyaric ML, de Almeida MV. Tetrahedron. 2009;65:3563–3572.
- 8.Andrews RS, Becker JJ, Gagné MR. Angew. Chem. Int. Ed. 2010;49:7274–7276. doi: 10.1002/anie.201004311. [DOI] [PubMed] [Google Scholar]
- 9.Compare the results reported in this table with the synthesis of the identical products reported in Table 5 of ref6.
- 10.This latter reaction exhibited low conversion of the N-phthalimidoyl oxalate and rapid consumption of methacrylonitrile, suggesting that methacrylonitrile was polymerized under these conditions.11
- 11.Similar photoredox catalysis conditions have been employed to initiate radical polymerization of methacrylates. See: Fors BF, Hawker CJ. Angew. Chem. Int. Ed. 2012;51:8850–8853. doi: 10.1002/anie.201203639.
- 12.Only a few radical addition-dimerization cascades of styrene derivatives are known in literature: Schäfer H. Angew. Chem. Int. Ed. 1970;9:158–159. Kambe N, Miyamoto M, Terao J, Watabe H. Bull. Chem. Soc. Jpn. 2003;76:2209–2214. Shen Z-L, Cheong H-L, Loh T-P. Tetrahedron Lett. 2009;50:1051–1054.
- 13.(a) Bockrath B, Bittner E, McGrewt J. J. Am. Chem. Soc. 1984;106:135–138. [Google Scholar]; (b) Mayer JM. Acc. Chem. Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wille U. Chem. Rev. 2013;113:813–853. doi: 10.1021/cr100359d. [DOI] [PubMed] [Google Scholar]; (d) Crich D, Grant D, Krishnamurthy V, Patel M. Acc. Chem. Res. 2007;40:453–463. doi: 10.1021/ar600020v. [DOI] [PubMed] [Google Scholar]
- 14.For other radical allylations realized using visible-light photocatalysis see: Larraufie M-H, Pellet R, Fensterbank L, Goddard J-P, Lacôte E, Malacria M, Ollivier C. Angew. Chem. Int. Ed. 2011;50:4463–4466. doi: 10.1002/anie.201007571. Dai X, Cheng D, Guan B, Mao W, Xu X, Li X. J. Org. Chem. 2014;79:7212–7219. doi: 10.1021/jo501097b.
- 15. Norcross BE, Klinedinst PE, Jr, Westheimer FH. J. Am. Chem. Soc. 1962;84:797–802. For recent use of 4,4-dideuterio-Hantzsch esters in studies of reactions promoted by visible-light photoredox catalysis see Neumann M, Zeitler K. Chem. Eur. J. 2013;19:6950–6955. doi: 10.1002/chem.201204573.
- 16.The position and amount of deuterium in product 9 was determined by integration of relevant signals by 1H NMR analysis; see the Experimental section for details.
- 17.The relative configuration was assigned on the basis of diagnostic nOe enhancements observed upon irradiation of the α-cyano-methine hydrogens of the two major diastereomers isolated from this reaction. See reference 6 for details.
- 18.Resubjection of 25 to the reaction conditions in Scheme 2 resulted in full recovery of 25 and no detection of 26, suggesting that 26 is not formed from 25 by base-promoted elimination.
- 19.Unreacted (N-acyloxy)phthalimide 4 was recovered in 60% yield.
- 20.Ru(bpy)3+ is the most reasonable electron-transfer agent under these conditions, as its reduction potential is much more negative (E1/2 = −1.33 V vs. SCE) than that of a tertiary amine (for Et3N, the corresponding E1/2 = +1.0 V vs. SCE)21 and more negative than that of the α-amino radical formed from i-Pr2NEt (for Et3N, the corresponding E1/2 = −1.12 V vs. SCE).22.
- 21.Wayner DDM, Dannenberg JJ, Griller D. Chem. Phys. Lett. 1986;131:189–191. [Google Scholar]
- 22.Wayner DDM, McPhee DJ, Griller D. J. Am. Chem. Soc. 1988;110:132–137. [Google Scholar]
- 23.Addition of a tertiary radical to 27 followed by homolysis of the C–Br bond is the most plausible mechanism. An alternative pathway involving coupling of the tertiary radical with an allylic radical derived from 27 is unlikely as products derived from allylic radical homodimerization were not observed.
- 24.Wang D, Liu Q, Chen B, Zhang L, Tung C, Wu L. Chin. Sci. Bull. 2010;55:2855–2858. [Google Scholar]
- 25.However, we have been unable to experimentally verify this suggestion: Exclusion of oxygen from a coupling reaction to the best of our ability by use of the freeze-pump-thaw degassing method did not affect the yield of product 9 obtained after a reaction time of 2 h.
- 26.This potential is reported as −1.11 V vs. ferrocene; see: Zhu X-Q, Tan Y, Cao C-T. J. Phys. Chem. B. 2010;114:2058–2075. doi: 10.1021/jp911137p.
- 27.Okada reports reduction potentials in the range of −1.28 V to −1.37 V (vs. SCE) for three (N-acyloxy)phthalimides in acetonitrile, see: Okada K, Okamoto K, Oda M. J. Am. Chem. Soc. 1988;110:8736–8738.
- 28.Savéant J-M. Advances in Physical Organic Chemistry. Vol. 35. New York: Tidwell T. T. Academic; 2000. Electron Transfer, Bond Breaking and Bond Formation; pp. 117–192. [Google Scholar]
- 29.(a) Owing to the low barrier for decarboxylation of carboxy radicals, K may well not be an intermediate, with decarboxylation occurring at the same time as fragmentation of the N–O bond. (b) The rate constant for the loss of CO2 from a carboxyl radical is estimated to be on the order of 109 s−1, see: Togo H. Advanced Free Radical Reactions for Organic Synthesis. Elsevier Science. 2004
- 30.Alternatively, although less likely in our view, electron transfer could be coupled with proton transfer to the phthalimide oxygen, avoiding the formation of an imide ketyl radical anion intermediate. For a comprehensive review of proton-coupled electron transfer (PCET), see: Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. (b) For a detailed mechanistic investigation establishing concerted PCET in a Ru(bpy)32+ photocatalyzed ketone-alkene coupling, see: Taratino KT, Liu P, Knowles RR. J. Am. Chem. Soc. 2013;135:10022–10025. doi: 10.1021/ja404342j.
- 31.Eey STC, Lear MJ. Org. Lett. 2010;12:5510–5513. doi: 10.1021/ol102390t. [DOI] [PubMed] [Google Scholar]
- 32.(a) Huang H, Liu X, Deng J, Qiu M, Zheng Z. Org. Lett. 2006;8:3359–3362. doi: 10.1021/ol0612399. [DOI] [PubMed] [Google Scholar]; (b) Kippo T, Fukuyama T, Ryu I. Org. Lett. 2011;13:3864–3867. doi: 10.1021/ol201395p. [DOI] [PubMed] [Google Scholar]
- 33.reference 32a. Young WG, Caserio FF, Jr, Brandon DD., Jr J. Am. Chem. Soc. 1960;82:6163–6168.
- 34.Ma S, Lu X, Li Z. J. Org. Chem. 1992;57:709–713. [Google Scholar]
- 35.Tripathi CB, Mukherjee S. Angew. Chem. Int. Ed. 2013;52:8450–8453. doi: 10.1002/anie.201304173. [DOI] [PubMed] [Google Scholar]
- 36.Hatch LF, Patton TL. J. Am. Chem. Soc. 1954;76:2705–2707. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.