

Abstract
Background
Cognitive deficits are prominent in schizophrenia and represent promising endophenotypes for genetic research.
Methods
The current study investigated the importance of two conceptually distinct genetic aggregates, one based on copy number variations (uncommon deletion burden), and one based on single nucleotide polymorphisms identified in recent risk studies (genetic risk score). The impact of these genetic factors, and their interaction, was examined on cognitive endophenotypes defined by principal component analysis (PCA) in a multi-center sample of 50 patients with schizophrenia and 86 controls. PCA was used to identify three different types of executive function (EF: planning, fluency, and inhibition), and in separate analyses, a measure general cognitive ability (GCA).
Results
Cognitive deficits were prominent among individuals with schizophrenia, but no group differences were evident for either genetic factor. Among patients the deletion burden measures predicted cognitive deficits across the three EF components and GCA. Further, an interaction was noted between the two genetic factors for both EF and GCA and the observed patterns of interaction suggested antagonistic epistasis. In general, the set of genetic interactions examined predicted a substantial portion of variance in these cognitive endophenotypes.
Limitations
Though adequately powered, our sample size is small for a genetic study.
Conclusions
These results draw attention to genetic interactions and the possibility that genetic influences on cognition differ in patients and controls.
Keywords: schizophrenia, genetics, executive function, mutation, cognitive, endophenotype
1. Introduction
In the search for the genetic roots of schizophrenia, the attempt to identify specific genetic influences on endophenotypes is increasingly common (Cannon and Keller, 2006). Cognitive endophenotypes such as general cognitive ability (GCA, or “g”) and executive function (EF), are (1) well measured, (2) clinically relevant, (3) heritable, and (4) from a cognitive neuroscience perspective, relatively well characterized (Langer et al., 2012; Miyake and Friedman, 2012). Greenwood and colleagues (Greenwood et al., 2013) explored genetic influences on several endophenotypes, including cognitive measures such as the California Verbal Learning Test and the Wisconsin Card Sorting Test. Linkage analysis across 296 families was unable to identify any single nucleotide polymorphism that significantly predicted performance on these cognitive tasks.
In the current report we investigate the genetic influences on GCA and EF, quantified through principal components analyses of many test scores, allowing reliable assessment of the relevant latent constructs while minimizing test-specific method variance (see Green et al., 2013, and Gold and Dickinson, 2013, for a recent discussion of GCA in schizophrenia). Genetic influences were represented by two conceptually distinct aggregate scores, not individual genetic loci. One aggregate was based on copy number variations (CNVs) and one was based on previously identified single nucleotide polymorphisms (SNPs). CNVs may represent either deletions or duplications of segments of DNA, and collectively, they account for many times more genetic variation in nucleotide sequences than SNPs (Girirajan et al., 2011). Because deletions are under greater negative selection pressure than duplications, and the population frequency of deletions is generally inversely related to their potential for harm (Zhang et al., 2009), one simple way to represent mutation load is by the total number of rare or uncommon deletions. We reported that a greater overall burden of uncommon deletions (less than 3% frequency) predicted lower GCA in patients with schizophrenia, but not controls (Yeo et al., 2013a). The specific deletions captured in this overall measure differ across individuals, constraining theoretical interpretations. The SNP aggregate used in the current study (genetic risk score, GRS), originally reported in Walton et al. (Walton et al., 2013), was derived from the empirical literature on alleles possibly distinguishing individuals with schizophrenia from controls. This measure combined the additive effects of 41 single nucleotide polymorphisms (SNPs) in 34 genes weighted by their odds ratios. The genes involved span many functions, most prominently, neurotransmission (32%) and neurodevelopment (26%).
