

Abstract
Tumor cells can be contained, but not eliminated, by traditional cancer therapies. A cell minor subpopulation is able to evade attack from therapies and may have cancer stem cell (CSC) characteristics, including self-renewal, multiple differentiation and tumor initiation (tumor initiating cells, or TICs). Thus, CSCs/TICs, aided by the microenvironment, produce more differentiated, metastatic cancer cells which the immune system detects and interacts with. There are three phases to this process: elimination, equilibrium and escape. In the elimination phase the immune system recognizes and destroys most of the tumor cells. Then the latency phase begins, consisting of equilibrium between immunological elimination and tumor cell growth. Finally, a minor attack-resistant subpopulation escapes and forms a clinically detectable tumor mass. Herein we review current knowledge of immunological characterization of CSCs/TICs. Due to the correlation between CTCs/TICs and drug resistance and metastasis, we also comment on the crucial role of key molecules involved in controlling CSCs/TICs properties; such molecules are essential to detect and destroy CSCs/TICs. Monoclonal antibodies, antibody constructs and vaccines have been designed to act against CSCs/TICs, with demonstrated efficacy in human cancer xenografts and some antitumor activity in human clinical studies. Therefore, therapeutic strategies that selectively target CSCs/TICs warrant further investigation. Better understanding of the interaction between CSCs and tumor immunology may help to identify strategies to eradicate the minor subpopulation that escapes conventional therapy attack, thus providing a solution to the problem of drug resistance and metastasis.
Keywords: Cancer stem cell (CSC), immunoresistance, immunotherapy
Introduction
Tumor immunosurveillance
Cancer is caused by an accumulation of genetic alterations in cells which drive activation or overexpression of proteins that promote cell cycle arrest and cell survival, while other proteins that promote cell cycle arrest or cell death are inactivated or downregulated (1). In normal circumstances, most of these lesions are repaired or the mutated cells are eliminated by control mechanisms such as DNA repair enzymes, tumor suppressor genes (2) and the immune system (3). Thus growth of tumor cells is prevented and innate immunity constitutes a first line of defense. Stress induces upregulation of ligands that activate natural killer (NK) cell receptors (4) and other immune stimulatory surface molecules that recognize and eliminate tumor cells. This response can activate an adaptive immune response against antigens specifically expressed by lysed tumor cells and lead to T cell-dependent tumor control. Key molecules for tumor immunosurveillance are interferon-gamma (5), interleukin-12 (IL-12) (6), perforin (7), TRAIL (8), DR4 and DR5 (9) and the recombination activating genes RAG1 (10), and RAG-2 (5). RAG1 and RAG-2 are required for cell development, as is the T cell receptor (11,12). Loss of any of these molecules results in more frequent or faster spontaneous or carcinogen-induced tumorigenesis. The ability of cells to evade destruction by the immune system is thus recognized as a hallmark of cancer (2).
Cell immune surveillance evasion
The immune system is able to maintain tumor growth in a dormant state for decades without completely eradicating all the malignant cells. Certain factors may reduce the ability of the anti-tumor immune system to detect and eliminate malignant cells: pre-established tolerance resulting from non-recognition of tumor antigens (13), generation of less immunogenic tumor cell subclones and immunosupressor molecules such as cytokines or hormones that cause NK and T cell suppression in the tumor microenvironment (14). Lower levels of activatory and/or higher levels of inhibitory NK cell receptor ligands may allow some malignant cells to survive (15). Aggressive tumors are often characterized by low levels of classical human leukocyte antigen (HLA) class I molecules. People with immune system deficiencies such as human immunodeficiency virus (HIV) (16), or who have undergone an organ transplant (17), and the very elderly run an increased risk of developing cancer (18).
Cancer stem cells (CSCs) hypothesis
Tumors are composed of heterogeneous cell subpopulations, defined by two different theories: the stochastic or clonal evolution model, and the hierarchical or CSC model. These theories appear to be mutually exclusive but new data suggest that neither should be discounted (19). In the stochastic model, all tumor cells are biologically equivalent, with a similar capacity for self-renewal and formation of new tumor cells. Cell heterogeneity arises from subclonal differences resulting from genetic and/or epigenetic changes during cancer development. In the hierarchical model, only a cell subpopulation—also known as tumor initiating cells (TICs) (20)—is able to initiate tumor growth. The hierarchical hypothesis defines CSCs as a minority cell tumor subpopulation endowed with properties such as self-renewal, differentiation and multi-potency. CSC-like properties may also be a function of cell type origin, signals from the stromal microenvironment, accumulated somatic mutations and stage of malignant progression (21). These cells display resistance to chemotherapy (22), radiotherapy (23) and immunotherapy (24) and are TICs (4).
Several mechanisms, such as quiescence, are involved in chemoresistance (22). Certain drug-resistant proteins also make stem cells more resistant to toxins that kill their terminally differentiated counterparts (25). For example, resistance is dependent on IL-4 signaling, since up-regulation of IL-4 may result in resistance to apoptosis (26). In addition, CSCs/TICs that have undergone an epithelial-mesenchymal transition (EMT) appear to be more resistant to chemotherapy (27). An increase in aldehyde dehydrogenase (ALDH) activity in these cells seems able to mediate resistance to some chemotherapic agents (28). B-cell lymphoma-2 (BCL-2) protein and its family members (29) also constitute another mechanism of chemoresistance. Therefore, CSCs/TICs possess different mechanisms of resistance to several therapies.
Exact characterization of markers that allow identification of CSCs/TICs in different tumors is still not possible, since no markers have been reported as being unique to CSCs/TICs. Markers such as CD166 have been defined for several tumors. For example, CD166 is a marker of CSCs/TICs in non-small cell lung cancer (NSCLC) (30). The diversity of markers associated with CSCs/TICs may be due to the existence within the tumor tissue of different subpopulations endowed with stem cell features but also with distinct biological properties (31) reflecting differences in patients’ genetic backgrounds and intra-and/or inter-cancer heterogeneity of the primary tumor (32).
CSCs/TICs and the immune system
Immune system and elimination of CSCs/TICs
The process by which the immune system detects and interacts with tumor cells, both before and after clinical detection of the tumor, is known as tumor immunoediting. This process has three phases: elimination, equilibrium and escape (33). In the elimination phase, the innate and the adaptive immune system recognize and destroy most of the tumor cells. However, some malignant cells escape and a latency phase begins, consisting of equilibrium between immunological elimination and growth of tumor cells that may persist for months, years or decades (34). During this period, the cells suffer genetic and epigenetic changes and some generate new immunogenic peptides, enabling the tumor to eliminate these cells. However, some of these changes generate a poorly immunogenic stem cell subpopulation that circumvents immune recognition and also these cells may manipulate the immune system to promote their own growth (35). However, the lack of a favorable microenvironment, and a low rate of cell division, still prevents the formation of a tumor mass (36). Finally, the less immunogenic CSCs/TICs, and the more aggressive clones, are able to form a clinically detectable tumor mass and initiate the escape phase. The reasons for this are as follows: (I) CSCs/TICs can produce immunosuppressive molecules that attenuate the immune system (34); (II) CSCs/TICs recruit cells that suppress the immune system (37); (III) immunology tolerance due to loss of tumor antigen expression, loss of antigen processing and presentation machinery, down-regulation major histocompatibility complex (MHC) class-I (MHC I) expression, and inhibition of co-stimulatory or MHC II molecule expression on antigen presenting cells (APCs) due to genetic alterations (38). Also, the immune system may be weakened by illness, aging or therapeutic immunosuppression. Certain signaling pathways, such as Notch, Wnt and Hedgehog, are able to promote CSC/TIC escape (39).
