

Abstract
Transcription factors are involved to varying extents in the health and survival of neurons in the brain and a better understanding of their roles with respect to the pathogenesis of Alzheimer’s disease (AD) could lead to the development of additional treatment strategies. Sp1 is a transcription factor that responds to inflammatory signals occurring in the AD brain. It is known to regulate genes with demonstrated importance in AD, and we have previously found it upregulated in the AD brain and in brains of transgenic AD model mice. To better understand the role of Sp1 in AD, we tested whether we could affect memory function (measured with a battery of behavioral tests discriminating different aspects of cognitive function) in a transgenic model of AD by pharmaceutical modulation of Sp1. We found that inhibition of Sp1 function in transgenic AD model mice increased memory deficits, while there were no changes in sensorimotor or anxiety tests. Aβ42 and Aβ40 peptide levels were significantly higher in the treated mice, indicating that Sp1 elevation in AD could be a functionally protective response. Circulating levels of CXCL1 (KC) decreased following treatment with mithramycin, while a battery of other cytokines, including IL-1α, IL-6, INF-γ and MCP-1, were unchanged. Gene expression levels for several genes important to neuronal health were determined by qRT-PCR, and none of these appeared to change at the transcriptional level.
Keywords: Alzheimer’s disease, neurodegeneration, transcription factor, Sp1, transgenic mice, cognitive function, neurons, gene expression
Introduction
There is currently no effective treatment for Alzheimer’s disease (AD). Development of the optimal therapeutic approach will require a better understanding of the cellular mechanisms involved in disease pathogenesis. One promising area of research involves the role of transcription factors in AD. Several pro-inflammatory transcription factors, such as c-Jun/AP-1, Nrf2, NF-κB, p53, and Sp1, have been shown to regulate the expression of genes known to be causal in AD, including amyloid precursor protein (APP) and β-secretase (BACE1) [1-4], yet little is understood about their role in the development of AD. We found that Sp1 and p53 expression increased several fold in the human AD brain and Sp1 was upregulated in the cortex and hippocampus of transgenic AD model mice [5]. Sp1 plays a significant role in the transcriptional regulation of APP [1,2] and APP induction by TGF-β [3], tau transcription [6], presenilin-2 upstream promoter transcription [7], and BACE1 regulation [4]. In addition, Sp1 also activates genes promoting neuronal survival, e.g., Bcl-2, Bcl-x, and IAPs like survivin [8].
To test the involvement of Sp1 in AD pathogenesis, we treated AD model mice with mithramycin, an Sp1 inhibitor, beginning 3 months after birth. Behavioral tests were performed at 6 and 13 months of age to measure memory performance. End point biochemical tests were performed to monitor changes in the AD brain. We found that treatment with mithramycin was not beneficial to mice and resulted in elevated soluble Aβ and further impaired memory in AD model mice.
Materials and methods
Mice and treatment
Model AD transgenic mice, B6C3-Tg (APPswe,PSEN1dE9) 85Dbo/J [9], were obtained from Jackson Laboratory (Bar Harbor, ME; strain 4462). Mice received daily i.p. injections of mithramycin (150 µg/kg body weight) or 100 µl of 1% DMSO in 0.9% saline vehicle beginning at 3.5 months of age until 6 months of age, at which age mice underwent behavioral testing for the following 6 weeks (no treatment was given during this behavioral testing period). Treatment was resumed at 12 months of age and continued until 17 months of age. At 13.5 months of age (1.5 months into resumed treatment) 10 days of additional behavioral testing was performed. Treatment was then continued for a final 3 months (until 17 months of age), when the mice were sacrificed for end point analysis. The four groups of this study were: WT controls (n=4), WT+mithramycin (n=4), Tg controls (n=6), and Tg+mithramycin (n=6). The mithramycin dosage was shown to be effective in other murine models of neurodegenerative disease [10]. All animal experiments were performed in accord with the guidelines of our institutions and approved by the Bay Pines VA and the University of South Florida Institutional Animal Care and Use committees.
Behavioral testing
The initial behavioral evaluation at 6 months of age consisted of our standard 6-week battery of sensorimotor, anxiety, and cognitive-based tasks, as previously described [11,12]. Tasks were performed according to the below protocol and in the order indicated. For the second test period at 13.5 months of age, the RAWM task was repeated for two 2-day blocks and the cognitive interference task of working memory was administered over two 2-day blocks (methodology for this later task is described at the end of the test battery).
Open field activity
To measure activity and exploration, crossings of horizontal and vertical lines 20 cm apart in an 81 cm square arena were counted for mice, placed one at a time, for five minutes.
