

SUMMARY
Acute myeloid leukemia (AML) is a heterogeneous and fatal disease with an urgent need for improved therapeutic regimens given that most patients die from relapsed disease. Irrespective of mutation status, the development of aggressive leukemias is enabled by increasing dependence on signaling networks. We demonstrate that a hyperactive signalosome drives addiction of AML cells to a tumor-specific Hsp90 species (teHsp90). Through genetic, environmental, and pharmacologic perturbations, we demonstrate a direct and quantitative link between hyperactivated signaling pathways and apoptotic sensitivity of AML to teHsp90 inhibition. Specifically, we find that hyperactive JAK-STAT and PI3K-AKT signaling networks are maintained by teHsp90 and, in fact, gradual activation of these networks drives tumors increasingly dependent on teHsp90. Thus, although clinically aggressive AML survives via signalosome activation, this addiction creates a vulnerability that can be exploited with Hsp90-directed therapy.
Graphical Abstract
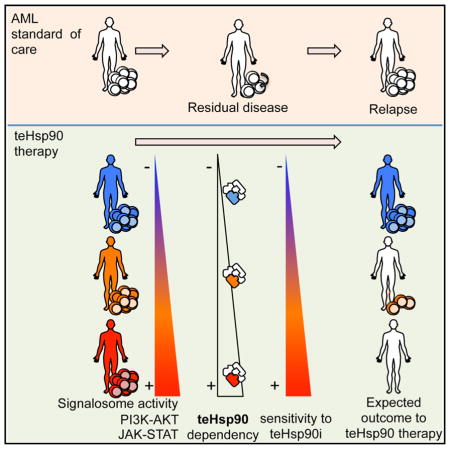
INTRODUCTION
Acute myelogenous leukemia (AML) is a rare but lethal malignancy (Siegel et al., 2012). Despite advances in targeted therapies for AML, the standard induction chemotherapy regimen has remained unchanged for the past three decades. Most adult patients achieve initial remission, although 85% relapse within 2 to 3 years (Burnett et al., 2011). Thus, it is critical to understand the molecular pathogenesis of AML to enable therapeutic advances.
Since the introduction of all-trans retinoic acid (ATRA) for the treatment of t(15;17) acute promyelocytic leukemia (APL) (Mi et al., 2012) and imatinib for chronic myeloid leukemia (CML), there has been an explosion of “targeted” compounds. The ensuing agents designed to address unique genetic and molecular lesions in AML (Perl and Carroll, 2007) have however generated disappointing results. Two main factors account for these failures. First, AML can result from the cooperation of several oncogenic events. Second, residual leukemia stem cells (LSCs) provide the cellular reservoir for relapse (Guzman and Allan, 2014). Available data suggest a reconfiguration of signaling pathways including PI3K, MAPK, and STAT and a continued requirement for an active “signalosome” to drive survival and mitogenic signals.
We hypothesize that, due to the inherent participation of chaperone proteins in the folding and activation of a variety of kinases, the oncogenic activation of the signalosome in leukemia may be regulated by a specialized tumor-enriched Hsp90 species (teHsp90). Whereas Hsp90 assists in the correct conformational folding of a variety of proteins involved in physiological housekeeping roles, specific modifications shunt Hsp90 to specialized functions in stress (Moulick et al., 2011; Nayar et al., 2013; Röhl et al., 2013; Taldone et al., 2014). Tumor-specialized Hsp90 species have been identified and referred to as “tumor-specific,” “oncogenic Hsp90,” or “tumor-enriched Hsp90” (Moulick et al., 2011; Nayar et al., 2013; Taldone et al., 2014); these species are often found with enhanced association to oncogenic proteins and can block their degradation (Bagatell and Whitesell, 2004; Moulick et al., 2011; Nayar et al., 2013; Röhl et al., 2013; Taldone et al., 2014). In diffuse large B cell lymphoma cells, teHsp90 forms a complex with Bcl6 at its target gene promoters and contributes to Bcl6-mediated gene silencing (Cerchietti et al., 2009); in CML cells, teHsp90 sustains Bcr-Abl activity and an active STAT5 signaling (Moulick et al., 2011); in virus-associated lymphomas, teHsp90 regulates viral proteins, blocking latent and lytic viral functions (Nayar et al., 2013). Therefore, whereas teHsp90 impacts numerous signaling pathways and cellular functions, the signaling pathways impacted do not overlap across all tumor types, nor is regulation of the signalosome behind its anti-apoptotic function in all tumor types. However, all tumor types demonstrate a physically modified teHsp90 with higher affinity for ATP and its mimetics, thus providing a basis for its specific pharmacologic targeting in the treatment of diseases (Kamal et al., 2003; Taldone et al., 2014).
To investigate the potential relationship between an activated signalosome in AML and teHsp90, here we use a combination of genetic and pharmacologic approaches. We start by deciphering the biological background of those AMLs more likely to depend upon teHsp90 for their survival, at the blast as well as the LSC level, using cells lines, primary specimens, and ex vivo and in vivo models. We then link, in a step-by-step mechanistic investigation, the vulnerability of the teHsp90-addicted AMLs to an active signalosome. Our data demonstrated that, as adverse-risk leukemias become dependent on greater numbers of signaling networks, they increase recruitment of tumor-specific chaperone functions and deepen their susceptibility to teHsp90 inhibitors.
RESULTS
Hsp90 Inhibition Induces Cell-type-Specific Cell Death in Leukemia Cell Lines and Primary Samples
To gain insights into the biological characteristics of teHsp90-dependent AML, we first investigated main drivers of sensitivity to Hsp90 inhibition by evaluating the effect of Hsp90 inhibitors in a spectrum of leukemia cell lines and primary samples with different cytogenetic abnormalities and genetic backgrounds (Tables S1 and S2). We used the small-molecule Hsp90 inhibitor PU-H71, an agent with preferential binding to the teHsp90 species. As PU-H71 has selective affinity for teHsp90, it can be used as a sensor to measure the dependency of a tumor upon teHsp90, in vitro and in vivo (Moulick et al., 2011; Nayar et al., 2013; Taldone et al., 2014). PU-H71 has been shown to have a potent anti-tumor activity in animal models for several types of tumors by preferably targeting teHSP90 (Caldas-Lopes et al., 2009; Marubayashi et al., 2010; Moulick et al., 2011; Nayar et al., 2013) and is currently in phase I clinical trial for metastatic solid tumors and lymphoma (http://www.clinicaltrials.gov, NCT01393509 and NCT01581541).
We found that, for each leukemia cell line tested, PU-H71 inhibited growth in a dose- and cell-dependent manner. PU-H71 treatment also induced cell killing, with notable differences observed among the cell lines (Figure 1A; cell killing represented by y axis values below 0%). Importantly, we did not observe a significant increase in sensitivity to PU-H71 in leukemia cell lines when grouped by mutations reported in the COSMIC database (Figure S1A). We found that the most-sensitive cell lines were MOLM-13, MV4-11, and M0-91 cells, in which over 95% of the initial cell population was killed at 72 hr by concentrations as low as 200 nM PU-H71. Other chemically distinct Hsp90 inhibitors, such as 17-AAG, CNF-2024 (BIIB021), and PU-DZ13 exhibited a similar activity spectrum (Figure S1B). Induction of apoptosis as analyzed by annexin-V/7-AAD staining (Figures 1B and S1C) indicated a similar trend, with M0-91, MOLM-13, and MV4-11 being most sensitive (LD50 < 500 nM); KG-1 and Kasumi-1 displayed moderate sensitivity (LD50 ranged between 1 and 2 μM), and HEL cells were least sensitive (LD50 > 2 μM). These observations were validated by PARP cleavage (Figure 1C) and activation of caspase 3,7 (Figures 1D and S1D). In the most-sensitive cells, apoptosis was associated with downregulation of anti-apoptotic molecules such as Bcl-xL (Figure 1E).
Figure 1. PU-H71 Is Cytotoxic in a Subset of AML Cells while Leaving Normal Blood Cells Unharmed.

(A) Growth of cells treated for 72 hr with PU-H71 (relative to untreated cells). PBL, peripheral blood lymphocytes.
(B) Cells were treated with or were not treated (control) with the Hsp90 inhibitor (Hsp90i), PU-H71. Percent annexin V+ cells relative to control after 48 hr treatment with PU-H71 is shown.
(C) Immunoblots showing cleaved PARP after 24 hr treatment with PU-H71.
(D) Percent increase in caspase-3, 7 activity assay after 24 hr treatment with PU-H71
(E) Immunoblots for Bcl-xL after 24 hr treatment with PU-H71
(F and G) Percent annexin V+ cells relative to control for primary AML cells (F) or cord blood mononuclear cells (MNCs) (G) after 48 hr treatment with PU-H71. (A, B, and D) Values denote mean ± SD (n = 4).
(F and G) Values denote SEM. See also Figure S1 and Tables S1 and S2.
The apoptotic response of primary AML cells to PU-H71 treatment was also variable (Figure 1F). Nearly 36% of the tested primary AML samples showed apoptosis of 50% or higher after 48 hr of treatment with 250 nM PU-H71. This fraction increased to 60% at 500 nM PU-H71 (Figure 1F, bottom panel). When compared to cytarabine (AraC), the cornerstone of induction therapy and consolidation therapy for AML (Figure S1E), PU-H71 was significantly more cytotoxic to primary AMLs.
