

α-O-Glycosides of N-acetylneuraminic acid (Neu5Ac, 1, Scheme 1) are often found terminating the oligosaccharide component of cell-surface glycoproteins and glycolipids. Neu5Ac is involved in a number of important biological events: intercellular interactions such as adhesion, aggregation, and agglutination; masking of antigenic oligosaccharides and suppressing undesired immune reactions (antirecognition phenomena); influencing the cell membrane permeability for ions, amino acids, and proteins; and protection of glycoproteins against proteolysis.1 Terminal Neu5Ac is an attachment site of pathogens to the cells, and often catabolic and inflammatory processes are initiated on the removal of this carbohydrate group.2 In general the “right” life time of a cell is a reflection of a delicate balance between the introduction and removal of terminal Neu5Ac or other sialic acids.
Scheme 1.
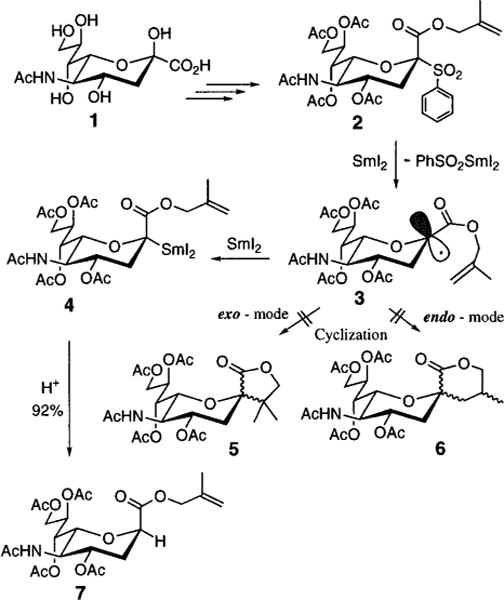
The glycosidic bond of Neu5Ac is cleaved in vivo by hydrolase type enzymes, called neuraminidases.3 Therefore, designing nonhydrolyzable analogs of Neu5Ac-α-O-glycosides is an attractive approach to control, at the molecular level, events of crucial importance to glycobiology and immunology. The replacement of the interglycosidic oxygen atom by a methylene group, for example, generates a class of hydrolytically and metabolically inert isosteres, the Neu5Ac C-glycosides. Despite several elegant methods for direct carbon–carbon (C–C) bond formation at the anomeric center in aldoses and ketoses,4 no major advances have been reported in the synthesis of Neu5Ac C-glycosides.5 The major problem confounding their synthesis is the requirement that the C–C bond being formed results in a quaternary C-atom.
Herewith, we report our findings of a general method for diastereocontrolled preparation of α-C-glycosides of Neu5Ac. This approach is tolerant of a wide variety of protecting groups. The reducing potential of SmI2 is exploited through the in situ generation of an N-acetylneuraminyl samarium(III) species and its coupling to carbonyl compounds under Barbier conditions.6
In model studies that led to this method, the SmI2-promoted generation of the anomeric capto-dative free radical 3 was attempted, employing an ester tethered Neu5Ac-sulfone 27 (Scheme 1). It was anticipated that 3 would collapse into a mixture of C-glycosides by cyclization through an exo- and/or endo-mode.8 Surprisingly, instead of the anticipated cyclic C-glycosides (5 and/or 6), the 2-deoxy compound 79 was isolated in excellent yield and stereoselectivity. No trace of the C-2-epimer having an equatorial carboxy function was observed. This exceptional stereoselectivity suggested an intermediate second electron transfer providing the organosamarium(III) derivative 4, in which the bulky I2Sm(III)-substituent adopts the more thermodynamically stable equatorial position.
A diastereocontrolled synthesis of α-linked C-disaccharides was designed using this C-2-samariated Neu5Ac derivative as a C9-nucleophile to react with a C-formyl sugar (a C7-electrophile) (Scheme 2). The proposed C9-nucleophile precursor 8 was obtained in four steps from Neu5Ac as previously described.7 A 2-pyridyl sulfone, similar to that suggested by Mazeas et al,8 replaced the phenyl sulfone moiety, decreasing the LUMO-energy level of the SO2Ar, facilitating one electron-transfer and homolytic fragmentation to the intermediate free radical of type 3. The C7-electrophile 9 was prepared in seven steps from methyl α-D-galactopyranoside as described by Schmidt et al.10
Scheme 2.