Balance beam
To evaluate motor capacity and balance, mice were placed perpendicularly on the center of an elevated beam (1.1 cm wide) with an escape platform at each end and observed for 1 minute. If the mouse fell (to a padded surface below) that time was recorded, otherwise 60 seconds was scored. For each mouse, three trials were performed and averaged.
String agility
Mice gripping a horizontal tight cotton string for a maximum of 60 seconds (with a padded area below) were scored between 0-5 as a measure of grip ability with their forepaws and their agility.
Elevated plus maze
To evaluate anxiety, mice were placed in the center of an 80 cm high maze formed by opposite open arms crossed by opposite closed arms. Beginning by facing the closed arm, the mice were observed for 5 minutes and the time spent in the open arms was recorded as an inverse measure of anxiety.
Y-maze
Basic memory function was tested over 5 minutes in a black Y-maze scoring spontaneous alternation as a percentage of arm choices that were different than the previous two choices divided by the total arm entries.
Morris water maze
Reference learning, acquisition, and memory retention was determined by placing mice in a 100-cm pool divided, with black lines at the bottom of the pool, into quadrants. The goal was a transparent 9-cm platform placed 1.5 cm under the water surface in the center of quadrant 2. Visual cues around the pool were visible to the mice. Each day, four 1 minute trials were performed placing mice in different quadrants but repeating the pattern through 9 days of acquisition divided into three equal testing blocks. The time seeking the platform was averaged for the four trials each day. The mouse was allowed to remain on the platform for 30 seconds. If the mouse was not successful, it was guided to the platform and scored as 60 seconds for that trial. After acquisition, the memory retention (probe) trial was performed on day 10 with the mouse placed in the quadrant opposite the platform and video analysis was used to determine the time in each quadrant, distance traveled, and velocity.
Circular platform
Spatial and reference learning was measured with a Barnes maze with 16 equal holes near the circumference of a 69 cm platform. An escape box, with bedding, was placed below one of the holes. Bright lights and a fan were placed above the platform to motivate the mice to find the escape box. Mice were acclimated to the platform for 5 minutes on day 1 and then on eight consecutive test days, mice were placed in the center facing away from an escape box location (randomly selected for each mouse) and the time to find the escape box was recorded up to a maximum of 5 minutes. Test data was collected into four blocks combining adjacent days.
Platform recognition
A cognitive-based task involved switching from the Morris maze spatial strategy to a recognition and identification strategy combined with a requirement to ignore spatial cues surrounding the pool. This was accomplished by measuring identification of a randomly placed 9 cm circular platform, with a distinct black design, elevated 0.8 cm above the water surface. The position of the platform was moved for each trial during which the mouse had 1 minute to find the platform and then remain for 30 seconds. Unsuccessful mice were guided to the platform. Four trials per day were performed for three consecutive days and then the escape latencies were averaged for the four trials on the fourth day.
Radial arm water maze (RAWM)
To monitor working/short-term memory, the pool was divided by aluminum inserts (reaching 5 cm above the surface) into six symmetric radial arms. Multiple visual cues surrounded the pool. Mice were tested with four consecutive acquisition trials and a 30-min delayed retention trial (T5) each day to find a platform submerged 1.5 cm near the end of one of the arms. Swimming down the incorrect arm required pulling the mouse back to the start arm and was recorded as an error, as was failure to move from the center in 20 seconds. Mice that were unsuccessful after 60 seconds were guided to the platform where they would remain for 30 seconds. The improvement, in terms of reduction in errors and in latency time to reach the platform, from the first trial (T1) to trials T4 and T5 reflected working memory and the final 2 day block was the most sensitive.
Cognitive interference task
This test was based on a Cognitive Interference task used to discriminate normal aged, MCI, and AD patients from one another [13]. This test was a stringent test for working memory for mice [14,15] and involved two difference Radial Arm Water Maze (RAWM) set-ups in two different rooms, each with different sets of visual cues. Mice were tested for their ability to remember a set of visual cues after they were exposed to a different set of cues (interference), and demonstrate that they could recall the initial set of cues for the Radial Arm Water Maze task. A set of four behavioral measures were comprised of A1-A3 (Composite three-trial recall score from first 3 trials performed in RAWM A), B (proactive interference measure attained from a single trial in RAWM B), A4 (retroactive interference measure attained during a single trial in RAWM A), and A5 (delayed-recall measure attained from a single trial in RAWM A following a 20 minute delay between A4 and A5). As a distracter between trials, animals were placed in a Y-maze and allowed to explore for 60 s between successive trials of the three-trial recall task, as well as during the proactive interference task. As with the standard RAWM task, this interference task involves the platform location being changed daily to a different arm for both of the RAWM set-ups utilized, and different start arms for each day of testing for both RAWM set-ups. For A1 and B trials, the animal was initially allowed 1 min to find the platform on their own before they were guided to the platform. Then the actual trial was performed in each case. As with the standard RAWM task, animals had 60 s to find the escape platform each time, with the number of errors recorded for each trial. Animals were tested for cognitive interference performance on four successive days, with statistical analysis performed for the two resultant 2-day blocks.
