

Abstract
β-Lactamase inhibitors (BLIs) restore the efficacy of otherwise obsolete β-lactams. However, commercially available BLIs are not effective against metallo-β-lactamases (MBLs), which continue to be disseminated globally. One group of the most clinically important MBLs is the VIM family. The discovery of VIM-24, a natural variant of VIM-2, possessing an R228L substitution and a novel phenotype, compelled us to explore the role of this position and its effects on substrate specificity. We employed mutagenesis, biochemical and biophysical assays, and crystallography. VIM-24 (R228L) confers enhanced resistance to cephems and increases the rate of turnover compared to that of VIM-2 (kcat/KM increased by 6- and 10-fold for ceftazidime and cefepime, respectively). Likely the R → L substitution relieves steric clashes and accommodates the C3N-methyl pyrrolidine group of cephems. Four novel bisthiazolidine (BTZ) inhibitors were next synthesized and tested against these MBLs. These inhibitors inactivated VIM-2 and VIM-24 equally well (Ki* values of 40–640 nM) through a two-step process in which an initial enzyme (E)–inhibitor (I) complex (EI) undergoes a conformational transition to a more stable species, E*I. As both VIM-2 and VIM-24 were inhibited in a similar manner, the crystal structure of a VIM-2–BTZ complex was determined at 1.25 Å and revealed interactions of the inhibitor thiol with the VIM Zn center. Most importantly, BTZs also restored the activity of imipenem against Klebsiella pneumoniae and Pseudomonas aeruginosa in whole cell assays producing VIM-24 and VIM-2, respectively. Our results suggest a role for position 228 in defining the substrate specificity of VIM MBLs and show that BTZ inhibitors are not affected by the R228L substitution.
Graphical abstract

Production of β-lactamases (EC 3.5.2.6) is the most common mechanism of resistance to β-lactam agents in clinically important Gram-negative bacteria.1,2 On the basis of conserved and distinguishing amino acid motifs, this group of enzymes is categorized into four molecular classes. Classes A, C, and D hydrolyze the β-lactam ring by forming an acyl-enzyme via an active site serine, whereas class B [the metallo-β-lactamases (MBLs)] achieve hydrolysis by using one or two divalent cations (Zn2+) to activate a coordinated water molecule to serve as the reactive nucleophile.3 MBLs can be further divided into three subclasses depending on the presence of specific binding motifs around the active site cavity associated with Zn2+ coordination: B1 (zinc binding to H116-H118-H196 and to D120-C221-H263), B2 (N116-H118-H196 and D120-C221-H263), and B3 (H/Q116-H118-H196 and D120-H121-H263).4 The first subclass (B1) is the most frequently reported and includes the clinically important SPM, NDM, SIM, GIM, IMP, and VIM MBLs, among others. Unlike most class A, C, and D enzymes, B1MBLs hydrolyze most β-lactams, including clavulanic acid, penicillanic acid sulfones, and carbapenems.5 Because of this feature, and the fact that the bla genes encoding B1MBLs are rapidly spreading worldwide,6 understanding substrate specificity and developing broad range MBL inhibitors are priorities.
VIM-type MBLs represent some of the most clinically important members of the B1 family. Since these enzymes were first discovered in Italy in 1999 in a carbapenem-resistant Pseudomonas aeruginosa clinical isolate,7 they have spread worldwide to other Gram-negative pathogens. At present, 41 different allotypes belonging to three sublineages have been identified: VIM-1 like, VIM-2 like, and VIM-7.8,9 Among these, the VIM-24 variant was first detected in a Klebsiella pneumoniae clinical isolate that displayed higher resistance toward ceftazidime and cefepime than to imipenem.10 VIM-24 differs from VIM-2 (and all previously reported VIM enzymes) by a G to T substitution at nucleotide 614, changing the amino acid from Arg to Leu [R228L (Figure 1)] at position 228 of the MBL standard numbering scheme.4 In the VIM MBLs, residue 228 is located at the entry channel of the active site and is either R, S, or L in currently known sequences.8,9 In B1MBLs, such as IMP-1 and NDM-1, the adjacent position 224 in so-called active site loop 10 (ASL-10) is a conserved lysine that plays a key role in substrate binding because of its proximity to the active site.11,12 In the VIM enzymes, position 224 has more allelic variants: histidine in VIM-1, -4, -7, -11, and -12; tyrosine in VIM-2; and leucine in VIM-5 and -13.13 Because the side chains of these residues are shorter and less basic than that of the lysine, R228, with its extended side chain, is hypothesized to replace K224 in interacting with the carboxylate moiety (C4 or C3) of β-lactam substrates.13,14 Accordingly, we advance that the change in VIM-24 from a large and positively charged amino acid (R) at position 228 to a shorter hydrophobic residue (L) may impact the catalytic activity, and possibly stability, of the enzyme. Therefore, our first goal was to investigate the structural and functional role of position 228 in binding of substrates by characterizing VIM-24 and a library of engineered variants at VIM-2 position 228.
Figure 1.

Active site superimposition of VIM-2 (PDB entry 2YZ3) and VIM-24 (modeled structure). Key residues in the active site are shown as sticks; position 228 is colored carnation and mustard (Arg in VIM-2 and Leu in VIM-24, respectively). ASL-3 and -10 are colored green. Zn2+ ions are represented as pink spheres.
We also recognized that diversity within and around the active site of MBLs, which is apparent both in the multiple variants of the IMP, NDM, and VIM enzymes and in differences among other members of the B1 subclass, also influences the interactions of MBLs with inhibitors. Several compound classes (e.g., thiols, cysteinyl peptides, biphenyl tetrazoles, 1β-methylcarbapenem derivatives, 2,3-disubstituted succinic acid derivatives, sulfonyl hydrazones, among others15) have shown activity as inhibitors of MBLs. However, with a few recent exceptions,16–18 the inhibitory activity of most of these compounds is limited to specific types of MBLs.19–22 As a consequence, the development of commercially available inhibitors for this class of enzymes is stalled.
The evaluation of novel bisthiazolidine (BTZ) compounds as inhibitors of a thioredoxin glutathione reductase, a flavoprotein, was recently reported.23 The BTZ scaffold was synthesized for use in dynamic combinatorial libraries of disulfides and belongs to a new compound class with a 5,5-fused bicyclic ring bearing carboxylic acid and mercaptomethyl substituents (Figure 2). Given the similarity of the bicyclic BTZ structure to the β-lactam scaffold, and the prediction that the BTZ carboxylate is able to replicate the interactions of the equivalent group at C3/C4 of β-lactams, our second goal in this work was to investigate the ability of a set of four BTZs to inhibit VIM-2 and VIM-24. Here we assessed the ability of BTZ compounds to act against VIM-2 and studied the effect of the R228L substitution upon inhibitor potency against the clinically significant VIM-24 variant.
Figure 2.

Chemical structures of bisthiazolidine (BTZ) inhibitors.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Cloning, and Mutagenesis
P. aeruginosa PA324 was used to amplify blaVIM-2 with and without the leader sequence (starting at codon 60; position S21). Both products were cloned into the pET-24a(+) vector (Novagen, Darmstadt, Germany) using NdeI and BamHI sites, yielding plasmids pET-24a(+)-blaVIM-2 and pET-24a(+)-blaVIM-2-S21. Plasmid pET-24a(+)-blaVIM-2-S21 was then used as a template to obtain plasmid pET-24a(+)-blaVIM-24-S21 using the Quick-Change XL Site-directed Mutagenesis Kit (Stratagene). VIM-2 and VIM-24 were expressed in Escherichia coli BL21(DE3) pLys cells (LifeTechnologies).
A forward primer containing the SacI restriction site (5′-AAA GAGCTC AAG AAG GAG ATA TAC ATA TG-3′) and a reverse primer with the BamHI restriction site (5′-CTC AGT CGT TGA GTA G GGATCC-3′) were used to amplify blaVIM-2, including the pET24a(+) ribosomal binding site, using plasmid pET-24a(+)-blaVIM-2 as a template. The product was then subcloned into phagemid pBCSK (−) (Agilent, Santa Clara, CA) using the restriction sites mentioned above, thus placing the blaVIM-2 open reading frame under control of the lac promoter, to yield phagemid pBCSK (−) blaVIM-2.
E. coli DH10B (Invitrogen, Carlsbad, CA) was used as a host strain for pBCSK (−) blaVIM-2 and to obtain a mutant library at position 228 by site saturation mutagenesis, using the QuickChange XL Site-directed Mutagenesis Kit (Stratagene). Briefly, primers that capture all 19 variants at MBL position 228 were designed, and mutagenic polymerase chain reactions were conducted using pBCSK (−) blaVIM-2 as a template; they were transformed into E. coli DH10B. Single colonies were selected for plasmid purification, and successful mutagenesis was verified by complete DNA sequencing.