Importantly, and potentially as an indication of its selective binding to teHsp90 over the normal cell housekeeping Hsp90, normal peripheral blood leukocytes (PBL) (Figure 1A) and mono-nuclear cells from cord blood (CB) cells showed only minor toxicity to PU-H71, even at the high concentration of 20 μM (Figure 1G). These data demonstrate that a subset of AMLs is highly dependent on the activity of teHsp90 but that no specific karyotype or transforming event can be directly associated with the sensitive subset (Figure 1A; Tables S1 and S2).
Inhibition of teHsp90 Depletes AML Oncoproteins, but this Effect Does Not Correlate with Apoptotic Sensitivity
Hsp90 is required for the stabilization of oncogenic clients such as BCR-ABL, NPM-ALK, and HER2 (Whitesell and Lindquist, 2005), and clinical translation of Hsp90 inhibitors to date has primarily focused on tumors driven by these and other known Hsp90 oncoclients (Moulick et al., 2011; Whitesell and Lindquist, 2005). Clinical activity observed for all Hsp90 inhibitors in HER2-positive breast tumors and NSCLC with ALK translocations validates this as a successful strategy for a subset of tumors (Jhaveri et al., 2014). The distinct apoptotic sensitivity of AML cell lines toward PU-H71 could be due to effective disruption of Hsp90 interactions with its oncoclients in certain cell lines, but not others. To test this hypothesis, we evaluated the effect of PU-H71 on two proteins demonstrated to be Hsp90 dependent in a majority of cancers, the RAF-1 and AKT kinases (Trepel et al., 2010). The Hsp90 inhibitor dose dependently and markedly reduced the steady-state levels of these proteins in all the tested cells, irrespective of their apoptotic sensitivity to Hsp90 inhibition (Figure 2A). In addition to these “pan-cancer” Hsp90 client proteins, PU-H71 also led to the degradation of leukemia-specific oncoproteins, such as mutant FLT3 in MOLM-13, TEL-TRKC in M0-91 (high Hsp90 sensitivity), AML1-ETO and mutant cKIT in Kasumi-1 and SKNO-1 (intermediate Hsp90 sensitivity), and mutant JAK2 in HEL (low Hsp90 sensitivity) and UKE-1 (high Hsp90 sensitivity; Figure 2A). These oncoproteins were previously reported to be degraded by Hsp90 inhibitors in AML or other transformed cells (Floris et al., 2011; Yang et al., 2007; Yao et al., 2003). In sum, PU-H71 depletes AML cells of key oncogenic driver proteins, but this effect fails to translate or be indicative of AML cells’ dependency status upon teHsp90.
Figure 2. PU-H71 Treatment Depletes AML Cells of Major Leukemogenesis-Driving Proteins Irrespective of Their Apoptotic Sensitivity.

(A–C) Immunoblot analysis for (A) AML (FLT3, TEL-TRKC, JAK2, c-KIT, and AML-ETO) and cancer (AKT and RAF-1) teHsp90 clients after 24 hr treatment with PU-H71; (B and C) phosphorylated or total NF-κB p65, STAT5, AKT, ERK1/2, and p38 for leukemia cells treated for 6 hr with 1 μM PU-H71 are shown.
(D) Protein expression levels indicated as mean fluorescence intensity (MFI) for phosphorylated (upper panel) or total (lower panel) AKT, NF-κB p65, and STAT5. Values denote mean ± SEM. ***p < 0.001.
(E) Pull-down assay for MV4-11 cells using PU-beads followed by immunoblotting for the indicated proteins. CB, control beads; PB, PU-H71-beads; WCL, whole cell lysate.
AML Cells with High Apoptotic Sensitivity to teHsp90 Inhibition Display Activation of Key Signaling Pathways
Because we found that cell death upon PU-H71 treatment did not correlate with degradation of specific oncogenic client proteins characteristic of AML subtypes, we next proceeded to investigate whether response to Hsp90 inhibitors was linked to key AML-signaling networks such as the MAPK, PI3K-AKT-mTOR, and JAK-STAT pathways (Echeverría et al., 2011; Moulick et al., 2011; Taipale et al., 2012). The rationale for the pathway versus client approach is provided by recent global proteomic analyses (Moulick et al., 2011; Nayar et al., 2013; Sharma et al., 2012). These studies suggest that response to Hsp90 inhibitors may be dependent on tumor-driving teHsp90 clientele networks and pathways as opposed to single oncogenic clients.
Indeed, in Hsp90-sensitive AML, we found constitutive activation of several signaling pathways, as indicated by the presence of phosphorylated forms of NF-κB p65, STAT5, AKT, and ERK1/2 (p44/42). These phosphoproteins were rapidly decreased by PU-H71 treatment (Figure 2B for MV-411 and MOLM13 cell lines and Figure 2C for a primary sample). The decrease was readily detectable as early as 2 hr (Figure S2), at a time the steady-state levels of the proteins were yet unchanged. These observations were also corroborated using flow cytometry assays. We found that PU-H71 treatment resulted in a significant downregulation of p-AKT, p-NF-κB p65, and p-STAT5 in both cell lines and primary samples (58.8%, 48.5%, and 62.0% in 1 μM PU-H71-treated MV4-11 cells and 65.2%, 65.3%, and 65.9%, respectively, in primary cells from AML17; Figure 2D). Because of its specificity for teHsp90, PU-H71 attached to a solid support can be used as a capture reagent to identify the teHsp90-interacting oncoproteins (Moulick et al., 2011; Nayar et al., 2013; Taldone et al., 2014). We thus performed PU-H71 pull-downs in AML cells with high sensitivity to Hsp90 inhibition and demonstrated that, indeed, p-p65, p-ERK, p-STAT5, and p-AKT are physically associated with teHsp90 (Figure 2E). Together, the data suggest that teHsp90 binds to and maintains the activity of numerous signaling proteins. AML cells may thus be dependent on preservation of these signaling pathways in a teHsp90-dependent manner.
To investigate this hypothesis, we next evaluated all the AML cells for their reliance on signaling pathways, specifically on the MAPK, PI3K-AKT-mTOR, and JAK-STAT pathways, which are known to play a role in AML survival (Perl and Carroll, 2007; Testa and Riccioni, 2007).
We observed that cell lines most dependent for survival on the activation of JAK-STAT, PI3K-AKT-mTOR, or MAPK pathways were also those most likely to die when teHsp90 was inhibited (Figures 3A–3C and S3A). Indeed, a significant correlation was observed between the sensitivity of these cells to PU-H71 and their sensitivity to JAKi (slope 0.51 ± 0.19; R2 = 0.495; p = 0.034), AKTi (slope 0.87 ± 0.21; R2 = 0.7034; p = 0.0047), and MEKi (slope 0.73 ± 0.09; R2 = 0.9052; p < 0.0001; Figure 3D). Similarly, phosphorylated forms of STAT5, ERK1/2, and AKT were significantly increased in primary AML patient samples with higher sensitivity to PU-H71 (2.97-, 2.82-, and 1.32-fold increase and p = 0.0477, 0.0490, and 0.0395, respectively; Figure 3E, top panels), and consistently, PU-H71-sensitive primary AML samples were also most sensitive to JAKi, MEKi, and AKTi (Figure 3E, bottom panels). Phosphorylation alone was insufficient to indicate the addiction of AML to a signaling molecule or its reliance on teHsp90; for example, whereas all Hsp90-sensitive AMLs expressed constitutive levels of p-p38, a molecule associated with leukemia proliferation (Birkenkamp et al., 1999), these phosphorylated species were insensitive to teHsp90 inhibition (Figures 2B, 2C, and S3B) and the viability of these cells was unaffected by p38 inhibition (Figures S3C and S3D).
Figure 3. Dependence of AML Cells on Activated PI3K/AKT, JAK/STAT Pathways and teHsp90 Overlaps.

(A–C) Percent cell death relative to untreated control evaluated at the indicated time points for a panel of leukemia cell lines treated with (A) 0.5 μM JAK1/2 inhibitor (JAKi), (B) 5 μM AKT inhibitor (AKTi), or (C) 20 μM MEK1/2 inhibitor (MEKi).
(D) The apoptotic sensitivity of AML cells lines to PU-H71 (x axis) and to AKTi, JAKi, or MEK1/2i (y axis).
(E) (Top) Levels of pSTAT5, pERK, and pAKT evaluated by phosphoflow in primary AML samples with low or high PU-H71 apoptotic sensitivity. (Bottom) Percent cell death after 48 hr treatment with JAKi, MEKi, or AKTi. Each symbol represents an individual sample. Values denote mean ± SEM. *p < 0.05 and **p < 0.01. See also Figure S3.
Collectively, our findings suggest an intrinsic link between the dependence of AML cells upon teHsp90-chaperoned signaling networks involved in AML survival and their addiction to teHsp90.
