

Abstract
We show how dynamic nuclear polarization (DNP) enhanced solid-state NMR spectroscopy can be used to characterize polymorphs and solvates of organic solids. We applied DNP to three polymorphs and one hydrated form of the asthma drug molecule theophylline. For some forms of theophylline, sample grinding and impregnation with the radical-containing solution, which are necessary to prepare the samples for DNP, were found to induce polymorphic transitions or desolvation between some forms. We present protocols for sample preparation for solid-state magic-angle spinning (MAS) DNP experiments that avoid the polymorphic phase transitions in theophylline. These protocols include cryogrinding, grinding under inert atmosphere, and the appropriate choice of the impregnating liquid. By applying these procedures, we subsequently demonstrate that two-dimensional correlation experiments, such as 1H–13C and 1H–15N HETCOR or 13C–13C INADEQUATE, can be obtained at natural isotopic abundance in reasonable times, thus enabling more advanced structural characterization of polymorphs.
Keywords: dynamic nuclear polarization, solid-state NMR, polymorphs, theophylline
Introduction
The development of a more complete understanding of polymorphism is an important goal for the solid-state chemist or process engineer, and is especially relevant to pharmaceuticals since each polymorphic form of a given compound can lead to different bioavailabilities.1,2 Characterization of polymorphs is often difficult, especially when single crystals suitable for X-ray diffraction (XRD) cannot be isolated. Consequently, a broad range of techniques, including “ab initio” crystal structure prediction (CSP),3−7 infrared spectroscopy,8,9 and powder X-ray diffraction (PXRD),10−14 are currently used to characterize organic powders in this context. However, these techniques often only provide partial details on atomic and molecular structure. Consequently, there is much interest in developing improved methods to characterize polymorphs.
Among these methods, magic angle spinning (MAS) solid-state NMR is an extremely powerful method for characterizing polymorphs. This is because 1H, 13C, and 15N chemical shifts are often diagnostic, both for “fingerprinting” polymorphs and/or for directly probing atomic structure.15,16 Recently, the combination of measured experimental NMR chemical shifts, CSP, and gauge-including projector augmented-wave (GIPAW) DFT calculations of chemical shifts has been very successful both for validation of polymorph trial structures and for complete structure determination.3,17−32 However, the main limitation of NMR methods is poor sensitivity. Indeed, 15N and many 2D solid-state NMR experiments at natural abundance often require very long experimental times, except for compounds with very favorable relaxation characteristics.15,33 Even in favorable cases, two-dimensional 13C–13C correlation experiments on samples at natural isotopic abundance typically require times on the order of days.
Dynamic nuclear polarization (DNP) is a technique that can increase the sensitivity of solid-state NMR by up to roughly 2 orders of magnitude at high fields.34,35 In a DNP experiment, microwave irradiation is used to drive the transfer of polarization from unpaired electrons to nuclei.34 DNP can theoretically provide a signal gain of up to a factor of 658 for 1H NMR and cross-polarization (CP) NMR experiments which involve 1H nuclei, although enhancements of 10–200 are typically obtained. We have recently shown how powdered organic solids can be efficiently polarized using DNP.36 In our approach, the organic solid is finely ground to reduce the particle sizes, and the powder is impregnated with a nonsolvent liquid that contains the DNP polarizing agents, usually a stable dinitroxide biradical.37 The DNP enhanced NMR experiments are typically carried out at around 105 K. Proton spin diffusion (exchange of 1H magnetization) then distributes the polarization throughout the sample, and the enhanced 1H polarization may be transferred to other desired nuclei via CP.38 With this impregnation DNP approach, we have shown that DNP enhancements between 5 and 200 can be obtained for fine powders of organic solids such as glucose, sulfathiazole, paracetamol, cetirizine dihydrochloride, histidine hydrochloride, polymers, etc.39−41 Substantial DNP sensitivity enhancements have previously enabled the rapid acquisition of 1H–13C/1H–15N heteronuclear correlation (HETCOR) and 13C–13C homonuclear scalar correlation (INADEQUATE) experiments in a few hours for a variety of organic solids.36,39,41−44 DNP also allows challenging multinuclear magnetic resonance experiments such as natural abundance 2H and 17O solid-state NMR and 14N overtone MAS NMR,41,45 including 17O solid-state NMR for surface-supported species.47 The sensitivity enhancement from DNP can also be used to overcome low loadings of active pharmaceutical ingredients (API) within formulated pharmaceuticals.39 Additionally, by analyzing the DNP enhanced NMR signal buildup rates with a numerical spin diffusion model it is possible to estimate macroscopic API particle sizes.36,39
This technique obviously has great potential for the rapid characterization of polymorphs and formulated pharmaceuticals, but there are open questions related to sample formulation for DNP experiments and how this may impact species which may display polymorphism or other changes such as (de)solvation. Before performing a DNP enhanced solid-state NMR experiment, the three main steps in sample preparation involve (i) grinding the sample into a very fine powder (note, this step may not be necessary for formulated pharmaceuticals), (ii) impregnation with an appropriate solution containing the electron polarization source, and (iii) rapidly cooling the sample to ca. 105 K within the NMR probe that will be used to acquire the NMR data. It is obvious that all three of these steps could potentially induce polymorphic transitions. Here we show how protocols can be developed to prepare samples for impregnation DNP while maintaining the desired polymorphic form of the sample. While we focus on the effects of grinding and impregnation on phase transitions, it is also well-known that a polymorphic transition can be induced by a reduction in temperature. However, we note that many crystal structures of polymorphs determined by single-crystal XRD are acquired at reduced temperatures. Interestingly, recent results indicate that DNP experiments at ambient temperatures could become feasible.48
Here, we present sample preparation protocols for impregnation DNP enhanced NMR of polymorphs. The protocols are demonstrated with theophylline as a model system (Scheme 1) for this study since it possesses several well-characterized polymorphs,6,49,50 as well as a hydrated form, and they interconvert quite easily. We note that Viel and co-workers have recently applied impregnation DNP techniques to a single polymorphic form of theophylline and showed the rapid acquisition of dipolar 13C–13C correlation spectra.51
Scheme 1. Molecular Structure of Theophylline.
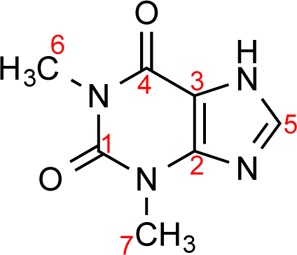
Red numbers correspond to carbon peak labeling associated with Figure 2.
Results and Discussion
Theophylline is known to crystallize as several different polymorphs. Here we study three anhydrous polymorphs of theophylline (Figure 1), along with theophylline-monohydrate (Form M).49 The kinetically stable form of theophylline under typical lab conditions is often referred to as Form II in the chemical literature,49 and is usually the form obtained from commercial suppliers. To obtain Form I, Form II was heated at 270 °C, close to its melting point (273 °C) for 2 h, and slowly cooled to room temperature, as outlined in the literature.6 The thermodynamically stable polymorph is Form IV.50 It is obtained by stirring a methanol solution containing powdered Form II for 15 days.50 Theophylline monohydrate (Form M) is obtained by the recrystallization of theophylline Form II in distilled water.49 More details are given in the Experimental Section.