Immunological characteristics of CSCs/TICs
The capacity of CSCs/TICs to present tumor antigens to T cells for immune recognition or to elicit immune response is determined by expression of antigen presentation molecules, such as MHC-I and MHC-II, as well as co-stimulatory (e.g., CD80, CD86) and co-inhibitory molecules [e.g., cytotoxic T-lymphocyte antigen 4 (CTLA4), B7-H2, B7-H3, programmed death receptor 1 (PD-1)/-1L] (where co-stimulatory molecule expression is negative for these cells and expression of co-inhibitory molecules is up-regulated) (40). CSCs/TICs subsequently show down-regulation of MHC-I and lack MHC-II molecule expression, resulting in downregulation of low molecular weight protein (LMP) antigen processing systems, a transporter associated with antigen processing (TAP), and beta macroglobulin which elicits escape from immune system attack (41).
CSCs/TICs have been shown to secrete cytokines such as transforming growth factor beta (TGF-β), IL-10 and IL-13 in vitro (42). In glioblastoma, CSC/TIC survival has been found to be dependent on secretion of associated angiogenic factors such as vascular endothelial growth factor (VEGF), macrophage-chemoattractant protein-1 (MCP-1), macrophage inhibitory factor (MIF), growth related oncogene alfa (GROα) and ecotaxin (43). Also, TGFβ, IL-6 and IL-8 expression are downregulated in CSCs/TICs (43). In addition, stromal fibroblasts of the tumor microenvironment may be involved in regulating CSC/TIC generation by release of CCL-2 (44). Breast cancer and glioblastoma CSCs/TICs secrete more TGFβ than normal cancer cells (45). Colon CSCs/TICs secrete IL-4, which promotes drug resistance and inhibits anti-tumor immune responses (46). CD200 is also expressed in CSCs/TICs and plays an important role in immune escape (47).
Anti-apoptotic molecules like bcl-2, bcl-xL and survivin protect cells against chemotherapy as well as conferring increased resistance to apoptosis-inducing immune effectors like T or NK cells (48). In a similar manner, the PI3K/Akt pathway mediates chemoresistance and tumor immune escape (49). HER2 interferes with antigen processing and presentation and is key to maintenance of CSCs in luminal breast cancer (50). In summary, CSCs/TICs express soluble and membrane-bound molecules that modulate immune responses and protect cells from immune system attack.
The STAT3 pathway plays an essential role in tumor-mediated immunosuppression by inhibiting macrophage activation (51). STAT3 pathway also reduces the cellular cytotoxicity of NK cells and neutrophiles as well as expression of MHC II, CD80, CD86 and IL-12 in dendritic cells (DCs), rendering them unable to activate T cells and initiate antitumor immunity (52). In addition, STAT3 regulates transcription of immunosuppressive factors such as IL-10, VEGF, PGE2 and TGF-β (53). It has been shown that STAT3 signaling is up-regulated in glioma CSC/TICs, and growth and self-renewal of this subpopulation is dependent on this pathway. CSCs/TICs also secrete some factors that induce STAT3 phosphorylation in immune cells (54).
Tumor-associated antigens (TAAs) expressed by CSCs/TICs
CSCs/TICs express TAAs, which characterize their condition of “stemness” and can be recognized by T cells. TAAs are classed as different subgroups of molecules (41,55) as follows:
Differentiation antigens from which the tumor derives and which could also be expressed by normal cells, i.e., carcino-embryonic antigen (CEA) in colon cancer, mucin-1 (MUC-1) in breast cancer, and gp100 and tyrosinase in melanoma (56);
hTERT and surviving antigens, and other apoptosis-inhibitory proteins expressed by non-stem cancer cells in addition to subsets of normal cells (57);
Cancet-testis (CT) antigens such as Melanoma-associated-antigen-A3 (MAGE-A3) and A4 and NY-ESO1 expressed in normal cells, tumor cells and CSCs/TICs (57);
Mutated antigens deriving from somatic point mutations in tumor cells that can result in entirely new epitopes recognizable by the immune system (58).
In melanoma, the CSC/TIC subpopulation that express ATP-binding cassette sub-family B member 5 (ABCB5) elicits tumor cell dissemination through mediation of chemotherapy resistance, has low levels of lineage-related and CT antigens (59). However, the CD133+ melanoma cell subpopulation has high expression of NY-ESO1 cancer testis antigen as well as susceptibility to specific T cells (60). The TAA DDX3X has been found in CD133+ CSCs/TICs in melanoma and many cancers, conferring immunogenicity on these cells and their ability to induce T-cell dependent protection against murine cancer growth in vivo (61). In contrast, the CD271+ CSC/TIC melanoma subpopulation is deficient in the expression of both lineage-related and CT antigens, making their removal by immune T cells difficult. This has been correlated with progression and metastasis of these cells. As such, melanoma cells offer a good example of multiple CSC/TIC subpopulations with different antigen expression patterns (62).
None of these potential TAAs seem to be a specific marker of CSCs/TICs since they may also be expressed in both tumoral and normal cells. However, T cell responses against TAAs are expressed by CSCs/TICs, such as IL-13Rα2, SOX2 and CD133 in gliomas (63), CEP55 and COA-1 in colorectal cancer (CRC) (64) and epithelial cell adhesion molecule (EpCAM) in retinoblastoma (65). A possible exception is TAAs resulting from somatic point mutations of tumor cells and their CSCs (66).
Immune targeting of lung CSCs/TICs
Introduction
There is a strong relationship between resistance to conventional therapies and intrinsic mechanisms of CSC/TIC resistance to chemo or radiotherapy. Direct targeting of CSCs/TICs or specific signaling pathways responsible for resistance can improve treatment benefit (67). Until recently, in contrast to tumors like melanoma, lung cancer was not thought to be immunogenic. Several immunotherapies, such as IL-2, interferon and bacille Calmette-Guerin, have been tried but have not proved successful to control the immune system in NSCLC patients. Therefore, immunotherapy for NSCLC was considered unsuccessful (68). However, immunotherapeutic approaches involving both stimulation of immune responses and inhibition of immune checkpoints have now been tested and could be combined with chemotherapy or targeted therapies with demonstrated efficiency in lung cancer (Figure 1). A body of evidence now suggests lung cancer is immunogenic. Lung cancer cells release growth factors, interleukins, cytokines and prostaglandins that inhibit T-cell response to the microenvironment, and also has been described that increased tumor-infiltrating lymphocytes (TILs), NK cells, DCs, cytotoxic T lymphocytes (CTLs) and T helper cells are associated with improved survival in NSCLC (69). Also, a high ratio of effector T-cells to regulatory T cells (T-reg) is associated with improved long-term survival (70). In addition, increased immunosuppressive T-regs as a proportion of total TILs are associated with poorer survival in lung cancer (69). MHC class I expression is reduced in NSCLC and these tumors can therefore escape routine antigen processing (71).
Figure 1.