Cell-Based Assays
To test the phenotypic effect of amino acid substitution, the minimal inhibitory concentrations (MICs) of ampicillin, cephalothin, ceftriaxone, ceftazidime, cefotaxime, cefepime, imipenem, and aztreonam were determined for each clone in the variant library by the agar dilution method using cation-adjusted Mueller-Hinton agar (MHA), following the recommendations of the Clinical and Laboratory Standards Institute.25 Ampicillin, cefotaxime, and cephalothin were purchased from Sigma (St. Louis, MO), and imipenem was purchased from U.S. Pharmacopeia (Rockville, MD). The effect of Zn2+ availability was also tested by agar dilution using MH supplemented with 250 μM ZnSO4 or 5 μM EDTA.26 Reported values are the modes of at least three biological replicates.
In Vitro Time-Kill Study
VIM-2-producing P. aeruginosa (imipenem MIC = 32 mg/L) and VIM-24-producing K. pneumoniae (imipenem MIC = 2 mg/L) were cultured overnight at 37 °C in Muller Hinton Broth (MHB) supplemented with 50 mg/L ampicillin. The following day, 1.5 μL of the overnight cultures was inoculated in 1 mL of MHB to obtain a bacterial suspension of approximately 106 colony-forming units/mL.
To examine the effects of the novel BTZ compounds on bacterial growth, the bacterial suspensions were grown at 37 °C under different conditions: MHB alone (growth control) or MHB supplemented with 0.4% DMSO (growth control), sublethal concentrations of imipenem (8 mg/L for P. aeruginosa and 0.5 mg/L for K. pneumoniae), each inhibitor at 100 mg/L, or a combination of imipenem and each inhibitor. Samples (10 μL) were removed after being exposed for 100, 300, and 500 min, and serial dilutions were performed on MHB. The number of viable cells was determined by spotting 20 μL of each dilution on MHA. The plates were incubated at 37 °C overnight, and the numbers of colonies were counted. Results shown are the means of three biological replicates.
Statistical analysis was performed using R version 3.1.1. Additional R packages lme4 and multcomp were utilized.27,28 A linear mixed-effects model was estimated to address the repeated measures performed within the sample. At each time point, post hoc pairwise tests were performed comparing imipenem to other conditions. This yielded a total of 12 pairwise tests per experiment. These p values were Bonferroni-adjusted.
Immunoblotting
E. coli DH10B cells carrying pBC SK (−) blaVIM-R228X plasmids were grown in LB broth with 20 mg/L chloramphenicol to midlog phase (OD600 = 0.7–0.8). Immunoblotting was performed as described previously,29 with two changes explained as follows. First, 1 mL of cells was pelleted, the supernatant removed, and the pellet frozen at −20 °C overnight. Next, pellets were resuspended in 50 μL of lysis buffer containing 50 mM Tris-HCl (pH 7.4), 0.04 mg/mL lysozyme, 1 mM MgSO4, and 5 units/mL benzonuclease (Novagen, Darmstadt, Germany). After incubation for 30 min at 25 °C, the solution was spun at 10000g, and 10 μL of the supernatant was loaded into each lane of a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel. Second, a 1:20000 dilution of anti-DNAK rabbit monoclonal antibody was added as a loading control (Enzo Life Sciences, Farmingdale, NY).
Protein Purification
E. coli BL21(DE3) pLys cells carrying pET-24a(+)-blaVIM-2-S21 or blaVIM-24-S21 phagemids were grown in LB containing 50 mg/L kanamycin for 18 h. Subsequently cells expressing the plasmid were grown in super optimal broth (SOB) while being constantly shaken at 200 rpm, until the OD600 reached 0.6, and then 2 mM ZnSO4 and 0.1 mM isopropyl β-D-thiogalactopyranoside (IPTG) were added for induction and the cells incubated for an additional 20 h at 20 °C while being constantly shaken at 200 rpm. Cells were harvested and frozen for 18 h at −20 °C. Pellets were resuspended in 50 mM Tris-HCl (pH 7.4) containing 200 mM NaCl and 2 mM ZnSO4 and lysed with 40 mg/L lysozyme and 1.0 unit/mL benzonase nuclease (Novagen). Cells were further lysed using a model 500 sonic dismembrator [three pulses of 30 s at 50% amplitude (Fisher Scientific, Waltham, MA)] and then centrifuged at 12000 rpm for 10 min to remove the cellular debris.
After an overnight dialysis against 50 mM Tris-HCl (pH 7.4) containing 2 mM ZnSO4, VIM-2 and VIM-24 were purified by anion-exchange chromatography (Q HiTrap Sepharose Fast Flow or Source 15Q, GE Healthcare) followed by gel filtration (Superdex 75 10/300 GL, GE Healthcare). Enzymes were stored at 4 °C in 50 mM Tris-HCl with 2 mM ZnSO4 (pH 7.4). Purity was assessed by SDS–PAGE with final acrylamide concentrations of 12 and 5% (w/v) for the resolving and stacking gels, respectively; Coomasie Blue staining was used for protein band visualization. Once 95% purity was achieved, the Zn2+ content was measured using the colorimetric reagent 4-(2-pyridylazo) resorcinol (PAR) under denaturing conditions.30
For X-ray crystallography, VIM-2 was expressed and purified as previously described31 and concentrated by centrifugal ultrafiltration to a final concentration of 15 mg/mL.
Electrospray Ionization (ESI) Mass Spectrometry (MS)
To confirm the molecular weight of the purified VIM-2 and VIM-24 MBLs, ESI-MS was performed on a Waters SynaptG2-Si quadrupole time-of-flight (Q-TOF) mass spectrometer equipped with a LockSpray dual electrospray ion source, using glu-1-fibrinopeptide B as the lock mass. The Synapt G2-Si instrument was calibrated with sodium iodide using a m/z 50–2000 mass range. For the experiments described herein, samples were desalted and concentrated using a C18 ZipTip (Millipore, Billerica, MA) according to the manufacturer’s protocol. Eluted protein samples were diluted with 50% acetonitrile and 0.2% formic acid and directly infused at a rate of 50 μL/min, and data were collected for 1 min. Lock mass spectra were collected prior to each sample in a similar manner. The tune settings for each data run were as follows: capillary voltage of 3.2 kV, sampling cone of 30, source offset of 30, source temperature of 100 °C, desolvation temperature of 450 °C, cone gas of 50 L/h, desolvation gas of 600 L/h, and nebulizer bar of 6.0. Spectra were analyzed using MassLynx version 4.1. Spectra were modified for lock mass deviations by applying a gain factor and deconvoluted using MaxEnt1.
Circular Dichroism (CD) Spectroscopy and Thermal Denaturation Analyses
CD experiments were performed in a Jasco (Easton, MD) J-815 spectrometer with a Peltier effect temperature controller. Quartz cells with a 0.1 cm path length were used for all experiments. For CD spectra, 10 μM VIM-2 or VIM-24 was incubated alone in 10 mM HEPES (pH 7.5) containing 0.2 M NaCl. CD spectra were obtained at 20 °C, and data points were recorded every 0.1 nm between λ200 and λ260 with a scan rate of 20 nm/min. For thermal denaturation, 10 μM VIM-2 or VIM-24 was monitored for helical content by CD at λ222 between 20 and 85 °C with a heating rate of 2 °C/min. Data were fit to a two-state model as previously described.32
Molecular Modeling
The crystal structure coordinates of VIM-2 (PDB entry 2YZ333) were used to construct a model of the VIM-24 enzyme using Discovery Studio 3.1 (DS 3.1, Accelrys, Inc., San Diego, CA). The crystallographic waters were taken out, and the enzyme structure was immersed in a water box (7 Å from any face of the box) using explicit periodic boundary conditions (PBC). The VIM-24 model was created using the Build module of DS by mutating the arginine at position 228 to leucine.
The molecule was prepared for energy minimization by adding hydrogen and setting the pH to 7.4. The system was minimized in several steps, with steepest descendent and conjugate gradient algorithms to reach the minimum convergence (i.e., 0.01 after 10000 iterations). All energy minimizations were conducted using CHARMm force field parameters. The particle mesh Ewald (PME) method was used to treat long range electrostatic interactions. Bonds that involved hydrogen atoms were constrained with the SHAKE algorithm.