Figure 1.
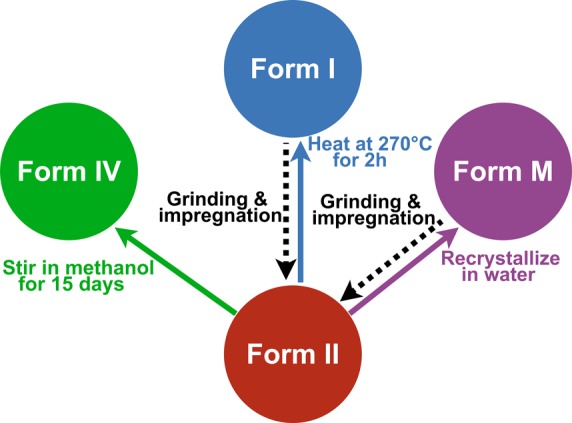
Schemes for the preparation and interconversion of the different theophylline-containing crystal structures considered herein. Form II of anhydrous theophylline was obtained commercially. Form IV is an anhydrous polymorph of theophylline obtained by stirring Form II in methanol for 15 days. Form I is an anhydrous polymorph of theophylline obtained by heating Form II at 270 °C for 2 h. Form M is a theophylline-containing hydrate obtained by recrystallizing Form II in water. Dashed arrows illustrate the conversions that are induced by grinding or impregnation.
The four theophylline-containing systems were synthesized (or in the case of Form II, used as received from the supplier), and 13C CPMAS NMR spectra were recorded at 105 K on the pristine powdered polymorphs (i.e., no grinding of the sample, and no impregnation with solvent), as shown in Figure 2. As expected, the 13C chemical shifts vary significantly between the forms. Comparison between 300 K spectra and 105 K spectra is presented in Figure S1. In all cases, the multiplicity of the carbon peaks is the same at both 105 and 300 K and the only difference is slight shifts in some of the peaks (less than a 2 ppm shift difference). Previously determined crystal structures for all polymorphs used diffraction data acquired at temperatures of less than 120 K,49,50 suggesting that there is no polymorphic phase transition at low temperature for all forms of theophylline considered herein. We note that Form IV possesses twice as many 13C peaks in its NMR spectrum as the other forms, which is consistent with the previously determined crystal structure, as it has two molecules in the asymmetric unit (Z′ = 2).501H MAS and 15N CPMAS NMR spectra of each form are also shown in Figures S2 and S3.
Figure 2.
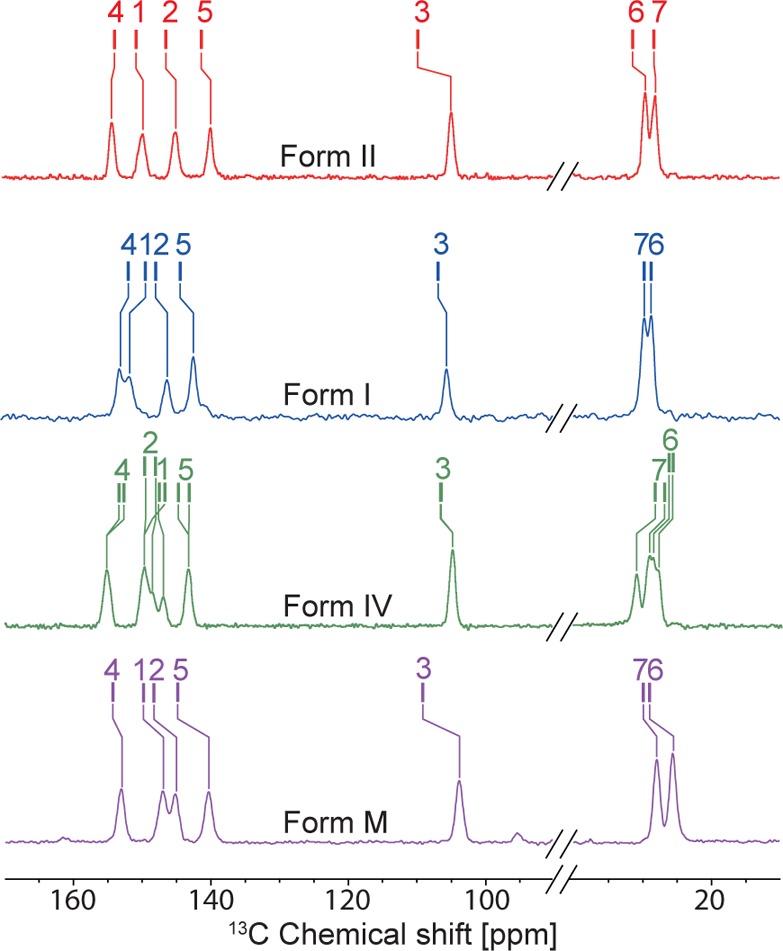
13C solid-state NMR spectra recorded at 105 K and B0 = 9.4 T without microwave irradiation and with 64 scans for all nonimpregnated polymorphic forms of theophylline studied here: Form II (red), Form I (blue), Form IV (green), and Form M (purple). The tick marks show the predicted shifts from GIPAW DFT calculations. 13C, 1H, and 15N solid-state NMR spectra can be found in Figures S1–S3. Additionally, comparison between 298 and 105 K 13C solid-state NMR spectra is presented in Figure S1.
Impregnation DNP Enhanced NMR Experiments on Form II
For DNP enhanced solid-state NMR experiments, a powder sample of each system was finely ground by hand in a mortar and pestle. The powder was then impregnated with a TCE:methanol-d4 (95:5) solution containing the biradical TEKPol as a polarization source.48 TCE was chosen as a solvent since theophylline was found to be insoluble in TCE, and it has previously been shown that this is a good impregnating medium for DNP.52 5% methanol was found to improve glass formation for TCE. For experiments on Form II, a high proton DNP enhancement (εH = εsolv) of 220 was obtained for the solvent (Figure 3).
Figure 3.
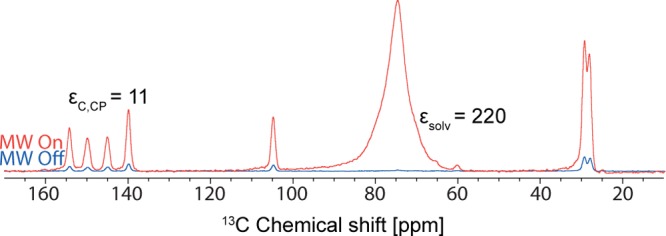
13C DNP enhanced solid-state NMR spectra of Form II of theophylline obtained at 105 K and B0 = 9.4 T (i.e., 1H resonance frequency of 400 MHz) with 16 scans for microwave on, 64 scans for microwave off, and 20 s recycle delay. The powder was finely ground in a mortar and pestle and impregnated with a solution of TCE:methanol-d4 (95:5) containing TEKPol as biradical.