As cognitive skills are typically conceptualized as hierarchical in nature (McGrew, 2009), with GCA at the apex and more specific skills such as EF as lower-order components, EF and GCA covary. EF can thus be represented as either a correlated trait sharing variance with GCA, or if GCA is covaried out, as an independent cognitive ability. Analyses were conducted both ways. Thus, the current report extends our prior study of uncommon deletion burden and GCA (Yeo et al., 2013a), to investigate deletion burden and GRS effects on both GCA and EF components.
2. Methods
2.1 Participants
Participants were recruited through the Mind Clinical Imaging Consortium (MCIC). This includes IRB approved research teams at the Mind Research Network and University of New Mexico, Massachusetts General Hospital, the University of Minnesota, and the University of Iowa (see Gollub et al., 2013, for additional details). From the original sample we included all participants who had high quality genetic data, structural MRI scans, and complete neuropsychological testing. The current analysis is limited to the subset of these individuals who stated their racial background was “white”. (See Liu et al., 2012, for additional details on the issue of population stratification in the MCIC sample.) The final sample included 50 individuals with schizophrenia (35 males, 15 females) and 86 controls (49 males, 37 females). The number of participants recruited from each site were: Albuquerque, NM (11 patients/15 controls), Boston, MA (12/11), Minneapolis, MN (9/14), and Iowa City, IA (18/46).
A comprehensive clinical diagnostic assessment included either the Structured Clinical Interview for the DSM IV (First et al., 1997) or the Comprehensive Assessment of Symptoms and History (CASH) (Andreasen et al., 1992). Symptoms were evaluated with the Scale for the Assessment of Positive Symptoms (Andreasen, 1984a) and the Scale for the Assessment of Negative Symptoms (Andreasen, 1984b). Healthy controls were recruited from the general community through medical clinics and advertisements in local newspapers. Exclusionary criteria for the control group were presence of a physical or neurologic disorder affecting brain function, and lifetime history of any Axis I disorder, including substance abuse or dependence. Parental socio-economic status (pSES) was calculated using the modified five-point Hollingshead-Redlich scale (1 = highest, 5 = lowest).
2.2 Cognitive assessment
Executive skills were assessed with a battery of six tests, yielding a total of 10 variables, and principal component analysis was used to reduce these variables to a smaller number of EF factors. Verbal fluency was assessed with the letter fluency (letters F, A, and S) and category fluency tests (animals, fruits) from the Delis-Kaplan Executive Functional System (Delis et al., 2001). Both total time and number of errors on the Trail Making Test B, a measure of processing speed, working memory, and sequencing, were also assessed. A computerized version of the Tower of London test was administered to assess planning and problem solving (Shallice, 1982). Three variables from this test were used: excess moves on the 3, 4, and 5 ring problems. The California Computerized Assessment Package (CalCap) taps processing speed, attention and executive skills (LaPointe et al., 2007). We included false positive errors from the Serial Pattern Matching 1 and Serial Pattern Matching 2 subtests.
A principal component analysis (PCA) with oblimin rotation (which allows for the emergence of correlated factors) was performed on the 10 executive function variables, from participants of both groups, to determine a smaller number of latent factors. This analysis was performed on the full sample (N = 237) described in (Yeo et al., 2013b), some of whom did not have genetic data, allowing for the emergence of a maximally stable factor structure. Given the pattern of results obtained, a follow-up analysis examined “overall EF”, which was determined by simply averaging the three components emerging from the original PCA; this overall measure correlated with a simple unweighted aggregrate of all EF variables at r = .99. GCA was operationally defined as the first component emerging from a principal component analysis of 25 variables from a comprehensive neuropsychological battery that included EF measures (Sponheim et al., 2010). The psychometric characteristics of the first principal component are quite robust to variations in the exact tests included in the PCA, as PCAs based on entirely different test correlate at r = .99 or better (Johnson et al., 2004), and in our sample, an unweighted composite of all cognitive tests employed correlated with the GCA derived from PCA at r = .99.