To model the complex with VIM-24, coordinates and restraints for unhydrolyzed cefepime were generated using JLigand34 and docked manually into the VIM-24 structure (generated as described above) using Coot.35 Docking was based upon the assumptions of interactions between the oxygen of the C8 carbonyl with Zn1 and the C3 carboxylate with Zn2, as previously proposed for binding of intact β-lactams to binuclear MBLs.19 Comparisons with the structure of NDM-1 bound to hydrolyzed meropenem (PDB entry 4EYL36) were useful in positioning the molecule relative to the metal ions. The resulting complex structure was minimized using the Yasara server (http://www.yasara.org/minimizationserver.htm37). For comparison with VIM-2, the complex was superimposed onto the crystal structure of the native enzyme (PDB entry 1KO313) using SSMSuperpose.38
Determination of Steady-State Kinetic Parameters
Steady-state reactions were followed using an Agilent model 8453 diode array spectrophotometer as previously described.39 Briefly, each assay was performed in 10 mM HEPES (pH 7.5) supplemented with 200 mM NaCl, 50 μM Zn2SO4, and 50 μg/mL bovine serum albumin (BSA) at 25 °C using nitrocefin (NCF), ampicillin, cephalothin, ceftazidime, cefepime, and imipenem as substrates at an excess molar concentration to establish pseudo-first-order kinetics. Excess metal is required in the buffer to maintain a constant active enzyme concentration and hence to ensure steady-state conditions, as there is metal dissociation during turnover.26 The following extinction coefficients were used: NCF, Δɛ482 = 17400 M−1 cm−1; ampicillin, Δɛ235 = −900 M−1 cm−1; cephalothin, Δɛ262 = −7660 M−1 cm−1; ceftazidime, Δɛ256 = −7600 M−1 cm−1; cefepime, Δɛ260 = −750 M−1 cm−1; and imipenem, Δɛ299 = −9000 M−1 cm−1.39 For velocity determinations for ampicillin, cephalothin, NCF, and imipenem, a 1 cm path length quartz cuvette was employed. For ceftazidime and cefepime, a 0.2 cm path length quartz cuvette was used. A nonlinear least-squares fit of the data (Henri Michaelis–Menten equation) using Enzfitter (Biosoft Corp., Ferguson, MO) was employed to obtain the steady-state kinetic parameters Vmax and KM according to eq 1.
| (1) |
To estimate the kcat values from the Vmax values obtained from the fits, the Zn2+ content of each protein preparation was taken into account to calculate the concentration of the active enzyme, as a small fraction of the MBL is found in an irreversibly inactive apo form, which remains inactive even in the presence of excess metal.40
In the case of cefepime and ceftazidime, where velocities of hydrolysis did not reach saturation under the conditions tested, the kcat/KM ratio was calculated by fitting hydrolysis curves to eq 241
| (2) |
where At is the absorbance at time t and Af and A0 represent the final and initial absorbance, respectively. In this equation, the observed first-order rate constant k = kcat/KM × [E].
Additionally, hydrolysis of cefepime and ceftazidime was assessed using periplasm preparations, as previously described.26 Briefly, periplasmic preparations of E. coli DH10B pBC SK (−) harboring blaVIM-2 or blaVIM-24 were obtained by osmotic shock with chloroform, and MBL production was assessed by immunoblotting. Initial velocities of hydrolysis at high substrate concentrations (800 μM for cefepime and 400 μM for ceftazidime) were measured using equal amounts of both β-lactamases in 15 mM HEPES (pH 7.5) and 200 mM NaCl, supplemented with 50 μM ZnSO4, at 25 °C.
Analysis of BTZ Inhibition Kinetics
Models involving one- or two-step slow binding were considered to analyze BTZ inhibition of NFC hydrolysis by VIM-2 and VIM-245 (Scheme 1).
| Scheme 1 |
where E represents the free enzyme, I is the free inhibitor, EI is the final enzyme–inhibitor complex for a single-step slow binding mechanism, and E*I is the enzyme–inhibitor complex formed after conformational changes in a two-step slow binding mechanism. Therefore, the dissociation constant for the EI complex (Ki), also called the inhibitor constant, is defined as Ki = k−1/k1; the dissociation constant for the E*I complex (Ki′) is defined as Ki′ = k−2/k2, and the overall dissociation constant for the two-step mechanism (Ki*) is defined as Ki* = Ki/(1 + k2/k−2).
The Ki for each inhibitor was determined by direct competition assays under steady-state conditions. The initial velocity was measured in the presence of a constant concentration of enzyme (3 nM for VIM-2 and 4 nM for VIM-24) with increasing concentrations of inhibitor (5–60 μM) against a fixed concentration (5KM) of the indicator substrate, NCF, as previously described.42–44 The reactions were started by addition of VIM-2 or VIM-24 to substrate or to mixtures of substrate and inhibitor. Data were linearized by plotting inverse initial steady-state velocities (1/v0) versus inhibitor concentration (I). Ki (observed) was determined by dividing the value for the y intercept by the slope of the line. Ki determinations were corrected to account for the affinity of NCF for the β-lactamase according to eq 3.45
| (3) |
Measurements of association rate constants for inhibitor binding were taken at 25 °C in 10 mM HEPES (pH 7.5) supplemented with 200 mM NaCl, 50 μM Zn2SO4, and 50 μg/mL BSA, in the presence of several concentrations of each compound. Reactions were then initiated by the addition of β-lactamase (3 nM VIM-2 or 4 nM VIM-24) to a mixture of 50 μM NCF and each BTZ without preincubation. Inhibitor association rate constants were estimated from linear extrapolation of the observed rate constant for inhibition (kobs), which was obtained by fitting progress curves of NFC hydrolysis to eq 4:
| (4) |
where A is the absorbance, v0 and vs are the initial and steady-state velocities of the reaction, respectively, and kobs is the apparent rate constant for formation of the enzyme–inhibitor complex.
As previously described, the linear dependence of kobs versus [I] at limiting I concentrations approximates second-order rate constant k2/Ki.46 The observed second-order rate constant also includes an adjustment for competition between inhibitor and substrate binding (eq 5).
| (5) |
Also, vs values calculated from the progress curves of NCF hydrolysis for each concentration of inhibitor were used to determine Ki*, according to the relationship shown in eq 6.5 The value reported is the mean of three velocities ± the standard deviation (SD).
| (6) |
To further test the time-dependent inhibition mechanism of inhibitor binding, assays using dilutions of preformed complexes were performed. For these experiments, 1 μM enzyme was incubated with varying concentrations of the inhibitor that allowed for complete inactivation of the enzyme during the 5 min incubation at 25 °C. The reaction mixtures were then diluted 2000-fold in 10 mM HEPES (pH 7.5) supplemented with 200 mM NaCl, 50 μM Zn2SO4, and 50 μg/mL BSA, which contained 50 μM NCF. Reactivation of the enzyme based on NCF hydrolysis was monitored over time. A control reaction with the free enzyme was performed in the same fashion.
Crystallization, Data Collection, and Structure Determination
Conditions for cocrystallization were identified by using a mixture of VIM-2, purified as described previously,31 and the BTZ compound L-CS319 (dissolved in dimethyl sulfoxide and added to the protein sample to a final concentration of 10 mM) in sparse-matrix crystallization screening experiments. The final DMSO concentration was maintained at ≤10% (v/v). Diffraction quality crystals were obtained from 0.1 M Bicine (pH 9.0) and 65% 2-methyl-2,4-pentanediol. Crystals were mounted in rayon loops and snap-frozen in liquid nitrogen prior to data collection. Diffraction data were collected on beamline I04-1 of the Diamond Light Source synchrotron (Didcot, U.K.) using a Pilatus detector. Images were integrated, scaled, and merged using XDS,47 POINTLESS, and SCALA48 as implemented in the XIA2 crystallography pipeline.49 The structure was determined by molecular replacement with PHASER50 using the native (unliganded) VIM-2 structure as a search model (PDB entry 1KO313) and subjected to alternate rounds of model building and refinement using Coot35 and Refmac551 as implemented in the CCP4 crystallography suite.52 The quality of the final model was assessed using the MolProbity server.53 Coordinates and structure factors have been deposited in the PDB as entry 4UA4.
RESULTS AND DISCUSSION
Effects of the R228 Substitution on the Phenotype and Expression of VIM-2 Variants Expressed in E. coli
To determine the effect of the R228L substitution and to investigate the role of position 228 in VIM MBLs, we generated all possible variants at this position by site saturation mutagenesis and assessed the impact of these substitutions on phenotype by antibiotic susceptibility testing against a variety of β-lactams (Table 1). Compared to that in VIM-2, the natural R228L substitution in VIM-24 increased resistance to cefepime (≤0.5 mg/L vs 4 mg/L) and ceftazidime (16 mg/L vs 64 mg/L), while the minimal inhibitory concentration (MIC) values for all the remaining antibiotics tested remained relatively unchanged. Results from the R228X library also showed this position to be tolerant of substitution as only two variants, R228K and R228P, displayed significant reductions in MIC, compared to those of VIM-2, for all the antibiotics tested. Remarkably, R228M and R228Q showed increased resistance toward all β-lactams, including imipenem, and thus represented “gain of function” mutations.
Table 1.
Cell-Based Assays Using E. coli DH10B pBCSK(−), Producing VIM-2, VIM-24 (R228L), and Other Variants
| MIC (mg/L)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| E. coli pBC SK(−) | VIM-2 (228R) | VIM-24 (R228L) | R228K | R228P | R228M | R228Q | R228Y | R228Xc | |
| ampicillin | 4 | 512 | 512 | 32 | 32 | 1024 | 1024 | 1024 | 64–512 |
| cephalothin | 8 | 256 | 256 | 16 | 16 | 256 | 1024 | 256 | 64–256 |
| ceftriaxone | 0.5 | 8 | 8 | 1 | 0.5 | 16 | 32 | 16 | 1–16 |
| cefotaxime | 0.06 | 8 | 16 | 1 | 1 | 16 | 32 | 16 | 2–32 |
| aztreonam | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 |
| ceftazidime | 0.06 | 16 | 64 | 1 | 1 | 128 | 128 | 64 | 16–128 |
| ceftazidime + Zna | ≤1 | 32 | 128 | ≤1 | 4 | 256 | 512 | 128 | 32–256 |
| ceftazidime + EDTAb | ≤1 | 16 | 32 | ≤1 | ≤1 | 64 | 128 | 64 | 8–64 |
| cefepime | 0.03 | 0.5 | 4 | 0.5 | 0.25 | 2 | 16 | 8 | 1–4 |
| cefepime + Zna | ≤0.25 | 0.5 | 4 | ≤0.25 | 0.5 | 2 | 32 | 8 | 1–4 |
| cefepime + EDTAb | ≤0.25 | 0.5 | 4 | ≤0.25 | ≤0.25 | 4 | 16 | 8 | 1–2 |
| imipenem | 0.25 | 0.5 | 0.5 | 0.25 | 0.5 | 2 | 4 | 1 | 0.25–1 |
| imipenem + Zna | 0.25 | 1 | 1 | 0.25 | 0.25 | 4 | >4 | 1 | 0.5–2 |
| imipenem + EDTAb | ≤0.125 | 0.5 | 0.5 | 0.25 | ≤0.125 | 1 | 4 | 1 | 0.25–1 |
MHA supplemented with a final concentration of 250 μM Zn2+.