The proton DNP enhancement for Form II of microcrystalline theophylline, as measured with 13C CPMAS experiments (εC,CP), was 11. The DNP enhanced proton signal buildup rate (TDNP) was measured with a DNP enhanced CP saturation recovery experiment. Usually, TDNP and the proton longitudinal relaxation time T1 are similar in magnitude. For the sake of convenience, we only measure in this study TDNP. For Form II, TDNP was measured to be ca. 12 s at ca. 105 K (we speculate this to be due to the presence of rotating methyl groups). Note however that a signal enhancement by a factor of 11 corresponds to an acceleration in acquisition times of a factor 121 (as the signal-to-noise ratio (S/N) scales with the square root of the number of transients acquired). The short TDNP is not critically limiting in the present case since, although it leads to lower enhancements, it also allows a relatively short recycle delay of ca. 20 s, which provides optimal sensitivity in this case. DNP enhanced 2D 1H–13C, 1H–15N HETCOR and 13C–13C INADEQUATE NMR spectra were recorded for Form II in 3, 5, and 2 days respectively, shown in Figures S4, S5, and S6. To obtain the INADEQUATE spectrum with similar conditions of compounds like this using conventional methods would typically take on the order of a week.22
Impregnation DNP Enhanced NMR Experiments on Form I
Form I was obtained by heating Form II to 270 °C, just below its melting point of 273 °C (see Experimental Section for details). A sample of powdered Form I was characterized by solid-state NMR at room temperature. The 13C CPMAS NMR spectrum showed 6 resolvable 13C peaks (in accord with the accepted crystal structure, as Z′ = 1).6 We observed different chemical shifts than those of Form II, as shown in Figures 2, S1, and S7.6Figure 4 shows 13C CPMAS NMR spectra of Form I before and after grinding. Grinding is known to supply mechanical energy to the sample, which can be sufficient to induce a polymorphic conversion or a desolvation if solvent is present in the lattice.53,54 When the powder of Form I was manually ground in a mortar and pestle at room temperature, solid-state 13C CPMAS NMR experiments confirmed that the sample partially transformed back to Form II.
Figure 4.

13C CPMAS NMR spectra recorded at 105 K and B0 = 9.4 T of what was initially powdered theophylline, Form I, subjected to different preparation procedures. From top to bottom: (a) pure powdered Form I before any procedure, (b) a mixture of Forms I and II obtained after manual grinding of pure Form I in air at room temperature, (c) Form I manually ground cold at −55 °C in a glovebox, (d) mixture of Forms II and I after impregnation with a 16 mM TCE:methanol-d4 (95:5) solution, (e) Form I manually ground cold at −55 °C in a glovebox and impregnated with a toluene-d8:decalin (i.e., Tol:dec) (90:10) solution, (f) pure Form II.
We found that the polymorphic transition back to Form II could be avoided by grinding the sample at reduced temperatures in the absence of atmospheric moisture. Two procedures were attempted: the first one was to use an automatic cryogrinder. To use an automatic cryogrinder, the powder was put into a water-tight tube immersed in liquid nitrogen and shaken in order to grind the powder. This procedure yielded a powder which, after appropriate sample preparation, did not lead to DNP NMR enhancements that were encouraging (i.e., εC,CP = 2), when one considers the eventual goal of efficient acquisition of 2D solid-state NMR data. This would suggest that the particles were not ground finely enough using this technique. SEM images were recorded before and after this procedure (see Figures S8 and S9) and allow us to conclude that the powder still contained some relatively big particles (dimensions of around 50 μm). Another possible simple way to cryogrind the sample was to cool the sample in a freezer and grind it while it was cold. This was performed by placing powdered Form I on a mortar and pestle in a −55 °C freezer inside a glovebox (argon atmosphere). The glovebox was used in order to avoid absorption of moisture on the cold sample. The powder and the mortar and pestle were allowed to rest in the freezer for 30 min to reduce their respective system temperatures. The Form I powder was then quickly ground into a fine powder while the powder (and mortar) were still cold. This procedure prevented the I → II transformation, as shown in Figure 4c. However, the 13C CPMAS NMR spectra of the cold ground samples show additional broadening of the 13C peaks. This could be explained by some disorder introduced in the lattice during grinding at cold temperature. A similar phenomenon was observed by Munson and co-workers.55
Once the problem of grinding was solved by grinding the cold sample in a glovebox, the powder was impregnated with a solution of TEKPol in TCE:methanol-d4 (95:5). However, impregnation with TCE:methanol-d4 solution also caused partial transformation of Form I to Form II. Figure 4d shows the 13C CPMAS NMR spectrum after impregnation with a 16 mM TEKPol TCE:methanol-d4 (95:5) solution. The polymorphic transformation could be due to the polarity of the solvent, which fosters the phase transition from Form I to II. Similar phenomena have already been mentioned by Carpenter and co-workers.56 Less polar solvents that are compatible with DNP were used for the impregnation step. Such solvents still were needed to dissolve the radical, while not dissolving the powdered theophylline-containing organic solid. We tried impregnation with ortho-terphenyl (OTP), 1,3-dibromobutane (DBB), and a mixed solution of toluene-d8:decalin (90:10), all of which are nonsolvents for theophylline and which have been demonstrated to be suitable for DNP.52,57 Impregnation with DBB resulted in partial conversion of Form I to Form II. Figure 4e shows that the toluene-d8:decalin mixture turns out to completely avoid any Form I to Form II transition.
In summary, for Form I, a protocol was developed involving grinding a cooled sample in a glovebox followed by impregnation with a toluene-d8:decalin mixture ready for DNP enhanced solid-state NMR spectra to be recorded without a change in the polymorphic form. In this case the 1H DNP enhancement of the solvent was around 16, and the 13C enhancement was measured to be approximately 4 for Form I, as shown in Figure 5. Here, the low DNP enhancement results from the poor performance of toluene-d8:decalin as a solvent for DNP. We are currently working on identifying additional solvents/mixtures for DNP on this system.
Figure 5.
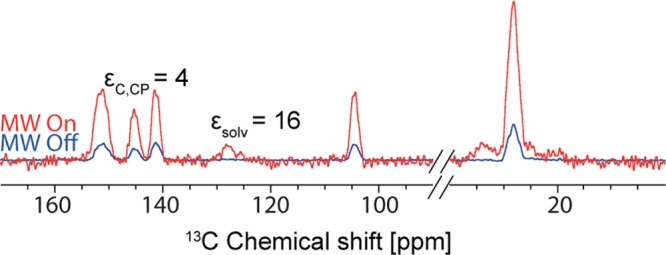
DNP enhanced 13C CPMAS NMR spectrum recorded at 105 K and B0 = 9.4 T of Form I of theophylline impregnated with a toluene-d8:decalin (90:10) solution containing 14 mM TEKPol with 4 scans for microwave on, 64 scans for microwave off, and 20 s recycle delay.