2.3 Genetic analyses
Details on the deletion burden measure, based on the number of uncommon deletions, were previously described (Yeo et al., 2013a) and details on the GRS were provided in Walton et al. (2013). DNA extracted from blood samples was genotyped using Illumina HumanOmin1-quad chip, including 1,140,419 markers. The number of uncommon deletions (i.e., those that occurred in 3% or fewer subjects in the combined sample with high quality CNV data) was summed for each subject. SNPs for the GRS were selected based on the continuously updated meta-analysis of genetic studies on schizophrenia, available at www.schizophreniaresearchforum.org as described in Walton et al. The GRS was weighted by multiplying the number of risk alleles with the logarithmized odds ratio of each SNP to take different effect sizes of SNPs into account.
2.4 Statistical Analysis
All statistical analyses were conducted in SPSS (v.21.0). A repeated measures ANOVA was performed, treating the three EF variables as repeated measures, with group as a between subjects factor and deletion burden and GRS as quantitative predictors, along with several covariates (age, sex, ethnicity [Anglo vs. Hispanic], pSES) often found to influence cognitive scores. The repeated measures analysis allowed us to evaluate whether genetic effects were specific to a given component or generalized across all EF components. A follow-up analysis additionally covaried GCA. For our primary GCA analysis, a univariate general linear model was conducted, with the same set of factors and covariates as above. In analyses including interactions, quantitative predictors were zero-centered.
3. Results
3.1 Descriptive statistics
We identified three EF factors with eigenvalues greater than one through principal components analysis. These were Fluency (35.14% of total variance accounted for), Planning (13.06% of variance), and Inhibition (11.16% variance). For additional details see 23. In the entire sample, partial correlations of GCA with the three EF factors, controlling for sex, age, and ethnicity, were r = 0.67 (Fluency), r = 0.71 (Planning), and r = 0.50 (Inhibition).
Table 1 provides descriptive statistics on demographic, genetic, and cognitive data (standard scores) for both groups. No group differences were evident in age, parent SES, GRS or deletion burden. The mean length of illness for patients with schizophrenia was 10.01 years (SD = 9.61, range 0 – 40). Symptom scores in the patient group were consistent with expectations (for positive symptoms, mean = 4.40, SD = 2.89), for negative symptoms mean = 8.32, SD = 3.91, and for disorganized symptoms mean = 1.84, SD = 1.99). In the entire sample, the two genetic factors were uncorrelated (r = 0.08, ns); a trend was noted in the patient group (r = 0.25, p = 0.08), but not controls (r = 0.003, ns).
Table 1.
Descriptive statistics for both groups, with significance testing of differences by independent samples t-tests. SES measures utilized the Hollingshead-Redlich Scale. Cognitive variables represent Z-scores
| Controls | Patients | ||||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | p | |
|
|
|
|
|
|
|
| Age (years) | 31.98 | 11.20 | 33.82 | 11.39 | ns |
| Education (years) | 15.36 | 1.98 | 13.40 | 2.15 | < 0.001 |
| Socioeconomic status | 2.63 | 0.55 | 3.47 | 0.96 | < 0.001 |
| Parent SES | 2.65 | 0.68 | 2.69 | 0.96 | ns |
| Deletion Burden (total number) | 11.93 | 4.64 | 12.06 | 4.02 | ns |
| Deletion Burden (mean number base pairs) | 117,972 | 176387 | 135,126 | 196,791 | ns |
| Genetic Risk Score | 6.88 | 0.81 | 6.88 | 0.70 | ns |