Biochemical changes
At 17 months of age, a blood sample was taken and animals were euthanatized; their brains were then immediately dissected into cortical and hippocampal tissues and frozen for later biochemical and molecular biological assays. ELISAs for cytokine determinations were performed with Bio-Plex reagents from Bio-Rad (Hercules, CA), and relative Aβ42 and Aβ40 levels were measured with capture ELISA assay (Invitrogen, Carlsbad, CA), according to manufacturer’s recommendations, with samples adjusted to 5 mg/ml protein. Bio-Plex assays measured interleukins 1α, 6, 10, 12, and 17, interferon γ, Interferon γ inducible protein 10 (IP-10/CXCL10), granulocyte-macrophage colony stimulating factor (GM-CSF/CSF2), granulocyte colony stimulating factor (G-CSF/CSF3), chemokine C-X-C motif ligand 1 (CXCL1/KC), monocyte chemotactic protein (MCP-1/CCL2), and tumor necrosis factor (TNF-α) in the hippocampus, posterior cortex, and plasma.
mRNA levels
Determinations of mRNAs were conducted with RNA extracted with Tri-reagent (Invitrogen) [16]. DNA was removed with DNase, and the RNA was further purified with RNeasy columns (Qiagen, Valencia, CA). mRNA levels were measured by qRT-PCR with QuantiTect SYBR reagents (Qiagen) and an MJ Research/BioRad Opticon fluorescent thermocycler. The following primers were used: IP-10 upstream 5’-GCC GTC ATT TTC TGC CTC AT, downstream 5’-GCT TCC CTA TGG CCC TCA TT; Bcl-2 upstream 5’-CCA GCG TGT GTG TGC AAG TGT AAA downstream 5’-ACA CTC CGG CTT CAC TGA GAA TGT; p53 upstream 5’-GGC AAC TAT GGC TTC CAC C downstream 5’-TGA CCC ACA ACT GCA CAG G; 18s upstream 5’-GCT CGC TCC TCT CCT ACT TG, downstream 5’-CCC TCT CCG GAA TCG AAC. Data was analyzed using the delta delta cT method.
Statistics
Mean values are depicted ± SEMs for behavioral tests and for biochemical assays. Groups were compared with ANOVA tests and a P<0.05 was considered significant.
Results
Sensorimotor and anxiety tasks
At 6 months of age and after 2.5 months of treatment, prior mithramycin administration did not result in any sensorimotor or anxiety changes to either AD transgenic (Tg) mice or normal wild-type (WT) mice (Table 1). There were no differences resulting from paired group-by-group comparisons, nor were there any overall treatment effects in open field activity, balance beam, or plus-maze anxiety level. In balance beam, the difference between control WT mice and Tg+Mithramycin mice approached significance (p=0.14). For String Agility (not shown), initial Kruskal-Wallis ANOVA indicated an H value of 1.61 (p=0.65), with follow-up pair-by-pair U-tests all being insignificant. Thus, any changes in cognitive performance induced by mithramycin cannot be attributed to changes in non-cognitive measures.
Table 1.
Behavioral Effects of Mithramycin in Wild Type (WT) and AD Transgenic (Tg) mice
| Test | Measure | WT Controls | WT+Mithramycin | Tg Controls | Tg+Mithramycin |
|---|---|---|---|---|---|
| Open Field | Line crossing/5 min | 62 ± 12 | 49 ± 6 | 56 ± 6 | 49 ± 10 |
| Balance Beam | Latency in seconds | 3.7 ± 0.1 | 15.2 ± 5.5 | 8.2 ± 2.2 | 16.7 ± 8.8 |
| Plus maze | % time in open arms | 4.3 ± 3.8 | 0.0 | 2.9 ± 1.9 | 1.1 ± 1.1 |
| Y-maze | % alternation | 72 ± 5 | 58 ± 7 | 74 ± 5 | 61 ± 5 |
| Morris Acquisition | Final block latency in sec | 11 ± 3 | 8 ± 2 | 11 ± 2 | 11 ± 1 |
| Morris Retention | % time in target quad | 42 ± 4 | 59 ± 8 | 54 ± 9 | 35 ± 9 |
| Circular Platform | Final block latency in sec | 112 ± 84 | 64 ± 40 | 52 ± 14 | 90 ± 44 |
| Platform Recognition | Final Day latency in sec | 10 ± 6 | 5 ± 1 | 13 ± 4 | 11 ± 1 |
Behavioral assessment was performed at 6 months of age, following 2.5 months of treatment. Data are expressed as mean ± SEM. Significant treatment effects for each genotype are in comparison to their respective controls (WT or Tg).