Impregnation DNP Enhanced NMR Experiments on Form IV
Form IV of anhydrous theophylline is the thermodynamically stable form of theophylline. It was obtained by stirring a solution of Form II in methanol for 15 days. For DNP enhanced NMR experiments, the powder isolated was ground and then impregnated with a TCE:methanol-d4 (95:5) solution containing the biradical TEKPol.48 A poor solvent enhancement (compared to usual enhancement obtained for a bulk solution)58 was obtained, εC,CP = 40, and the 13C enhancement for Form IV was even lower, εC,CP = 2 (Figure 6). The low enhancements are likely due to the very short TDNP (and by extension proton T1’s) measured for Form I, which were around only 1.5 s.
Figure 6.
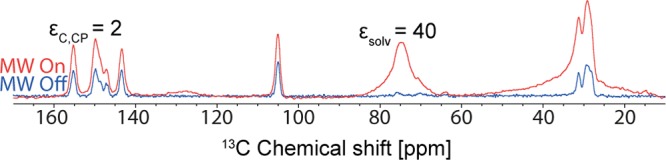
DNP enhanced 13C CPMAS NMR spectra recorded at 105 K and B0 = 9.4 T of Form IV of theophylline impregnated with a TCE:methanol-d4 (95:5) solution containing 16 mM TEKPol, with 4 scans for microwave on, 16 scans for microwave off, and 20 s recycle delay.
Impregnation DNP Enhanced NMR Experiments on Form M
The monohydrated form of theophylline is obtained by a simple recrystallization of Form II in water. Grinding Form M at room temperature causes dehydration of the sample and partial reconversion to Form II, as shown in Figure 7.
Figure 7.
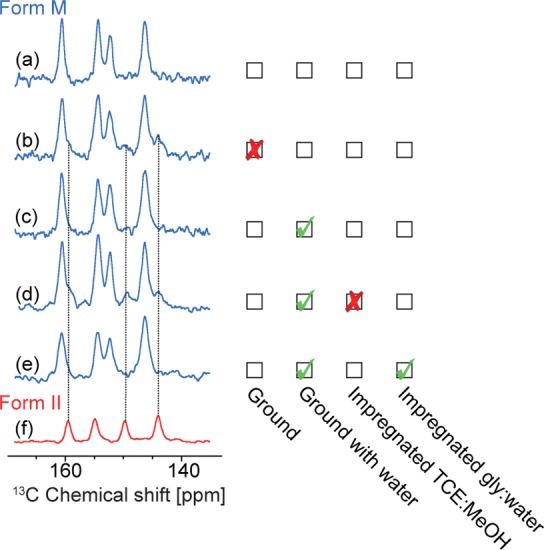
Comparison of 13C CPMAS solid-state NMR spectra as a function of the grinding procedure and the solvent used for impregnation of Form M. All spectra were recorded with a 16.4 T spectrometer at ca. 300 K. (a) Pure Form M before any procedure. (b) Mixture of Forms M and II obtained after grinding at room temperature. (c) Form M after grinding with a drop of water. (d) Mixture of Forms M and II after grinding with a drop of water and impregnation with TCE:methanol-d4 (95:5) solution. (e) Form M after grinding with a drop of water and impregnated with a glycerol-d8:D2O:H2O (60:30:10) solution. (f) Pure Form II.
To potentially avoid this, ca. 40 mg of powder was ground in the presence of several drops of distilled water, to prevent dehydration. This technique worked well, as shown in the resulting 13C CPMAS NMR spectrum in Figure 7c. An alternative is to grind the powder of Form M while it is completely immersed in liquid nitrogen, and let it warm under atmospheric air so that the cold powder absorbs the moisture of the air and prevents Form M from dehydrating. When the ground powder of Form M was impregnated with TCE:methanol-d4 (95:5) (as done with some of the polymorphs of anhydrous theophylline), partial conversion of Form M to Form II was observed. However, impregnation with a glycerol-d8:D2O:H2O (60:30:10) solution did not cause any measurable transformation to Form II, as shown in Figure 7e. Unfortunately, relatively low DNP enhancements of εC,CP = 2 were obtained from Form M, even though the enhancement of the solvent was, εC,CP, was 170 (Figure 8). The low enhancement for the crystalline phase could be due to aggregation of particles during grinding with water, which would explain the high ratio between the solvent and the crystal enhancement. 2D HETCOR 1H–13C and 1H–15N NMR spectra have also been recorded in order to confirm the structure and the presence of hydrogen bonding between the 2 molecules of theophylline and a water molecule, as predicted by the crystal structure (Figures S10 and S11).
Figure 8.
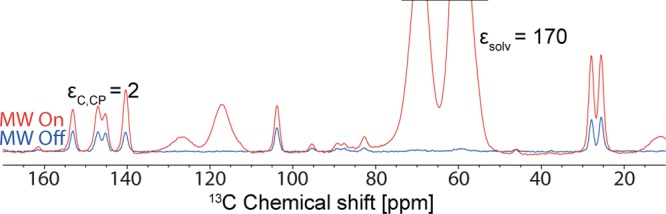
DNP enhanced 13C CPMAS NMR spectra of theophylline/water Form M ground with a drop of water, and impregnated with a glycerol-d8:D2O:H2O (60:30:10) solution containing AMUPol.59 Spectra were recorded at 105 K and B0 = 9.4 T with 4 scans for microwave on, 16 scans for microwave off, and 20 s recycle delay.
Conclusion
We investigated the formulation for DNP enhanced NMR experiments of powdered samples of three polymorphs of theophylline and the monohydrate form. We observed that the grinding and impregnation procedures may cause polymorphic transitions or dehydration in certain situations. For each form we were able to determine a procedure for grinding, and an appropriate solvent for impregnation that prevented the conversion to Form II. DNP enhanced NMR was then applied to these polymorphs. However, DNP enhancements were relatively low in most theophylline polymorphs, which is speculated as being due to the presence of methyl group rotation inducing short TDNP times. Natural abundance 2D correlation experiments such as 1H–15N, 1H–13C HETCOR or 13C–13C INADEQUATE were acquired in reasonable times, with a factor over 100 reduction in acquisition times in the best case (i.e., Form II). This will have a direct impact on the efficiency of NMR crystallography protocols for polymorphs. Finally, we note that the methods outlined here can most probably also be adapted to other approaches to formulating samples for DNP experiments in organic solids, such as recrystallization with polarizing agents,43 or swelling (polymers) with radical-containing solutions.60−62
Experimental Section
Geometry optimizations and magnetic shielding calculations were performed using the Cambridge Serial Total Energy Package (CASTEP) density functional theory (DFT) program,63 which uses a plane wave basis set to describe the valence electrons, and pseudopotentials to describe the core electrons. The GIPAW method64 provides an efficient method to calculate magnetic shielding values in crystalline solids.65 This method uses ultrasoft Vanderbilt-type pseudopotentials in the core.66 Geometry optimizations, starting from the XRD reference structures, were carried out using the generalized gradient approximation (GGA) functional of Perdew, Burke, and Ernzerhof (PBE).67 A maximum plane wave energy cutoff for the valence electrons of 700 eV was applied. A Monkhorst–Pack grid of k-points68 was used and corresponded to a maximum spacing of 0.05 Å–1 in reciprocal space. During the geometry optimizations, only the hydrogen positions were relaxed, since the unit cell and all heavy atoms (C, N, O) were fixed. The magnetic shielding values were calculated using the same functional and parameters as those used for the geometry optimization. The magnetic shielding values were then converted into calculated chemical shifts using standard procedures.69 For the chemical shielding calculation the rotational dynamics of the methyl group was taken into account by averaging over the three shifts. Geometry optimization and magnetic shielding calculations were performed at the Pôle Scientifique de Modélisaiton Numérique (PSMN) at the ENS Lyon.