| General Cognitive Ability | 0.63 | 0.41 | -0.51 | 0.92 | < 0.001 |
| Executive Function (overall) | 0.39 | 0.34 | -0.31 | 0.71 | < 0.001 |
| Fluency | 0.42 | 0.77 | -0.46 | 0.86 | < 0.001 |
| Planning | 0.43 | 0.43 | -0.29 | 1.28 | < 0.001 |
| Inhibition | 0.32 | 0.46 | -0.18 | 0.98 | < 0.001 |
3.2 The impact of genetic factors on EF
A repeated measures ANOVA examined the impact of genetic factors on the three EF factors, controlling for other sources of variance. Sex, age, ethnicity (Hispanic vs. Anglo), pSES, and group (patients vs. controls) were entered as covariates, as were several two-way interactions (group × GRS, group × deletion burden, group × pSES, GRS × deletion burden) and one three-way interaction (group × GRS × deletion burden). Preliminary analyses revealed no sex by group interaction on EF variables. The genetic factors did not interact with the repeated EF measures factor; thus, all genetic effects reflect impact on overall EF functioning. The significant effects of pSES and group × pSES were as identified in a similar analysis reported in (Yeo et al., 2013b). There was no main effect of GRS on EF (F(1,117) = 3.04, p = 0.08, partial eta squared = 0.02), though there was a main effect for deletion burden (F(1,117) = 9.06, p = 0.003, partial eta squared = 0.07). The interaction of group with deletion burden was also significant (F(1,117) = 17.01, p < 0.001, partial eta squared = 0.13). The pattern was the same as noted in our prior study of GCA, as increased deletion burden was associated with poorer performance only in the patient group. The interaction of GRS with group was not significant (F(1,117) = 0.31, partial eta squared = 0.003), but importantly, the interaction of GRS with deletion burden was significant (F(1,117) = 20.42, p < 0.001, partial eta squared = 0.15), as was the three-way interaction of GRS, deletion burden, and group (F(1,117) = 15.11, p < 0.001, partial eta squared = 0.11). To evaluate the robustness of our EF observations, we conducted two additional analyses with different overall EF measures, one based on an unweighted aggregate of all EF scores, and one based on forcing a single-factor solution to the PCA; results were unchanged.
Follow-up analyses of the two-way interaction of GRS with deletion burden indicated that the adverse effects of each genetic variable on EF was greatest when scores on the other genetic variable was relatively low. Alternatively stated, when either genetic score was high (i.e., lots of rare deletions or many high-risk alleles), the adverse impact of the other genetic factor was reduced. The three-way interaction revealed that these genetic interactions were more pronounced in the patient group.
In another follow-up analysis the interaction between GRS and deletion burden was significant in patients (F(1,38) = 13.00, p = 0.001, partial eta squared = 0.26), but not controls (F(1,76) = 1.85, ns). Thus, among patients only, we examined the overall level of EF (after adjusting for the effects of group, parent SES, age, sex, and ethnicity) in four groups, defined by median splits on both genetic variables (i.e., high GRS/high deletion burden; low GRS/low deletion burden; high GRS/low deletion burden; and low GRS/high deletion burden), as shown in Figure 1. Low scores on each genetic variable was beneficial for EF (the low/low group performed best), whereas at higher levels of either variable (or both variables) EF did not differ.
Figure 1.
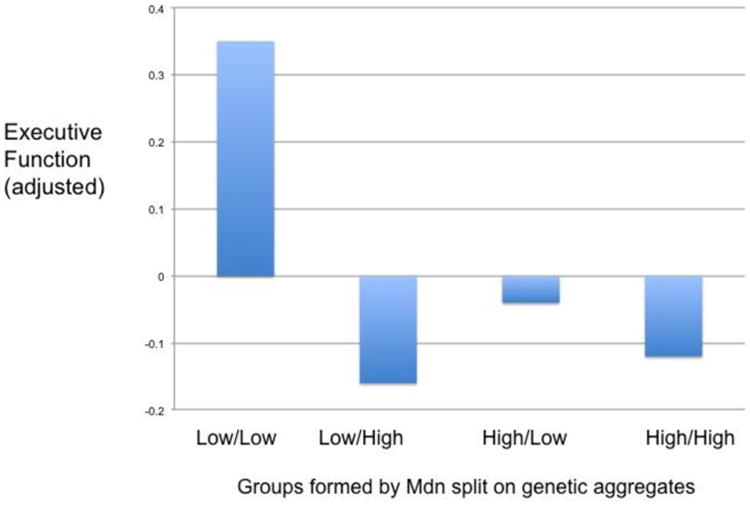
Interaction of the two genetic factors in the patient sample for executive function (the average of the three components, adjusted for demographic variables).