Addiction to teHsp90 Is a Result of Additive Dependence on Activated Signaling Pathways
To quantify the contribution of each pathway toward teHsp90 addiction, we used cellular models in which the activation of signaling pathways may be fine-tuned by several means. First, we made use of FL5.12-derived isogenic cell lines. FL5.12 is interleukin-3 (IL-3)-dependent for growth, proliferation, and survival. The level of p-STAT5, but not of p-AKT, is dependent on the presence of IL-3, and constitutive activation of AKT can be found in derivatives resistant to IL-3 withdrawal (Karnauskas et al., 2003).
To investigate the effect of STAT5 activation on sensitivity to teHsp90 inhibition, we cultured parental FL5.12 cells in 1 ng/ml IL-3-containing media. As expected (Karnauskas et al., 2003), STAT5 was active in these cells, as evidenced by increased p-STAT5 levels (Figures 4A and 4B). Both the JAKi and PU-H71, but not the AKTi, significantly decreased IL-3-stimulated STAT5 activity (Figures 4A and 4B). When STAT5 activity was increased by the addition of IL-3 to the culture media, cells became more prone to die when treated with the JAKi or the teHsp90 inhibitor (Figure 4C).
Figure 4. Activation of JAK, AKT, and ERK Pathways Renders AML Cells Addicted to teHsp90.

(A and B) Immunoblot (A) and flow cytometry (B) of FL5.12 cells after 24 hr treatment with JAKi, AKTi, or PU-H71 (PU) in the presence or absence of IL-3.
(C) Percent dead cells in FL5.12 cells after 48 hr treatment with JAKi, AKTi, or PU.
(D and E) Immunoblots for pAKT (D) and percent apoptotic cells (E) of vector control- or mAKT-transfected cells treated with PU-H71, AKTi ± doxycycline (DOX) for 24 hr or 48 hr, respectively.
(F) Percentage of mCherry+ cells KG-1/N51 or KG-1/WT over 30-day cultures.
(G) Percentage of apoptotic cells after 48 hr treatment with PU-H71.
(H and I) Phosphorylated protein levels in cells evaluated by immunoblot (H) or flow cytometry (I) (fold change of MFI for KG-1/N51 relative to KG-1/WT).
(J) Immunoblots of Hsp90-interacting proteins as isolated by PU-beads.
(K) Immunoblots for Hsp90 and FLT3 immunoprecipitated with an anti-FLT3 antibody.
(L) Correlation of the apoptotic sensitivity to PU-H71 (x axis) and to AKTi, JAKi, MEK1/2i, or p38i (y axis) in cells treated for 48 hr.
(M) Percent cell death for FLT3-ITD− or FLT3-ITD+ primary AML cells after 48 hr exposure to PU-H71.
(N) Hsp90 expression of cells from (M) as measured by flow cytometry. Each symbol represents an individual sample.
Values denote mean ± SEM. *p < 0.05; **p < 0.01;***p < 0.001. See also Figure S4.
Next, we incorporated an additional layer of activation of the signalosome by turning on AKT. To do so, we used FL5.12 cells transfected with a constitutively activated, doxycycline (DOX)-inducible myristylated form of AKT (mAKT) (Karnauskas et al., 2003). Activation of AKT using mAKT was corroborated by the increased phosphorylation of AKT observed by immunoblot (Figure 4D). Thus, in addition to STAT5 activation provided by the IL-3 in the media, these cells have a constitutively activated AKT. Increasing the activity of AKT by turning on mAKT led to an increased reliance of these cells for survival on activated AKT (Figure 4E, compare mAKT+AKTi to mAKT+DOX+AKTi; p = 0.015) and, in turn, in a greater dependence on teHsp90 (Figure 4E, compare mAKT+PU to mAKT+DOX+PU; p = 0.035).
We thus conclude that the higher apoptotic effect observed for PU-H71 versus AKTi or JAKi alone suggests a cooperation of both the STAT and AKT pathways to the survival and proliferation of these cells and indicate that their sensitivity to teHsp90 inhibition may stem from combined, and potentially additive, addiction to the AKT and the JAK/STAT pathways. Indeed, combination of AKTi and JAKi resulted in significantly higher apoptosis in FL5.12 cells supplemented with IL-3 (Figure S4A).
To further test this observation, we employed an AML oncogenic driver, FLT3-ITD. To this end, we utilized a leukemia cell line with moderate sensitivity to PU-H71 (KG-1) and transduced it with lentiviral vectors (pLVX-EF1α-IRES-mCherry) containing either wild-type FLT3 (WT-FLT3) or FLT3 internal-tandem duplication (ITD) (N51-FLT3) genes (Figure S4B). FLT3-ITD results in ligand-independent constitutive activation of FLT3, and its downstream pathways include PI3K-AKT and JAK-STAT (Brandts et al., 2005; Thiede et al., 2002). We found that the additional transformation event with FLT3-ITD mutation resulted in a growth advantage over the parental cells (Figure 4F; N51-FLT3 [KG-1/N51], red squares; WT-FLT3 [KG-1/WT], green crossed squares).
KG-1/N51 cells were more sensitive to teHsp90 inhibition compared to the parental KG-1 or KG-1/WT cells (Figure 4G). The percent annexin V+ cells after treatment of parental KG-1, KG-1/WT, and KG-1/N51 with 1 μM PU-H71 for 48 hr were 34.7, 25.4, and 52.9, respectively. Higher sensitivity to teHsp90 correlated with increased activation of survival signaling networks, such as the JAK-STAT, PI3K-AKT, and MAPK pathways. Immunoblots demonstrated that the phosphorylated form for each of STAT5, AKT, ERK1/2, and NF-κB p65 was higher, with their total protein levels remaining unchanged, in KG-1/N51 cells (Figure 4H). By phospho-flow, we found 1.6-, 1.4-, 2.1-, 1.5-, and 2.6-fold increase in the phosphorylated ERK1/2, NF-κB p65, STAT5, AKT, and S6 species, respectively, in KG-1/N51 cells compared to KG-1/WT (Figure 4I). Phosphorylation of p38 MAPK was not changed in KG-1/N51 cells, demonstrating the specificity of the transduction with FLT3-ITD in the activation of JAK-STAT-, PI3K-AKT-, and ERK1/2-signaling pathways in KG-1 cells.
Furthermore, to determine whether upregulation of JAK-STAT-, PI3K-AKT-, and ERK1/2-signaling pathways by FLT3-ITD results in increased dependence on teHsp90 chaperoning, we performed pull-down assay using the solid-support immobilized PU-H71 (PU-beads). We confirmed increased association of teHsp90 with phosphorylated STAT5, AKT, ERK1/2, and NF-κB p65 in KG-1/N51, but not in KG-1/WT cells, when compared to parental KG-1 cells (Figure 4J), indicating that these proteins are clients of teHsp90 and that the increased activation of these proteins resulting from the transduction with FLT3-ITD require teHsp90. Immunoprecipitation assays were also performed using an anti-FLT3 antibody to demonstrate an increased association between FLT3-ITD and Hsp90 in KG-1/N51, but not in KG-1/WT cells (Figure 4K). Importantly, in these cells, we found that apoptosis induction by JAKi or AKTi significantly correlated with cell death by the teHsp90 inhibitor (Figure 4L; JAKi R2 = 0.979, p = 0.090; AKTi R2 = 0.99, p = 0.048; MEKi p = 0.161; p38i = 0.349).
In sum, these data indicate that additive upregulation of signaling networks results in increased dependence upon teHsp90, and thus, leukemic cells in this biologic state may develop a higher vulnerability to teHsp90 inhibition.
Because FLT3-ITD is one transforming event capable of simultaneously activating these signaling pathways, we next tested primary AML cells bearing FLT3-ITD mutations. We found indeed that induction of apoptosis in FLT3-ITD+ primary AML cells by 500 nM PU-H71 treatment was significantly higher than in FLT3-ITD− primary AML cells (p = 0.02; Figure 4M). Importantly, the total levels of Hsp90 were not significantly different in FLT3-ITD+ or FLT3-ITD− primary AML samples (p = 0.12; Figure 4N). Of note, we still observed some differences in sensitivity to PU-H71 (Figure 4M), for example, one FLT3-ITD-negative sample with relative sensitivity to PU-H71 showing a 60% cell death and one FLT3-ITD-positive sample with less sensitivity to PU-H71 (40% cell death upon Hsp90 inhibition), suggesting that FLT3-ITD alone would not be sufficient to predict for teHsp90-addicted AML.
Taken together, our data strongly suggest that, in AML, additive addiction conferred by the simultaneous activation of PI3K-AKT- and JAK-STAT- and potentially ERK1/2-signaling pathways results in an increased dependence on teHsp90 for survival. The more the AML cell increases its signaling activity through these pathways, the more the key components of these pathways become associated with teHsp90. This increased dependence on teHsp90 increases their vulnerability to Hsp90 inhibition.
