

Abstract
Immunoconjugates are specific, highly effective, minimally toxic anticancer therapies that are beginning to show promise in the clinic. Immunoconjugates consist of three separate components: an antibody that binds to a cancer cell antigen with high specificity, an effector molecule that has a high capacity to kill the cancer cell, and a linker that will ensure the effector does not separate from the antibody during transit and will reliably release the effector to the cancer cell or tumour stroma. The high affinity antibody–antigen interaction allows specific and selective delivery of a range of effectors, including pharmacologic agents, radioisotopes, and toxins, to cancer cells. Some anticancer molecules are not well tolerated when administered systemically owing to unacceptable toxicity to the host. However, this limitation can be overcome through the linking of such cytotoxins to specific antibodies, which mask the toxic effects of the drug until it reaches its target. Conversely, many unconjugated antibodies are highly specific for a cancer target, but have low therapeutic potential and can be repurposed as delivery vehicles for highly potent effectors. In this Review, we summarize the successes and shortcomings of immunoconjugates, and discuss the future potential for the development of these therapies.
Introduction
Immunoconjugates represent an important development in the armamentarium of anticancer therapies. These molecules consist of antibodies capable of recognizing cancer antigens with high specificity, a cytotoxic agent and a linker that will ensure the effector does not separate from the antibody. These drugs function by delivering the therapeutic agents to specific tumour targets.
At the turn of the twentieth century, Nobel laureate Paul Ehrlich envisioned the development of a therapeutic molecule, such as an antibody, that could deliver a toxic agent directly to a cancer cell.1 However, useful implementation of this concept required nearly a century of technological developments. Among cancer therapies, chemotherapy agents have been developed to arrest cellular replication, with a relative degree of specificity for malignant cells. Such agents can be highly effective; however, the administered doses typically must be limited to minimize the risk of lethal toxicity, at the expense of efficacy. Other cancer therapies, such as radiation therapy and cytokine therapies, have encountered similar limitations.2
Further research has led to the development of an increasing number of highly cancer-specific unconjugated antibodies that are used as anticancer agents. However, while these antibodies effectively target cancer cells, they typically have limited clinical activity; for example, cetuximab that targets EGFR, demonstrated an improvement in overall survival of only 4 months for treatment of patients with colorectal cancer.3,4 Moreover, while cetuximab does have single agent activity, the efficacy of some antibodies, such as bevacizumab, is observed only when administered in conjunction with chemotherapy.5,6
Immunoconjugates possess complementary properties that link the potent anticancer effects of cytotoxic agents with the highly specific cancer targeting properties of monoclonal antibodies. Several immunoconjugates, including brentuximab vedotin and trastuzumab emtansine, have proven highly effective in the management of Hodgkin lymphoma, large-cell lymphoma, and breast cancers.7–9 We discuss the structure, targets, therapeutic entities as well as future perspectives of such molecules in terms of cancer therapy.
Immunoconjugate targets
The choice of the antigen to be targeted is of prime importance, and the selection of unique targets on the cancer cell is not always straightforward. Most antigens that are abundantly expressed by cancer cells are not tumour-specific.10 The ability to develop therapeutic antibodies typically relies upon the identification of antigens that are distinguishable from their host cell counterparts (tumour-specific) and/or that are expressed at higher levels than by host cells (tumour-selective).11 Furthermore, the efficiency of internalization of the delivered therapeutic structure also needs to be considered.12,13 A number of useful tumour-selective antibody targets have been exploited in cancer medicine, including several B-cell antigens (such as CD20, CD19, CD22, CD70) for the treatment of haematological malignancies, and HER2 and EGFR for the treatment of certain solid tumours, including breast, gastric, colon, lung, and head and neck cancers.14–17 These validated targets are potentially attractive antigens for further exploitation with immunoconjugate therapy. Indeed, several of the available monoclonal antibodies currently employed in clinical practice have been further developed as immunoconjugates.7,9 As discussed above, an ideal target for immunotherapy is highly and specifically expressed on malignant cells, rapidly and completely internalized by the targeted cells, and its internalization would adversely affect cell proliferation, survival and resistance to the cytotoxic agent. No such ideal target has been identified yet, but several immunoconjugate-based approaches have been developed. For example, HER2 is highly expressed in a subset of human breast cancers and the protein has an important role in the proliferation and survival of HER2-amplified breast cancer cells. This antigen can be rapidly internalized; however, HER2 is also expressed by some normal cells, such as cardiac myocytes, and therefore does not represent a highly specific target. Nonetheless, HER2 can be targeted effectively by immunoconjugates by linking an antibody to HER2 to a therapeutic entity, for highly specific delivery to the HER2 expressing tumour cell.10,18,19
Immunoconjugate structure
Three components are essential for the construction of an effective immunoconjugate: the antibody, the therapeutic payload and the linker that joins the two components (Figure 1).20 These components are described below.