Cognitive-based tasks
In the behavioral battery’s first four cognitive-based tasks (Y-maze, Morris maze, circular platform, and platform recognition) administered at 6-7 months of age and beginning a week after cessation of treatment, control Tg mice were unimpaired in any measure compared to control WT mice (Table 1). Moreover, there was no effect of prior mithramycin treatment in any cognitive measure evaluated in either Tg mice or WT mice. For the Morris maze task (memory retention phase), there were also no group differences in annulus crossings or swim speed. Comparisons of control Tg mice vs. treated Tg mice approached significance in Y-maze (p=0.09) and Morris maze retention (p=0.12), as did comparisons between control WT+Mith vs. Tg+Mith mice in Morris maze retention (p=0.08) and Tg controls vs. WT+Mith in both Y-maze (p=0.08) and Platform recognition (p=0.09). It is important to underscore that this study involved group sizes of 4-6 mice, so relatively large differences were required to get significant differences.
Because it is possible that the aforementioned cognitive-based tasks are not very sensitive and/or do not evaluate short-term (working) memory, we followed these tasks with a complex task of working memory - the radial arm water maze task. As shown in Figure 1 for the last of three 2-day blocks of testing, Tg control mice were also unimpaired in this sensitive task of working memory. However, Tg mice that had been given mithramycin treatment up until 1 month earlier were impaired (vs. control Tg mice) during working memory Trials 4 and 5 (T4 and T5) for both measures of escape latency and error numbers. Prior mithramycin treatment to WT mice had no such deleterious effect on the excellent performance of WT mice in this task. Thus, a prominent memory retention deficit was evident in treated Tg mice in comparison to all three other groups due to the increased memory burden provided by the 30-minute time delay between the final acquisitional trial (T4) and the retention trial (T5).
Figure 1.
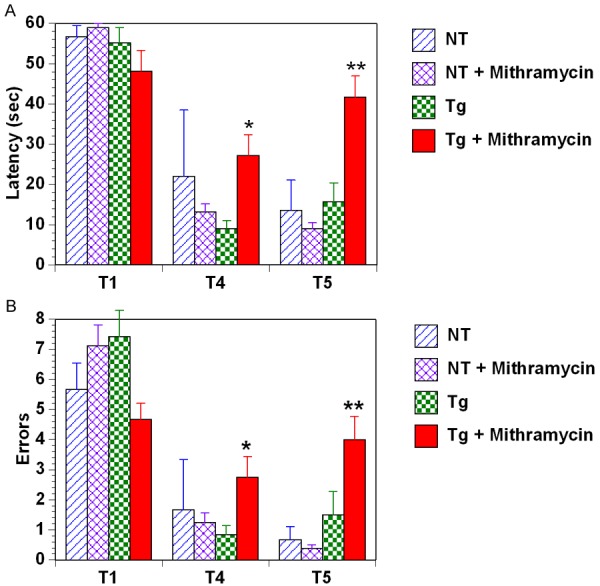
Working Memory performance. The final block of radial arm water maze testing performed at 7 Months of age and approximately one month after cessation of 2.5 months of mithramycin treatment. RAWM testing reveal that Tg control mice were not impaired in this task compared to WT groups. However, prior mithramycin treatment resulted in clear memory impairment in Tg mice for both Trial 4 (T4) and Trial 5 (T5), and for both latency (A) and error (B) measures. *p<0.05 vs. Tg controls, **p<0.02 vs. all other groups.