Commercially available theophylline Form II was obtained from Sigma-Aldrich and was used without further purification. Form I was obtained by heating Form II in a tube furnace.6 Form II was loaded into a quartz tube and was then heated to 373 K with a 753 K/h ramp, stabilized for 5 min, then heated to 523 K with a 753 K/h ramp, then stabilized for 30 min, and heated to 543 K (270 °C) with a 373 K/h ramp. The sample was then left at 543 K for 2 h and then cooled down to room temperature with a 373 K/h ramp. The resulting compact light yellow powder was collected from inside the tube. Form IV was obtained by stirring a methanol solution containing theophylline powder for 15 days.50 Theophylline monohydrate, called Form M,49 was obtained by recrystallization of theophylline in distilled water.
A scanning electron microscope MSE-636 was used to capture images of the automatically cryoground Form II. In a typical procedure, the powder was first spread onto a carbon tape. The powder was then put under vacuum and covered with a 12 nm gold layer using a Cressington sputter coater based on a plasma vapor deposition technique. The covered powder was then subjected to a 10–5 bar vacuum for analysis.
For DNP enhanced NMR experiments, the powder was finely ground by hand in a mortar and pestle for several minutes. Other system-dependent conditions are specified as part of the Results and Discussion. Incipient wetness impregnation (IWI) was used to uniformly wet the surface of particles with radical-containing solution.48,70 In a typical preparation, 10–20 μL of 16 mM biradical solution in a suitable solvent was added to ca. 30–40 mg of finely ground powdered solid. The powder and solvent were then mixed with a glass stirring rod, transferred to a sapphire rotor, and capped with a polyfluoroethylene insert. For theophylline Form IV, a 16 mM solution of TEKPol48 in 1,1,2,2-tetrachloroethane (TCE):methanol-d4 (95:5) solution was used. For Form I, different radical-containing solutions were tested, as detailed in the Results and Discussion. For Form M, a 16 mM AMUPol59 solution in glycerol-d8:D2O:H2O (60:30:10) was used.
DNP solid-state NMR experiments were performed on wide bore 400 MHz (i.e., B0 = 9.4 T) Bruker Avance I (EPFL, Lausanne) and Avance IIIHD (ENS-Lyon) spectrometers equipped with a 263 GHz gyrotron, a low temperature cooling cabinet, and a triple resonance 3.2 mm low temperature probe. The sample temperature for DNP experiments was approximately 105 K. The field sweep coil of the main magnetic fields was set so that microwave irradiation occurred at the same position as the positive enhancement maximum for 1-(TEMPO-4-oxy)-3-(TEMPO-4-amino)propan-2-ol (TOTAPOL). Additional solid-state NMR experiments were performed on a 700 MHz Bruker Avance III solid-state NMR spectrometer (B0 = 16.4 T). Room temperature experiments using the 700 MHz spectrometer employed a 3.2 mm triple resonance HCN probe and 3.2 mm zirconia rotors.
CPMAS NMR experiments were performed with a contact pulse on 1H which was linearly ramped from ν1 = 66 to 93 kHz and from 58 to 82 kHz for 13C and 15N experiments, respectively; 13C and 15N CP spin lock rf field amplitudes of 57 and 42 kHz were used, respectively. The SPINAL-64 heteronuclear decoupling scheme was applied during acquisition with 1H rf fields of ca. 105 kHz. The number of scans is specified in the figure captions. The contact time for CP spin lock was 2500 μs. CP-HETCOR experiments were recorded for 1H–13C and 1H–15N correlation spectra. During the 1H indirect evolution period (t1), e-DUMBO-122 homonuclear 1H decoupling was applied with an RF field strength of approximately 100 kHz to improve the resolution in the 1H dimension, followed by cross-polarization to the heteronucleus transition and signal detection.71 The 2D refocused 13C–13C INADEQUATE NMR spectra was recorded with a triple resonance 3.2 mm low temperature probe at 12500 kHz MAS. The sample temperature for DNP experiments was approximately 105 K. The SPINAL-64 sequence at a proton nutation frequency ν1 of 80 kHz was used for heteronuclear decoupling. 128 increments of 64 transients each were acquired with a repetition delay of 20 s, resulting in a total experimental time of 2 days.
Acknowledgments
This work was supported by ERC Advanced Grant No. 320860. C.M.W. would like to acknowledge NSERC for a postdoctoral fellowship. We are grateful to Prof. P. Tordo, Dr. O. Ouari and Dr. G. Casano (Aix-Marseille Université, France) for providing the biradicals used in the DNP NMR experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.5b00610.
1H, 13C, and 15N NMR spectra and SEM images (PDF)
Author Present Address
§ C.M.W.: Department of Chemistry, Durham University, DH1 3LE Durham, United Kingdom.
The authors declare no competing financial interest.