Figure 1.
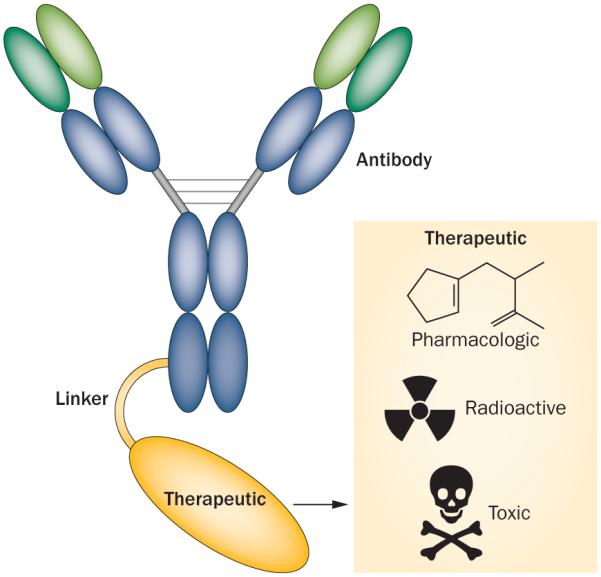
Immunoconjugate modules. Immunoconjugates are composed of three different components: a monoclonal antibody, a therapeutic entity, and a linker that joins the antibody and the therapeutic agent. Using this basic immunoconjugate model, different therapeutic entities, including pharmacologics, radioactive agents, and toxins, can be delivered to the cancer cells.
The antibody moiety
Antibodies of the IgG class are most commonly employed for cancer therapy, and their structural properties have been well described.6,15 The binding of antibodies to their target antigens occurs via the Fab domains, whereas the Fc domain interacts with cellular Fc receptors that regulate antibody-directed binding to immune and endothelial cells (Figure 2a).
Figure 2.

Immunoconjugates structure. a | IgG is the typical antibody component of the immunoconjugate. The Fa domain of the antibody is responsible for the antigen binding. Through the use of various display technologies, the Fv molecules, composed of variable-heavy (VH) and variable-light (VL) chains, can be designed to achieve high antigen affinity. The Fc-region glycan, through point mutation or glycan modification, can be modified in immunoconjugate synthesis to link a therapeutic molecule for delivery to a cancer cell. b | Enzymatically cleaved antibody fragments such as Fab' or F(ab')2 and their recombinantly designed counterparts are smaller than typical IgG, and thus are cleared more rapidly. c | Genetically engineered antibody-based molecules based on a single chain Fv (scFv) composed of the variable domains (VH and VL) of an antibody, combined or altered to construct a wide array of recombinant molecules including (scFV)2, minibody, diabody, triabody and tetrabody. Many of these constructs are small enough to achieve highly selective tumour targeting, with potentials for both therapy and imaging applications.
The identification of novel cancer antigens has been accompanied by the design of antibodies that can be used in immunoconjugate-based therapy and other therapies.21 Some of these antibodies retain canonical IgG structures, but several binding structures have been designed and employed to facilitate highly selective tumour targeting. The biodistribution properties of conventionally structured IgG molecules have been extensively described, and even the most-effective targeting molecules exhibit surprisingly low selectivity of tumour binding.11 In fact, in general no more than 30% of the injected dose of targeting IgG antibodies is retained by tumours in experimental murine models.22,23 In clinical trials, the delivery of radiolabelled IgG immunoconjugates is typically less than 0.1% of injected dose per gram of tumour, owing to the fact that the molecule is diluted in a much larger volume of distribution.24,25 To address this issue, many groups have developed high-affinity antibody variants to improve antibody off-rates and prolong selective tumour retention. However, such an approach had surprisingly modest effects, in part because antibodies could be prevented from penetrating tumours owing to their successful binding to the target antigen, a concept known as binding site barrier.26 Two other aspects to consider are that high-affinity antibodies are rapidly internalized and degraded, and that high concentrations of shed tumour antigens in the intratumoral space can block antibody binding and retard the targeting of the tumour cell.27–32 Hence, during the design process, the affinity of antibody–antigen binding requires careful consideration depending on the nature of the target.33 Achieving high antibody–antigen affinity can result in an increase in therapeutic activity and thus reduce the amount of treatment needed.21 This design process has been facilitated by the use of phage display, yeast display and even the use of non-antibody scaffold approaches.10,14,34–37 Typically, these display technologies create single-chain Fv molecules composed of paired variable-heavy and variable-light chains, joined together by an amino acid linker (Figure 2a). The prediction of the key residues necessary for high-affinity binding is difficult, and might ultimately only be possible through random mutagenesis.21,38,39 Nonetheless, computational design has improved prediction and selection of these residues.40,41
Antibody properties other than affinity can influence tumour-targeting efficacy. For example, antibody size and valence can regulate tumour antigen engagement, in vivo biodistribution and in vivo tumour targeting. Proteins with molecular weight >55 kDa, such as IgG (that typically are 150 kDa) do not undergo first pass elimination through the kidneys and thus exhibit prolonged in vivo biodistribution.23,42 Enzymatically cleaved fragments, such as Fab and their recombinantly designed counterparts, are smaller and are cleared more rapidly (Figure 2b).43 This smaller size has the consequences of improving the selectivity of tumour targeting due to more rapid antibody clearance from the body, but at the expense of quantitative retention of the antibody fragment. While such selectivity would be highly desirable, quantitative delivery is required for efficacy, and the vast majority of successful immunoconjugates have been based on molecular species larger than 55 kDa. Nonetheless, a wide array of recombinant small high-selective antibody-based molecules with potential for both therapy and imaging applications, have been generated. One such platform, the diabody, has been created by covalently linking two recombinant single-chain Fv fragments to create a divalent binding structure that can target tumours with high in vivo specificity (Figure 2c).44 Building on these observations, a HER2-specific diabody has been generated to support treatment selection and monitoring of therapeutic effects by immuno-PET imaging.45,46