A second period of cognitive assessment was performed at 13.5 months of age, after reinitiation of treatment had occurred for 1.5 months. For this testing, treatment was continued during the 10-day period of testing in the RAWM task (re-testing) and cognitive interference task. In RAWM re-testing, the now much older Tg mice showed an overall impairment (across both blocks of testing) for working memory trial T5 (data not shown). For T4 overall, no differences in performance between WT controls vs. Tg controls were seen, although Tg mice that were being given mithramycin treatment exhibited poorer working memory than Tg controls (latency to find platform: 52 ± 3 vs. 38 ± 2 sec.; p<0.02). During the first of two 2-day blocks of cognitive interference testing, Tg+mithramycin mice performed much worse than all three of the other groups on three-trial recall for both errors and latency. During the second 2-day block, mithramycin treated mice were nearly impaired (p=0.06) in latency for the delayed-recall measure (A5).
Reduced memory from mithramycin treatment may result from increased Aβ
Soluble Aβ peptides were measured in the Tg+Mithramycin and untreated Tg mice at 17 months of age. Aβ1-40 peptides were found increased about 2 fold in mice treated with mithramycin compared to transgenic controls in the hippocampus and cortex (p=0.012, p=0.013 respectively) (Figure 2). Similarly, increased Aβ1-42 levels were observed in hippocampus (p=0.042). Wild-type mice were not measured. No significant differences were observed for either peptide in plasma.
Figure 2.
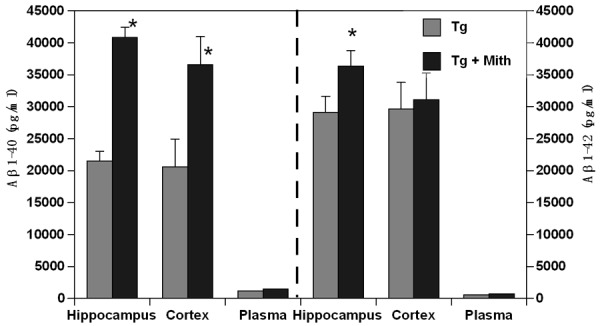
Aβ Levels. Aβ1-40 levels were significantly elevated in both the hippocampus and cortex of mithramycin-treated Tg mice, as were Aβ1-42 levels in hippocampus. Plasma levels of both Aβ isoforms were not affected by mithramycin treatment. Peptide levels were determined with capture ELISA and displayed ± s.d (*p<0.013).
Mithramycin treatment alters cytokines in the wt mouse
To determine whether mithramycin altered inflammation in AD model mouse brains end point cytokine levels were monitored in wt and Tg mice treated with either mithramycin or vehicle. Cytokines were measured in the hippocampus, posterior cortex, and plasma at 17 months of age (Figure 3A-C).
Figure 3.
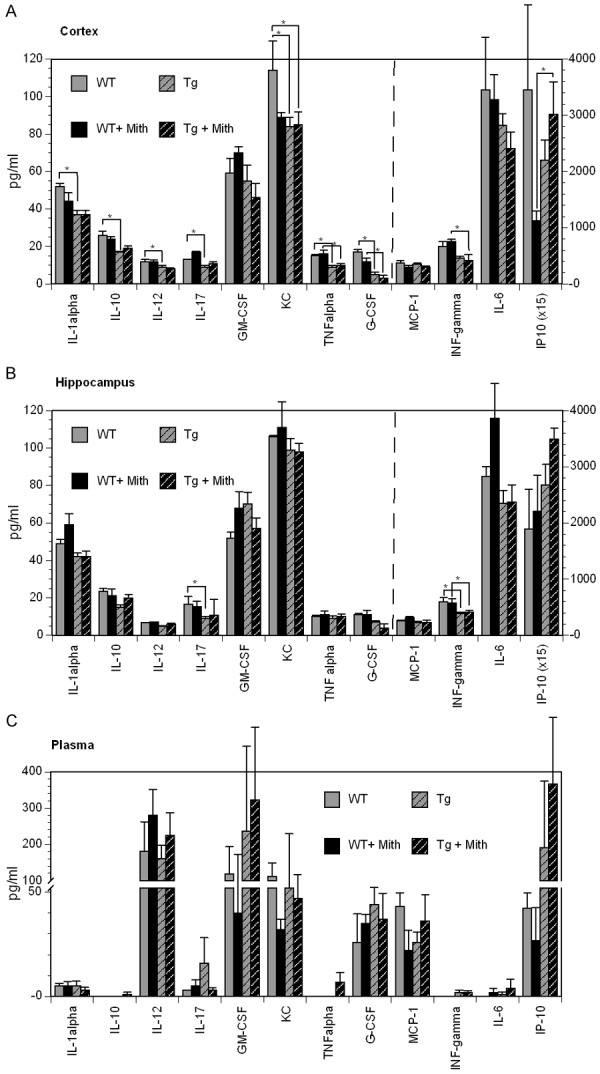
Cytokine levels. ELISA panels were used to determine cytokine levels within the hippocampus, posterior cortex and plasma samples. Genotype and the mithramycin treatment did result in some altered cytokine levels (*p<0.05).