Supplementary Material
References
- Bernstein J.Polymorphism in Molecular Crystals; Oxford University Press: Oxford, 2002. [Google Scholar]
- Polymorphism, 1st ed.; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- Day G. M. Current approaches to predicting molecular organic crystal structures. Crystallogr. Rev. 2011, 17, 3–52. 10.1080/0889311X.2010.517526. [DOI] [Google Scholar]
- Price S. L. The computational prediction of pharmaceutical crystal structures and polymorphism. Adv. Drug Delivery Rev. 2004, 56, 301–319. 10.1016/j.addr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Vasileiadis M.; Pantelides C. C.; Adjiman C. S. Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule. Chem. Eng. Sci. 2015, 121, 60–76. 10.1016/j.ces.2014.08.058. [DOI] [Google Scholar]
- Smith E. D. L.; Hammond R. B.; Jones M. J.; Roberts K. J.; Mitchell J. B. O.; Price S. L.; Harris R. K.; Apperley D. C.; Cherryman J. C.; Docherty R. The determination of the crystal structure of anhydrous theophylline by X-ray powder diffraction with a systematic search algorithm, lattice energy calculations, and C-13 and N-15 solid-state NMR: A question of polymorphism in a given unit cell. J. Phys. Chem. B 2001, 105, 5818–5826. 10.1021/jp002060x. [DOI] [Google Scholar]
- Baias M.; Dumez J.-N.; Svensson P. H.; Schantz S.; Day G. M.; Emsley L. De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. 10.1021/ja4088874. [DOI] [PubMed] [Google Scholar]
- Bauer J.; Spanton S.; Henry R.; Quick J.; Dziki W.; Porter W.; Morris J. Ritonavir: An extraordinary example of conformational polymorphism. Pharm. Res. 2001, 18, 859–866. 10.1023/A:1011052932607. [DOI] [PubMed] [Google Scholar]
- Threlfall T. L. Analysis of Organic Polymorphs - a Review. Analyst 1995, 120, 2435–2460. 10.1039/an9952002435. [DOI] [Google Scholar]
- Tiwari M.; Chawla G.; Bansal A. K. Quantification of olanzapine polymorphs using powder X-ray diffraction technique. J. Pharm. Biomed. Anal. 2007, 43, 865–872. 10.1016/j.jpba.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Caira M.; Alkhamis K. A.; Obaidat R. M. Preparation and crystal characterization of a polymorph, a monohydrate, and an ethyl acetate solvate of the antifungal fluconazole. J. Pharm. Sci. 2004, 93, 601–611. 10.1002/jps.10541. [DOI] [PubMed] [Google Scholar]
- Crowley K. J.; Zografi G. Cryogenic grinding of indomethacin polymorphs and solvates: Assessment of amorphous phase formation and amorphous phase physical stability. J. Pharm. Sci. 2002, 91, 492–507. 10.1002/jps.10028. [DOI] [PubMed] [Google Scholar]
- Lu G. W.; Hawley M.; Smith M.; Geiger B. M.; Pfund W. Characterization of a novel polymorphic form of celecoxib. J. Pharm. Sci. 2006, 95, 305–317. 10.1002/jps.20522. [DOI] [PubMed] [Google Scholar]
- Qiu J.-b.; Li G.; Sheng Y.; Zhu M.-r. Quantification of febuxostat polymorphs using powder X-ray diffraction technique. J. Pharm. Biomed. Anal. 2015, 107, 298–303. 10.1016/j.jpba.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Harris R. K. NMR studies of organic polymorphs & solvates. Analyst 2006, 131, 351–373. 10.1039/b516057j. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Wasylishen R. E.; Duer M. J.. NMR Crystallography; John Wiley & Sons Ltd.: 2009; pp 387–413. [Google Scholar]
- Potrzebowski M. J.; Tekely P.; Dusausoy Y. Comment to C-13-NMR studies of alpha and gamma polymorphs of glycine. Solid State Nucl. Magn. Reson. 1998, 11, 253–257. 10.1016/S0926-2040(98)00028-9. [DOI] [PubMed] [Google Scholar]
- Brown S. P.; Schaller T.; Seelbach U. P.; Koziol F.; Ochsenfeld C.; Klarner F. G.; Spiess H. W. Structure and dynamics of the host-guest complex of a molecular tweezer: Coupling synthesis, solid-state NMR, and quantum-chemical calculations. Angew. Chem., Int. Ed. 2001, 40, 717–720. 10.1002/1521-3773(20010216)40:4<717::AID-ANIE7170>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Ochsenfeld C.; Brown S. P.; Schnell I.; Gauss J.; Spiess H. W. Structure assignment in the solid state by the coupling of quantum chemical-calculations with NMR experiments: A columnar hexabenzocoronene derivative. J. Am. Chem. Soc. 2001, 123, 2597–2606. 10.1021/ja0021823. [DOI] [PubMed] [Google Scholar]
- Goward G. R.; Sebastiani D.; Schnell I.; Spiess H. W.; Kim H. D.; Ishida H. Benzoxazine oligomers: Evidence for a helical structure from solid-state NMR spectroscopy and DFT-based dynamics and chemical shift calculations. J. Am. Chem. Soc. 2003, 125, 5792–5800. 10.1021/ja029059r. [DOI] [PubMed] [Google Scholar]
- Yates J. R.; Dobbins S. E.; Pickard C. J.; Mauri F.; Ghi P. Y.; Harris R. K. A combined first principles computational and solid-state NMR study of a molecular crystal: flurbiprofen. Phys. Chem. Chem. Phys. 2005, 7, 1402–1407. 10.1039/b500674k. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Joyce S. A.; Pickard C. J.; Cadars S.; Emsley L. Assigning carbon-13 NMR spectra to crystal structures by the INADEQUATE pulse sequence and first principles computation: a case study of two forms of testosterone. Phys. Chem. Chem. Phys. 2006, 8, 137–143. 10.1039/B513392K. [DOI] [PubMed] [Google Scholar]
- Mifsud N.; Elena B.; Pickard C. J.; Lesage A.; Emsley L. Assigning powders to crystal structures by high-resolution H-1-H-1 double quantum and H-1-C-13 J-INEPT solid-state NMR spectroscopy and first principles computation. A case study of penicillin G. Phys. Chem. Chem. Phys. 2006, 8, 3418–3422. 10.1039/b605227d. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Cadars S.; Emsley L.; Yates J. R.; Pickard C. J.; Jetti R. K. R.; Griesser U. J. NMR crystallography of oxybuprocaine hydrochloride, Modification II degrees. Phys. Chem. Chem. Phys. 2007, 9, 360–368. 10.1039/B614318K. [DOI] [PubMed] [Google Scholar]
- Heider E. M.; Harper J. K.; Grant D. M. Structural characterization of an anhydrous polymorph of paclitaxel by solid-state NMR. Phys. Chem. Chem. Phys. 2007, 9, 6083–6097. 10.1039/b711027h. [DOI] [PubMed] [Google Scholar]