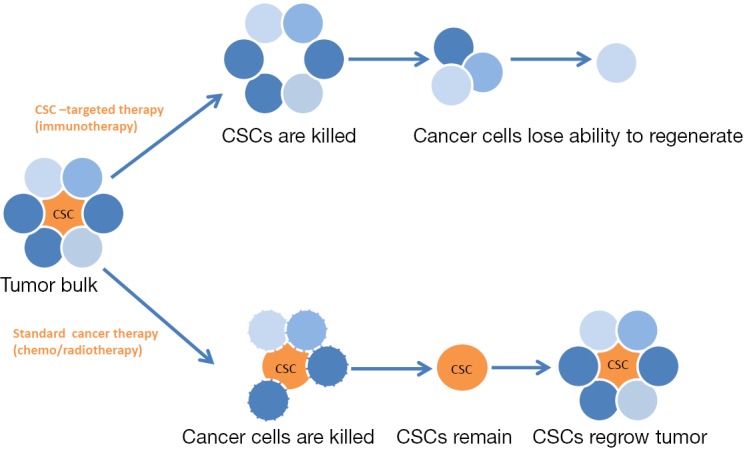
Heterogeneous subpopulation of cancer cells could be treated with standard therapy, as chemo- or radio-therapy. This treatment eliminates cancer cells but not CSCs and tumor grows back. If the cancer cells are treated with CSC-targeted therapy, as monoclonal antibodies or vaccines, the immune system could be stimulated or immune checkpoints inhibited, and then CSCs are killed and tumor loses its ability to generate new cancer cells. CSC, cancer stem cell.
Immunotherapy tends to produce durable responses in small subpopulations of patients. The challenge currently facing investigators is to identify biomarkers predictive of response. Good examples so far are CTLA4 and PD-1 and its ligand (72). Immune targeting of stem cells carries some risks, one obvious one being that pathways are shared with normal adult stem cells, and autoimmunity could carry toxicity to these normal cells. Therefore, it is crucial to identify markers exclusive to CSCs (73). Other obstacles could also limit immune responses, such as a variety of defense mechanisms like soluble mediators TGF-β and COX-2 which make prostaglandin E, IL-10 and arginase. Also defensive molecules such as Fas ligand, B7-H1, nonconventional HLA molecules, lack of MHC class I and recruitment of suppressor type cells (74). Very low levels of expression of these molecules limit detection and elimination of CSCs/TICs.
Cancer cells express many antigens that can be recognized and presented to T cells, leading to T cell activation and elimination of these tumoral cells. This T cell immune response is modulated by negative regulatory molecules such as the immune checkpoint molecules CTLA-4, PD-1, killer cell immunoglobulin-like receptor (KIR) and lymphocyte-activation gene 3 (LAG3); these molecules prevent overstimulation of immune responses. The T cell immune response could be also modulated by co-stimulatory molecules such as glucocorticoid-induced tumor necrosis factor receptor (GITR), OX-40, CD28 and CD137 (75). Deregulation of these molecules in the tumor leads to tolerance of the tumor by the immune system and cancer cell escape from surveillance. A description of the different compounds tested to target these regulatory molecules follows (Table 1).
Table 1. Immune checkpoint blockade.
| Compound | Target |
|---|---|
| Ipilimumab | CTLA4 |
| Tremelimumab | CTLA4 |
| Nivolumab | PD-1 |
| Pembrolizumab | PD-1 |
| BMS-936559 | PD-L1 |
| MPDL3280A | PD-L1 |
| MEDI4736 | PD-L1 |
| Bec2 | GD3 |
| Bevacizumab | VEGF |
| Urelumab | CD137 |
| TRX518 | GITR |
| Anti-OX40 | OX40 |
| Anti-CD40 | CD40 |
| Solitomab | EpCAM |
| Anti-CD133 | CD133 |
| Lirilumab | KIR |
| BMS-9896016 | LAG-3 |
| Racotumomab | N-glycolil-GM3 ganglioside |
CTLA4, cytotoxic T-lymphocyte antigen 4; PD-1, programmed death receptor 1; PD-L1, programmed death ligand 1; VEGF, vascular endothelial growth factor; GITR, glucocorticoid-induced tumor necrosis factor receptor; EpCAM, epithelial cell adhesion molecule; KIR, killer cell immunoglobulin-like receptor; LAG-3, lymphocyte-activation gene 3.
Immune checkpoint blockade
Immune checkpoints are inhibitory pathways crucial for maintaining self-tolerance and to escape to immune system control by the tumor (76). It has observed that inhibitory ligands and receptors are usually overexpressed in cancer cells or their microenvironment (77). Inhibition of these immune checkpoints releases the brakes on the immune system, resulting in antigen-specific T-cell responses. Such inhibition of immune checkpoints relies on the presence of TILs. Stimulation of TILs and/or modulation of the tumor microenvironment could weaken immune responses (78). In lung cancer, targeting the immune checkpoint molecules, CTLA4, PD-1 and its ligand PD-L1 has achieved promising and durable responses but it remains unclear why some patients have only transient or no response (79). One strategy is to target CSCs/TICs with monoclonal antibodies targeting antigens that are differentially overexpressed in these cells. These could be used alone as unmodified antibodies to allow antibody-dependent cytotoxicity (ADCC) to occur, or used with radioisotopes, chemotherapy, cytokines or enzymes to target cancer. A problem of this treatment is that stem cells could escape the cytotoxic effect of specific antibodies by decreasing expression of surface antigen, developing chemotherapy resistance or acquiring multiple mutations. Therefore, antibody treatment is used in combination with conventional cancer therapies (80).
CTLA4
CTLA4 is an immunomodulatory molecule expressed in T cells which plays a role in regulating T-cell activity at early stages of activation; its expression on T cells increases after exposure to an antigen. Binding of the CTLA-4 receptor to CD80/86 expressed on APCs has a co-inhibitory effect on T cells. By competing with the CD28 molecule for the same ligands, albeit with a higher binding affinity than CD28, CTLA-4 inhibits T-cell activation (68). Negative signals are delivered to T cells upon binding to APC CD80/CD86 molecules via CTLA4, T cell function is inhibited and T cells can then be eliminated via apoptosis. Lung cancer could stimulate abnormal expression of CTLA-4 in T cells and these T cells exhibit an anergic phenotype (81). There are currently several clinical trials in lung cancer with human monoclonal antibodies against CTLA4 like ipilimumab or tremelimumab. To date, the response rate is low but these responses are more durable than with cytotoxic therapies (82).
Ipilimumab is a human monoclonal antibody that blocks binding of CTLA-4 to its ligand. As a single agent it has virtually no effect (83) but does seem to provide modest benefit in NSCLC and small cell lung cancer (SCLC) patients in combination with chemotherapy. A phase II study of chemotherapy, paclitaxel and carboplatin with and without ipilimumab in stage IV NSCLC showed a significant improvement in progression-free survival (PFS) when ipilimumab was given after chemotherapy (5.7 vs. 4.6 months) (84). Patients with squamous histology showed better response than non-squamous histology. Now are several clinical trials ongoing. A phase III trial is currently comparing ipilimumab to placebo in SCLC patients receiving platinum and etoposide, and another phase II is comparing ipilimumab to pemetrexed in non-squamous NSCLC. Ipilimumab is also being evaluated with the anti-KIR antibody BMS-986015 that recognizes KIR in NSCLC, castration-resistant prostate cancer (CRPC) and melanoma (85). These antibodies must be used carefully as they can cause autoimmunity and other severe side effects that limit their use (86). Tremelimumab, which also targets CTLA4, has been tested as maintenance therapy compared with observation in patients with stable or responding disease after first line chemotherapy, however, no improvement in PFS was seen (87).