A second repeated measures analyses added GCA as a covariate to the demographic variables described above, allowing assessment of EF independent of the shared variance with GCA. No significant main effect was noted for the GRS or deletion burden variables. However, a two-way interaction was found for GRS and deletion burden (F(1,116)= 12.72, p = 0.001, partial eta squared = 0.099, and for the three-way interaction of group with GRS and deletion burden (F(1,116) = 9.60, p = 0.002; partial eta squared = 0.076). The direction of all these effects was the same as described above. A non-significant trend was noted for the interaction of GRS with group (F(1,116) = 3.33, p = 0.071). Again, follow-up analyses in each group separately revealed that the interaction of the two genetic variables was significant only in patients.
3.3 The impact of genetic factors on general cognitive ability
Finally, we examined the impact of each genetic factor and their interaction on GCA, with a univariate GLM procedure using the same demographic covariates as above. A main effect of deletion burden was found (F(1,120) = 6.93, p = 0.01, partial eta squared = 0.055), as well as interactions of deletion burden with GRS (F(1,120) = 6.19, p = 0.014, partial eta squared = 0.049) and group (F(1,120) = 10.33, p = 0.002, partial eta squared = 0.079), and GRS and group (F(1,120) = 4.36, p = 0.039, partial eta squared = 0.035). All effects were in the direction as noted above for EF.
Table 2 provides a summary of the variance accounted for by significant genetic effects for each type of cognitive endophenotype examined (overall EF, EF with GCA partialed out (termed “EF-Unique”), and GCA). Of note, the three-way interaction of deletion burden with GRS and group captured a substantial amount of variance for each endophenotype.
Table 2.
Summary of percent of variance accounted for by significant genetic effects and their interactions across three cognitive endophenotypes: Executive Function (EF), Executive Function - Unique (EF-U, with General Cognitive Ability covaried), and General Cognitive Ability (GCA).
| Cognitive Endophenotype | |||
|---|---|---|---|
| EF | EF-U | GCA | |
|
|
|
|
|
| Deletion burden | 7.2 | 8.3 | |
| Genetic Risk Score (GRS) | |||
| Deletions × GRS | 14.9 | 9.9 | 6.9 |
| Deletions × group | 12.7 | 11.2 | |
| GRS × group | |||
| Deletions × GRS × group | 11.4 | 7.6 | 5.2 |
| TOTAL | 46.2 | 17.5 | 31.6 |
4. Discussion
Though patients and controls did not differ on GRS or deletion burden variables, these genetic factors were important for both EF and GCA in patients, but not controls. The variance captured by the genetic interactions was substantial. The absence of genetic interactions with the repeated measures factor (specific types of EF) suggests that the assessed genetic factors are important for EF in general, rather than one or two specific components. The predictors of EF and GCA were quite similar. For each, there was a main effect for deletion burden, an interaction of deletion burden with group, an interaction of deletion burden with GRS, and a three-way interaction of group, deletion burden, and GRS. When EF was examined after controlling for GCA, the total amount of variance captured by genetic terms was relatively smaller, with significant effects being limited to the interaction of deletion burden and GRS, and the three-way interaction of these two factors with group. These results draw attention to the importance of interacting genetic influences for cognitive endophenotypes in schizophrenia.