The linker
Immunoconjugates can be effective only if the toxic payload is successfully delivered to the cancer cell, thus necessitating the design of a linker that liberates the therapeutic entity after delivery and efficiently releases the payload.16,47 Some linkers, such as thioether bonds, require the complete degradation of the antibody once the immunoconjugate has been internalized into the cancer cell (Figure 3a).33 More commonly, the linkers are designed to allow the cleavage (at the linker site) upon delivery to the target cancer cell and release of the antibody from the therapeutic entity, (Figure 3b).33 A number of cleavable linkers have been developed, taking advantage of differences between the plasma and intracellular environments to allow cleavage upon delivery to the target cell. For example, hydrazone bond linkers remain stable in the neutral pH of the plasma, but are degraded once internalized and exposed to the acidic pH of lysosomes.47 Disulfide bonds can similarly function as linkers that remain stable in the plasma but are readily cleaved intracellularly, owing to the higher concentrations of intracellular glutathione, which reduces the disulfide bridge in the linker.49 Peptides, including specific protease sites, can also function as linkers, which are then cleaved by intracellular lysosomal proteases.50
Figure 3.

Internalization process. a | Immunoconjugates with non-cleavable linkers are internalized once the antibody binds to the antigen on the cell surface. Release of the therapeutic entity requires complete digestion of the antibody inside the lysosome. A thioether bond (see inset) is an example of a non-cleavable linker. b | Immunoconjugates with cleavable linkers are internalized once the antibody binds to the antigen on the cell surface. Release of the therapeutic is achieved by cleavage of the linker. Hydrazone bonds and disulfide bonds (see inset) are examples of commonly employed cleavable linkers. c | Immunoconjugates with cleavable linkers in some cases will undergo cleavage extracellularly, once bound to the cancer cell antigen, with the therapeutic entity undergoing internalization after being released.
The functionality of these different linker types necessitates the development of immunoconjugates that have a high rate of cellular internalization in addition to a strong antigen binding affinity. Immunoconjugates that are poorly internalized can still be effective if their linkers are cleaved in the extracellular space of the tumour environment, with a subsequent high rate of cellular internalization of the released effector agent (Figure 3c).33 Of note, this approach of extracellular cleavage necessitates consideration of toxic effects associated with any systemic leakage of the effector molecules once they are released from the immunoconjugates, but before cancer cell internalization. When effective, this method eliminates the need for high rates of internalization. Hence, linkers that are specifically designed to be extracellularly cleaved might be preferable in some circumstances, as they can overcome the limitations of a poorly internalized immunoconjugate.33
The importance of linker stability and antibody specificity for immunoconjugate efficacy has become clear. For example, the agent BR96-DOX employed an acid-labile hydrazine bond and thioether bond to link the epithelial tumour cell-directed antibody BR96 to doxorubicin.51 Patients treated in a phase I study of this agent did not experience the toxicity typically associated with doxorubicin administration, suggesting that high doses of doxorubicin could be delivered to a tumour in this manner. However, linker instability and antibody targeting of the healthy gut and pancreas, with consequent toxicity in the targeted organs prompted the discontinuation of this agent’s development, providing important lessons for the design of future immunoconjugates.52
The therapeutic moiety
The therapeutic entity employed in immunoconjugate construction must be highly toxic to the cancer cell to which it is delivered. In theory, any number of effective anticancer drugs could be linked to the antibody. However, it is sensible to use potent anticancer agents that induce unacceptable host toxicity when used alone. Clinically-tested conjugates are listed in Tables 1–3. The different types of therapeutic entities are described below.