Dependence of AML Signaling Networks on teHsp90 Is Conserved at the Leukemic Progenitor and Stem Cell Level
One of the challenges to AML therapy is the failure of current chemotherapeutic drugs to eliminate LSCs (Felipe Rico et al., 2013; Guzman and Allan, 2014). Thus, we also evaluated whether the sensitivity of primary AML samples to PU-H71 will extend to leukemia stem and progenitor cells. First, we evaluated sensitivity of LSCs and lymphocyte populations defined by immunophenotype from AML patient samples (Guzman and Allan, 2014). We found that LSCs had higher sensitivity than normal lymphocytes from the same patient sample to PU-H71 (p = 0.0005; Figure 5A). As observed in bulk AML blasts and cell lines, LSCs also presented heterogeneity in their response to PU-H71. In order to determine whether their sensitivity to PU-H71 is also dependent on an active signalosome, we evaluated the basal levels of p-STAT5 in LSCs, as it has been reported that the JAK/STAT pathway plays a critical role in malignant stem cell growth (Cook et al., 2014). Indeed, we found that sensitive LSC samples had significantly higher levels of basal phosphorylation of STAT5 (Figure 5B; R2 = 0.4900; p = 0.0242). Our findings suggest an increased addiction of LSCs to teHsp90 in subsets with constitutive STAT5 signaling, as we observed for leukemic blasts.
Figure 5. Dependence of AML Signaling Networks on teHsp90 Is Conserved at the Leukemic Progenitor and Stem Cell Level.

(A) Percent annexin-V-positive leukemia stem cells (LSCs) and lymphocytes in primary AML specimens treated with 500 nM PU-H71 for 48 hr. Each color represents an individual sample.
(B) Correlation between p-STAT5 (y axis) and sensitivity of LSCs to PU-H71 (x axis).
(C) Percent colony-forming units (CFUs) relative to untreated control (UT) for primary AML cells or CD34+ cord blood (CB) after 48 hr treatment with 500 nM PU-H71. Myeloid colonies are shown for normal CB cells.
(D) Schematic representation for the ex vivo engraftment assay to evaluate the effect of PU-H71 treatment on the ability of LSCs to initiate disease in immunodeficient mice.
(E and F) Percent engraftment of human AML cells (E) and CD34+ CB cells from two units (CBa and CBb) (F) after ex vivo treatment with PU-H71 for 24 hr. Each symbol represents an individual mouse.
Values denote mean ± SEM.
To determine the effect of inhibition of teHsp90 in the function of progenitor cells, we evaluated the ability of AML progenitor cells to form progeny after exposure to PU-H71 using colony-forming assays. We found a significant decrease in colony formation after exposure to 500 nM PU-H71 (75%–98% reduction; Figure 5C, circles). In contrast, PU-H71 demonstrated less impairment of normal myeloid colony-forming ability of the normal CD34+ progenitor cells obtained from umbilical CB cells (Figure 5C, squares), demonstrating selectivity for malignant progenitor cells.
To further determine whether PU-H71 has an effect on the ability of LSCs to initiate disease, we performed xenotransplantation assays after in vitro treatment with 500 nM PU-H71 (Figure 5D) of primary AML samples with constitutive p-STAT5 and known in vitro sensitivity for bulk tumor populations. We found that PU-H71 in vitro treatment resulted in a significant decrease in engraftment of human AML samples compared to untreated controls (Figure 5E), demonstrating the ability of PU-H71 to target LSCs. In contrast, the engraftment ability of normal CD34+ CB cells was not significantly affected by PU-H71 in vitro treatment (Figure 5F), demonstrating that normal HSCs have little, if any, expression and/or dependence on teHsp90.
Constitutive Activation of teHsp90-Dependent Signaling Networks Determines PU-H71 In Vivo Efficacy
Unlike experiments performed in culture where cells can be under steady drug pressure and thus constant saturation of the target, tumor cells in vivo are exposed to gradient drug levels, with high concentrations soon after injection that decrease in a time-dependent fashion as the drug is cleared from both the organism and the tumor. Because pathways most dependent on teHsp90 are also most sensitive to its pharmacologic inhibition, they remain affected for the longest period of time upon single-dose administration of PU-H71. Thus, in vivo studies enable a kinetic analysis of individual teHsp90-dependent pathways and permit correlation between individual signaling pathway suppression and in vivo apoptosis induction. We performed such correlative analyses in tumors generated from M0-91 cells (high apoptotic sensitivity in vitro) and HEL cells (low apoptotic sensitivity in vitro). In M0-91 tumors that harbor constitutively elevated p-AKT and p-STAT5 levels, PU-H71 treatment resulted in a marked induction of apoptosis, determined by an increase in cleaved PARP (Figure 6A). Cleaved PARP in M0-91 was observed up to 96 hr post-administration of one dose of PU-H71, mirroring the parallel potent inhibition of both AKT and STAT pathways (Figure 6A, left panel). The highest level of cleaved PARP was observed in the interval of 12–72 hr post-PU-H71 administration, when both p-AKT and p-STAT5 levels were reduced by 70%–100% (Figure 6A, left panel). It declined by 96 hr, when p-AKT, but not p-STAT5, recovered to baseline levels (Figure 6A, middle panel).
Figure 6. PU-H71 Ablates In Vivo LSCs of Primary AML Samples with High pSTAT5 Levels.

(A) Immunoblots (left) and graphed summary (right; means ± SEM) for the indicated proteins in M0-91 or HEL cells-engrafted mice after one dose of PU-H71 (75 mg/kg).
(B) Schematic representation for the in vivo treatment of MOLM-13-GFP-Luc xenografts.
(C) Whole-body luciferase imaging of MOLM-13-GFP-Luc xenografts before and during treatment with PU-H71.
(D) Top panel: representative plots for mouse (mCD45) and human (hCD45) BM cells harvested from MOLM-13-GFP-Luc xenografts after 2 weeks of treatment with PU-H71. Bottom panel: percent human cells in xenografts treated with either PBS or PU-H71 (PU) is shown.
(E) Schematic representation for the in vivo AML-patient-derived xenograft (PDX) treatments.
(F) Correlations between p-STAT5 (y axis; left panel) and pS6 (y axis; right panel) and decreased tumor burden after PU-H71 treatment (x axis).
(G) Percent decrease in tumor burden after treatment with PU-H71 for seven PDX-AML xenograft cohorts grouped by their relative levels of pSTAT5 and pS6.
(H) Percent leukemia engraftment in secondary transplants for the indicated PDX-AML xenograft cohorts grouped by their relative levels of pSTAT5 and pS6.
(I) CFUs of murine bone marrow cells evaluated from disease-free C57BL6 mice treated for 3 weeks with either PBS or PU-H71.
Tumors derived from HEL cells (low apoptotic sensitivity; Figure 1B) were less sensitive than the M0-91 tumors to teHsp90 inhibition (Figure 6A). Nonetheless, and in contrast to tissue culture, when xenografted in nude mice, HEL cells demonstrated low to moderate expression level of p-AKT. This is not surprising, as it has been reported that AKT activity can be stimulated in AML cells by the environment. Consistent with our hypothesis, elevation of AKT activity in xenografted HEL cells appears to be necessary for tumor survival as PU-H71 induces markedly higher apoptosis in xenografted HEL tumors (Figure 6A) than in cultured HEL cells (Figure 1C). As with M0-91 tumors, highest level of cleaved PARP was observed when both p-AKT and p-STAT5 levels were reduced by PU-H71 by 70%–100% (in the interval of 12–48 hr post-PU-H71 administration). PARP cleavage diminished significantly when p-AKT recovered to baseline levels (72 hr; Figure 6A, right and middle panels).
These data show that activation of key survival signaling molecules in AML in vivo remains modulated by the teHsp90. The endogenous levels of these activated signalosome members, the kinetics and potency of suppression of these pathways by teHsp90 inhibition, and the gradual decline in PARP cleavage that parallels the recovery of individual key signaling networks further support our hypothesis that additive addiction of AML to the teHsp90-regulated signaling networks is directly proportional with the addiction of AML to teHsp90.
To further test whether the in vivo effectiveness of PU-H71 in primary AML cell is dependent on the status of the teHsp90-dependent signaling pathways, we used MOLM-13 cells as they have both AKT and STAT pathways active. We transduced these cells with CMV-GFP-T2A-luciferase lentiviral vectors to express GFP and luciferase (MOLM-13-GFP-Luc). MOLM-13-GFP-Luc cells were injected into NSG mice in order to visualize tumor burden using the IVIS in vivo imaging system (Figure 6B). The in vivo activation levels of p-STAT5 and p-S6 (as a surrogate indicator of the AKT pathway activity) were evaluated in MOLM-13-GFP-Luc cells using flow cytometry (Figure S5A). Animals were evaluated for tumor burden prior the treatment with 75 mg/kg PU-H71 or saline three times a week for 2 weeks when a high disease burden was confirmed by imaging (see Figure 6B). We found that PU-H71 treatment resulted in vivo ablation of MOLM-13 cells reflected by the absence of luciferase signal in the treated mice (Figure 6C). Eradication of leukemia cells was confirmed by flow cytometry using GFP and human CD45 (Figure 6D).