- Salager E.; Stein R. S.; Pickard C. J.; Elena B.; Emsley L. Powder NMR crystallography of thymol. Phys. Chem. Chem. Phys. 2009, 11, 2610–2621. 10.1039/b821018g. [DOI] [PubMed] [Google Scholar]
- Webber A. L.; Emsley L.; Claramunt R. M.; Brown S. P. NMR Crystallography of Campho[2,3-c]pyrazole (Z ′ = 6): Combining High-Resolution H-1-C-13 Solid-State MAS NMR Spectroscopy and GIPAW Chemical-Shift Calculations. J. Phys. Chem. A 2010, 114, 10435–10442. 10.1021/jp104901j. [DOI] [PubMed] [Google Scholar]
- Webber A. L.; Elena B.; Griffin J. M.; Yates J. R.; Pham T. N.; Mauri F.; Pickard C. J.; Gil A. M.; Stein R.; Lesage A.; Emsley L.; Brown S. P. Complete H-1 resonance assignment of beta-maltose from H-1-H-1 DQ-SQ CRAMPS and H-1 (DQ-DUMBO)-C-13 SQ refocused INEPT 2D solid-state NMR spectra and first principles GIPAW calculations. Phys. Chem. Chem. Phys. 2010, 12, 6970–6983. 10.1039/c001290d. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Hodgkinson P.; Zorin V.; Dumez J. N.; Elena-Herrmann B.; Emsley L.; Salager E.; Stein R. S. Computation and NMR crystallography of terbutaline sulfate. Magn. Reson. Chem. 2010, 48, S103–S112. 10.1002/mrc.2636. [DOI] [PubMed] [Google Scholar]
- Abraham A.; Apperley D. C.; Gelbrich T.; Harris R. K.; Griesser U. J. NMR crystallography - Three polymorphs of phenobarbital. Can. J. Chem. 2011, 89, 770–778. 10.1139/v11-011. [DOI] [Google Scholar]
- Brouwer D. H.; Langendoen K. P.; Ferrant Q. Measurement and calculation of C-13 chemical shift tensors in alpha-glucose and alpha-glucose monohydrate. Can. J. Chem. 2011, 89, 737–744. 10.1139/v11-017. [DOI] [Google Scholar]
- Webber A. L.; Masiero S.; Pieraccini S.; Burley J. C.; Tatton A. S.; Iuga D.; Pham T. N.; Spada G. P.; Brown S. P. Identifying Guanosine Self Assembly at Natural Isotopic Abundance by High-Resolution H-1 and C-13 Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2011, 133, 19777–19795. 10.1021/ja206516u. [DOI] [PubMed] [Google Scholar]
- Althaus S. M.; Mao K. M.; Stringer J. A.; Kobayashi T.; Pruski M. Indirectly detected heteronuclear correlation solid-state NMR spectroscopy of naturally abundant N-15 nuclei. Solid State Nucl. Magn. Reson. 2014, 57–58, 17–21. 10.1016/j.ssnmr.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Ni Q. Z.; Daviso E.; Can T. V.; Markhasin E.; Jawla S. K.; Swager T. M.; Temkin R. J.; Herzfeld J.; Griffin R. G. High Frequency Dynamic Nuclear Polarization. Acc. Chem. Res. 2013, 46, 1933–1941. 10.1021/ar300348n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj V. S.; Farrar C. T.; Hornstein M. K.; Mastovsky I.; Vieregg J.; Bryant J.; Elena B.; Kreischer K. E.; Temkin R. J.; Griffin R. G. Dynamic nuclear polarization at 9T using a novel 250 GHz gyrotron microwave source. J. Magn. Reson. 2003, 160, 85–90. 10.1016/S1090-7807(02)00192-1. [DOI] [PubMed] [Google Scholar]
- Rossini A. J.; Zagdoun A.; Hegner F. S.; Schwarzwälder M.; Gajan D.; Copéret C.; Lesage A.; Emsley L. Dynamic Nuclear Polarization NMR Spectroscopy of Microcrystalline Solids. J. Am. Chem. Soc. 2012, 134, 16899–16908. 10.1021/ja308135r. [DOI] [PubMed] [Google Scholar]
- Matsuki Y.; Maly T.; Ouari O.; Karoui H.; Le Moigne F.; Rizzato E.; Lyubenova S.; Herzfeld J.; Prisner T.; Tordo P.; Griffin R. G. Dynamic Nuclear Polarization with a Rigid Biradical. Angew. Chem., Int. Ed. 2009, 48, 4996–5000. 10.1002/anie.200805940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines A.; Waugh J. S.; Gibby M. G. Proton-Enhanced Nuclear Induction Spectroscopy C-13 Chemical Shielding Anistropy in Some Organic Solids. Chem. Phys. Lett. 1972, 15, 373–376. 10.1016/0009-2614(72)80191-X. [DOI] [Google Scholar]
- Rossini A. J.; Widdifield C. M.; Zagdoun A.; Lelli M.; Schwarzwälder M.; Copéret C.; Lesage A.; Emsley L. Dynamic Nuclear Polarization Enhanced NMR Spectroscopy for Pharmaceutical Formulations. J. Am. Chem. Soc. 2014, 136, 2324–2334. 10.1021/ja4092038. [DOI] [PubMed] [Google Scholar]
- Blanc F.; Chong S. Y.; McDonald T. O.; Adams D. J.; Pawsey S.; Caporini M. A.; Cooper A. I. Dynamic Nuclear Polarization NMR Spectroscopy Allows High-Throughput Characterization of Microporous Organic Polymers. J. Am. Chem. Soc. 2013, 135, 15290–15293. 10.1021/ja408074v. [DOI] [PubMed] [Google Scholar]
- Rossini A. J.; Schlagnitweit J.; Lesage A.; Emsley L. High-resolution NMR of hydrogen in organic solids by DNP enhanced natural abundance deuterium spectroscopy. J. Magn. Reson. 2015, 259, 192–198. 10.1016/j.jmr.2015.08.020. [DOI] [PubMed] [Google Scholar]
- Takahashi H.; Lee D.; Dubois L.; Bardet M.; Hediger S.; De Paëpe G. Rapid Natural-Abundance 2D 13C–13C Correlation Spectroscopy Using Dynamic Nuclear Polarization Enhanced Solid-State NMR and MatrixFree Sample Preparation. Angew. Chem. 2012, 124, 11936–11939. 10.1002/ange.201206102. [DOI] [PubMed] [Google Scholar]
- Takahashi H.; Viverge B.; Lee D.; Rannou P.; De Paepe G. Towards Structure Determination of Self-Assembled Peptides Using Dynamic Nuclear Polarization Enhanced Solid-State NMR Spectroscopy. Angew. Chem., Int. Ed. 2013, 52, 6979–6982. 10.1002/anie.201210093. [DOI] [PubMed] [Google Scholar]
- Takahashi H.; Hediger S.; De Paepe G. Matrix-free dynamic nuclear polarization enables solid-state NMR C-13-C-13 correlation spectroscopy of proteins at natural isotopic abundance. Chem. Commun. 2013, 49, 9479–9481. 10.1039/c3cc45195j. [DOI] [PubMed] [Google Scholar]
- Blanc F.; Sperrin L.; Jefferson D. A.; Pawsey S.; Rosay M.; Grey C. P. Dynamic nuclear polarization enhanced natural abundance (17)o spectroscopy. J. Am. Chem. Soc. 2013, 135, 2975–2978. 10.1021/ja4004377. [DOI] [PubMed] [Google Scholar]
- Perras F. A.; Kobayashi T.; Pruski M. Natural Abundance 17O DNP Two-Dimensional and Surface-Enhanced NMR Spectroscopy. J. Am. Chem. Soc. 2015, 137, 8336–8339. 10.1021/jacs.5b03905. [DOI] [PubMed] [Google Scholar]