In contrast to what is observed in humans we observed a decrease in IL-1α (p=0.013), IL-10 (p=0.002), IL-12 (p=0.041), IL-17 (p=0.019), CXCL1 (p=0.021), TNFα (p=0.025), and G-CSF (p=0.002) in the cortex of Tg mice compared to wt mice. In the hippocampus we observed a decrease in INFγ (p=0.030) and IL-17 (p=0.044) in transgenic mice compared to WT mice. No differences were found in circulating cytokines between Tg and WT mice.
Following treatment with mithramycin we did not observe any significant differences in cytokines in the posterior cortex, hippocampus, or plasma of Tg mice. We did however notice a treatment effect in the posterior cortex of wt mice with an increase in IL-17 (0.009), and a decrease in CXCL1 (0.0458), and IP-10 (0.031) in the posterior cortex. While there was no treatment effect in Tg animals, wt animals treated with mithramycin decreases CXCL1 in the posterior cortex and in circulating CXCL1 (Figure 3A and 3C).
Mithramycin does not alter IP-10 at the transcriptional level
To determine whether the increased IP-10 protein expression was the consequence of direct SP-1 transcriptional regulation, we measured IP-10 mRNA levels in the hippocampus using SYBR qRT-PCR. Significant differences were not found between either genotype or treatment (Table 2). We also measured expression of some of the genes that can be affected by SP-1 that are important in neuronal health and observed no treatment induced changes in p53 or Bcl-2 mRNA.
Table 2.
Gene expression changes
| Gene | Genotype | Treatment | Relative expression | Sem |
|---|---|---|---|---|
| IP-10 | Control | Control | 1.00 | 0.35 |
| Control | Mithramycin | 0.24 | 0.03 | |
| Transgenic | Control | 2.42 | 1.31 | |
| Transgenic | Mithramycin | 0.88 | 0.10 | |
| p53 | Control | Control | 1.00 | 0.13 |
| Control | Mithramycin | 0.42 | 0.05 | |
| Transgenic | Control | 2.08 | 0.94 | |
| Transgenic | Mithramycin | 0.64 | 0.21 | |
| Bcl-2 | Control | Control | 1.00 | 0.06 |
| Control | Mithramycin | 0.70 | 0.09 | |
| Transgenic | Control | 1.80 | 0.24 | |
| Transgenic | Mithramycin | 1.40 | 0.16 |
mRNA levels in the hippocampus were examined for IP-10, p53, andbcl-2 by qRT-PCR, normalized to 18S rRNA, and are depicted relative to the untreated, non-transgenic mice. Significant differences were not detected.
Discussion
We have previously identified elevated expression of the transcription factor Sp1 in AD brains [5], suggesting that transcription factors may play a key role in disease pathogenesis. In addition to its role upregulating APP, Sp1 can regulate COX-2 [17], which can affect APP processing and amyloid formation [18]. Furthermore, the protective genes, Bcl-2, Bcl-x, and IAPs, can be activated by Sp1 [8]. With AD model transgenic mice carrying the APP and PS1 mutations, we tested the relationship between transcription factor dysregulation and the course of AD neurodegenerative processes.
We found that treatment with mithramycin impaired the memory of model AD mice beginning at 6 months of age with some additional impairment at 12 months in the radial arm water maze. Both soluble Aβ1-40 and Aβ1-42 were increased in Tg AD mice treated with mithramycin. This suggests that an increase in SP-1 activity may be protective with respect to Aβ in the AD brain and may be responsible for the SP-1 inhibitor-induced memory degradation.
The mithramycin treatment had little effect on the cytokines measured. Cytokines that did not change with mithramycin treatment included interleukins IL-1α, IL-6, IL-10, IL-12, IL-17, interferon γ (IFNγ), CXCL1/KC, monocyte chemotactic protein-1 (MCP-1), TNFα, and granulocyte colony-stimulating factor (G-CSF). IP-10 is known to be expressed in the brain [19], human AD CSF [20,21] and has been shown to be upregulated, by immunohistochemistry, in human AD [22] and late stage transgenic AD model brains [23]. Reactive astrocytes are the most commons source of IP-10 in the brain and this chemokine is known to be a key regulator of inflammatory responses. We did observe an increase in IP-10 mRNA in the AD transgenic mice.