MHA supplemented with 5 μM EDTA.
Values shown in this column are the ranges of the MIC values obtained for the other variants. A complete list of the MICs is provided as Supporting Information.
MBLs are exported to the bacterial periplasm as unfolded polypeptides, and metal incorporation takes place in the periplasmic space where Zn2+ availability is limited.54 Thus, MBLs with reduced capability to bind Zn2+ are deficient in conferring resistance.26 Accordingly, we determined the MIC values of our R228X library in E. coli cells in media containing excess or limiting concentrations of Zn2+ using cefepime, ceftazidime, and imipenem as representative antibiotics. As shown in Table 1, the effect of Zn2+ availability on resistance was especially noticeable for ceftazidime, for which VIM-24, but also 90% of the in vitro generated variants, displayed higher MICs. Increments of up to two doubling dilutions (MIC increases from 128 to 512 mg/L) were detected under conditions that included high Zn2+ availability. Of note, consistent decreases in resistance, especially in restricted Zn2+ media, were observed for only two variants, R228P and R228K. Furthermore, the activity of the R228K variant was not improved in Zn2+-supplemented agar.
An additional factor that can affect antibiotic resistance is the expression level of the individual VIM variants. Therefore, we next evaluated the steady-state expression of each of the variants expressed from pBCSK (−) in E. coli DH10B cells, by immunoblotting. The results on whole cell extracts showed all variants to be produced at either the same level of wild-type VIM-2 or a lower level (Figure 3A). For instance, the R228K and R228Q variants are expressed at a level comparable to that of the wild type, whereas the level of expression of the R228M variant is lower. We assert that phenotypic differences arise from a combination of inherent properties (sequence and/or structure stability) that affects catalytic activity. The deficient resistance conferred by the R228K enzyme may then be explained by structural changes that disrupt the conformation of active site loop 10 (ASL-10) and may also indirectly reduce the affinity for Zn2+. In the case of the “gain of function” variants, R228M and R228Q, despite the absence of a strongly charged side chain at position 228, both the Met and Gln substitutions clearly retain the ability to bind substrate in a productive orientation for hydrolysis. These data imply that, in the absence of a significant stabilizing interaction with the R228 side chain, the C3 or C4 carboxylate of the substrate can make alternative contacts with the enzyme active site that are still compatible with efficient hydrolysis. More detailed studies of the R228Q, R228M, and R228K variants will thus provide insights into the basis of substrate specificity, and the catalytic mechanism, of VIM enzymes.
Figure 3.

Characterization of proteins expressed in E. coli DH10B and E. coli BL21(DE3) pLys cells. Steady-state expression of the R228X library in E. coli DH10B was analyzed by immunoblotting, as described in Experimental Procedures. (A) Proteins were detected from whole cell lysates. VIM-2 and VIM-24 were overexpressed in E. coli BL21(DE3) pLys cells and analyzed by electrospray ionization mass spectrometry (ESI-MS) and circular dichroism (CD). (B) Mass spectra of purified VIM-2 show one major peak at 25602 ± 2 amu, which is consistent with cleavage of the leader sequence after residue 25. (C) Mass spectra of purified VIM-24 show two major peaks of 26206 and 25987 ± 2 amu, which correspond to cleavage after residues 20 and 21, respectively. (D) Far-UV CD spectra of VIM-2 (black line) and VIM-24 (gray line). (E) Thermal denaturation of VIM-2 and VIM-24 followed by ellipticity at 222 nm. Solid lines are best fits to a two-state denaturation model.
Consequences of the R228L Substitution for VIM-2 Structure and Catalytic Activity
Because the R228L mutant (VIM-24) was detected in a clinical isolate and thus represents a natural mutation in response to selective pressure, we characterized the consequences of this substitution for VIM structure and function. Purified VIM-2 and VIM-24 both bound approximately two Zn2+ atoms as measured by the PAR method. Mass spectrometry further confirmed protein identity and revealed one major peak at 25602 ± 2 amu for VIM-2. This value is consistent with cleavage of the protein’s leader sequence after residue 25. On the other hand, pure VIM-24 showed two main peaks at 26206 and 25987 ± 2 amu, which correspond to cleavage after residue 20 (plus an additional Met residue on the N-terminus, as the codon for this residue is introduced with the NdeI restriction site used to clone the gene into the vector) and residue 21, respectively (Figure 3B,C). Comparison of the CD scans revealed that VIM-2 and VIM-24 exhibit very similar CD spectra (Figure 3D). On thermal denaturation, both proteins displayed a single cooperative transition to an unfolded state with calculated Tm values of 62 °C for VIM-2 and 63 °C for VIM-24 (Figure 3E). However, the unfolding transition for VIM-24 is less steep than that for VIM-2, suggesting a more gradual unfolding process. Crystal structures of VIM-2 have revealed that Arg228 hydrogen bonds with a terminally Zn-coordinated water molecule. The presence of this water molecule has been linked to a cooperative metal binding, meaning that the binding of one atom of Zn2+ facilitates the binding of the second.55 Likewise, the unbinding of one Zn2+ leads to the unbinding of the second, which, we hypothesize, results in a faster unfolding process. Because the presence of the Leu residue at position 228 in VIM-24 does not allow the coordination of this terminally Zn-coordinated water molecule, the Zn binding–unbinding process is not cooperative. This may lead to the presence of apo, monozinc, and dizinc forms of the enzyme, resulting in a slower unfolding process. Taken together, our results suggest that the R to L substitution does not introduce significant changes into the overall metal or secondary structure content or global stability of the protein.
To improve our understanding of the impact of the R228L change on kinetic behavior, the steady-state kinetic parameters of VIM-2 and VIM-24 were determined with a series of substrates. VIM-24 displays a lower catalytic efficiency against the majority of the substrates tested (Table 2). For ampicillin and cephalothin, the decreased kcat/KM is due to the higher KM values, with no concomitant change in kcat. For imipenem, a lower kcat is observed with little change in KM, consistent with slower turnover. For ceftazidime and cefepime, we could not reach saturating concentrations of substrate under the experimental conditions used. However, clear differences were evident in the steady-state hydrolysis of these two substrates by VIM-2 and VIM-24 (Figure 4A,B) and were manifest in the kcat/KM ratio calculated by fitting progress curves to eq 2. For ceftazidime, kcat/KM was 1 × 104 M−1 s−1 for VIM-2 and 6 × 104 M−1 s−1 for VIM-24. For cefepime, the values were 3 × 104 M−1 s−1 for VIM-2 and 1 × 105 M−1 s−1 for VIM-24.
Table 2.
Steady-State Kinetic Data of VIM-2 and VIM-24a
| substrate | VIM-2
|
VIM-24
|
||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) | KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) | |
| nitrocefin | 13 ± 1 | 430 ± 20 | 3.3 × 107 | 11 ± 1 | 171 ± 4 | 1.6 × 107 |
| ampicillin | 23 ± 4 | 217 ± 9 | 9.9 × 106 | 97 ± 6 | 217 ± 5 | 2.2 × 106 |
| cephalothin | 17 ± 2 | 168 ± 8 | 9.8 × 106 | 33 ± 5 | 150 ± 9 | 4.5 × 106 |
| ceftazidime | NMc | NMc | 1 × 104b | NMc | NMc | 6 × 104b |
| cefepime | NMc | NMc | 3 × 104b | NMc | NMc | 1 × 105b |
| imipenem | 12 ± 1 | 45 ± 1 | 3.6 × 106 | 5.0 ± 0.7 | 8.8 ± 0.3 | 1.7 × 106 |
Values reported are averages ± standard deviations from triplicate experiments.
Calculated from the progress curves of reaction, as described in Experimental Procedures.
Not measured.
Figure 4.

Leu at position 228 enhaces ceftazidime and cefepime hydrolysis by VIM-24. Hydrolysis of 500 μM of (A) ceftazidime and (B) cefepime was monitored at λ256 and λ260, respectively, using 70 nM purified VIM-2 and VIM-24. Chemical structures of the substrates are shown at the top right side of each plot. (C) Production of VIM-2 and VIM-24 expressed in E. coli DH10B assessed from periplasmic preparations by immunoblotting. Two independent periplasmic extractions are presented. A standard curve of purified VIM-2 was used to quantify the proteins from each preparation.