Table 1.
Pharmacological immunoconjugates
| Agent | Diseases treated | Pharmacologic agent |
Linker | Antibody type | Developmental stage |
|---|---|---|---|---|---|
| Brentuximab vedotin7,10,59,61 |
Relapsed/refractory Hodgkin lymphoma or Anaplastic large-cell lymphoma |
MMAE | Internally cleaved | Mouse monoclonal IgG1 targeting CD30 |
FDA approved, under investigation for other disease types |
|
| |||||
| Trastuzumab emtansine62–66 |
Pretreated, HER2-positive breast cancer |
DM1 | Noncleavable linker |
Humanized IgG1 targeting HER2/neu |
FDA approved in 2013 |
|
| |||||
| Gemtuzumab ozogamicin69,70 |
Relapsed acute myeloid leukaemias, in patients over 60 years old and not candidates for further cytotoxics |
Calicheamicin | Internally cleaved |
Humanized IgG4 targeting CD33 |
Accelerated FDA approval in 2000; withdrawn in 2010 due to fatal toxicity concerns |
|
| |||||
| Inotuzumab ozogamicin69,71–74 |
B-cell non-Hodgkin lymphoma or Acute lymphoid leukaemias |
Calicheamicin | Internally cleaved |
Humanized IgG4 targeting CD22 |
In phase I/II investigation In phase III investigation |
|
| |||||
| Glembatumumab vedotin78,81 |
Metastatic breast cancer or Unresectable melanoma |
MMAE | Internally cleaved |
Humanized IgG2 targeting transmembrane glycoprotein NMB |
In phase II investigation |
|
| |||||
| Lorvotuzumab mertansine84 |
Ovarian, Merkel cell, and small-cell lung cancers |
DM1 | Internally cleaved |
Humanized IgG1 targeting CD56 |
In phase I investigation |
|
| |||||
| DNIB0600A86 | NaPi2b-expressing ovarian and NSCLC |
MMAE | Internally cleaved |
Humanized IgG targeting NaPi2b |
In phase II investigation |
|
| |||||
| ABT-41485 | Glioblastoma | MMAF | Internally cleaved |
Humanized IgG targeting EGFR |
Phase II investigation pending |
|
| |||||
| SAR341975–77 | B-cell non-Hodgkin lymphoma or Acute lymphoid leukaemia |
DM4 | Internally cleaved |
Humanized IgG1 targeting CD19 |
In phase II investigation |
Abbreviations: MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; NMB, non-metastatic B; NSCLC, non small cell lung cancer.
Table 3.
Toxin immunoconjugates in development
| Agent | Diseases treated | Toxin | Linker | Antibody type | Developmental stage |
|---|---|---|---|---|---|
| 260F9104,105 | Breast cancer | Ricin A | Internally cleaved |
Mouse monoclonal IgG1 targeting Mr 55,000 breast cancer antigen |
Not pursued due to toxicity |
|
| |||||
| Moxetumomab pasudotox110 |
Relapsed/refractory hairy-cell leukaemia Relapsed/refractory ALL ALL/NHL |
Pseudomonas exotoxin A |
Internally cleaved |
Fv antibody fragment targeting CD-22 |
Phase III trial Phase I/II trial Phase I trial |
|
| |||||
| SS1P55 | Mesothelioma | Pseudomonas exotoxin A |
Internally cleaved |
Fv antibody fragment targeting mesothelin |
Early phase trials |
Abbreviations: ALL, acute lymphocytic leukaemia; NHL, non-Hodgkin lymphoma.