PD-1
PD-1, like CTLA-4, is a member of the CD28 family. PD-1 is expressed in T cells and inhibits their survival, proliferation and immune function through interaction with its ligands PD-L1 and L2. PD-1 is also expressed in B cells and in some myeloid cells (88). Interactions between PD-1 and its ligands attenuate immune responses (89) and serve to protect tumor cells from cytotoxic T cells since T cells become triggered for apoptosis upon signal transduction with PD-1 family proteins (90). Clinical trials with humanized monoclonal antibodies against PD-1 have shown good antitumor activity in subsets of patients with metastasis disease with a good safety profile (80). Several PD-1 antibody trials are ongoing and one study has found a strong correlation between pretreatment tumor expression and responses (72).
Nivolumab, a human monoclonal antibody that binds to PD-1, has been tested in several clinical studies in NSCLC and in two trials specifically for primary squamous cell carcinoma (SQCC), either as a single agent or in combination with chemotherapy or ipilimumab (68). In other studies it was combined with anti-KIR antibody (91). In a phase I clinical study it was administered to 306 patients with different tumor types, including 129 NSCLCs. Overall response rate (ORR) of this study was 17% and the median duration of response was 47 weeks. Another 10% of patients showed stable disease for 6 months with median survival of 9.6 months. Thirty seven patients who received nivolumab at doses of 3 mg/kg showed 24% response rate and 14.9 months median survival (68). Ongoing phase III studies are comparing nivolumab vs. docetaxel in the second-line setting and a phase III first line trial of nivolumab vs. standard chemotherapy in PD-L1 positive metastatic NSCLC is currently recruiting (92). An ongoing phase I clinical trial is combining nivolumab plus ipilimumab with an ORR of 22% at time of interim analysis (93).
Another anti-PD-1 antibody similar to nivolumab, pembrolizumab (also known as MK-3475 or lambrolizumab) is a humanized IgG4 antibody that contains a mutation at C228P designed to prevent Fc-mediated ADCC. In a phase I study, 38 NSCLC patients were treated with pembrolizumab, achieving 24% of lasting responses in previously treated patients. Pembrolizumab is now being examined in the relapsed/refractory setting (NCT01905657) and in combination with first-line chemotherapy (NCT01840579) (85,86).
PD-L1 (B7-H1)
Another therapeutic strategy is inhibition of PD-L1. PD-L1 is overexpressed in around 50% of NSCLC patients and is associated with poor prognosis. Its overexpression induces T-cell anergy and circumvents recognition and processing of tumor antigens by APCs (90). A potential advantage of this approach is lack of interference with T-cell PD-1 receptor interaction with APCs via other ligands, such as B7-H2 (94). A human anti-PD-L1 antibody, BMS-936559, has been tested in a phase I trial and showed promising clinical activity and good safety profile in NSCLC with partial response in 5 of 49 patients (68,95). Other antibodies in clinical development are MPDL3280A (RG7446), a human IgG1-kappa anti PD-L1 monoclonal antibody with a single amino acid substitution in its Fc region that docks with Fc receptors in circulating immune cells, thus preventing ADCC and inadvertent killing of bystander immune cells that also express PD-L1, such as activated T cells. In a phase I trial, 85 NSCLC patients received MPDL3280A as a single agent, with 23% best overall response and 24-week PFS of 46% (96,97). Another IgG1-kappa PD-L1 inhibitor is the antibody MEDI4736, engineered with a triple mutation in the Fc domain that also avoids ADCC as does MPDL3280A. MEDI4736 is currently being tested in a phase I clinical trial. In this study, of 11 NSCLC patients evaluated for efficacy, three achieved partial response, two showed stable disease and one had disease progression (68).
In conclusion, a few patients have good responses to anti-PD-L1 antibodies like nivolumab, MPDL3280A or MEDI4736, despite the absence of PD-L1 expression by immunohistochemistry. However, robust predefined cut-points or independent external validation methodology are not available in the literature. In addition, use of fresh or paraffin-embedded tumor samples could affect results in fresh samples due to the influence of cytokines, such as IFN-α, that upregulate PD-L1 expression (86,98).
GD3
GD3 is a cell surface ganglioside highly expressed in SCLC but not in NSCLC. Bec2/bacille Calmette-Guerin is an anti-idiotypic antibody that binds to the idiotype of the antibody against GD3. Therefore, Bec2/bacille Calmette-Guerin is thought to mimic GD3. In a phase III clinical trial in 515 limited stage patients, use of Bec2/bacille Calmette-Guerin showed no improvement in survival, PFS, or quality of life in the vaccination arm compared with control arm (median survival 16.4 vs. 14.3 months, respectively) (99,100). 1E10 is an anti-idiotypic antibody against Neu-glycosylated sialic acid ganglioside (NeuGc-GM3). It was used in clinical trials in SCLC and NSCLC, and a survival benefit of about 6 months was noted in those patients that developed immunity to NeuGc-GM3 (101).
Vascular endothelial growth factor (VEGF)
Bevacizumab is an anti-VEGF antibody that plays a role in tumor angiogenesis and inhibition of immune response by switching off the action of DCs. A phase III clinical trial in metastatic NSCLC demonstrated improved PFS and overall survival (12.5 vs. 10.2 months) (102).
Other immunotherapy compounds
CD137, GITR and OX40 are positive regulatory molecules of T cell immune responses. Now we describe some compounds that target these molecules.
Urelumab (BMS-663513) is a human IgG4 monoclonal antibody that targets CD137 receptor of the tumor growth factor alpha (TNFα) family and acts as co-stimulatory molecule of T cell activation. Urelumab activates a component of the TNF receptor expressed on the cell membrane of activated white blood cells, subsequently activating CD137-expressing immune cells and stimulating a cytotoxic T cell response against tumor cells. Clinical development in NSCLC has been stopped but is continuing in other cancers (NCT014712109) (85,86).
GITR is a member of the TNF receptor family. GITR co-stimulates CD4+ and CD8+ naïve T cells, leading to T cell proliferation and effector function (85,103). TRX518 is an anti-GITR antibody currently being tested in a phase I trial in melanoma (NCT01239134).
OX-40 (CD134) is also a member of the TNF receptor family. Like CD137 and GITR (101), OX-40 is a co-stimulatory molecule in activated T cells at sites of inflammation and regulates antigen-specific T-cell expansion, survival and cytokine production (IL-2, IL-4, IL-5, IFN-gamma) (104). In a phase I trial, 30 patients with solid tumors were treated with an anti-OX-40 antibody with tumor reduction in 12 patients and enhanced humoral and cellular immunity (75,105).
CD40, a member of the TNF receptor family, is expressed in APCs and its ligand is expressed in T cells. Binding of both enhances APC ability to present antigens and activate T cells. Preclinical studies have demonstrated that anti-CD40 antibodies have the potential to suppress tumor growth and metastasis (106).
Racotumomab (1E10) is an anti-idiotype murine monoclonal antibody against the human monoclonal antibody for N-glycolil-GM3 ganglioside. N-glycolil-GM2 is a glycolipid present within gangliosides, sulfatides, and other antigens expressed in some solid tumors which seems to correlate with survival and suppression of immune activity in NSCLC. A phase III clinical trial (NCT01460472) is currently ongoing with a planned accrual of 1,018 participants (85).