To correlate the MIC results with the kinetic activity of VIM-2 and VIM-24, we measured the hydrolysis of cefepime and ceftazidime in periplasma. Determination of enzymatic activity in periplasma has been shown to parallel the resistance profile, as it more closely resembles conditions in vivo.26,56 Periplasmic MBL levels were quantified by immunoblotting (Figure 4C), and initial velocities were determined at 800 μM cefepime or 400 μM ceftazidime (maximal concentrations at which velocities could be measured). For VIM-24, the rate of cefepime hydrolysis (5.3 × 10−7 M s−1) increased compared to that of VIM-2 (2.9 × 10−7 M s−1). VIM-24 hydrolyzed ceftazidime at a rate of 4 × 10−7 M s−1, whereas measurable rates of hydrolysis were not obtained using VIM-2. These results clearly demonstrate that substitution of Arg for Leu at position 228 changes the substrate specificity of VIM enzymes to favor hydrolysis of third- and/or fourth-generation cephalosporins.
The VIM-2 R228L Substitution Relieves Steric Clashes with Bound Cephems
In MBLs such as VIM-2, the extended guanidine side chain of Arg228 may replicate the role of Lys224 in interacting with the C3/C4 carboxylate of β-lactam substrates,13,14 while docking experiments implicate Arg228 (in addition to residues at the ASL-3) in positioning the substrate through interactions with the large, charged C3 substituents (R2 groups) of hydrolyzed cephalosporins such as ceftazidime.57 For VIM-1, VIM-19, and related enzymes, activity against such substrates may be enhanced by the presence of Ser at position 228, resulting in a more “open” active site.14,57,58 In line with these observations, screening of VIM-2 variants at position 228 determined that small residues (Val, Ala, Ser, Thr, and Gly), and those with nonpolar side chains [Ile and Leu (VIM-24)] all increased the MICs for cefepime and ceftazidime (complete MIC data provided as Supporting Information). We consider the most likely explanation for these findings to be that both steric and charge–charge interactions involving position 228 of the VIM MBLs and the β-lactam R2 substituent group can orient and position substrates within the active site. To test this notion, we used the crystal structure of VIM-2 (PDB entry 2YZ3) to derive a molecular model of VIM-24.
A docking of unhydrolyzed cefepime was modeled into the active site of VIM-24 and minimized the resulting complex using YASARA.37 As shown in Figure 5, the presence of Leu228 creates an active site groove in VIM-24 large enough to accommodate the bulky C3 N-methylpyrrolidine group of cefepime without requiring significant conformational rearrangement (Figure 5A). By way of contrast, the guanidinium side chain of VIM-2 Arg228 protrudes sufficiently into the substrate binding cleft to create a clear steric clash (Figure 5B). In this conformation, the Arg is ≈8 Å from the carboxylate group of the substrate and completely unable to form any hydrogen bond with it. We anticipate the interaction between Arg228 and the R2 substitution of ceftazidime to be equally unfavorable, not only because of the structural similarity but because of the fact that in this case the R2 substitution is positively charged. Therefore, we conclude that the shorter Leu side chain facilitates access of both cephems to the active site of VIM-24, increasing turnover of what are still poor substrates.
Figure 5.
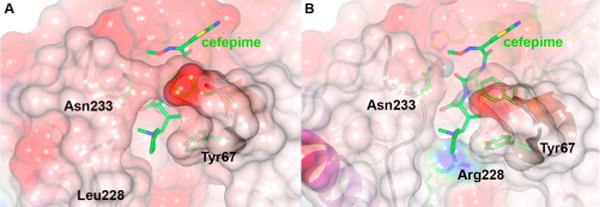
Interactions of cefepime with VIM-24 and VIM-2. (A) Intact cefepime was manually docked into the model of VIM-24 and the complex minimized using the YASARA server.37 (B) The resulting orientation was superimposed onto the crystal structure of native VIM-2 (PDB entry 1KO313) using SSMSuperpose.38 Note the steric clash between the N-methylpyrrolidine group at cefepime C3 and the guanidino side chain of VIM-2 Arg228. This figure was generated using CCP4mg;64 the molecular surface is colored by electrostatic charge from −0.5 V (red, negative) to 0.5 V (blue, positive).
BTZs Are Slow Binding Inhibitors of VIM-2 and VIM-24 with Micromolar Affinity
In addition to interacting with substrates (especially cephems), VIM-2 Arg228 is also positioned to form part of a binding site for potential MBL inhibitors. Thus, we investigated the interaction of VIM-2 and VIM-24 with four candidate BTZ inhibitors comprising two pairs of stereoisomers. Interestingly, under steady-state conditions, BTZs were effective inhibitors of both VIM-2 and VIM-24, with observed Ki values (determined using NCF as a reporter substrate) between 3.7 and 14 μM (Table 3). All four compounds increased the apparent KM but exert essentially no effect on kcat, consistent with a competitive mode of inhibition. Although the Ki values were in general slightly higher for VIM-24, the maximal effect was <2-fold. On this basis, we conclude that unlike the effects seen with β-lactams, interaction with Arg228 does not make a major contribution to the affinity of BTZ for the VIM MBLs. This has significant implications for further development of this class of BTZ inhibitors.
Table 3.
Inhibition of VIM-2 and VIM-24 by BTZsa
| compound | VIM-2
|
VIM-24
|
||||||
|---|---|---|---|---|---|---|---|---|
| Ki (μM) | Ki* (μM) | k2 (s−1) | k−2 (s−1) | Ki (μM) | Ki* (μM) | k2 (s−1) | k−2 (s−1) | |
| L-CS319 | 3.7 ± 0.3 | 0.04 ± 0.01 | (0.03 ± 1) × 10−3 | (0.0003 ± 1) × 10−4 | 6.0 ± 0.7 | 0.12 ± 0.02 | (0.06 ± 2) × 10−3 | (0.001 ± 1) × 10−3 |
| D-CS319 | 5.4 ± 0.4 | 0.18 ± 0.02 | (0.01 ± 1) × 10−3 | (0.0003 ± 1) × 10−4 | 11 ± 0.8 | 0.64 ± 0.01 | (0.07 ± 4) × 10−3 | (0.004 ± 1) × 10−3 |
| L-VC26 | 3.8 ± 0.2 | 0.25 ± 0.01 | (0.02 ± 1) × 10−3 | (0.001 ± 1) × 10−3 | 4.9 ± 0.3 | 0.13 ± 0.02 | (0.04 ± 2) × 10−3 | (0.001 ± 1) × 10−3 |
| D-VC26 | 14 ± 1 | 0.10 ± 0.03 | (0.09 ± 4) × 10−3 | (0.0006 ± 4) × 10−4 | 12 ± 3 | 0.08 ± 0.01 | (0.01 ± 3) × 10−3 | (0.00007 ± 1) × 10−5 |
Ki determined from the initial rate of hydrolysis (v0). Ki* and k2 were calculated from the analysis of the change in vs with inhibitor concentration, in the slow binding model.
More detailed investigations identified that BTZ inhibition of both VIM-2 and VIM-24 arises from a slow binding two-step process, as previously described for neutral thiol inhibitors of IMP-1,5 captopril inhibition of angiotensin converting enzyme, and some thiol inactivators of leucine amino peptidase.59–61 At increasing inhibitor concentrations, NCF hydrolysis proceeds in two phases transitioning from a rapid initial (v0) to a slower steady-state (vs) velocity (Figure 6A). This behavior is consistent with initial formation of a relatively weak inhibitor complex (EI) that resolves to a more stable species (E*I) during the course of the reaction. According to this model of inhibition, Ki is the measure of the affinity of the initial complex, EI, whereas the true overall dissociation constant of an inhibitor that conforms to this mechanism is defined by the dissociation constant for the final high-affinity conformation of the enzyme–inhibitor complex Ki* [Ki* = Ki/(1 + k2/k−2)].62
Figure 6.

BTZ-induced inhibition of hydrolysis. (A) Reaction was initiated by addition of 3 nM VIM-2 to a mixture of buffer, L-CS319, and 50 μM nitrocefin. (B) kobs vs [I] plot for L-CS319 onset of VIM-2 inhibition. Data were fit as described in Experimental Procedures. (C) Reaction was initiated by addition of 0.5 nM VIM-2 preincubated for 5 min with L-CS319 to a mixture of buffer and 50 μM nitrocefin.
To verify that this model correctly describes the observed behavior, the kinetic data were fit to alternative models as described above (Scheme 1). The best fit was obtained with a two-step model in which both v0 and vs decrease with an increasing inhibitor concentration (Figure 6A), as evidenced by a fit to the data more consistent than, and 95% confidence intervals narrower than, those that could be obtained with a simpler, one-step scheme in which v0 did not vary with inhibitor concentration (statistical analysis for the fitting of L-CS319-induced inhibition of NCF hydrolysis by VIM-2 is provided as Supporting Information). From these data, the overall equilibrium dissociation constant for inhibitor binding, Ki*, could be calculated by fitting the final vs determined at different inhibitor concentrations to eq 6. These values are summarized in Table 3. In agreement with the conclusion that inhibition occurs by a slow binding two-step process, we found that Ki ≫ Ki*, as the values of Ki* were observed to be significantly lower (0.04–0.6 μM) than the Ki values obtained by analysis of the inhibitor dependence of v0 (4–14 μM).