We extended our evaluation by establishing AML patient-derived xenografts (PDX) (see Figure 6E for schematic representation), in which primary AML samples were selected with different levels of p-S6 and p-STAT5 (n = 7; Figure S5B). Once the PDX models were established, we first re-evaluated the status of p-S6 and p-STAT5 in the human AML cells obtained from the PDX animals (Figure S5C; PDX-AML; n = 7). We classified samples based on their levels of activation of PI3K/AKT and JAK/STAT pathways, where we defined pS6 high level (>20×), pS6 mid-level (<5×; >20×), pS6 low level (<5×), pSTAT5 high level (>3×), pSTAT5 mid-level (<3×; >1.5×), and pSTAT5 low level (<1.5×). We found that neither the activation levels of pS6 alone nor pSTAT5 alone were sufficient to correlate with decrease in tumor burden after treatment with PU-H71 (Figure 6F). However, samples with high/mid levels of activation for both pS6 and pSTAT5 had the greatest decrease in AML tumor burden (Figure 6G), demonstrating that the highest level of PU-H71 sensitivity occurred in PDX-AML samples, demonstrating hyperactivation of both PI3K/AKT and JAK/STAT pathways. For example, PDX-AML4 with high activation of both PI3K/AKT and JAK/STAT pathways demonstrated the largest decrease in total tumor burden (90% decrease relative to vehicle control; p = 0.004; Figures 6G and S5C). PDX-AML5 with mid-activation for both PI3K/AKT pathway and JAK/STAT pathway demonstrated an intermediate decrease in total tumor burden (48% decrease relative to vehicle control; Figures 6G and S5C), whereas PDX-AML9 that demonstrated mid-activation of PI3K/AKT pathway but no STAT-pathway activity resulted in only a 29.1% decrease in tumor burden (p = 0.0371) after treatment with PU-H71 (Figures 6G and S5C). These data strongly demonstrate that, with additive increased activation of survival signaling networks, there is an increased dependence on teHSP90.
Importantly, to determine the ability of PU-H71 to target LSCs in vivo, we evaluated the ability of the human cells harvested from the treated PDXs to initiate leukemia by performing secondary transplants (using equal numbers of human AML cells obtained from the PU-H71-treated and vehicle control PDX mice). We found a significant decrease in leukemia engraftment in the secondary transplants derived from PDX-AML4 and PDX-AML2 (96.3% and 68.4% [p = 0.02] decrease, respectively; Figures 6H and S5C), suggesting that PU-H71 effectively targets LSCs in the samples with activation of both PI3K/AKT and JAK/STAT pathways. Unfortunately, we were unable to evaluate PDX-AML5 in the secondary transplant due to a lack of overall engraftment. In contrast, the secondary transplants derived from PDX-AML9 demonstrated a 1.7-fold increase in leukemia cells after the animals were exposed to PU-H71, suggesting that the treatment did not eliminate the LSCs from PDX-AML samples with the activation of only one pathway (Figures 6H and S5C). PDX-AML37, PDX-AML16, and AML-PDX33 were not evaluated in secondary transplants. These data support the concept that primary AML samples with constitutive activation of survival networks present higher dependence on teHsp90. Importantly, the anti-AML effects of PU-H71 occurred with no toxicity to normal hematopoietic cells as demonstrated by no changes in colony formation of bone marrow hematopoietic stem and progenitor cells following chronic treatment with PU-H71 (Figure 6I). This safety profile finding concords with that seen on the phase 1 clinical study of PU-H71 in patients with previously treated solid tumors, lymphoma, and myeloproliferative neoplasms (abstract by Gerecitano et al., 2015).
DISCUSSION
Despite the large number of targeted agents entering clinical evaluation every year, only 5%–8% may reach registration. There is an estimated 50% failure rate for oncology agents in phase 3 studies. Such failures are especially expensive and deprive many patients of potentially more-effective treatments, highlighting the urgent need to implement means for patient selection and trial enrichment. Our study aims to address this problem in AML. Targeted therapies designed to induce selective apoptosis in leukemic cells are believed to be most promising in leukemias, and we therefore focused our study on apoptotic sensitivity (Testa and Riccioni, 2007).
To begin, we tested the teHsp90-specific inhibitor, PU-H71, in a panel of genetically defined leukemia cell lines and primary AML samples. Specifically, we evaluated a large panel of cell lines and primary AML samples for sensitivity to Hsp90 inhibition and correlated the response to somatic genetic aberrations, expression of leukemia-associated proteins, and the specific activated signaling pathways in different AML cells. We found that teHsp90 inhibition resulted in a heterogeneous response across leukemia cell lines and primary AML samples. This heterogeneity could not be attributed to the ability of PU-H71 to inhibit several distinct and specific oncogenic targets in genetically diverse leukemia subsets as there was downregulation and inhibition of numerous different leukemia driver proteins of AML that were sensitive as well as resistant to PU-H71.
Interestingly, we found apoptotic sensitivity to teHsp90 inhibition to be a reflection of the cellular dependence for survival on specific hyperactivated signaling pathways. Constitutive activation of multiple signaling networks in AML patients has been shown to confer poor clinical outcomes (Kornblau et al., 2006). These pathways include PI3K/AKT (Kornblau et al., 2006, 2011; Xu et al., 2003, 2005), JAK-STAT (Brady et al., 2012; Yoshimoto et al., 2009), and RAS/Raf/MEK/ERK (Kornblau et al., 2006), all of which have been implicated in the pathogenesis of the disease (Burnett et al., 2011; Cook et al., 2014; Tamburini et al., 2007). Importantly, as the number of activated signaling networks increased, patient survival was accumulatively reduced (Kornblau et al., 2006), suggesting cooperativity among pathways for transformation. Furthermore, activation of these signaling networks is not always associated with FLT3 or JAK2 mutations (Brady et al., 2012; Kornblau et al., 2006). JAK/STAT and PI3K/AKT pathways have been implicated in the survival and self-renewal of malignant stem cells from CMLs and AMLs, apart from their role in disease progression (Cook et al., 2014; Xu et al., 2003, 2005). Together, these suggest that AML treatment strategies should aim to simultaneously target multiple signaling networks and provide an explanation to the improved effectiveness of drugs that abrogate individual signaling networks when combined with other agents. Our data indicate that the engaging of these pathways in leukemia cells, whereas advantageous to their aggressiveness, drives these cells to become reliant on teHsp90. We hypothesized that, as more than one pathway is activated, the dependence on teHsp90 also increases. Thus, the higher the activity of the JAK-STAT and PI3K-AKT pathways, the greater the association of key effectors of these pathways with teHsp90 and the larger the sensitivity of AML to Hsp90 inhibition.
We manipulated, in isogenic leukemia cell lines, the activation status of these pathways by exogenous or endogenous means. Turning on STAT5 and/or AKT activity by exogenous IL3, activating oncogenic pathways by the introduction of a mAKT or an “always on” receptor (FLT3 containing the ITD mutation), or changing the microenvironment (HEL cells in tissue culture versus xenograft) all resulted in the activation of JAK-STAT and PI3K-AKT pathways; this in turn led to increased apoptotic sensitivity to PU-H71. Among these three activators, the FLT3-ITD mutation was more likely to be associated with the activation of these signaling pathways; we also demonstrated higher sensitivity of primary AML cells when compared to those harboring the WT receptor. We found moderate sensitivity/resistance in AML samples within FLT3-ITD-negative/positive groups, respectively, consistent with reports for AML patients where activation of multiple signaling networks was not always associated with FLT3-ITD (Kornblau et al., 2006, 2009). These data support the concept that teHsp90 dependence can be dictated by other genetic and epigenetic events that modify the extent of activation of JAK-STAT- and PI3K-AKT-signaling networks.
Further, we went on to probe our proposed mechanism by pharmacologically modulating the activity of the JAK-STAT-and PI3K-AKT-signaling pathways. We found that reducing the activity of these pathways in native leukemia cells using small-molecule inhibitors translated directly in cell killing; a close relationship existed between the ability of these pharmacologic pathway suppressors and that of PU-H71 to induce apoptosis. This relationship was also evident in xenotransplanted AML tumors. By manipulating the kinetics of inhibition of these signaling pathways by the teHsp90 inhibitor, we demonstrated that tumor apoptosis was quantitatively and temporally linked to suppression of one or both of these pathways.
To demonstrate that such a role for teHsp90 is also observed in vivo, we utilized a cell line with high levels of both JAK-STAT-and PI3K-AKT-signaling pathways (MOLM-13) and found that PU-H71 treatment effectively eliminated the tumor cells after 2 weeks of treatment. Furthermore, to evaluate the effect on malignant stem and progenitor cells, we used established AML patient-derived xenotransplants (PDX-AML). Here, we evaluated samples with different levels of activation of JAK-STAT- and PI3K-AKT-signaling networks as measured by phospho-flow (i.e., levels of p-STAT5 and p-S6 in the transplanted human AML cells). We found that samples with activation of both p-STAT5 and p-S6 were sensitive to PU-H71 treatment in vivo at the stem cell level, resulting in a decreased ability to initiate disease in secondary transplants.
Important from a therapeutic perspective, concentrations of PU-H71 that we report here to be toxic in the sensitive AML subset are achievable in clinic at safe doses; in an ongoing phase 1 study, concentrations of PU-H71 of over 500 nM were recorded in tumor cells at 24 hr after administration of doses much lower than the maximum tolerated dose (abstract by Gerecitano et al., 2013).