- Zagdoun A.; Casano G.; Ouari O.; Schwarzwälder M.; Rossini A. J.; Aussenac F.; Yulikov M.; Jeschke G.; Copéret C.; Lesage A.; Tordo P.; Emsley L. Large Molecular Weight Nitroxide Biradicals Providing Efficient Dynamic Nuclear Polarization at Temperatures up to 200 K. J. Am. Chem. Soc. 2013, 135, 12790–12797. 10.1021/ja405813t. [DOI] [PubMed] [Google Scholar]
- Fucke K.; McIntyre G. J.; Wilkinson C.; Henry M.; Howard J. A. K.; Steed J. W. New Insights into an Old Molecule: Interaction Energies of Theophylline Crystal Forms. Cryst. Growth Des. 2012, 12, 1395–1401. 10.1021/cg201499s. [DOI] [Google Scholar]
- Khamar D.; Pritchard R. G.; Bradshaw I. J.; Hutcheon G. A.; Seton L. Polymorphs of anhydrous theophylline: stable form IV consists of dimer pairs and metastable form I consists of hydrogen-bonded chains. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2011, 67, O496–O499. 10.1107/S010827011104786X. [DOI] [PubMed] [Google Scholar]
- Mollica G.; Dekhil M.; Ziarelli F.; Thureau P.; Viel S. Quantitative Structural Constraints for Organic Powders at Natural Isotopic Abundance Using Dynamic Nuclear Polarization Solid-State NMR Spectroscopy. Angew. Chem., Int. Ed. 2015, 54, 6028–6031. 10.1002/anie.201501172. [DOI] [PubMed] [Google Scholar]
- Zagdoun A.; Rossini A. J.; Gajan D.; Bourdolle A.; Ouari O.; Rosay M.; Maas W. E.; Tordo P.; Lelli M.; Emsley L.; Lesage A.; Copéret C. Nonaqueous Solvents for DNP Spectroscopy. Chem. Commun. 2012, 48, 654–656. 10.1039/C1CC15242D. [DOI] [PubMed] [Google Scholar]
- Hedoux A.; Guinet Y.; Paccou L.; Danede F.; Derollez P. Polymorphic transformation of anhydrous caffeine upon grinding and hydrostatic pressurizing analyzed by low-frequency raman spectroscopy. J. Pharm. Sci. 2013, 102, 162–170. 10.1002/jps.23346. [DOI] [PubMed] [Google Scholar]
- Grillo D.; Polla G.; Vega D. Conformational polymorphism on imatinib mesylate: Grinding effects. J. Pharm. Sci. 2012, 101, 541–551. 10.1002/jps.22772. [DOI] [PubMed] [Google Scholar]
- Lubach J. W.; Xu D.; Segmuller B. E.; Munson E. J. Investigation of the effects of pharmaceutical processing upon solid-state NMR relaxation times and implications to solid-state formulation stability. J. Pharm. Sci. 2007, 96, 777–787. 10.1002/jps.20684. [DOI] [PubMed] [Google Scholar]
- Trask A. V.; Shan N.; Motherwell W. D. S.; Jones W.; Feng S. H.; Tan R. B. H.; Carpenter K. J. Selective polymorph transformation via solvent-drop grinding. Chem. Commun. 2005, 880–882. 10.1039/b416980h. [DOI] [PubMed] [Google Scholar]
- Ong T. C.; Mak-Jurkauskas M. L.; Walish J. J.; Michaelis V. K.; Corzilius B.; Smith A. A.; Clausen A. M.; Cheetham J. C.; Swager T. M.; Griffin R. G. Solvent-Free Dynamic Nuclear Polarization of Amorphous and Crystalline ortho-Terphenyl. J. Phys. Chem. B 2013, 117, 3040–3046. 10.1021/jp311237d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicki D. J.; Rossini A. J.; Purea A.; Zagdoun A.; Ouari O.; Tordo P.; Engelke F.; Lesage A.; Emsley L. Amplifying Dynamic Nuclear Polarization of Frozen Solutions by Incorporating Dielectric Particles. J. Am. Chem. Soc. 2014, 136, 15711–15718. 10.1021/ja5088453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvee C.; Rosay M.; Casano G.; Aussenac F.; Weber R. T.; Ouari O.; Tordo P. Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency. Angew. Chem., Int. Ed. 2013, 52, 10858–10861. 10.1002/anie.201304657. [DOI] [PubMed] [Google Scholar]
- Fernandez-de-Alba C.; Takahashi H.; Richard A.; Chenavier Y.; Dubois L.; Maurel V.; Lee D.; Hediger S.; De Paepe G. Matrix-Free DNP-Enhanced NMR Spectroscopy of Liposomes Using a Lipid-Anchored Biradical. Chem. - Eur. J. 2015, 21, 4512–4517. 10.1002/chem.201404588. [DOI] [PubMed] [Google Scholar]
- Ouari O.; Phan T.; Ziarelli F.; Casano G.; Aussenac F.; Thureau P.; Gigmes D.; Tordo P.; Viel S. Improved Structural Elucidation of Synthetic Polymers by Dynamic Nuclear Polarization Solid-State NMR Spectroscopy. ACS Macro Lett. 2013, 2, 715–719. 10.1021/mz4003003. [DOI] [PubMed] [Google Scholar]
- Le D.; Casano G.; Phan T. N. T.; Ziarelli F.; Ouari O.; Aussenac F.; Thureau P.; Mollica G.; Gigmes D.; Tordo P.; Viel S. Optimizing Sample Preparation Methods for Dynamic Nuclear Polarization Solid-state NMR of Synthetic Polymers. Macromolecules 2014, 47, 3909–3916. 10.1021/ma500788n. [DOI] [Google Scholar]
- Clark S. J.; Segall M. D.; Pickard C. J.; Hasnip P. J.; Probert M. J.; Refson K.; Payne M. C. First principles methods using CASTEP. Z. Kristallogr. - Cryst. Mater. 2005, 220, 567–570. 10.1524/zkri.220.5.567.65075. [DOI] [Google Scholar]
- Pickard C. J.; Mauri F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B: Condens. Matter Mater. Phys. 2001, 63, 245101. 10.1103/PhysRevB.63.245101. [DOI] [Google Scholar]
- Yates J. R.; Pickard C. J.; Mauri F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 76, 024401. 10.1103/PhysRevB.76.024401. [DOI] [Google Scholar]
- Vanderbilt D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B: Condens. Matter Mater. Phys. 1990, 41, 7892–7895. 10.1103/PhysRevB.41.7892. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- Baias M.; Widdifield C. M.; Dumez J.-N.; Thompson H. P. G.; Cooper T. G.; Salager E.; Bassil S.; Stein R. S.; Lesage A.; Day G. M.; Emsley L. Powder crystallography of pharmaceutical materials by combined crystal structure prediction and solid-state (1)H NMR spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 8069–8080. 10.1039/c3cp41095a. [DOI] [PubMed] [Google Scholar]
- Lesage A.; Lelli M.; Gajan D.; Caporini M. A.; Vitzthum V.; Mieville P.; Alauzun J.; Roussey A.; Thieuleux C.; Mehdi A.; Bodenhausen G.; Copéret C.; Emsley L. Surface Enhanced NMR Spectroscopy by Dynamic Nuclear Polarization. J. Am. Chem. Soc. 2010, 132, 15459–15461. 10.1021/ja104771z. [DOI] [PubMed] [Google Scholar]
- van Rossum B. J.; de Groot C. P.; Ladizhansky V.; Vega S.; de Groot H. J. M. A method for measuring heteronuclear (H-1-C-13) distances in high speed MAS NMR. J. Am. Chem. Soc. 2000, 122, 3465–3472. 10.1021/ja992714j. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.