Other types of neurodegeneration and other neuronal loss phenomena, such as spinal cord injury, involve altered transcription factor regulation [24,25] and there has been evidence of Sp1 participation in Parkinson’s disease [26] and Alzheimer’s disease [27,28]. Another Sp1 modulator, tofenamic acid, has been reported to inhibit Sp1, improve memory performance, and reduce Aβ1-40 and Aβ1-42 levels after 1 month of oral treatment [29]. In the current study, the longer term of treatment with mithramycin, known to have additional influences beyond Sp1, produced detrimental effects with respect to neurodegeneration.
In summary, we found that several months of mithramycin injections, which among other effects, inhibits Sp1, resulted in greater memory impairment in AD mice. Amyloid β peptide levels were increased by the mithramycin treatment. This indicated that Sp1 and/or other factors, modulated by long term treatment with mithramycin, rather than playing a protective role, can contribute to neurodegeneration in AD and that, in the context of optimizing neuroprotection, future studies should seek to elucidate these underlying processes as potential therapeutic targets.
Acknowledgements
This study was supported by the Alzheimer’s Association, the NIH-designated Florida Alzheimer’s Disease Research Center (FADRC), the Department of Veterans Affairs (Veterans Health Administration, Office of Research and Development, Biomedical Laboratory and Rehabilitation Research and Development), the Florida Department of Health James and Esther King Biomedical Research Program, and the Bay Pines Foundation.
Disclosure of conflict of interest
None.
Disclaimer
The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Lukiw WJ, Rogaev EI, Wong L, Vaula G, McLachlan DR, St George Hyslop P. Protein-DNA interactions in the promoter region of the amyloid precursor protein (APP) gene in human neocortex. Brain Res Mol Brain Res. 1994;22:121–131. doi: 10.1016/0169-328x(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 2.Querfurth HW, Jiang J, Xia W, Selkoe DJ. Enhancer function and novel DNA binding protein activity in the near upstream betaAPP gene promoter. Gene. 1999;232:125–141. doi: 10.1016/s0378-1119(99)00091-8. [DOI] [PubMed] [Google Scholar]
- 3.Docagne F, Gabriel C, Lebeurrier N, Lesne S, Hommet Y, Plawinski L, Mackenzie ET, Vivien D. Sp1 and Smad transcription factors co-operate to mediate TGF-beta-dependent activation of amyloid-beta precursor protein gene transcription. Biochem J. 2004;383:393–399. doi: 10.1042/BJ20040682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Mol Cell Biol. 2004;24:865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Citron BA, Dennis JS, Zeitlin RS, Echeverria V. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J Neurosci Res. 2008;86:2499–2504. doi: 10.1002/jnr.21695. [DOI] [PubMed] [Google Scholar]
- 6.Heicklen-Klein A, Ginzburg I. Tau promoter confers neuronal specificity and binds Sp1 and AP-2. J Neurochem. 2000;75:1408–1418. doi: 10.1046/j.1471-4159.2000.0751408.x. [DOI] [PubMed] [Google Scholar]
- 7.Renbaum P, Beeri R, Gabai E, Amiel M, Gal M, Ehrengruber MU, Levy-Lahad E. Egr-1 upregulates the Alzheimer’s disease presenilin-2 gene in neuronal cells. Gene. 2003;318:113–124. doi: 10.1016/s0378-1119(03)00766-2. [DOI] [PubMed] [Google Scholar]
- 8.Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 10.Ferrante RJ, Ryu H, Kubilus JK, D’Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, Kristal BS, Stavrovskaya IG, Hewett S, Rubinsztein DC, Langley B, Ratan RR. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF. Multimetric behavioral comparison of APPsw and P301L models for Alzheimer’s disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 2004;1012:29–41. doi: 10.1016/j.brainres.2004.02.081. [DOI] [PubMed] [Google Scholar]
- 12.Arendash GW, Jensen MT, Salem N Jr, Hussein N, Cracchiolo J, Dickson A, Leighty R, Potter H. A diet high in omega-3 fatty acids does not improve or protect cognitive performance in Alzheimer’s transgenic mice. Neuroscience. 2007;149:286–302. doi: 10.1016/j.neuroscience.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Loewenstein DA, Acevedo A, Czaja SJ, Duara R. Cognitive rehabilitation of mildly impaired Alzheimer disease patients on cholinesterase inhibitors. Am J Geriatr Psychiatry. 2004;12:395–402. doi: 10.1176/appi.ajgp.12.4.395. [DOI] [PubMed] [Google Scholar]