Several limitations should be noted. Our sample size was small for a genetic study, though not underpowered for analysis of two genetic variables. The absence of group differences on either genetic variable may reflect difficulty in detecting very small effect sizes in this sample. Replication is critically important in psychiatric genetics, especially for novel effects such as those reported here. There is also room for improvement in both of our genetic measures. A detailed item analysis of the GRS measure (and additional SNPs) could possibly identify better weightings that could capture more variance. Similarly, our measure of deletion burden could perhaps be refined by distinguishing deletions near “hot spots” for recombination (Gangestad et al., 2011), i.e., those that may be in chromosomal regions disproportionately contributing to human brain development (Marques-Bonet et al., 2009).
The nature of the observed interactions is of great interest. For EF, a group by deletion burden interaction was found, such that the adverse effect of deletions was limited to the schizophrenia group. Possible mechanisms and implications of this type of interaction were reviewed in our earlier report (Yeo et al., 2013a). However, this specific interaction appears to largely reflect the influence of deletion burden on the shared variance between GCA and EF, as the group by deletion burden interaction term was not significant when GCA was added as a covariate (i.e., for EF-unique). The interaction between the two genetic components for EF was significant only in the patient group. As shown in Figure 1, low scores on both genetic variables were clearly associated with higher EF scores, but little difference was noted among the other three groups shown in the figure, i.e., individuals who were high on one and not the other, or those high on both.
This pattern of interaction between the two genetic variables suggests antagonistic epistasis (Jarosz et al., 2010), i.e., no additional adverse effects are seen when both types of genetic risks are high, as compared to when a single one is high. One possible reason for this pattern is that the two genetic factors may act on similar neurodevelopmental pathways. High scores on either factor appear to be sufficient to induce abnormality in the pathways affected. But, what are these pathways? Is it the reduced buffering capacity typically linked with deletion burden, an effect consistent with evidence for greater sensitivity among patients to environmental perturbations such as obstetric complications (McNeil et al., 2000) and low parent SES (Yeo et al., 2013b) ? Is it abnormal neural development and synaptic functioning, as suggested by the ontology of the GRS constituents (Walton et al., 2013)? Of course, the critical pathways may differ across patients and aggregate genetic measures such as those used here can only suggest broad directions. Future research efforts may benefit from attempting to identify developmental/metabolic processes that mediate the link between genetic interactions and cognitive phenotypes.
An important part of the original motivation for endophenotype research was its potential for revealing the genetic contributions to disease. Cognitive endophenotypes in particular seem relevant for schizophrenia (Kahn & Keefe, 2013) and psychosis (Caspi et al., 2014). An underlying assumption of the endophenotype research strategy has been that it would identify genetic factors that (1) differed in level across controls and affected individuals, and (2) this genetic difference would account for group differences on the endophenotype. Our data raise questions about this assumption. Genetic variation does indeed contribute to cognitive endophenotype scores, but only in patients, and levels of this factor do not differ across groups. Our results suggest that a single genetic vector, leading to variation on the endophenotype, independent of diagnosis, is untenable. Schizophrenia, and the endophenotype of cognition within affected individuals, must represent an interaction of genetic factors or an interaction of genetic and environmental factors. Thus, our application of the endophenotype strategy may have revealed possible causal genetic influences, but the effective genetic influences constitute interactions, not main effects.
All effects of deletion burden, GRS, and their interaction were limited to the schizophrenia group. This observation has interesting implications. One concerns the nature of the mutation-selection balance. As schizophrenia is relatively rare in the population, negative selection pressure on the specific SNPs and CNVs comprising our aggregates is much lower in the total population than it would be if the adverse effects were seen in both patients and controls. This observation helps account for the maintenance of many potentially harmful genetic variants in the human gene pool. Also, SNP effects may well depend on genetic background variables (Chandler et al., 2013), such as the level of deletion burden, raising concerns about human studies and experimental models that do not specifically address such issues. The current results also raise the possibility that genetic influences on cognitive endophenotypes differ from those that distinguish groups. Speculatively, the underlying causal factor(s) for schizophrenia may be distinct from the types of genetic influence we examined in this study. This hypothetical causal factor might lead to two different neurodevelopmental trajectories, one normal, and one psychosis-prone. In the latter trajectory alone, the sorts of genetic variation studied here might moderate the risk of developing the full phenotype.