Many small molecules of variable selectivity and affinity for the teHsp90 (Beebe et al., 2013; Moulick et al., 2011) have been designed to inhibit Hsp90 in an ATP competitive manner, some of which have been tested in clinical trials (Jhaveri et al., 2014), including 17-DMAG and STA-9090 in leukemias. In AML, Hsp90 inhibitors have been evaluated in vitro and in phase I clinical trials (Lancet et al., 2010; Reikvam et al., 2009). The phase I clinical trial for the Hsp90 inhibitor KOS-1022 (17-DMAG) reported three complete remissions out of 17 patients evaluated (Lancet et al., 2010). This 18% response potentially indicates a subpopulation of AML patients with high dependence on teHsp90 who may be more likely to benefit from the incorporation of Hsp90 inhibitory therapy in their treatment regimen. Our study proposes this population to be characterized, in part, by a hyperactive signalosome.
The role of teHsp90 in maintaining dependence of tumors on signaling networks is likely to be context dependent or unique to each tumor type. Indeed, a recent study in multiple myeloma cells found that IL-6-dependent activation of downstream signaling networks in CD45+ cells resulted in increased sensitivity to the Hsp90 inhibitor 17-DMAG (Lin et al., 2013). Thus, the identification of the Hsp90-dependent proteomic signature specific to each tumor type will be critical to the success of teHsp90 inhibitory therapies in the clinic.
In summary, our work describes how hyperactivated signaling networks drive a robust dependence on teHsp90 for survival of AML cells. Our findings have significant clinical implications as Hsp90 inhibitors can be evaluated clinically for AML therapy. Our data demonstrate varied sensitivities to inhibition of teHsp90 in AML; we conclude that not all patients will benefit from an Hsp90 drug. There is an urgent need to identify the responder group in the patient population; our manuscript aims to present the biological makeup of these patients. We demonstrated that PU-H71 eliminates tumor burden as well as LSC in cell lines and PDX models identified with “high” biomarker level (i.e., with a hyperactive signalosome). Our data also show that the lower the AML is on our biomarker classification curve (i.e., lower signalosome activity), the smaller the effect of PU-H71 (i.e., lower cytotoxicity). Our manuscript suggests that teHsp90 inhibitory therapy will likely benefit a subset of AML patients with constitutive activation of PI3K-AKT/JAK-STAT-signaling pathways, and thus clinical trials targeting teHsp90 should aim to enroll patients with hyperactivation of these networks.
EXPERIMENTAL PROCEDURES
Cell Lines and Primary Cells
Primary AML cells were obtained with informed consent and IRB approval from Weill Cornell Medical College-New York Presbyterian Hospital or University of Rochester. Cell lines were purchased from ATCC or DSMZ. FL5.12 cell lines were cultured as described previously (Karnauskas et al., 2003). Additional details are provided in Supplemental Experimental Procedures.
Animal Studies
Experiments were carried out under an Institutional Animal Care and Use Committee (IACUC)-approved protocol, and institutional guidelines for the proper and humane use of animals in research were followed.
Xenografted Cell Lines
Four- to six-week-old nu/nu athymic female mice were obtained from Taconic Farms. HEL and M0-91 were subcutaneously implanted in the right flank of mice and allowed to reach 6–8 mm in diameter. Mice bearing tumors were treated i.p. with 75 mg/kg of PU-H71. Tumors were homogenized and proteins analyzed by western blot as previously described (Caldas-Lopes et al., 2009).
Primary AML Xenotransplantation Assays
NOD/SCID or NSG mice were obtained from National Cancer Institute (NCI) or Jackson Laboratories (JAX). Primary human cells (AML or CB; 2.5 × 106 cells per mouse) were treated with 500 nM PU-H71 overnight and then injected i.v. into sub-lethally irradiated with 2.7 Gy mice. Human engraftment was evaluated 6–8 weeks after transplant as previously described (Hassane et al., 2010). To evaluate the in vivo anti-leukemic activity of PU-H71, xenotransplants were established by injecting 1 × 106 human primary AML cells into sub-lethally irradiated NOD/SCID or NSG mice. Three weeks later, the mice were treated with either PBS or 75 mg/kg PU-H71 three times a week for 3 weeks. To further investigate the LSC activity after treatments, equal numbers of human AML cells were transplanted to secondary recipient mice. The engraftment in the secondary mouse was evaluated 8–16 weeks posttransplantation.
In Vivo Imaging
MOLM-13 cells were transduced with lentivirus carrying MSCV-GFP-T2A-luciferase lentivector (System Biosciences). GFP+ cells were sorted using FACSAria II cell sorter (BD Biosciences). NSG mice were transplanted with 1 × 106 MOLM-13-GFP-Luc cells and were treated with 75 mg/kg PU-H71 1 week postengraftment for a total of 2 weeks. The in vivo images were captured before treatment (1 week) and 1 week after the treatment using the IVIS imaging system (PerkinElmer) and were analyzed using the Living Image software (Caliper Life Sciences).
Additional details are provided in Supplemental Experimental Procedures.
Cell Viability Assays
Cell viability assays were performed using annexin V/7-AAD, alamar blue, or acridine orange/ethidium bromide staining as previously described (Caldas-Lopes et al., 2009; Hassane et al., 2010). Apoptosis was determined by measuring caspase 3,7 activation or PARP cleavage. For primary AML samples, cells were stained with CD34, CD38, and CD45 (BD Biosciences) prior to annexin V/7-AAD staining. Levels of phosphorylated and total proteins were determined by immunoblotting or by flow analysis, as indicated. Colony-forming assays were performed as described (Guzman et al., 2007).
Chemical precipitation/PU beads pull-down assays were performed as previously described (Moulick et al., 2011).
Additional details are provided in Supplemental Information.
SUPPLEMENTAL METHODS
Reagents
PU-H71, PU-H71 coupled to agarose beads (PU-beads) and PU-DZ13 were synthesized and used as previously described(Moulick et al., 2011). The AKT inhibitor VIII, MEK inhibitor PD98059, and the pan-JAK inhibitor I were purchased from Calbiochem. The Hsp90 inhibitors 17-AAG and BIIB021 were purchased from Selleck Chemicals. These compounds were used at concentrations as indicated by the vendor to result in inhibition of their target pathways. All compounds, except for in vivo studies, were used as DMSO stocks.
Cell lines and primary cell culture
Primary cryopreserved AML samples were thawed and cultured for 1 hour at 37°C followed by treatment as described previously (Hassane et al., 2010). MV4-11, THP-1, Kasumi-1, KG-1, HL-60, U937, TUR and Kasumi-4 leukemia cell lines were purchased from the American Type Culture Collection (ATCC), SKNO-1, M0-91, HEL and OCI-AML3 cell lines were obtained from DSMZ and were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM; Life technologies) supplemented with 10–20% fetal bovine serum (FBS) according to culture conditions indicated by ATCC and 1% penicillin/streptomycin (Pen/Strep; Life Technologies).
For the generation of FL5.mAKT the myristylated AKT was cloned in the doxycycline inducible vector pRevTRE (Clontech) (this clone is designated mAKT). As a control, cell were transfected with vector alone (this clone is designated Vector). To activate AKT, FL5.12-derived cell lines were pre-treated with 1 g/ml doxycycline for 18h prior to treatment with inhibitors. These lines were cultured as described previously (Karnauskas et al., 2003) using RPMI-1640 media supplemented with 20mM HEPES, 55 μM beta-mercaptoethanol (β-ME), 10% FBS media containing 0 or 1 ng/mL of murine IL-3 (Fitzgerald Industries International).
Animal Studies
Experiments were carried out under an Institutional Animal Care and Use Committee-approved protocol, and institutional guidelines for the proper and humane use of animals in research were followed.
Xenografted cell lines
Four- to 6-week-old nu/nu athymic female mice were obtained from Taconic Farms. HEL and M0-91 (1×107 cells) were subcutaneously implanted in the right flank of mice using a 20-gauge needle and allowed to reach 6–8 mm in diameter before treatment. Mice bearing M0-91 and HEL tumors were administered intraperitoneally 75 mg/kg of PU-H71 hydrochloride formulated in sterile PBS. Animals were sacrificed by CO2 euthanasia at 12, 24, 48, 72, and 96h post administration of PU-H71. Tumors were homogenized and proteins analyzed by western blot as previously described(Caldas-Lopes et al., 2009).
Evaluation of engraftment in NOD/SCID xenotransplants
The evaluation of human engraftment was performed as previously described (Hassane et al., 2010). Briefly, Animals were sacrificed and the bone marrow (BM) cells were harvested. The BM cells were stained with PE-Cy5 conjugated mouse CD45 (mCD45), APC-H7 conjugated human CD45 (hCD45), PE conjugated human CD33 (hCD33), Alexa Fluor 700 conjugated human CD19 (hCD19), APC conjugated human CD34 (hCD34) and PE-Cy7 conjugated human CD38 (hCD38) antibodies at room temperature for 15 min. Cells were then washed, resuspended in FACS buffer containing 1 μg/ml DAPI and analyzed by for the presence of viable human leukemic cells (DAPI−/mCD45−/hCD45+) by flow cytometry.