EpCAM is a transmembrane glycoprotein overexpressed in most human carcinomas (107). Solitomab (MT110) is a single-chain bispecific T-cell engager (BiTE) antibody targeting EpCAM (108) which has been tested in dose escalation phase I clinical trials in patients with locally advanced, recurrent or metastatic lung cancer (109). CD133 is reported in CSC/TICs in lung cancer (110). A bispecific antibody against CD3 and CD133 has been designed to eradicate CD133+ cancer cells (111).
KIR and LAG3 are negative regulatory molecules of T cell immune responses, like PD1 and CTLA-4. Several monoclonal antibodies are designed to target these molecules.
KIR is a receptor on NK cells that downregulates NK cytotoxicity activity (86). Lirilumab (IPH2102), an anti-KIR human monoclonal antibody, was used in combination with nivolumab and demonstrated efficacy in preclinical models. A clinical trial in 32 NSCLC patients is ongoing (NCT01714739) as is another combining lirilumab plus ipilimumab in 20 NSCLC patients (NCT01750580) (86).
LAG3 (CD223) is a receptor expressed with PD-1 on tolerant T cells and T-regs which suppresses APC activation by binding with MHC II (112) and becoming an inhibitory molecule of T cell activation in the same manner as KIR. A clinical trial is ongoing with BMS-9896016, an anti-LAG3 monoclonal antibody, alone and in combination with nivolumab (NCT01968109) (86).
Vaccines
TAAs contain more than 70 proteins, including CT antigens such as MAGE-A3, and antigens like MUC-1 that are overexpressed in tumor cells. Using protein or peptide vaccines such as Stimuvax (tecemotide or L-BPLP25) and GSK1572932, TAAs can be targeted for subsequent killing of tumor cells (113).There are many TAAs expressed by tumors not identified, and to recognize them whole tumor vaccines were designed. Vaccines such as Lucanix can be harvested from the patient’s own tumor (autologous) or from established cancer cell lines (allogeneic) and express many TAAs found in patient tumors, theoretically generating an immune response to the tumor (113).
Adaptative T-cell therapy is a passive strategy that involves the transfusion of T-lymphocytes to attack cancer cells in the patient. NY-ESO-1 is one such vaccines (113).
Vaccines currently in clinical trials in lung cancer (Table 2).
Table 2. Vaccines.
| Compound | Target |
|---|---|
| Stimuvax | MUC-1 |
| Anti-NY-ESO1 | NY-ESO 1 |
| GSK1572932 | MAGE-A3 |
| CimaVax | EGF |
| Anti-WT-1 | WT-1 |
| Anti cyclophilin B | Cyclophilin B |
| Lucanix | TGF-β2 |
| IDM-2101 | CEA, p53, HER2, MAGE 2 and 3 |
| Dendritic vaccine | p53 |
| TG4010 | MUC-1 |
| Anti-IDO | IDO |
| GV1001 | Telomerase |
MUC-1, mucin-1; MAGE-A3, melanoma associated antigen A3; EGF, epidermal growth factor; WT-1, Wilms tumor antigen-1; TGF-β2, transforming growth factor beta 2; CEA, carcino embryonic antigen; IDO, indoleamine-2,3-dioxigenase.
MUC1 is a highly glycosylated transmembrane protein overexpressed and abnormally glycosylated in many cancers including NSCLC (114). High levels of MUC1 could enhance immunosuppression and predict poor prognosis in patients with adenocarcinoma (115). Stimuvax is a 25-aminoacid MUC-1 peptide formulated into liposomes targeting MUC1 (116). Several clinical trials have already been performed, including a phase IIb study in stage IIIB and IV NSCLC (117). Median survival time in patients receiving Stimuvax was 17.2 vs. 13.0 months for those receiving best supportive care. Three year survival was 31% with Stimuvax vs. 17% for supportive care (118). Following this study, a phase III clinical trial in NSCLC was carried out (START trial) in 1,513 patients with median overall survival of 25.6 months for patients treated with Stimuvax and 22.3 with placebo. Therefore, the trial did not achieve its primary endpoint of improvement in overall survival. However, analysis of treatment with Stimuvax plus chemotherapy and radiotherapy did show an improvement in median overall survival of 30.8 months compared to 20.6 months for placebo. Inspired by these results, a new phase III clinical study is currently ongoing (START 2 trial) with a primary end-point of overall survival in patients receiving Stimuvax plus chemotherapy and radiotherapy (113). Similar to the START trial, a phase III clinical trial in Asian NSCLC patients is ongoing (INSPIRE) comparing Stimuvax with placebo. In another phase III–IV trial, NSCLC patients were treated with Stimuvax plus bevacizumab following chemotherapy. In a stage III–IV trial, 16 of 65 patients showed a T-cell immune response and had median survival of 30.6 months compared to 13.3 months for best supportive care (85,119).
NY-ESO-1 is a fusion protein vaccine currently being tested in NSCLC (120). In a clinical trial with other tumors a measurable response rate of 66% (four of six patients) was reported in synovial cell sarcomas and 45% in melanoma (five of eleven patients) (120).
MAGE-A3 is an antigen present in about 35% to 55% of NSCLC patients. GSK1572932 is a recombinant DNA vaccine composed of MAGE-A3 and immunoadjuvant AS15. In a phase II clinical trial, 182 stage I and II patients were enrolled with a 27% improvement in time to progression and disease-free survival in patients receiving the vaccine. A phase III clinical trial is ongoing studying the combination of the vaccine with adjuvant chemotherapy in 2,270 NSCLC patients (121).
Mutations in the epidermal growth factor receptor (EGFR) gene are associated with cell proliferation, apoptosis, angiogenesis and metastasis. The epidermal growth factor (EGF) ligand is often overexpressed in lung cancer and its receptors frequently mutated (122). The CimaVax vaccine is a humanized recombinant EGF fusion protein that targets the EGF ligand circulating to prevent EGFR activation. Circulating anti-EGF antibody titers increased as a result of vaccination. These findings were then correlated with decreased levels of serum EGF and patient survival. A phase II trial included 80 patients with NSCLC (stage IIIB or IV) after first-line chemotherapy and demonstrated a decrease in EGF concentration in patient serum. A strong correlation was found between antibody titer and reduction in EGF concentration. Reduction of EGF concentration to below 168 pg/mL is associated with prolongation of overall survival (13 months with 168 pg/mL or less vs. 5.6 months above 168 pg/mL). High initial concentration is a predictive factor of vaccine response and an adverse prognostic factor for non-vaccinated patients. A phase III clinical trial is ongoing (85,123).
A phase I trial in stage III–IV NSCLC is investigating vaccines targeting indoleamine-2,3-dioxigenase (IDO), an immune regulatory protein that suppresses activity of CD8+ cytotoxic T cells. To date, long-lasting clinical benefits have been demonstrated in almost half of the patients (124).
GV1001 is a telomerase-based vaccine used in clinical trials in NSCLC patients previously treated with chemotherapy and radiotherapy (85). In a phase II trial (CTN-2006), 23 stage III patients received radiotherapy and docetaxel followed by GV1001 vaccination. Long-term immunomonitoring showed durable responses in 13 patients. Immune responders achieved a median of 371 days survival, compared with 182 days for non-responders. In another clinical trial (CTN-2000), 26 patients were vaccinated with two telomerase peptides (GV1001 and I540). Thirteen developed a GV1001 response and achieved increased survival compared with non-responders (median survival 19 vs. 3.5 months, respectively) (125).