Acknowledgments
Role of Funding Source: This work was supported by grants from the Department of Energy under Award Number DE-FG02-08ER64581, and the National Institute Health (grants 5P20RR021938, 1RC1MH089257 and R01EB005846.
Footnotes
Contributors: None
Conflict of Interest: The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreasen NC. The Scale for the Assessment of Positive Symptoms. The University of Iowa; Iowa City, IA: 1984a. [Google Scholar]
- Andreasen NC. Scale for the Assessment of Negative Symptoms. University of Iowa; Iowa City, IA: 1984b. [Google Scholar]
- Andreasen NC, Flaum M, Arndt S. The Comprehensive Assessment of Symptoms and History (CASH) - an instrument for assessing diagnosis and psychopathology. Arch Gen Psychiatry. 1992;49:615–623. doi: 10.1001/archpsyc.1992.01820080023004. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol. 2006;2:267–90. doi: 10.1146/annurev.clinpsy.2.022305.095232. [DOI] [PubMed] [Google Scholar]
- Caspi A, Houts RM, Belsky DW, Goldman-Mellor HH, Israel S, Meier MH, Ramrakha S, Shalev I, Poulton R, Moffitt TE. The p factor: One general psychopathology factor in the structure of psychiatric disorders? Clin Psychol Sci. 2014;2:119–137. doi: 10.1177/2167702613497473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler CH, Chari S, Dworkin I. Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution. Trends Genet. 2013;29:358–366. doi: 10.1016/j.tig.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delis D, Kaplan E, Kramer J. The Delis-Kaplan Executive Function System. San Antonio, TX: 2001. [Google Scholar]
- First M, Spitzer RL, Gibbon M, Williams JB. Structured Clinical Interview for DSM-IV-TR Axis I Disorders. American Psychiatric Press, Inc.; Washington, DC: 1997. [Google Scholar]
- Gangestad SW, Yeo RA, Liu J. Rare deletions predict general cognitive ability, brain neurometabolite concentrations, and schizophrenia phenotype. Human Behavior and Evolution Society 2011 [Google Scholar]
- Girirajan S, Campbell CD, Eichler EE. Human copy number variation and complex genetic disease. Annu Rev Genet. 2011;45:203–26. doi: 10.1146/annurev-genet-102209-163544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold JM, Dickinson D. “Generalized cognitive deficit” in schizophrenia: overused or underappreciated?”. Schizophr Bull. 2013;39:263–5. doi: 10.1093/schbul/sbs143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollub RL, Shoemaker JM, King MD, White T, Ehrlich S, Sponheim SR, Clark VP, Turner JA, Mueller BA, Magnotta V, O'Leary D, Ho BC, Brauns S, Manoach DS, Seidman L, Bustillo JR, Lauriello J, Bockholt J, Lim KO, Rosen BR, Schulz SC, Calhoun VD, Andreasen NC. The MCIC collection: a shared repository of multi-modal, multi-site brain image data from a clinical investigation of schizophrenia. Neuroinformatics. 2013;11:367–88. doi: 10.1007/s12021-013-9184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF, Horan WP, Sugar CA. Has the generalized deficit become the generalized criticism? Schizophr Bull. 2013;39:257–62. doi: 10.1093/schbul/sbs146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Swerdlow NR, Gur RE, Cadenhead KS, Calkins ME, Freedman R, Green MF, Gur RC, Lazzeroni LC, Nuechterlein KH, Olincy A, Radant AD, Ray A, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Sugar CA, Ph D, Tsuang MT, Light GA, Braff DL. Genome-wide linkage analysis 12 endophenotypes for schizophrenia from the consortium on the genetics of schizophreniaof Schizophrenia. Am J Psychiatry. 2013;170:521–532. doi: 10.1176/appi.ajp.2012.12020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Johnson W, Bouchard TJ, Krueger RF, McGue M, Gottesman II. Just one g: conistent results from three test batteries. Intelligence. 2004;32:85–107. [Google Scholar]