Mouse Colony Forming Assays
C57BL/6 mice were treated with 75 mg/kg PU-H71 by i.p. twice a week for 3 weeks. Mouse bone marrow (BM) cells were collected, and 2×104 BM cells were plated in Methocult M3434 medium (Stemcell Technologies). Two weeks later, the images of the colonies were captured by STEMvision (Stemcell Technologies). The colony forming units (CFU) derived from PU-H71 treated mouse BM cells were compared to those from PBS treated control mice.
Cell viability assays
Annexin V/7-AAD staining. AML cell lines were stained with annexin V–FITC (BD Biosciences) and 7-aminoactinomycin (7-AAD, Life technologies). For primary AML samples, cells were stained with CD34, CD38, and CD45 (BD Biosciences) prior to annexin V/7-AAD staining. At least 2×104 events for cell lines or 2×105 events for primary AML samples were collected per condition on a BD LSR II flow cytometer. Data analysis was performed using FlowJo 9.3 software for Mac OS X (TreeStar). Cells that were negative for annexin V and 7-AAD were scored as viable, and the viability was represented as the percent relative to untreated controls. Acridine orange/ethidium bromide staining. Cells were co-stained with a mixture of acridine orange (100 mg/ml) and ethidium bromide (100 mg/ml) added in a 1:1 ratio, followed by cell visualization and counting using a fluorescent microscope (Zeiss Axiovert). Growth Inhibition. Growth inhibition studies were performed using the alamar blue assay as previously described (Caldas-Lopes et al., 2009; Rodina et al., 2007). In summary, experimental cultures were plated at 20,000 cells/well in microtiter plates (Corning # 3603). One column of wells was left without cells to serve as the blank control. Cells were allowed to grow overnight. The following day, growth medium having either drug or DMSO at four times the desired concentration was added to another microtiter plate (Nunc 167008) in triplicate and was serially diluted at a 1:1 ratio in the plate. The diluted drug or DMSO was then added to the plated cells in a 1:1 ratio. After 72 h, the cell growth in treated versus control wells was estimated by adding alamar blue (10% v/v of 440 μM solution of resazurin (Sigma #R7017)) and measuring fluorescence intensity after six hours. The IC50 was calculated as the drug concentration that inhibits cell growth by 50% compared with control growth. Y-axis values below 0% represent cell death of the starting population. All experimental data were analyzed using SOFTmax Pro 4.3.1 and plotted using Prism 4.0 (Graphpad Software Inc., San Diego, CA).
Caspase 3,7 Activation Assay
Caspase 3,7 activation studies were performed as previously described(Rodina et al., 2007). Following a 24h or 48h exposure of cells to Hsp90 inhibitors, 100 μL buffer containing 10 mM HEPES, pH 7.5, 2 mM EDTA, 0.1% CHAPS, 0.1 mg/mL PMSF, Complete Protease Inhibitor mix (#1697498; Roche), and the caspase substrate Z-DEVD-R110 (#R22120; Molecular Probes) at 25 μM was added to each well. Plates were placed on an orbital shaker to promote cell lysis and reaction. The fluorescence signal of each well was measured in an Analyst GT (Molecular Devices) microplate reader (excitation 485 nm; emission at 530 nm). The percentage increase in caspase-3,7 activity was calculated by comparison of the fluorescence reading obtained from treated versus control cells. All experimental data were analyzed using SOFTmax Pro and plotted using Prism (Graphpad Software Inc., San Diego, CA).
Immunoblots
Cells were lysed in protein lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 15 mM MgCl2, 5 mM EDTA, 5 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, and 1% protease inhibitor cocktail (Calbiochem)) on ice for 30 minutes. Twenty micrograms of protein lysates were resolved on a 10% Bis-Tris gels (Life technologies) and transferred to an Immobilon-FL polyvinylidene difluoride (FL-PVDF) or PVDF membrane (Millipore). Membranes were probed with the following primary antibodies: Anti-AKT from rabbit (1:500, 9272; Cell Signaling), anti-phospho-AKT (Ser 473) from rabbit (1:500, 9271S; Cell Signaling), anti-RAF-1 from rabbit (1:300, sc-133; Santa Cruz Biotechnology), anti-PARP (p85 fragment) from rabbit (1:250, G7341; Promega), anti-Bcl-xL from rabbit (1:1,000, 2762; Cell Signaling), anti-JAK2 from rabbit (1:500, 3773; Cell Signaling), anti-c-KIT from mouse (1:1,000, 3308; Cell Signaling), anti-AML1 from rabbit (1:500, 4334; Cell Signaling), anti-FLT3 from rabbit (1:1,000, 3462; Cell Signaling), anti-TRKC from rabbit (1:1,000, ab43078; Abcam), and anti-β-Actin from mouse (1:3,000, ab8227–50; Abcam). anti-ERK1/2 (1:2000, 4695), anti-phospho-ERK1/2(Thr202/Tyr204; 1:2000, 4370), anti-p38 (1:2000, 9212), anti-phospho-p38(Thr180/Tyr182; 1:2000, 4511), anti-phospho-NF-κB p65(Ser536; 1:1000, 3033), were obtained from Cell Signaling. Anti-phospho-STAT5(Tyr694; 1:1000, 611964) and anti-STAT5 (1:1000, 610191) antibodies were obtained from BD Biosciences. Anti-Hsp90 (1:3000, SC-13119), anti-NF-κB p65 (1:2000, SC-372) antibodies were obtained from Santa Cruz Biotech. Hsp70 (1:2000, SPA-810) and β-actin (1:50000, A5441) antibodies were purchased from Stressgen and Sigma-Aldrich, respectively. The membranes were then incubated with either a 1:3,000 dilution of a perioxidase-conjugated corresponding secondary or IRDye 680 goat anti-rabbit or IRDye 800CW goat anti-mouse secondary antibodies (Li-COR) at room temperature for 30 min. antibody and proteins were detected via ECL-Enhanced Chemiluminescence Detection System (Amersham Biosciences) or scanned using the Odyssey infrared imaging system (Li-COR).
Phosphoflow analysis
Phosphoflow assay was performed as described with minor modifications (Schulz et al., 2012). Cells were fixed with 4% formaldehyde (EM-grade; Electron Microscopy Sciences) at room temperature for 10 min, pelleted by centrifugation, and permeabilized by resuspending in cold PBS containing 0.1% Triton X-100 and then placed on ice for 20 min. Cells were then washed with FACS buffer (0.5% FBS in PBS) twice, and stained for 1 h with antibodies against p-AKT(pS473)-Alexa Fluor 488, p-ERK1/2(pT202/pY204)-AlexaFluor-488, p-p65(pS529)-AlexaFluor-647, p-STAT5(pY694)-PE, p-p38(pT180/pY182)-Pacific Blue antibodies (BD Biosciences), NF-kBp65-AlexaFluor-647, AKT-AlexaFluor-488 and/or p-S6(S235/236)-AlexaFluor-488 antibodies
Lentiviral transduction
Wild type FLT3 (WT-FLT3) and FLT3-ITD mutant (N51-FLT3) cDNA were PCR amplified from MSCV-WTFLT3-IRES-GFP and MSCV-N51FLT3-IRES-GFP (kindly provided by Dr. Craig Jordan) respectively. pLVX-WTFLT3-mCherry and pLVX-N51FLT3-mCherry plasmids were generated by subcloning WT-FLT3 and N51-FLT3 cDNA to the SpeI/BamHI restriction sites of pLVX-EF1-IRES-mCherry vector (Clontech). Lentiviral particles were prepared, and lentiviral infection in KG-1 cells was performed according the manufacturer’s manual. Cells were sorted for mCherry+ population at 72 h following lentiviral transduction.
Immunoprecipitation
Ten million cells were lysed in Felts buffer (20 mM HEPES, 50 mM KCl, 5 mM MgCl2, 0.01% (w/v) NP-40, freshly prepared 20 mM Na2MoO4 (pH 7.2–7.3), 5 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, and 1% protease inhibitor cocktail (Calbiochem)) at 4 °C for 30 min. Total protein concentration was determined using the BCA kit (Pierce) according to the manufacturer’s instructions. Immunoprecipitation was performed by mixing 500 μg total cell lysates with 3 μg FLT3 antibody or normal rabbit IgG and 50 μl Protein A/G PLUS-agarose beads (Santa Cruz) at 4 °C overnight. The beads were washed five times with Felts lysis buffer and separated by SDS-PAGE followed by immunoblotting.
Chemical precipitation/PU-beads pulldowns
Cell lysates were prepared as described in immunoprecipitation section. PU-beads or control beads containing an Hsp90 inactive chemical (2-methoxyethylamine) conjugated to agarose beads were washed three times in Felts buffer. Unless otherwise indicated, the bead conjugates (80 μl) were then incubated with cell lysates at 4 °C overnight. Following incubation, bead conjugates were washed five times with the lysis buffer and proteins in the pull-down were analyzed by western blotting.
Statistical analyses
Analyses and graphs were derived using GraphPad Prism software to evaluate significance. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Supplementary Material
Highlights.