The Wilms tumor antigen-1 (WT-1) is found in most NSCLC and SCLC patients (126) and a clinical trial tested a 9-mer of WT-1 in several tumor types. Three of 10 lung cancer patients showed an immunological response and one patient continues to survive following repeated vaccinations over more than 2 years (127). WT2725 is a peptide vaccine derived from Wilms tumor protein; a clinical trial in SCLC is also ongoing (85).
Cyclophilin B is found in lung cancer patients and can be a target of CTLS (128). A cyclophilin-based vaccine is being tested in a phase I trial, though no significant increases in cellular response have been observed.
TGF-β2 is released by tumor cells in their microenvironment to protect themselves from immune system. Expression of TGF-β2 has been correlated with poor prognosis in NSCLC (129). Lucanix (Belagenpumatucel-L) is a vaccine consisting of allogeneic NSCLC cell lines transfected with an antisense plasmid to TGF-α2, designed to block TGF-β secretion. A phase II clinical trial in 75 NSCLC patients (stages II–IV) has been completed. The estimated probability of surviving 1 or 2 years was 39% and 20% for patients receiving a low dose of the vaccine and 68% vs. 52% for the higher doses. Estimated median survival time for patients on the low dose was 252 vs. 581 days for the high dose (129). This vaccine in now in a phase III study (STOP) with 532 patients enrolled. This trial did not meet its primary endpoint, with median overall survival of 20.3 months in vaccine-treated patients treated vs. 17.8 months in the group control, but a marked improvement in survival has been detected in specific subgroups of patients (85,113).
The IDM-2101 peptide vaccine is based upon ten different HLA-A2 restricted epitopes against five different antigens (CEA, p53, HER2, MAGE-2 and MAGE-3 antigens along with a pan-DR epitope). A phase II study has been completed and demonstrated immune response (130).
DC vaccines: most smoking-related cancers have p53 mutations and DC vaccines are based on infecting DCs with p53 adenoviruses (131). In in vitro experiments, when these transfected DCs are activated they can generate CTLs against p53 (132). In SCLC patients, a significant immune response is induced and patients are sensitized to chemotherapy (133). Cyclophosphamide followed by vaccinations with tumor-antigen-loaded, DC-derived exosomes inhibits Treg functions, restoring T and NK cell effector functions and activating cell immunity. This is currently being studied in phase I trials (85).
Conclusions
The study of two different scientific fields such as stem cell research and cancer immunology and the links between the two could be crucial to develop new therapeutic approaches to prevent metastasis and development of therapy resistance. CSCs/TICs are characterized by low immunogenicity and immunosuppressive activity. They defend themselves from the immune system and adapt to modifications in the tumor microenvironment caused by chemotherapy or radiotherapy. After chemotherapy and radiotherapy, some resistant cells remain that could be detected and partially killed by the immune system. Equilibrium subsequently occurs between immunological elimination and growth of cancer cells and during this period cells may suffer some changes, giving rise to a poorly immunogenic stem cell subpopulation that is not recognized by the immune system. The molecular identification of immunomodulating agents that can reverse or inhibit CSC/TIC escape from immunosurveillance should allow design of new immunotherapy protocols targeting CSCs/TICs. Immune checkpoint blockade has shown promising results in clinical trials in lung cancer. Responses tend to be durable, but there are problems with inter-patient heterogeneity of responses and appropriate patient subpopulations need to be identified. The tumor microenvironment could play a major role in modulating immune response. The success of immunotherapeutic approaches will depend on a better understanding of the basic biology of immune responses and, in particular, the role that tumor microenvironment plays in shaping immune responses. Vaccines targeting stem cells genes, however, are not without potential risks and adverse effects. The most obvious risks relate to pathways shared with normal stem cells. Research into combination of CSC/TIC-targeting antibodies and/or vaccines with conventional cancer therapies at the optimum moment during the course of the disease, and the identification of suitable biomarkers could improve cancer treatment, is therefore crucial.
Acknowledgements
We are grateful for the diligent revision of our manuscript by Kate Williams and Sònia Guil-Domènech.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Lodish H, Berk A, Kaiser CA, et al. Molecular Cell Biology, 5th edition. New York: W. H. Freeman, 2000. [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70. [DOI] [PubMed] [Google Scholar]
- 3.Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res 1970;13:1-27. [DOI] [PubMed] [Google Scholar]
- 4.Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol 2009;9:568-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001;410:1107-11. [DOI] [PubMed] [Google Scholar]
- 6.Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007;450:903-7. [DOI] [PubMed] [Google Scholar]
- 7.van den Broek ME, Kägi D, Ossendorp F, et al. Decreased tumor surveillance in perforin-deficient mice. J Exp Med 1996;184:1781-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda K, Smyth MJ, Cretney E, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell Immunol 2001;214:194-200. [DOI] [PubMed] [Google Scholar]
- 9.Finnberg N, Klein-Szanto AJ, El-Deiry WS. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest 2008;118:111-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nepal RM, Zaheen A, Basit W, et al. AID and RAG1 do not contribute to lymphomagenesis in Emu c-myc transgenic mice. Oncogene 2008;27:4752-6. [DOI] [PubMed] [Google Scholar]
- 11.Girardi M, Oppenheim DE, Steele CR, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001;294:605-9. [DOI] [PubMed] [Google Scholar]
- 12.Guerra N, Tan YX, Joncker NT, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008;28:571-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willimsky G, Czéh M, Loddenkemper C, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med 2008;205:1687-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campoli M, Ferrone S. Tumor escape mechanisms: potential role of soluble HLA antigens and NK cells activating ligands. Tissue Antigens 2008;72:321-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iguchi-Manaka A, Kai H, Yamashita Y, et al. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med 2008;205:2959-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dal Maso L, Serraino D, Franceschi S. Epidemiology of AIDS-related tumours in developed and developing countries. Eur J Cancer 2001;37:1188-201. [DOI] [PubMed] [Google Scholar]
- 17.Ulrich C, Schmook T, Sachse MM, et al. Comparative epidemiology and pathogenic factors for nonmelanoma skin cancer in organ transplant patients. Dermatol Surg 2004;30:622-7. [DOI] [PubMed] [Google Scholar]
- 18.Bruttel VS, Wischhusen J. Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front Immunol 2014;5:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011;469:356-61. [DOI] [PubMed] [Google Scholar]
- 20.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012;22:457-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009;15:1010-2. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res 2011;17:4936-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao S, Wu Q, Li Z, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res 2008;68:6043-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irvin DK, Jouanneau E, Duvall G, et al. T cells enhance stem-like properties and conditional malignancy in gliomas. PLoS One 2010;5:e10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochedlinger K, Yamada Y, Beard C, et al. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005;121:465-77. [DOI] [PubMed] [Google Scholar]
- 26.Francipane MG, Alea MP, Lombardo Y, et al. Crucial role of interleukin-4 in the survival of colon cancer stem cells. Cancer Res 2008;68:4022-5. [DOI] [PubMed] [Google Scholar]
- 27.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 2011;18:1414-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maccalli C, Volontè A, Cimminiello C, et al. Immunology of cancer stem cells in solid tumours. A review. Eur J Cancer 2014;50:649-55. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 2012;72:576-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeda N, Jain R, LeBoeuf MR, et al. Interconversion between intestinal stem cell populations in distinct niches. Science 2011;334:1420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329-60. [DOI] [PubMed] [Google Scholar]