Immunoconjugate therapeutic entities
Immunoconjugates can be divided into three different classes, based upon the mechanism of action of the therapeutic agent conjugated to the antibody. The first class, which is perhaps the most thoroughly explored, employs a pharmacological agent as the therapeutic, allowing for the delivery of a high concentration of a potentially cytotoxic agent directly to the tumour site. The second class consists of radionucleotides.53 Several such agents have been developed, but have not been used widely despite their clinical efficacy. Finally, catalytic toxins have been explored extensively with promising results across several diseases, including hair cell leukemia and mesothelioma.54,55
Pharmacological immunoconjugates
Approved pharmacological immunoconjugates
Pharmacological agents that block tubulin polymerization (such as auristatin and maytansinoids) or induce DNA strand scission (such as calicheamicin) have been successfully explored as therapeutic immunoconjugates in cancer therapy.56 The two most efficacious immunoconjugates to date are brentuximab vedotin and trastuzumab emtansine, which are both approved by the FDA for the treatment of certain lymphomas and certain breast cancers, respectively. Brentuximab vedotin is composed of an anti-CD30 antibody linked via a cleavable peptide linker to the antimicrotubule agent monomethyl auristatin E (MMAE). This immunoconjugate is internalized when the antibody binds the CD30 antigen and enters into the lysosome, where MMAE is cleaved and released. MMAE can then block tubulin polymerization and interrupt the microtubule network, inducing apoptosis of the CD30-expressing tumour cell.57 This agent has proven clinical benefit.4 In a study of patients with anaplastic large-cell lymphoma refractory to at least one prior therapy, treatment with brentuximab vedotin demonstrated an 86% objective response rate, with 57% of patients achieving complete remission; median duration of these responses were 12.6 months and 13.2 months, respectively.4 A separate study evaluated the effect of brentuximab vedotin in patients who had relapsed Hodgkin disease despite autologous stem-cell transplantation. An overall response rate of 75% was observed, with 34% of patients achieving a complete remission, with a median duration of 20.5 months.8 Brentuximab vedotin received FDA approval for the treatment of these two haematological malignancies, and continues to be investigated for use in other diseases and lines of treatment, including CD30-expressing germ-cell solid tumours.58–60 A phase I/II study of an unconjugated anti-CD30 antibody in the treatment of Hodgkin lymphoma and anaplastic large-cell lymphoma demonstrated that the treatment was well-tolerated, but yielded few clinical responses.61 The efficacy and safety results obtained with the use of the anti-CD30–MMAE immunoconjugate were sufficient to warrant FDA approval after phase II clinical trials,7,8 demonstrating the value of repurposing a clinically ineffective antibody.
Trastuzumab emtansine is an immunoconjugate of the anti-HER2 monoclonal antibody trastuzumab and the maytansinoid DM1, which inhibits tubulin polymerization.62–64 Phase II studies demonstrated robust clinical activity of trastuzumab emtansine in patients with HER2-positive breast cancer who had been heavily pretreated, including with anti-HER2 therapy.65,66 These results were further explored in the EMILIA phase III study, which compared the efficacy of trastuzumab emtansine to the combination of lapatinib and capecitabine in patients with HER2-positive breast cancer whose disease was refractory to trastuzumab therapy.9 This study demonstrated a superior progression-free survival (PFS, 9.6 months versus 6.4 months) and an improved overall survival (30.9 months versus 25.1 months) for the trastuzumab emtansine arm when compared with the lapatinib–capecitabine arm. Based on these results, in February 2013, the FDA approved the use of trastuzumab emtansine for patients with HER2-positive late stage metastatic breast cancer.67
Pharmacological immunoconjugates in development
The use of trastuzumab emtansine demonstrates the potential for repurposing a monoclonal antibody with validated single-agent anticancer efficacy as a delivery vehicle for a second anticancer entity, in order to achieve continued anticancer effect beyond the mechanism of action of the antibody itself. Extending this concept, a number of novel immunoconjugate entities are in various phases of development that exploit known specific surface markers as targets for therapeutic delivery. Table 1 summarizes the pharmacological immunoconjugates that have been approved for use or are undergoing investigation. The immunoconjugate gemtuzumab ozogamicin, which links calicheamicin to a CD33 targeting antibody, received accelerated FDA approval in 2000 for the treatment of patients with relapsed acute myeloid leukemia.68 This accelerated approval was granted because the agent, in early-phase studies, appeared to be well tolerated and effective, whereas conventional chemotherapy is often poorly tolerated and ineffective.69 However, in a regulatory agency-required post-approval study, gemtuzumab ozogamicin did not demonstrate clinical benefit and was associated with higher rates of toxicity compared to conventional chemotherapy.69,70 Gemtuzumab ozogamacin was withdrawn in 2010. Other calicheamicin immunoconjugates have been developed and tested. For example, inotuzumab ozogamicin links calicheamicin to a humanized anti-CD22 antibody.69 This agent is being studied in a phase III multicentre trial in patients with relapsed or refractory acute lymphoblastic leukaemia.71 Additionally, two early phase studies are evaluating the safety and activity of this agent in patients with B-cell non-Hodgkin lymphoma, and a phase I/II study is recruiting patients to evaluate the use of this immunoconjugate as part of a preparative regimen for stem-cell transplantion.72–74