AML cells with hyperactivated signaling networks become dependent on teHsp90
This AML addiction results in vulnerability to inhibitors of teHsp90 function
Leukemias with hyperactivated networks are likely candidates for teHsp90i therapy
Acknowledgments
We thank Drs. Craig T. Jordan and Alex Kentsis for their input in the manuscript writing. M.L.G. is funded by the NIH through the NIH Director’s New Innovator Award Program, 1 DP2 OD007399-01, Leukemia and Lymphoma Foundation (LLS 6427-13) and the Hirschl/Weill-Caulier Research Award. G.C. and M.L.G. are funded by R01 CA172546 and LLS 6330-11. G.C. and A.M. are funded by R01 CA155226 and P50 CA192937. J.K. is funded by F32 CA192786. A.G., E.C.-L., E.M.G.-D., A.R., G.C., M.L.G., and G.J.R. were funded by grants UL1RR024996 and UL1TR00457 of the Clinical and Translation Science Center at Weill Cornell Medical College. S.D.N. is funded by R01 CA-166835.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.10.073.
AUTHOR CONTRIBUTIONS
H.Z., A.G., and E.C.-L. designed research, performed experiments, developed methods, analyzed data, and wrote the paper; T.T. and S.O.O. provided PU-H71 and its derivatives; E.S., S.B., S.S., and E.M.G.-D. performed experiments; M.W.B. and G.J.R. provided patient specimens; and C.M.R., J.K., and A.R. provided critical reagents. A.M., R.L.L., S.D.N., G.C., and M.L.G. designed research, analyzed data, and wrote the paper.
CONFLICTS OF INTEREST
Memorial Sloan Kettering Cancer Center holds the intellectual rights to PU-H71. Samus Therapeutics, of which G.C. has partial ownership, has licensed PU-H71.
References
- Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3:1021–1030. [PubMed] [Google Scholar]
- Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, Taldone T, Pullen L, Zierer BK, Lee MJ, et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4:1065–1074. doi: 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenkamp KU, Esselink MT, Kruijer W, Vellenga E. Differential effects of interleukin-3 and interleukin-1 on the proliferation and inter-leukin-6 protein secretion of acute myeloid leukemic cells; the involvement of ERK, p38 and STAT5. Eur Cytokine Netw. 1999;10:479–490. [PubMed] [Google Scholar]
- Brady A, Gibson S, Rybicki L, Hsi E, Saunthararajah Y, Sekeres MA, Tiu R, Copelan E, Kalaycio M, Sobecks R, et al. Expression of phosphorylated signal transducer and activator of transcription 5 is associated with an increased risk of death in acute myeloid leukemia. Eur J Haematol. 2012;89:288–293. doi: 10.1111/j.1600-0609.2012.01825.x. [DOI] [PubMed] [Google Scholar]
- Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwäble J, Buerger H, Muller-Tidow C, Choudhary C, McMahon M, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005;65:9643–9650. doi: 10.1158/0008-5472.CAN-05-0422. [DOI] [PubMed] [Google Scholar]
- Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci USA. 2009;106:8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchietti LC, Lopes EC, Yang SN, Hatzi K, Bunting KL, Tsikitas LA, Mallik A, Robles AI, Walling J, Varticovski L, et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat Med. 2009;15:1369–1376. doi: 10.1038/nm.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AM, Li L, Ho Y, Lin A, Li L, Stein A, Forman S, Perrotti D, Jove R, Bhatia R. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123:2826–2837. doi: 10.1182/blood-2013-05-505735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverría PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS ONE. 2011;6:e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felipe Rico J, Hassane DC, Guzman ML. Acute myelogenous leukemia stem cells: from Bench to Bedside. Cancer Lett. 2013;338:4–9. doi: 10.1016/j.canlet.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris G, Sciot R, Wozniak A, Van Looy T, Wellens J, Faa G, Normant E, Debiec-Rychter M, Schöffski P. The novel HSP90 inhibitor, IPI-493, is highly effective in human gastrostrointestinal stromal tumor xenografts carrying heterogeneous KIT mutations. Clin Cancer Res. 2011;17:5604–5614. doi: 10.1158/1078-0432.CCR-11-0562. [DOI] [PubMed] [Google Scholar]
- Guzman ML, Allan JN. Concise review: Leukemia stem cells in personalized medicine. Stem Cells. 2014;32:844–851. doi: 10.1002/stem.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman ML, Li X, Corbett CA, Rossi RM, Bushnell T, Liesveld JL, Hébert J, Young F, Jordan CT. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8) Blood. 2007;110:4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassane DC, Sen S, Minhajuddin M, Rossi RM, Corbett CA, Balys M, Wei L, Crooks PA, Guzman ML, Jordan CT. Chemical genomic screening reveals synergism between parthenolide and inhibitors of the PI-3 kinase and mTOR pathways. Blood. 2010;116:5983–5990. doi: 10.1182/blood-2010-04-278044. [DOI] [PubMed] [Google Scholar]
- Jhaveri K, Ochiana SO, Dunphy MP, Gerecitano JF, Corben AD, Peter RI, Janjigian YY, Gomes-DaGama EM, Koren J, 3rd, Modi S, Chiosis G. Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Expert Opin Investig Drugs. 2014;23:611–628. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Karnauskas R, Niu Q, Talapatra S, Plas DR, Greene ME, Crispino JD, Rudin CM. Bcl-x(L) and Akt cooperate to promote leukemogenesis in vivo. Oncogene. 2003;22:688–698. doi: 10.1038/sj.onc.1206159. [DOI] [PubMed] [Google Scholar]
- Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, Estey EH, Andreeff M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108:2358–2365. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblau SM, Tibes R, Qiu YH, Chen W, Kantarjian HM, Andreeff M, Coombes KR, Mills GB. Functional proteomic profiling of AML predicts response and survival. Blood. 2009;113:154–164. doi: 10.1182/blood-2007-10-119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblau SM, Covey T, Putta S, Cohen A, Woronicz J, Fantl WJ, Gayko U, Cesano A. Signaling changes in the stem cell factor-AKT-S6 pathway in diagnostic AML samples are associated with disease relapse. Blood Cancer J. 2011;1:e3. doi: 10.1038/bcj.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancet JE, Gojo I, Burton M, Quinn M, Tighe SM, Kersey K, Zhong Z, Albitar MX, Bhalla K, Hannah AL, Baer MR. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010;24:699–705. doi: 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- Lin H, Kolosenko I, Björklund AC, Protsyuk D, Österborg A, Grandér D, Tamm KP. An activated JAK/STAT3 pathway and CD45 expression are associated with sensitivity to Hsp90 inhibitors in multiple myeloma. Exp Cell Res. 2013;319:600–611. doi: 10.1016/j.yexcr.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, Caldas-Lopes E, Ross KN, Gönen M, Gozman A, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–3593. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi JQ, Li JM, Shen ZX, Chen SJ, Chen Z. How to manage acute promyelocytic leukemia. Leukemia. 2012;26:1743–1751. doi: 10.1038/leu.2012.57. [DOI] [PubMed] [Google Scholar]
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes Da, Gama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood. 2013;122:2837–2847. doi: 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AE, Carroll M. Exploiting signal transduction pathways in acute myelogenous leukemia. Curr Treat Options Oncol. 2007;8:265–276. doi: 10.1007/s11864-007-0043-z. [DOI] [PubMed] [Google Scholar]
- Reikvam H, Ersvaer E, Bruserud O. Heat shock protein 90 - a potential target in the treatment of human acute myelogenous leukemia. Curr Cancer Drug Targets. 2009;9:761–776. doi: 10.2174/156800909789271486. [DOI] [PubMed] [Google Scholar]
- Röhl A, Rohrberg J, Buchner J. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci. 2013;38:253–262. doi: 10.1016/j.tibs.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, Hartl FU. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol Cell Proteomics. 2012;11:M111014654. doi: 10.1074/mcp.M111.014654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Ochiana SO, Patel PD, Chiosis G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci. 2014;35:592–603. doi: 10.1016/j.tips.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburini J, Elie C, Bardet V, Chapuis N, Park S, Broët P, Cornillet-Le-febvre P, Lioure B, Ugo V, Blanchet O, et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood. 2007;110:1025–1028. doi: 10.1182/blood-2006-12-061283. [DOI] [PubMed] [Google Scholar]
- Testa U, Riccioni R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica. 2007;92:81–94. doi: 10.3324/haematol.10279. [DOI] [PubMed] [Google Scholar]
- Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M, Neubauer A, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–980. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106:4261–4268. doi: 10.1182/blood-2004-11-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Thompson MA, Brandt SJ, Hiebert SW. Histone deacetylase inhibitors induce the degradation of the t(8;21) fusion oncoprotein. Oncogene. 2007;26:91–101. doi: 10.1038/sj.onc.1209760. [DOI] [PubMed] [Google Scholar]
- Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson WA, Kersey JH. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003;9:4483–4493. [PubMed] [Google Scholar]
- Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, Iino T, Rocnik JL, Kikushige Y, Mori Y, Shima T, Iwasaki H, Takenaka K, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood. 2009;114:5034–5043. doi: 10.1182/blood-2008-12-196055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.