For a two-step time-dependent onset of inhibition model, the dependence of kobs on the concentration of inhibitor at limiting [I] reflects the second-order rate constant k2/Ki*[I]/(1 + [S]/Km), where k2 is the rate constant for formation of E*I and Ki is the equilibrium binding constant for the first binding step (eq 5).46 The observed values for k2, estimated from the slope obtained from the plot of the calculated kobs versus inhibitor concentration, range from 0.01 to 0.09 s−1 (Figure 6B). Given the observed values of Ki (4–14 μM) and Ki* (0.04–0.6 μM), the equilibrium constant for the conformational change step (defined as Ki′ = k−2/k2) must lie in the range of 0.1–0.01 for both VIM-2 and VIM-24 [because Ki* = Ki/(1 + k2/k−2)].62 As a consequence of the slow rate constants observed for formation of E*I, the rate constant for breakdown of the E*I complex (k−2) is also very slow (Table 3).
To further test this model, the kinetics of reactions in which the enzyme was preincubated with inhibitor prior to the addition of substrate were analyzed. For a time-dependent onset of inhibition mechanism, a preincubation step should eliminate the biphasic kinetics observed in reactions where inhibitor and substrate are added at the same time. Consistent with this conclusion, reactions after preincubation of enzyme and inhibitor show simple, linear, steady-state kinetics. Moreover, the observed initial rates obtained from enzyme preincubated with inhibitor are consistent with the tighter inhibitor binding constant Ki* (Figure 6C and Table 3). Taken together, the kinetic data are consistent with a model in which rapid but relatively weak binding of the inhibitor in the substrate-binding pocket is followed by conformational changes to a more stable enzyme–inhibitor complex.
Crystallography Reveals Key Interactions between VIM-2 and L-CS319
To further investigate the interaction of VIM enzymes and BTZ inhibitors, we sought to obtain a crystal structure for a BTZ–VIM-2 complex. Initial experiments in which the most potent inhibitor, L-CS319, was soaked into a previously described VIM-2 crystal form13 failed to yield interpretable electron density. Hence, we used cocrystallization to generate a new crystal form [space group P21, two molecules in the asymmetric unit (Table 4)], diffracting to near-atomic resolution (1.25 Å). The structure was determined by molecular replacement and refined as described above, with the final model containing 230 and 234 amino acids in chains A and B, respectively.
Table 4.
Crystallographic Data Collection and Refinement Statistics
| data set | VIM-2:CS319 |
| processing | XDS/SCALA |
| beamline | DLS I04-1 |
| space group | P21 |
| cell dimensions | a = 52.89 Å, b = 61.40 Å, c = 68.92 Å, β = 100.9° |
| wavelength (Å) | 0.9200 |
| resolution (Å)a | 29.64–1.25 (1.32–1.25) |
| total no. of reflectionsa | 302755 (38004) |
| no. of unique reflectionsa | 115461 (15946) |
| completeness (%)a | 96.7 (91.8) |
| redundancya | 2.6 (2.4) |
| I/(σI)a | 11.1 (2.3) |
| Rmerge (%)a | 0.040 (0.395) |
| refinement | REFMAC5 |
| total no. of reflections | 109653 |
| resolution (Å) | 29.64–1.25 |
| Rcryst (%) | 14.5 |
| Rfree (%)b | 16.4 |
| root-mean-square deviation for bond lengths (Å) | 0.0064 |
| root-mean-square deviation for bond angles (deg) | 1.4591 |
| no. of protein atoms | 3578 |
| no. of water molecules | 355 |
| % residues in Ramachandran regions (favored/allowed/disallowed) | 98.5/1.5/0 |
| Wilson B factor | 11.3 |
| B factors (protein) | 15.6 (15.4, 16.0)c |
| B factors (ligand) | 26.7,d 22.7,e 14.7f |
| B factors (water molecules) | 27.6 |
| PDB entry | 4UA4 |
Data for the highest-resolution shell are in parentheses.
Rfree was calculated with 5% of the reflections omitted.
Values for chains A and B, respectively.
Major conformation, chain B (occupancy of 0.6).
Minor conformation, chain B (occupancy of 0.3).
Dimer, chain A.
Difference electron density maps gave clear evidence of bound ligand in both molecules of the asymmetric unit but showed the nature of the ligand to differ between the two subunits. In molecule A, high-quality electron density extending along the active site groove indicated a ligand significantly larger than a single L-CS319 molecule, which was modeled as a dimeric species with two L-CS319 monomers connected by a disulfide bond (data not shown). However, as assays in solution demonstrated that prolonged exposure to oxidative conditions abolishes the inhibitory activity of L-CS319 and other BTZs toward MBLs (data not shown), we conclude that this is most likely an artifact structure generated by oxidation of the L-CS319 ligand during crystallization and does not represent the inhibitory VIM-2–L-CS319 complex.
In contrast, the active site of molecule B contained positive electron density of a size and shape expected for a monomer of L-CS319 (Figure 7A). Although this positive density was more diffuse than would be expected at this resolution, a strong peak between the two zinc ions was considered to indicate the inhibitor thiol group in the “bridging” position and a second strong peak close to the Asn233 backbone amide the inhibitor carboxylate. On this basis, L-CS319 could be fit into the difference map and refined with an occupancy of 0.6 (Table 4). However, the best results were obtained when a second inhibitor molecule, with an occupancy of 0.3, was fit into the additional diffuse electron density. This low-occupancy conformation shares a common location for the carboxylate group but differs in the precise orientation of the BTZ core and in the positioning of the thiol, which notably is not intercalated between the two Zn2+ ions. The description below focuses on the high-occupancy conformation. We note, however, that observation of two conformations for bound L-CS319 in the crystal structure is consistent with the presence of multiple enzyme–inhibitor complexes in solution as observed in the inhibition assays described above.
Figure 7.

Crystal structure of the VIM-2–L-CS319 complex. (A) Stereoview of the active site of chain B showing L-CS319 overlaid with |Fo| − |Fc|Φcalc density (green, contoured at 2.5σ) calculated from refinement of the final model, from which the ligand was removed, against experimental diffraction data. The major conformation of bound inhibitor (occupancy of 0.6) is rendered as solid sticks (cyan); the minor conformation (occupancy of 0.3) is rendered semitransparent. (B) Orientation of the major conformation of L-CS319 bound in the VIM-2 active site. Hydrogen bonds and zinc coordination bonds are shown as black dashes. Inhibitor L-CS319 interacts with both zinc ions via its sulfhydryl group; the inhibitor carboxylate group interacts with Asn233 amide and a bound water molecule (Wat). (C) Overlay of the VIM-2–L-CS319 complex (green) with structures of unliganded VIM-2 (slate blue, PDB entry 1KO313), the mercaptocarboxylate inhibitor (M5P) complex (magenta, PDB entry 2YZ333), and the rhodanine fragment (ML302F) complex (gray, PDB entry 4PVO16). For all panels, carbon atoms are colored green (protein, other than where specified for panel C), zinc ions gray, water molecules red, and other atoms as standard. This figure was generated using PyMol (www.pymol. org).
L-CS319 is positioned with the BTZ ring system lying at approximately 45° with respect to the floor of the active site groove (Figure 7B). Key interactions with VIM-2 involve the thiol, which adopts the bridging position between the two zinc ions seen in other MBL–thiol complexes (for example, phenylC3SH,33 rhodanine,16 and Tiopronin18), and the carboxylate, which forms hydrogen bonds to the Asn233 backbone amide nitrogen and to a bound water molecule (Figure 7B, Wat) that is positioned by the Cys221 backbone carbonyl. Notably, the inhibitor carboxylate is too far from the Zn2 (Cys-His-Asp site) metal ion to make a direct interaction, unlike the situation for complexes of other B1MBLs with hydrolyzed β-lactams.36,63 Interestingly, although the Arg228 guanidino moiety is within hydrogen bonding distance of the inhibitor carboxylate, the two groups are not well-oriented for interaction. Weak electron density for the Arg228 guanidino group, which was refined in two conformations, also argues against a strong interaction between this residue and bound inhibitor. This apparent lack of involvement of Arg228 in BTZ binding is consistent with the similar levels of BTZ inhibition of VIM-2 and VIM-24, supporting the conclusion that Arg228 does not substantially contribute to inhibitor affinity. L-CS319 binding does involve hydrophobic contacts, namely, edge–face interactions between the two rings of the fused BTZ system and the side chains of Phe61 and Tyr67 at the base of ASL-3 (residues 60–67), and with the more distant side chain of the conserved Trp87. In addition, the side chain of the zinc ligand His263 sits beneath and approximately parallel to the inhibitor scaffold.