In 2014, SAR3419, an immunoconjugate of a CD19 antibody and the maytansinoid DM4 was reported to be well-tolerated and active for the treatment of relapsed or refractory B-cell non-Hodgkin lymphoma.75 Phase II studies assessing SAR3419 in the treatment of relapsed or refractory diffuse large B-cell lymphoma and in relapsed or refractory acute lymphocytic leukaemia are ongoing.76,77
A number of additional antibody targets in solid tumours are also actively being investigated in the development of pharmacological immunoconjugates. Glycoprotein non-metastatic b (GPNMB) is overexpressed in certain solid tumours, including triple-negative breast cancer and melanoma, and is the target for the immunoconjugate glembatumumab vedotin.78 In a phase II study using glembatumumab vedotin, patients with metastatic, heavily pre-treated, breast cancer experienced a comparable overall survival to investigator’s choice of chemotherapy.79 However, in the subset of patients with triple-negative breast cancer and high GPNMB expression, a statistically significant improvement in overall survival was observed with the use of glembatumumab vedotin over investigator’s choice of chemotherapy (10.0 months versus 5.5 months, P = 0.003).79,80 Activity and tolerability were also reported for the use of this immunoconjugate in patients with advanced-melanoma, with preliminary data suggesting an increase in PFS in patients with tumours with a high expression of GPNMB.81 Glembatumumab vedotin is currently being tested in a phase II trial enrolling patients with triple-negative breast cancer.82
Several other antibody–drug immunoconjugates have shown promise, but remain in early phases of development. For example, lorvotuzumab mertansine, which links DM1 with an anti-CD56 targeting antibody, is being tested in a phase I study in patients with advanced-stage ovarian, Merkel cell, and small-cell lung cancers.83 Early reports for the clinical activity of this immunoconjugate in CD56-expressing Merkel-cell tumours led to its orphan status approval by the FDA for the treatment of Merkel-cell carcinoma.84 An ongoing study of the ABT-414 immunoconjugate, linking auristatin to a EGFR antibody shows promise in patients with glioblastoma.85 Furthermore, studies of DNIB0600A, which links MMAE to an antibody targeting the membrane transporter NPT2B, are underway to evaluate its efficacy following encouraging phase I data in the treatment of ovarian and non-small-cell lung cancers.86 As these and other studies develop, they will add to the therapeutic toolbox while contributing to the development of future pharmacological immunoconjugates.
Radionucleotide immunoconjugates
Approved radionucleotide immunoconjugates
Effective immunoconjugates can also be obtained by linking an antibody to radionucleotide, such as radioactive iodine.87 The radioimmunoconjugate ibritumomab tiuxetan showed important antitumour activity in patients with rituximab-refractory follicular lymphoma.88 Ibritumomab is a monoclonal antibody that binds the CD20 antigen. Tiuxetan is a linker molecule that allows the antibody to deliver the radioactive isotope Yttrium-90 to the tumour cell. In patients with non-Hodgkin lymphoma who were refractory to therapy with rituximab and who were heavily pretreated with systemic therapies, the administration of ibritumomab tiuxetan produced an overall response rate of 74%, with 15% of patients achieving a complete response.88 The median time to progression (TTP) reported was 6.8 months, and reached 8.7 months among those patients whose disease demonstrated response to treatment. Ibritumomab tiuxetan has been approved by the FDA for the management of these haematological malignancies for over a decade.89 It is currently under investigation as a treatment for other malignancies as well, including aggressive B-cell lymphomas and as a part of a transplant preparative regimen.90,91 Despite its efficacy, this agent has not been used widely in the clinic, in part because of the complexity of its administration and owing to the efficacy of unconjugated rituximab.
Another radionucleotide immunoconjugate approved by the FDA, iodine tositumomab, encountered unusual challenges that prompted its manufacturer to remove it from the market in February 2014. Developed in the late 1990s, iodine tositumomab delivers radioactive 131-iodine to CD20-expressing tumour cells via an anti-CD20 monoclonal antibody, and is effective in the treatment of follicular lymphoma.92 Reported median survival with this agent is 22.8 months.92 However, also in this case, the practitioner familiarity with the use of rituximab and the complicated administration of this radioimmunoconjugate impacted negatively on its use.93 Medical oncologists needed to refer patients to a nuclear medicine pharmacy for administration of the single dose of the radioimmunoconjugate. Additional problems included a fragile supply chain for the radioactive component from Canada to the USA and inadequate Medicare reimbursement for the cost of the expensive agent. Ultimately, the lack of a practical niche for this immunoconjugate led to its discontinuation.