- 34.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007;7:834-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565-70. [DOI] [PubMed] [Google Scholar]
- 36.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005;438:820-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sceneay J, Smyth MJ, Möller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 2013;32:449-64. [DOI] [PubMed] [Google Scholar]
- 39.Takebe N, Harris PJ, Warren RQ, et al. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011;8:97-106. [DOI] [PubMed] [Google Scholar]
- 40.Comber JD, Philip R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther Adv Vaccines 2014;2:77-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Tomaso T, Mazzoleni S, Wang E, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res 2010;16:800-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salem ML, El-Badawy AS, Li Z. Immunobiology and signaling pathways of cancer stem cells: implication for cancer therapy. Cytotechnology 2015;67:749-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsuyada A, Chow A, Wu J, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 2012;72:2768-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007;11:259-73. [DOI] [PubMed] [Google Scholar]
- 46.Todaro M, Alea MP, Di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007;1:389-402. [DOI] [PubMed] [Google Scholar]
- 47.Kawasaki BT, Mistree T, Hurt EM, et al. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun 2007;364:778-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med 2013;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni J, Cozzi P, Hao J, et al. Cancer stem cells in prostate cancer chemoresistance. Curr Cancer Drug Targets 2014;14:225-40. [DOI] [PubMed] [Google Scholar]
- 50.Seliger B, Kiessling R. The two sides of HER2/neu: immune escape versus surveillance. Trends Mol Med 2013;19:677-84. [DOI] [PubMed] [Google Scholar]
- 51.Lang R, Patel D, Morris JJ, et al. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol 2002;169:2253-63. [DOI] [PubMed] [Google Scholar]
- 52.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005;11:1314-21. [DOI] [PubMed] [Google Scholar]
- 53.Steinbrink K, Wölfl M, Jonuleit H, et al. Induction of tolerance by IL-10-treated dendritic cells. J Immunol 1997;159:4772-80. [PubMed] [Google Scholar]
- 54.Wu A, Wei J, Kong LY, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 2010;12:1113-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parmiani G, Russo V, Marrari A, et al. Universal and stemness-related tumor antigens: potential use in cancer immunotherapy. Clin Cancer Res 2007;13:5675-9. [DOI] [PubMed] [Google Scholar]
- 56.Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res 2008;68:2419-26. [DOI] [PubMed] [Google Scholar]
- 57.Hirohashi Y, Torigoe T, Inoda S, et al. Cytotoxic T lymphocytes: Sniping cancer stem cells. Oncoimmunology 2012;1:123-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008;68:889-92. [DOI] [PubMed] [Google Scholar]
- 59.Schatton T, Schütte U, Frank NY, et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res 2010;70:697-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gedye C, Quirk J, Browning J, et al. Cancer/testis antigens can be immunological targets in clonogenic CD133+ melanoma cells. Cancer Immunol Immunother 2009;58:1635-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koshio J, Kagamu H, Nozaki K, et al. DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 3, X-linked is an immunogenic target of cancer stem cells. Cancer Immunol Immunother 2013;62:1619-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boiko AD, Razorenova OV, van de Rijn M, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010;466:133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown CE, Starr R, Aguilar B, et al. Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res 2012;18:2199-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Inoda S, Hirohashi Y, Torigoe T, et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am J Pathol 2011;178:1805-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitra M, Kandalam M, Harilal A, et al. EpCAM is a putative stem marker in retinoblastoma and an effective target for T-cell-mediated immunotherapy. Mol Vis 2012;18:290-308. [PMC free article] [PubMed] [Google Scholar]
- 66.Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012;4:136ra68. [DOI] [PubMed]
- 67.Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther 2011;89:491-502. [DOI] [PubMed] [Google Scholar]
- 68.Yoon SH. Immunotherapy for non-small cell lung cancer. Tuberc Respir Dis (Seoul) 2014;77:111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Al-Shibli KI, Donnem T, Al-Saad S, et al. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res 2008;14:5220-7. [DOI] [PubMed] [Google Scholar]
- 70.Koyama K, Kagamu H, Miura S, et al. Reciprocal CD4+ T-cell balance of effector CD62Llow CD4+ and CD62LhighCD25+ CD4+ regulatory T cells in small cell lung cancer reflects disease stage. Clin Cancer Res 2008;14:6770-9. [DOI] [PubMed] [Google Scholar]
- 71.Derniame S, Vignaud JM, Faure GC, et al. Alteration of the immunological synapse in lung cancer: a microenvironmental approach. Clin Exp Immunol 2008;154:48-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dhodapkar MV. Immunity to stemness genes in human cancer. Curr Opin Immunol 2010;22:245-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gomez GG, Kruse CA. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene Ther Mol Biol 2006;10:133-46. [PMC free article] [PubMed] [Google Scholar]
- 75.Sundar R, Soong R, Cho BC, et al. Immunotherapy in the treatment of non-small cell lung cancer. Lung Cancer 2014;85:101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol 2006;90:297-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ascierto PA, Kalos M, Schaer DA, et al. Biomarkers for immunostimulatory monoclonal antibodies in combination strategies for melanoma and other tumor types. Clin Cancer Res 2013;19:1009-20. [DOI] [PubMed] [Google Scholar]
- 79.Karachaliou N, Cao MG, Teixidó C, et al. Understanding the function and dysfunction of the immune system in lung cancer: the role of immune checkpoints. Cancer Biol Med 2015;12:79-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kurtz JE, Dufour P. Adecatumumab: an anti-EpCAM monoclonal antibody, from the bench to the bedside. Expert Opin Biol Ther 2010;10:951-8. [DOI] [PubMed] [Google Scholar]
- 81.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734-6. [DOI] [PubMed] [Google Scholar]
- 82.Sapoznik S, Hammer O, Ortenberg R, et al. Novel anti-melanoma immunotherapies: disarming tumor escape mechanisms. Clin Dev Immunol 2012;2012:818214. [DOI] [PMC free article] [PubMed]
- 83.Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol Res 2013;1:85-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 2012;30:2046-54. [DOI] [PubMed] [Google Scholar]
- 85.Shtivelman E, Hensing T, Simon GR, et al. Molecular pathways and therapeutic targets in lung cancer. Oncotarget 2014;5:1392-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Creelan BC. Update on immune checkpoint inhibitors in lung cancer. Cancer Control 2014;21:80-9. [DOI] [PubMed] [Google Scholar]
- 87.Zatloukal P, Heo DS, Park K, et al. Randomized phase II clinical trial comparing tremelimumab (CP-675,206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol 2009;27:15s. [Google Scholar]
- 88.Agata Y, Kawasaki A, Nishimura H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996;8:765-72. [DOI] [PubMed] [Google Scholar]
- 89.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793-800. [DOI] [PubMed] [Google Scholar]
- 91.Sanborn RE, Sharfman WH, Segal NH, et al. A phase I dose-escalation and cohort expansion study of lirilumab (anti-KIR; BMS-986015) administered in combination with nivolumab (anti-PD-1; BMS-936558; ONO-4538) in patients (Pts) with advanced refractory solid tumors. J Clin Oncol 2013;31:abstr TPS3110.
- 92.Anagnostou VK, Brahmer JR. Cancer immunotherapy: a future paradigm shift in the treatment of non-small cell lung cancer. Clin Cancer Res 2015;21:976-84. [DOI] [PubMed] [Google Scholar]
- 93.Antonia SJ, Gettinger SN, Chow LQ, et al. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) and ipilimumab in first-line NSCLC: Interim phase I results. J Clin Oncol 2014;32:5s. [Google Scholar]
- 94.Bennett F, Luxenberg D, Ling V, et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J Immunol 2003;170:711-8. [DOI] [PubMed] [Google Scholar]
- 95.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Herbst RS, Gordon MS, Fine GD, et al. A study of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic tumors. J Clin Oncol 2013;31:abstr 3000.