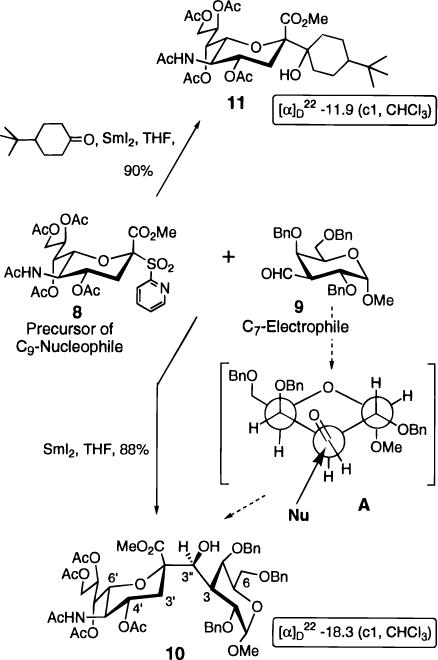
Treatment of a neat mixture of sulfone 8 and aldehyde 9 (1.5 equiv) in inert atmosphere with 3.1 equiv of freshly prepared 0.1 M SmI2 solution in THF at 20 °C gave a nearly instantaneous conversion to the C-disaccharide 10 in excellent yield.
Addition of the aldehyde immediately after the SmI2 solution does not lead to a condensation with 8 clearly demonstrating the Barbier conditions of the reaction. Under these conditions, only protonation (presumably from THF) of the intermediate organosamarium(III) species was observed.
The structural assignment of 10 was based on 1D and 2D 1H-NMR. The formation of the α-anomer was confirmed using empirical rules for the determining of the anomeric configuration of Neu5Ac glycosides.12 The chemical shift of H-4′ (4.90 ppm), the J7′,8′-value (7.7 Hz) and the Δδ/H-9′A-H-9′B/-value (0.26 ppm) clearly indicated the α-configuration of the Neu5Ac residue in 10. The 1H–1H ROESY spectra (τm = 700 or 100 ms) showed negative NOEs between H-4′, H-6′ and the protons of the methyl ester group and between H-3′ax, H-3′eq and H-3, confirming the α-configuration. The same spectra were used for indirect assignment of the stereochemistry at the newly formed hydroxymethylene bridge. The lack of any NOE between H-4 and H-3′eq indicated a restricted mobility around both the interglycosidic bonds and the negative cross peaks between the proton at the bridging carbon atom, and both the C-6 protons of the galacto moiety showed that they are spatially close.
The observed diastereoselectivity of the reaction could be rationalized based on the Felkin-Anh model13 for predicting the stereochemical outcome of a kinetically controlled addition of a nucleophile to a chiral aldehyde (Scheme 2, A). The bulkiest ligand α to the carbonyl group in 9 is the C-2 atom containing an equatorial benzyloxy group and attached to the C-1 atom bearing an axial α-OMe glycosidic substituent. This ligand has a perpendicular relationship to the plane of the carbonyl group and is anticlinal to the Bürgi-Dunitz trajectory14 of the incoming nucleophile. Only traces of other diastereomers (<1% based on 1H-NMR) were observed after silica gel separation of product 10 and unreacted aldehyde 9.
The coupling of a ketone with 8 was also investigated to establish the scope of this reaction. An excellent yield of C-glycoside 11 was obtained (Scheme 2).
These preliminary results suggest the future incorporation of the C-glycosidic pseudodisaccharide fragment 10 into larger, biologically important oligosaccharides, affording carbon bridged sialyl Lewis X derivatives.
Supplementary Material
Acknowledgments
We thank the Professor Toshihiko Toida for his assistance in the NMR experiments.
Footnotes
Supporting Information Available: Experimental procedures for the synthesis of 10 and 11, supporting 1H-NMR spectra and 1H–1H ROSY spectra for 10 and analytical data (7 pages). See any current masthead page for ordering information and Internet access instructions.