Radionucleotide immunoconjugates in development
While radionucleotide immunoconjugate management of haematological malignancies has demonstrated clinical efficacy, parallel success in solid tumour management has been slow. To date, no radionucleotide immunoconjugates have been approved for solid tumour management, or are beyond phase II clinical testing as monotherapies.94 Solid tumours are characteristically less radiosensitive than haematological cancers. Radiolabelled antibodies typically do not deliver adequate radiation to be therapeutically valuable in this setting, suggesting that intrinsic antitumour activity of the antibody component of the radioimmunoconjugate might be necessary to achieve therapeutic activity in solid tumours.95 One important way to overcome this limitation might be through the combination with other therapies. In a xenograft model, Sharkey et al.96 demonstrated that a radionucleotide and pharmacological immunoconjugate combination could improve efficacy while avoiding additional toxicity. This work has led to phase II and III clinical trials of radioimmunotherapy combined with conventional chemotherapy or radiation therapy, which are currently ongoing.97,98
In addition to the development of new agents and novel combinations, a number of approaches to optimize radionucleotide immunoconjugate therapy have been explored. One particular approach employs a pretargeting strategy with an unlabelled antibody to localize the tumour;99,100 a radionucleotide–hapten complex is subsequently administered, which reacts with the pre-targeted antibody to concentrate the radionucleotide to the tumour site. Preclinical evaluation of this method demonstrated promising results when targeting CD-20.99 However, in a phase II study of patients with metastatic colorectal cancer treated with pre-targeted radioimmunotherapy based on a biotin–streptavidin binding system, results were disappointing. Both haematological and nonhaematological adverse events were observed, with 32% of patients experiencing grade 3–4 diarrhea; the overall response rate was only 8%.100
Efforts to optimize the pre-targeting approach are ongoing, and are expanding with the investigation of other radioisotopes.101 The work of Brechbiel and colleagues has focused on the use of targeted α-therapy, in which α-particles are delivered by appropriate vectors to tumour cells.102 Exquisitely cytotoxic and exhibiting a short path length, the α-particles are particularly suited in the context of minimal residual disease or micrometastases.102 Limitations to the use of targeting α-therapy include high costs and radionucleotide availability, which could be addressed in the future with the development of additional sources and more-efficient production, but also through improved isotope delivery.103 Table 2 summarizes the radionucleotide immunoconjugates that have been approved for use or are undergoing investigation. In the wake of the discontinuation of iodine tositumomab production, it remains unclear if such agents will have a greater use in the treatment of human cancers. Nevertheless, from the clinical point of view, several effective therapies have been developed for lymphoma, and future radioimmunoconjugates might yet find appropriate niches for their use.
Table 2.
Radionucleotide immunoconjugates
| Agent | Diseases treated | Radionucleotide | Linker | Antibody type | Developmental stage |
|---|---|---|---|---|---|
| Ibritumomab tiuxetan88,89 |
Relapsed/refractory B-cell non-Hodgkin lymphoma |
Yttrium-90 or Indium-111 |
Not cleaved | Mouse monoclonal IgG1 targeting CD20 |
FDA approved in 2002 |
|
| |||||
| Iodine tositumomab92,93 |
Follicular lymphoma | Iodine-131 | Not cleaved |
Mouse monoclonal IgG2 targeting CD20 |
FDA approved in 2003 Discontinued by manufacturer in 2014 |
|
| |||||
| 90Y-DOTA-biotin with NR-LU-10 pretargeting antibody100 |
Colorectal cancer | Yttrium-90 | NA | Mouse monoclonal IgG2 targeting Ep-CAM (expressed by epithelial tumours) |
Proof of concept, proved too toxic and ineffective |
|
| |||||
| Targeted α-therapy99,100 |
NA | α-Particle | NA | NA | In preclinical development |
Abbreviation: NA, not applicable.
Toxin immunoconjugates
Immunoconjugates that employ a catalytic toxin as the therapeutic entity are interesting, relatively under-explored and potentially useful. As a systemic treatment, an intact bacterial or other pathological toxin cannot be readily used as a cancer treatment, owing to the significant collateral toxicity to healthy tissue. However, if such molecules can be reliably delivered to the tumour with high selectivity, then those toxins have the potential to be extremely effective. Several toxin immunoconjugates are under development; however, to date, none has achieved FDA approval for standard use.
One early attempt to develop a therapeutic immunotoxin consisted of the 260F9 antibody directed against a breast cancer associated antigene conjugated to a recombinant ricin A chain. In a phase I study, this ricin A-260F9 immunoconjugate was used for the treatment of patients with metastatic breast cancer. These patients experienced severe toxicities, including fluid overload and sensorimotor neuropathies. Such adverse events were attributed to the antibody targeting the Schwann cells, resulting in demyelination.104,105 These results illustrate the delicate balance that must be considered when targeting extraordinarily potent toxins using tumour targeting antibodies. The identification of tumour-specific antigens by modern sequencing technologies creates important new opportunities for the safe use of toxins in cancer therapy.