The overall conformation of VIM-2 in the L-CS319 complex differs little from that of the free enzyme13 and complexes with mercaptocarboxylate33 and rhodanine16 inhibitors (Figure 7C). The main differences are in ASL-3, which adapts its position, in particular the orientation of the Phe61 side chain, to accommodate the different inhibitors. However, these differences are far less marked than in ligand complexes of other B1 enzymes, such as IMP-1 or NDM-1, where much more profound changes in the ASL-3 conformation have been observed. This may arise from the location of aromatic residues likely to be important to ligand binding at the base of VIM ASL-3 (Phe61 and Tyr67) rather than at its apex, as is the case for Trp64 or Phe64 in the IMP and NDM MBLs, respectively. Repositioning of the side chain at position 64 on ligand binding is likely to require much greater movement of ASL-3 in both of these enzymes. Comparison of the mode of binding of L-CS319 and the rhodanine fragment ML302F shows that the bridging thiol, multiple conformations for the Arg228 side chain, and interactions with the Asn233 backbone amide and the Cys221-bound water molecule are common to binding of both inhibitors. As this water molecule is present in an identical position in all but one available VIM-2 structure, while the conformation of the Arg228 side chain varies, it is possible that in VIM-2 it is this water molecule, rather than Arg228, that makes the major contribution to binding the C3/C4 carboxylate of β-lactam substrates and thus replicates the role of Lys224 in other B1MBLs.
BTZs Restore Imipenem Activity against VIM-2 and VIM-24, Producing Gram-Negative Pathogens
To assess the capacity of the BTZ inhibitors to restore the efficacy of imipenem, we performed microbiological assays using two clinical isolates: VIM-2 harboring P. aeruginosa (imipenem MIC = 32 μg/mL) and VIM-24 producing K. pneumoniae (imipenem MIC = 1 μg/mL). Viable cell counts following exposure to sublethal concentrations of imipenem in the presence of inhibitors (Figure 8) showed that the BTZs are able to inhibit both MBLs within bacteria, as evidenced by the significant reductions in bacterial count (p < 0.05). The inhibitors do not have any antimicrobial effect on their own, as differences could not be detected in viable cell number between BTZ-exposed cells and broth-only controls (data not shown).
Figure 8.

BTZs restore the in vitro activity of imipenem against (A and B) VIM-24-producing K. pneumoniae and (C) VIM-2-producing P. aeruginosa. (A) Bacteria were grown at sublethal concentrations of imipenem alone (8 mg/L for P. aeruginosa and 0.5 mg/L for K. pneumoniae) or in combination with each BTZ at 100 mg/L. Viable cells were recovered at 100, 300, and 500 min. (B and C) Results shown are the means of three biological replicates ± SD. Adjusted p values are illustrated on the figures: 0.05 (one asterisk), 0.01 (two asterisks), and 0.001 (three asterisks).
CONCLUDING REMARKS
In this study, we present a detailed characterization of the effect of substitutions at VIM-2 position 228, including the R228L substitution found in the naturally occurring variant VIM-24, upon antibiotic susceptibility of recombinant E. coli, substrate, and inhibitor binding. Our results emphasize the impact of this position on hydrolysis of third- and/or fourth-generation cephalosporin substrates and thus provide further evidence of the functional importance of residue 228 in defining the profile of the VIM enzymes. In some ways, this natural variant (R228L) recapitulates the clinical observation made in class A β-lactamases such as TEM or SHV, in which a single amino acid substitution results in an “extended spectrum” phenotype. However, our data show that binding of other β-lactam classes and BTZ inhibitors is little affected by the substitution, arguing against an essential role for Arg228 in ligand binding by VIM-2. This conclusion is supported by the absence of interactions between Arg228 and the inhibitor carboxylate in our crystal structure of a VIM-2–BTZ complex. Thus, VIM enzymes, in which differences at residue 228 can affect interaction of VIM variants with some substrates but high-affinity binding can be tolerant of substitutions, differ from other clinically important B1MBLs in which the importance of the conserved Lys224 is well-established. These differences highlight the challenge of designing inhibitors effective across the whole range of MBL targets. The structural, biochemical, and mechanistic information presented here will provide a starting point for such optimizations. Lastly, our results emphasize the importance of identifying a mechanism of inhibition to allow both assessment of the true affinity of a compound for its target and the interpretation of structure–activity relationships.
Supplementary Material
Acknowledgments
We thank Dr. Shahriar Mobashery for kindly providing us nitrocefin and Dr. William Merrick for critical reading of the manuscript.
Funding
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health via Grants R01AI072219 (R.A.B.), R01AI063517 (R.A.B.), and R01AI100560 (A.J.V., J.S., and R.A.B.). K.M.P.-W. is supported by a Veterans Affairs Career Development Award. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, Veterans Affairs Merit Review Program Award 1I01BX001974, and Geriatric Research, Education and Clinical Center VISN 10 to R.A.B.
ABBREVIATIONS
- BLI
β-lactamase inhibitor
- MBL
metallo-β-lactamase
- SPM
Sao Paulo metallo-β-lactamase
- SIM
Seoul imipenemase
- GIM
German imipenemase
- IMP
imipenemase
- NDM
New Delhi metallo-β-lactamase
- VIM
Verona integron-encoded metallo-β-lactamase
- MIC
minimal inhibitory concentration
- BTZ
bisthiazolidine
- ASL-3
active site loop 3 (residues 60–67)
- ASL-10
active site loop 10 (residues 223–242)
- PDB
Protein Data Bank
Footnotes
Supporting Information
Full description of the organic synthesis of the BTZs, complete MIC data, and statistical analysis of the fitting. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00106.
Notes
The authors declare no competing financial interest.
References
- 1.Bebrone C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol. 2007;74:1686–1701. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 2.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z, Fast W, Valentine AM, Benkovic SJ. Metallo-β-lactamase: Structure and mechanism. Curr Opin Chem Biol. 1999;3:614–622. doi: 10.1016/s1367-5931(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 4.Garau G, García-Sáez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frère JM, Dideberg O. Update of the standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother. 2004;48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siemann S, Clarke AJ, Viswanatha T, Dmitrienko GI. Thiols as classical and slow-binding inhibitors of IMP-1 and other binuclear metallo-β-lactamases. Biochemistry. 2003;42:1673–1683. doi: 10.1021/bi027072i. [DOI] [PubMed] [Google Scholar]
- 6.Walsh TR, Toleman MA, Poirel L, Nordmann P. Metallo-β-lactamases: The quiet before the storm? Clin Microbiol Rev. 2005;18:306–325. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauretti L, Riccio ML, Mazzariol A, Cornaglia G, Amicosante G, Fontana R, Rossolini GM. Cloning and characterization of blaVIM, a new integron-borne metallo-β-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob Agents Chemother. 1999;43:1584–1590. doi: 10.1128/aac.43.7.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bush K, Jacoby GA. β-Lactamase Classification and Amino Acid Sequences for TEM, SHV and OXA Extended-Spectrum and Inhibitor Resistant Enzymes. 2014 ( http://www.lahey.org/studies)
- 9.Meini MRG, Lisandro J, Vila AJ. Antibiotic resistance in Zn(II)-deficient environments: Metallo-β-lactamase activation in the periplasm. Future Microbiol. 2013;8:2. doi: 10.2217/fmb.13.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montealegre MC, Correa A, Briceño DF, Rosas NC, De La Cadena E, Ruiz SJ, Mojica MF, Camargo RD, Zuluaga I, Marin A. Novel VIM metallo-β-lactamase variant, VIM- 24, from a Klebsiella pneumoniae isolate from Colombia. Antimicrob Agents Chemother. 2011;55:2428–2430. doi: 10.1128/AAC.01208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Materon IC, Beharry Z, Huang W, Perez C, Palzkill T. Analysis of the context dependent sequence requirements of active site residues in the metallo-β-lactamase IMP-1. J Mol Biol. 2004;344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 12.Yuan Q, He L, Ke H. A potential substrate binding conformation of β-lactams and insight into the broad spectrum of NDM-1 activity. Antimicrob Agents Chemother. 2012;56:5157–5163. doi: 10.1128/AAC.05896-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Saez I, Docquier JD, Rossolini G, Dideberg O. The three-dimensional structure of VIM-2, a Zn-β-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form. J Mol Biol. 2008;375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Lassaux P, Traoré DA, Loisel E, Favier A, Docquier JD, Sohier JS, Laurent C, Bebrone C, Frère JM, Ferrer JL. Biochemical and structural characterization of the subclass B1 metallo-β-lactamase VIM-4. Antimicrob Agents Chemother. 2011;55:1248–1255. doi: 10.1128/AAC.01486-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drawz SM, Bonomo RA. Three decades of β-lactamase inhibitors. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brem J, van Berkel SS, Aik W, Rydzik AM, Avison MB, Pettinati I, Umland KD, Kawamura A, Spencer J, Claridge TD. Rhodanine hydrolysis leads to potent thioenolate mediated metallo-β-lactamase inhibition. Nat Chem. 2014;6:1084–1090. doi: 10.1038/nchem.2110. [DOI] [PubMed] [Google Scholar]
- 17.King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature. 2014;510:503–506. doi: 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klingler FM, Wichelhaus TA, Frank D, Bernal JC, El-Delik J, Müller HF, Sjuts H, Göttig S, Koenigs A, Pos KM. Approved Drugs containing Thiols as Inhibitors of Metallo-β-Lactamases: A Strategy to Combat Multidrug-Resistant Bacteria. J Med Chem. 2015;58:3626–3630. doi: 10.1021/jm501844d. [DOI] [PubMed] [Google Scholar]
- 19.Crowder MW, Spencer J, Vila AJ. Metallo-β-lactamases: Novel weaponry for antibiotic resistance in bacteria. Acc Chem Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 20.Faridoon NUI. An update on the status of potent inhibitors of metallo-β-lactamases. Sci Pharm. 2013;81:309–327. doi: 10.3797/scipharm.1302-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González JM, Buschiazzo A, Vila AJ. Evidence of adaptability in metal coordination geometry and active-site loop conformation among B1 metallo-β-lactamases. Biochemistry. 2010;49:7930–7938. doi: 10.1021/bi100894r. [DOI] [PubMed] [Google Scholar]
- 22.Spencer J, Clarke AR, Walsh TR. Novel mechanism of hydrolysis of therapeutic β-lactams by Stenotrophomonas maltophilia L1 metallo-β-lactamase. J Biol Chem. 2001;276:33638–33644. doi: 10.1074/jbc.M105550200. [DOI] [PubMed] [Google Scholar]
- 23.Saiz C, Castillo V, Fontán P, Bonilla M, Salinas G, Rodríguez-Haralambides A, Mahler SG. Discovering Echinococcus granulosus thioredoxin glutathione reductase inhibitors through site-specific dynamic combinatorial chemistry. Mol Diversity. 2014;18:1–12. doi: 10.1007/s11030-013-9485-3. [DOI] [PubMed] [Google Scholar]
- 24.Szabó D, Szentandrássy J, Juhász Z, Katona K, Nagy K, Rókusz L. Imported PER-1 producing Pseudomonas aeruginosa, PER-1 producing Acinetobacter baumanii and VIM-2-producing Pseudomonas aeruginosa strains in Hungary. Ann Clin Microbiol Antimicrob. 2008;7:1–5. doi: 10.1186/1476-0711-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-second Informational Supplement M100-S22. Clinical and Laboratory Standards Institute; Wayne, PA: 2012. [Google Scholar]
- 26.González JM, Meini MR, Tomatis PE, Martín FJM, Cricco JA, Vila AJ. Metallo-β-lactamases withstand low Zn(II) conditions by tuning metal-ligand interactions. Nat Chem Biol. 2012;8:698–700. doi: 10.1038/nchembio.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bates D, Maechler M. lme4: Linear mixed-effects models using Eigen and S4. R package, version 1.1-7. 2014 http://cran.r-project.org/web/packages/lme4/index.html.