- 14.Arendash GW, Sanchez-Ramos J, Mori T, Mamcarz M, Lin X, Runfeldt M, Wang L, Zhang G, Sava V, Tan J, Cao C. Electromagnetic field treatment protects against and reverses cognitive impairment in Alzheimer’s disease mice. J Alzheimers Dis. 2010;19:191–210. doi: 10.3233/JAD-2010-1228. [DOI] [PubMed] [Google Scholar]
- 15.Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, Norden M, Padmanabhan J, Neame P, Wefes I, Sanchez-Ramos J, Arendash GW, Potter H. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimers Dis. 2010;21:507–518. doi: 10.3233/JAD-2010-091471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 17.Xu Q, Ji YS, Schmedtje JF Jr. Sp1 increases expression of cyclooxygenase-2 in hypoxic vascular endothelium. Implications for the mechanisms of aortic aneurysm and heart failure. J Biol Chem. 2000;275:24583–24589. doi: 10.1074/jbc.M003894200. [DOI] [PubMed] [Google Scholar]
- 18.Xiang Z, Ho L, Yemul S, Zhao Z, Qing W, Pompl P, Kelley K, Dang A, Teplow D, Pasinetti GM. Cyclooxygenase-2 promotes amyloid plaque deposition in a mouse model of Alzheimer’s disease neuropathology. Gene Expr. 2002;10:271–278. doi: 10.3727/000000002783992352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang D, Han Y, Rani MR, Glabinski A, Trebst C, Sorensen T, Tani M, Wang J, Chien P, O’Bryan S, Bielecki B, Zhou ZL, Majumder S, Ransohoff RM. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol Rev. 2000;177:52–67. doi: 10.1034/j.1600-065x.2000.17709.x. [DOI] [PubMed] [Google Scholar]
- 20.Galimberti D, Schoonenboom N, Scarpini E, Scheltens P Dutch-Italian Alzheimer Research Group. Chemokines in serum and cerebrospinal fluid of Alzheimer’s disease patients. Ann Neurol. 2003;53:547–548. doi: 10.1002/ana.10531. [DOI] [PubMed] [Google Scholar]
- 21.Galimberti D, Schoonenboom N, Scheltens P, Fenoglio C, Venturelli E, Pijnenburg YA, Bresolin N, Scarpini E. Intrathecal chemokine levels in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2006;66:146–147. doi: 10.1212/01.wnl.0000191324.08289.9d. [DOI] [PubMed] [Google Scholar]
- 22.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108:227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 23.Duan RS, Yang X, Chen ZG, Lu MO, Morris C, Winblad B, Zhu J. Decreased fractalkine and increased IP-10 expression in aged brain of APP(swe) transgenic mice. Neurochem Res. 2008;33:1085–1089. doi: 10.1007/s11064-007-9554-z. [DOI] [PubMed] [Google Scholar]
- 24.Kane MJ, Citron BA. Transcription factors as therapeutic targets in CNS disorders. Recent Pat CNS Drug Discov. 2009;4:190–199. doi: 10.2174/157488909789104820. [DOI] [PubMed] [Google Scholar]
- 25.Zhang F, Jiang L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2015;11:243–256. doi: 10.2147/NDT.S75546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen T, Hou R, Li C, Wu C, Xu S. MPTP/MPP+ suppresses activation of protein C in Parkinson’s disease. J Alzheimers Dis. 2015;43:133–142. doi: 10.3233/JAD-140126. [DOI] [PubMed] [Google Scholar]
- 27.Villa C, Ridolfi E, Fenoglio C, Ghezzi L, Vimercati R, Clerici F, Marcone A, Gallone S, Serpente M, Cantoni C, Bonsi R, Cioffi S, Cappa S, Franceschi M, Rainero I, Mariani C, Scarpini E, Galimberti D. Expression of the transcription factor Sp1 and its regulatory hsa-miR-29b in peripheral blood mononuclear cells from patients with Alzheimer’s disease. J Alzheimers Dis. 2013;35:487–494. doi: 10.3233/JAD-122263. [DOI] [PubMed] [Google Scholar]
- 28.Ramanan VK, Saykin AJ. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am J Neurodegener Dis. 2013;2:145–175. [PMC free article] [PubMed] [Google Scholar]
- 29.Subaiea GM, Adwan LI, Ahmed AH, Stevens KE, Zawia NH. Short-term treatment with tolfenamic acid improves cognitive functions in Alzheimer’s disease mice. Neurobiol Aging. 2013;34:2421–2430. doi: 10.1016/j.neurobiolaging.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]