- 97.Gordon MS. MO18.01 - An analysis of the relationship of clinical activity to baseline EGFR status, PD-L1 expression and prior treatment history in patients with non-small cell lung cancer (NSCLC) following PD-L1 blockade with MPDL3280A (anti-PDL1). Available online: http://library.iaslc.org/search-speaker?search_speaker=22102
- 98.Mühlbauer M, Fleck M, Schütz C, et al. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol 2006;45:520-8. [DOI] [PubMed] [Google Scholar]
- 99.Grant SC, Kris MG, Houghton AN, et al. Long survival of patients with small cell lung cancer after adjuvant treatment with the anti-idiotypic antibody BEC2 plus Bacillus Calmette-Guérin. Clin Cancer Res 1999;5:1319-23. [PubMed] [Google Scholar]
- 100.Giaccone G, Debruyne C, Felip E, et al. Phase III study of adjuvant vaccination with Bec2/bacille Calmette-Guerin in responding patients with limited-disease small-cell lung cancer (European Organisation for Research and Treatment of Cancer 08971-08971B; Silva Study). J Clin Oncol 2005;23:6854-64. [DOI] [PubMed] [Google Scholar]
- 101.Hernández AM, Toledo D, Martínez D, et al. Characterization of the antibody response against NeuGcGM3 ganglioside elicited in non-small cell lung cancer patients immunized with an anti-idiotype antibody. J Immunol 2008;181:6625-34. [DOI] [PubMed] [Google Scholar]
- 102.Jain RK, Duda DG, Clark JW, et al. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 2006;3:24-40. [DOI] [PubMed] [Google Scholar]
- 103.Ronchetti S, Nocentini G, Riccardi C, et al. Role of GITR in activation response of T lymphocytes. Blood 2002;100:350-2. [DOI] [PubMed] [Google Scholar]
- 104.Gramaglia I, Weinberg AD, Lemon M, et al. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol 1998;161:6510-7. [PubMed] [Google Scholar]
- 105.Curti BD, Kovacsovics-Bankowski M, Morris N, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res 2013;73:7189-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.von Scheidt B, Leung PS, Yong MC, et al. Combined anti-CD40 and anti-IL-23 monoclonal antibody therapy effectively suppresses tumor growth and metastases. Cancer Res 2014;74:2412-21. [DOI] [PubMed] [Google Scholar]
- 107.van der Gun BT, Melchers LJ, Ruiters MH, et al. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis 2010;31:1913-21. [DOI] [PubMed] [Google Scholar]
- 108.Brischwein K, Schlereth B, Guller B, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol 2006;43:1129-43. [DOI] [PubMed] [Google Scholar]
- 109.Fiedler WM, Wolf M, Kebenko M, et al. A phase I study of EpCAM/CD3-bispecific antibody (MT110) in patients with advanced solid tumors. J Clin Oncol 2012;30:abstr 2504.
- 110.Grosse-Gehling P, Fargeas CA, Dittfeld C, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol 2013;229:355-78. [DOI] [PubMed] [Google Scholar]
- 111.Huang J, Li C, Wang Y, et al. Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133(high) cancer stem cells in vitro and in vivo. Clin Immunol 2013;149:156-68. [DOI] [PubMed] [Google Scholar]
- 112.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A 2010;107:7875-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramlogan-Steel CA, Steel JC, Morris JC. Lung cancer vaccines: current status and future prospects. Transl Lung Cancer Res 2014;3:46-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ho SB, Niehans GA, Lyftogt C, et al. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res 1993;53:641-51. [PubMed] [Google Scholar]
- 115.Agrawal B, Krantz MJ, Reddish MA, et al. Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nat Med 1998;4:43-9. [DOI] [PubMed] [Google Scholar]
- 116.Samuel J, Budzynski WA, Reddish MA, et al. Immunogenicity and antitumor activity of a liposomal MUC1 peptide-based vaccine. Int J Cancer 1998;75:295-302. [DOI] [PubMed] [Google Scholar]
- 117.Butts C, Murray N, Maksymiuk A, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol 2005;23:6674-81. [DOI] [PubMed] [Google Scholar]
- 118.Butts C, Maksymiuk A, Goss G, et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol 2011;137:1337-42. [DOI] [PubMed] [Google Scholar]
- 119.Palmer M, Parker J, Modi S, et al. Phase I study of the BLP25 (MUC1 peptide) liposomal vaccine for active specific immunotherapy in stage IIIB/IV non-small-cell lung cancer. Clin Lung Cancer 2001;3:49-57; discussion 58. [DOI] [PubMed] [Google Scholar]
- 120.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011;29:917-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ulloa-Montoya F, Louahed J, Dizier B, et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 2013;31:2388-95. [DOI] [PubMed] [Google Scholar]
- 122.Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169-81. [DOI] [PubMed] [Google Scholar]
- 123.Reissmann PT, Koga H, Figlin RA, et al. Amplification and overexpression of the cyclin D1 and epidermal growth factor receptor genes in non-small-cell lung cancer. Lung Cancer Study Group. J Cancer Res Clin Oncol 1999;125:61-70. [DOI] [PubMed] [Google Scholar]
- 124.Iversen TZ, Engell-Noerregaard L, Ellebaek E, et al. Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res 2014;20:221-32. [DOI] [PubMed] [Google Scholar]
- 125.Brunsvig PF, Kyte JA, Kersten C, et al. Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res 2011;17:6847-57. [DOI] [PubMed] [Google Scholar]
- 126.Oji Y, Miyoshi S, Maeda H, et al. Overexpression of the Wilms' tumor gene WT1 in de novo lung cancers. Int J Cancer 2002;100:297-303. [DOI] [PubMed] [Google Scholar]
- 127.Oka Y, Tsuboi A, Taguchi T, et al. Induction of WT1 (Wilms' tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci U S A 2004;101:13885-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gomi S, Nakao M, Niiya F, et al. A cyclophilin B gene encodes antigenic epitopes recognized by HLA-A24-restricted and tumor-specific CTLs. J Immunol 1999;163:4994-5004. [PubMed] [Google Scholar]
- 129.Nemunaitis J, Dillman RO, Schwarzenberger PO, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol 2006;24:4721-30. [DOI] [PubMed] [Google Scholar]
- 130.Barve M, Bender J, Senzer N, et al. Induction of immune responses and clinical efficacy in a phase II trial of IDM-2101, a 10-epitope cytotoxic T-lymphocyte vaccine, in metastatic non-small-cell lung cancer. J Clin Oncol 2008;26:4418-25. [DOI] [PubMed] [Google Scholar]
- 131.Antonia SJ, Mirza N, Fricke I, et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res 2006;12:878-87. [DOI] [PubMed] [Google Scholar]
- 132.Ciernik IF, Berzofsky JA, Carbone DP. Human lung cancer cells endogenously expressing mutant p53 process and present the mutant epitope and are lysed by mutant-specific cytotoxic T lymphocytes. Clin Cancer Res 1996;2:877-82. [PubMed] [Google Scholar]
- 133.Chiappori AA, Soliman H, Janssen WE, et al. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin Biol Ther 2010;10:983-91. [DOI] [PMC free article] [PubMed] [Google Scholar]