- Kahn RS, Keefe RSE. Schizophrenia is a cognitive illness: Time for a change. JAMA Psychiatry. 2013;70:1107–1112. doi: 10.1001/jamapsychiatry.2013.155. [DOI] [PubMed] [Google Scholar]
- Langer N, Pedroni A, Gianotti LRR, Hänggi J, Knoch D, Jäncke L. Functional brain network efficiency predicts intelligence. Hum Brain Mapp. 2012;33:1393–406. doi: 10.1002/hbm.21297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe LL, Heald GR, Stierwalt JaG, Kemker BE, Maurice T. Effects of auditory distraction on cognitive processing of young adults. J Atten Disord. 2007;10:398–409. doi: 10.1177/1087054706293221. [DOI] [PubMed] [Google Scholar]
- Liu J, Ulloa A, Perrone-Bizzozero N, Yeo RA, Chen J, Calhoun VD. A pilot study on collective effects of 22q13.31 deletions on gray matter concentration in schizophrenia. PLoS One. 2012;7:e52865. doi: 10.1371/journal.pone.0052865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Girirajan S, Eichler EE. The origins and impact of primate segmental duplications. Trends Genet. 2009;25:443–54. doi: 10.1016/j.tig.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrew KS. CHC theory and the human cognitive abilities project: Standing on the shoulders of the giants of psychometric intelligence research. Intelligence. 2009;37:1–10. [Google Scholar]
- McNeil TF, Cantor-Graae E, Weinberger DR. Relationship of obstetric complications and differences in size of brain structures in monozygotic twin pairs discordant for schizophrenia. Am J Psychiatry. 2000;157:203–12. doi: 10.1176/appi.ajp.157.2.203. [DOI] [PubMed] [Google Scholar]
- Miyake A, Friedman NP. The nature and organization of individual differences in executive functions: Four general conclusions. Curr Pirections Psychol Sci. 2012;21:8–14. doi: 10.1177/0963721411429458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shallice T. Specific impairments of planning. Philos Trans R Soc London Ser B-Biological Sci. 1982;298:199–209. doi: 10.1098/rstb.1982.0082. [DOI] [PubMed] [Google Scholar]
- Sponheim SR, Jung RE, Seidman LJ, Mesholam-Gately RI, Manoach DS, O'Leary DS, Ho BC, Andreasen NC, Lauriello J, Schulz SC. Cognitive deficits in recent-onset and chronic schizophrenia. J Psychiatr Res. 2010;44:421–8. doi: 10.1016/j.jpsychires.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton E, Turner J, Gollub RL, Manoach DS, Yendiki A, Ho BC, Sponheim SR, Calhoun VD, Ehrlich S. Cumulative genetic risk and prefrontal activity in patients with schizophrenia. Schizophr Bull. 2013;39:703–711. doi: 10.1093/schbul/sbr190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo RA, Gangestad SW, Liu J, Ehrlich S, Thoma RJ, Pommy JM, Mayer AR, Schulz SC, Wassink TH, Morrow EM, Bustillo JR, Sponheim SR, Ho BC, Calhoun VD. The impact of copy number deletions on general cognitive ability and ventricle size in patients with schizophrenia and healthy control subjects. Biol Psychiatry. 2013a;73:540–5. doi: 10.1016/j.biopsych.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo RA, Matrinez D, Pommy J, Ehrlich S, Schulz SC, Ho BC, Bustillo JR, Calhoun VD. The impact of parent socioeconomic status on executive functioning and cortical morphology in individuals with schizophrenia and healthy controls. Psychol Med. 2013b doi: 10.1017/S0033291713001608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–81. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]