- 28.Hothorn T, Bretz F, Westfall P. Simultaneous Inference in General Parametric Models. Biom J. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 29.Pegg KM, Liu EM, George AC, LaCuran AE, Bethel CR, Bonomo RA, Oelschlaeger P. Understanding the determinants of substrate specificity in IMP family metallo-β-lactamases: The importance of S262. Protein Sci. 2014;10:1451–1460. doi: 10.1002/pro.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fast W, Wang Z, Benkovic SJ. Familial mutations and zinc stoichiometry determine the rate-limiting step of nitrocefin hydrolysis by metallo-β-lactamase from Bacteroides fragilis. Biochemistry. 2001;40:1640–1650. doi: 10.1021/bi001860v. [DOI] [PubMed] [Google Scholar]
- 31.van Berkel SS, Brem J, Rydzik AM, Salimraj R, Cain R, Verma A, Owens RJ, Fishwick CWG, Spencer J, Schofield CJ. Assay plattform for clinically relevant metallo-β-lactamases. J Med Chem. 2013;56:8. doi: 10.1021/jm400769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc. 2006;1:2527–2535. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J-i, Shibata N, Arakawa Y, Kurosaki H. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J Med Chem. 2007;50:6647–6653. doi: 10.1021/jm701031n. [DOI] [PubMed] [Google Scholar]
- 34.Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. 2012;D68:431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King DT, Worrall LJ, Gruninger R, Strydnadka NC. New Delhi Metallo-β-lactamase: Structural insights into β-lactam recognition and inhibition. J Am Chem Soc. 2012;134:11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 37.Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77:114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 39.Levitt PS, Papp-Wallace KM, Taracila MA, Hujer AM, Winkler ML, Smith KM, Xu Y, Harris ME, Bonomo RA. Exploring the role of a conserved class A residue in the Ω-loop of KPC-2 β-lactamase: A mechanism for ceftazidime hydrolysis. J Biol Chem. 2012;287:31783–31793. doi: 10.1074/jbc.M112.348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llarrull LI, Tioni MF, Kowalski J, Bennett B, Vila AJ. Evidence for a dinuclear active site in the metallo-β-lactamase BcII with substoichiometric Co(II). A new model for metal uptake. J Biol Chem. 2007;282:30586–30595. doi: 10.1074/jbc.M704613200. [DOI] [PubMed] [Google Scholar]
- 41.Toth M, Vakulenko V, Antunes NT, Frase H, Vakulenko SB. Class A carbapenemase FPH-1 from Francisella philomiragia Antimicrob Agents Chemother. 2012;56:2852–2857. doi: 10.1128/AAC.00223-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papp-Wallace KM, Taracila M, Hornick JM, Hujer AM, Hujer KM, Distler AM, Endimiani A, Bonomo RA. Substrate selectivity and a novel role in inhibitor discrimination by residue 237 in the KPC-2 β-lactamase. Antimicrob Agents Chemother. 2010;54:2867–2877. doi: 10.1128/AAC.00197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, Bonomo RA. Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: Exploring the roles of Arg220 and Glu276. Antimicrob Agents Chemother. 2012;56:4428–4438. doi: 10.1128/AAC.05769-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C. Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J Med Chem. 2013;56:1084–1097. doi: 10.1021/jm301490d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Meester F, Joris B, Reckinger G, Bellefroid-Bourguignon C, Frère JM, Waley SG. Automated analysis of enzyme inactivation phenomena: Application to β-lactamases and DD-peptidases. Biochem Pharmacol. 1987;36:2393–2403. doi: 10.1016/0006-2952(87)90609-5. [DOI] [PubMed] [Google Scholar]
- 46.Ehmann DE, Jahić H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci USA. 2012;109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. 2010;D66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans P. Scaling and assessment of data quality. Acta Crystallogr. 2005;D62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 49.Winter G. xia2: An expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2010;43:1260–1273. [Google Scholar]
- 50.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 52.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr. 2011;D67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB, Arendall BW, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2009;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morán-Barrio J, Limansky AS, Viale AM. Secretion of GOB metallo-β-lactamase in Escherichia coli depends strictly on the cooperation between the cytoplasmic DnaK chaperone system and the Sec machinery: Completion of folding and Zn(II) ion acquisition occur in the bacterial periplasm. Antimicrob Agents Chemother. 2009;53:2908–2917. doi: 10.1128/AAC.01637-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aitha M, Marts AR, Bergstrom A, Møller AJ, Moritz L, Turner L, Nix JC, Bonomo RA, Page RC, Tierney DL. Biochemical, mechanistic, and spectroscopic characterization of metallo-β-lactamase VIM-2. Biochemistry. 2014;53:7321–7331. doi: 10.1021/bi500916y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.González LJ, Moreno DM, Bonomo RA, Vila AJ. Host-specific enzyme-substrate interactions in SPM-1 metallo-β-lactamase are modulated by second sphere residues. PLoS Pathog. 2014;10:e1003817. doi: 10.1371/journal.ppat.1003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borra PS, Leiros HKS, Ahmad R, Spencer J, Leiros I, Walsh TR, Sundsfjord A, Samuelsen Ø. Structural and computational investigations of VIM-7: Insights into the substrate specificity of VIM metallo-β-lactamases. J Mol Biol. 2011;411:174–189. doi: 10.1016/j.jmb.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez-Martinez JM, Nordmann P, Fortineau N, Poirel L. VIM-19, a metallo-β-lactamase with increased carbapenemase activity from Escherichia coli and Klebsiella pneumoniae. Antimicrob Agents Chemother. 2010;54:471–476. doi: 10.1128/AAC.00458-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bienvenue DL, Bennett B, Holz RC. Inhibition of the aminopeptidase from Aeromonas proteolytica by l-leucinethiol: Kinetic and spectroscopic characterization of a slow, tight-binding inhibitor–enzyme complex. J Inorg Biochem. 2000;78:43–54. doi: 10.1016/s0162-0134(99)00203-2. [DOI] [PubMed] [Google Scholar]
- 60.Huntington KM, Bienvenue DL, Wei Y, Bennett B, Holz RC, Pei D. Slow-binding inhibition of the aminopeptidase from Aeromonas proteolytica by peptide thiols: Synthesis and spectroscopic characterization. Biochemistry. 1999;38:15587–15596. doi: 10.1021/bi991283e. [DOI] [PubMed] [Google Scholar]
- 61.Reynolds CH. Kinetics of inhibition of angiotensin converting enzyme by captopril and by enalapril diacid. Biochem Pharmacol. 1984;33:1273–1276. doi: 10.1016/0006-2952(84)90180-1. [DOI] [PubMed] [Google Scholar]
- 62.Copeland RA. Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmacologists. John Wiley & Sons; New York: 2013. [PubMed] [Google Scholar]
- 63.Zhang H, Hao Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 2011;25:2574–2582. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 64.McNicholas S, Potterton E, Wilson K, Noble M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. 2011;D67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.