References
- 1.Schauer R, editor. Sialic Acids. Springer-Verlag; New York: 1985. [Google Scholar]; Rosenberg A, Shengrund C, editors. Biological Roles of Sialic Acid. Plenum; New York: 1976. [Google Scholar]; Sharon N. Complex Carbohydrates. Addison-Wesley; London: 1975. [Google Scholar]; Varki A. Glycobiology. 1992;2:25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharon N, Lis H. Science. 1989;246:227–234. doi: 10.1126/science.2552581. [DOI] [PubMed] [Google Scholar]; Sharon N, Lis H. Lectins. Chapman and Hall; London: 1989. [Google Scholar]
- 3.Air GM, Laver WG. Proteins: Structure, Function and Genetics. 1989;6:341–356. doi: 10.1002/prot.340060402. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.Postema M. C-Glycoside Synthesis. CRC Press; Boca Raton, FL: 1995. [Google Scholar]; Recent reviews:; Postema M. Tetrahedron. 1992;48:8545–8599. [Google Scholar]; Herscovici J, Antonakis K. In: Studies in Natural Products Chemistry. Atta-ur-Rahman, editor. Vol. 10. Elsevier; Amsterdam: 1992. pp. 337–403. [Google Scholar]
- 5.Only the trivial alkyl and hydroxymethyl C-glycosides of Neu5Ac have been synthesized:; Paulsen H, Matschulat P. Liebigs Ann Chem. 1991:487–495. [Google Scholar]; Walliman K, Vasella A. Helv Chim Acta. 1991;74:1520–1532. [Google Scholar]; Nagy J, Bednarski M. Tetrahedron Lett. 1991;32:3953–3956. [Google Scholar]
- 6.Examples and mechanistic studies of the Barbier reaction with organo- and glycosyl samarium compounds:; Namy J, Collin J, Bied C, Kagan H. Synlett. 1992:733–734. [Google Scholar]; Curran D, Fevig T, Jasperse C, Totleben M. Synlett. 1992:943–961. [Google Scholar]; Molander G, McKie J. J Org Chem. 1991;56:4112–4120. [Google Scholar]; de Pouilly P, Chénédé A, Mallet J-M, Sinaÿ P. Bull Soc Chim Fr. 1993;130:256–265. [Google Scholar]; Mazeas D, Skrydstrup T, Beau J-M. Angew Chem Int Ed Engl. 1995;34:909–912. [Google Scholar]; Excellent reviews on the application of SmI2:; Molander G, Harris C. Chem Rev. 1996;96:307–338. doi: 10.1021/cr950019y. [DOI] [PubMed] [Google Scholar]; Molander G. Chem Rev. 1992;92:29–68. [Google Scholar]
- 7.The corresponding 2-thiophenylglycoside was prepared using the following method:; Cao S, Meuneir S, Andersson F, Letellier M, Roy R. Tetrahedron: Asymmetry. 1994;5:2303–2312. [Google Scholar]; It was oxidized to a sulfone according to the following:; Marra A, Sinaÿ P. Carbohydr Res. 1989;187:35–42. [Google Scholar]
- 8.A temporary acetal tether as an attractive approach for governing the stereoselectivity in the prostaglandin synthesis was published:; Stork G, Sher P, Chen HJ. Am Chem Soc. 1986;108:6384–6387. [Google Scholar]; Applying this idea in the carbohydrate area resulted in a number of elegant syntheses of β-O-mannosides:; Stork G, Kim G. J Am Chem Soc. 1992;114:1087–1088. [Google Scholar]; Stork G, La Clair J. J Chem Soc. 1996;118:247–248. [Google Scholar]; Barresi F, Hindsgaul O. J Am Chem Soc. 1991;113:9376–9377. [Google Scholar]; Barresi F, Hindsgaul O. Can J Chem. 1994;72:1447–1465. [Google Scholar]; Ito Y, Ogawa T. Angew Chem Int Ed Engl. 1994;33:1765–1967. [Google Scholar]; Dan A, Ito Y, Ogawa T. J Org Chem. 1995;60:4680–4681. [Google Scholar]; Exploiting a similar intramolecular aglycon delivery strategy through silicon tethers, initial examples of C-glycoside construction were proposed:; Stork G, Suh H, Kim G. J Am Chem Soc. 1991;113:7054–7066. [Google Scholar]; Xin YC, Mallet JM, Sinaÿ P. J Chem Soc, Chem Commun. 1993:864. [Google Scholar]; Vauzeilles B, Cravo D, Mallet JM, Sinaÿ P. Synlett. 1993;522 [Google Scholar]; Chénédé A, Perrin E, Rekaï E, Sinaÿ P. Synlett. 1994;420 [Google Scholar]; Mazeas D, Skrydstrup T, Doumeix O, Beau J-M. Angew Chem, Int Ed Engl. 1994;33:1383–1386. [Google Scholar]; Mallet A, Mallet J-M, Sinaÿ P. Tetrahedron: Asymmetry. 1994;5:2593–2608. [Google Scholar]
- 9.1H-NMR data of 7 match published:; Schmid W, Christian R, Zbiral E. Tetrahedron Lett. 1988;29:3643–3646. [Google Scholar]
- 10.Schmidt R, Beyerbach A. Liebigs Ann Chem. 1992:983–986. [Google Scholar]
- 11.Girard P, Namy J, Kagan H. J Am Chem Soc. 1980;102:2693–2698. [Google Scholar]
- 12.Kanie O, Kiso M, Hasegawa A. J Carbohydr Chem. 1988;7:501–506. [Google Scholar]
- 13.Anh N, Eisenstein O. Nouv J Chim. 1977;1:61–70. [Google Scholar]
- 14.Bürgi H, Dunitz J, Shefter E. J Am Chem Soc. 1973;95:5065–5067. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.