Pastan and colleagues chemically and genetically linked Pseudomonas exotoxin A and its fragments to antibodies targeting CD22 antigens in B-cell malignancies, CD25 in T-cell malignancies, and mesothelin in several epithelial tumours.106,107 In the attempt to bring the toxin-immunotherapy into the clinical setting, Kreitman et al.54 completed an early phase trial of moxetumomab pasudotox to treat patients with relapsed and/or refractory hairy-cell leukaemia by delivering Pseudomonas exotoxin A to the CD22 expressing leukaemic cells.54 This treatment was well-tolerated without the development of dose-limiting toxicities. An 86% overall response rate was observed, and 46% of patients achieved a complete remission.54 These encouraging results have moved forward into a phase III multicentre study that is currently enrolling patients in order to confirm promising findings of the efficacy of moxetumomab pasudotox in the management of this disease.108 Treatment with moxetumomab pasudotox is being explored in other disease types as well, such as relapsed and/or refractory acute lymphocytic leukaemia and for the treatment of children, adolescents, and young adults with acute lymphocytic leukaemia or non-Hodgkin lymphoma.109,110
In a separate trial, the SS1P anti-mesothelin immunotoxin is being administered to patients with mesothelioma along with T-cell and B-cell depleting chemotherapies to minimize anti-toxin antibody responses.55 Major tumour regressions were reported in 3 of the 10 patients with mesothelioma who were treated with this regimen, supporting the further development of this therapy in solid tumours.55 Given the number of other mesothelin-expressing epithelial cancers—cancers of the pancreas, ovaries, lungs, and bile ducts—that remain in need of new, more-effective treatments, additional uses for this agent are anticipated. A list of toxin-immunoconjugates under development is summarized in Table 3.
Despite their potency, in some cases, immunotoxins or other therapies are ineffective owing to cellular resistance to apoptosis, mediated by endogenous prosurvival proteins.111 Traini et al.112 have targeted the apoptosis regulator Bcl-2 using the protein kinase inhibitor enzastaurin in conjunction with the immunotoxin SS1P, resulting in a synergistic effect on cellular apoptosis caused by a complete loss of Bcl-2 as well as other mechanisms.113,114 Although enzastaurin likely targets other kinases and some off-target effect is possible, this synergistic effect merits continued investigation.
In general, the use of a non-mammalian toxin as the therapeutic entity presents some challenges. The toxin is recognized by the host immune system as foreign, and induces the production of neutralizing antibodies, limiting the doses that can be administered to the patient, which potentially impacts efficacy.115 In the mesothelioma trial described above, this induction of neutralizing antibodies was partially mitigated by the use of the immunosuppressive agents cyclophosphamide and pentostatin.55 This approach to immunotoxin therapy that neutralizes the host immune system adds complexity and potential toxicity to the host. Recently, major B-cell epitopes of the SS1 immunotoxin have been identified and silenced by introducing point mutations into the toxin structure. The resulting mutated toxins retain their antitumour potencies but have less immunogenicity.115,116 Other efforts to suppress toxin-directed immune responses have been reported.117–119 For example, five patients treated with the immunotoxin LMB-1 also underwent immunosuppression with rituximab, which had no effect in suppressing their immune-response to the toxin. In an approach analogous to the silencing of B-cell epitopes, engineering out T-cell epitopes on the toxin is also being investigated, and suggests that selective modification of identified amino acid residues can allow for creation of immunotoxins that are highly cytotoxic, but do not stimulate host T-cell response.120 Pegylation has also been developed as a modification to decrease the immunogenicity of therapeutic proteins; however, pegylation of immunotoxins might also reduce the anticancer efficacy of the toxin.121,122 These novel approaches require further investigation, and when optimized, have the potential to facilitate the use of toxin immunoconjugate agents without the risk of added morbidity from the use of the additional cytotoxic agents necessary to suppress the host’s immune response.
Future outlooks
At the time of this writing this article, ClinicalTrials.gov lists 15 ongoing or pending clinical trials investigating immunoconjugate therapy for solid and haematological tumours. This number is surprisingly low, given the impressive recent clinical results of antibody–drug conjugates. The scientific community and pharmaceutical industry have been rather slow to recognize the potential of immunoconjugates. Countless tumour-directed antibodies have been produced in academia and industry, and many could be repurposed as immunoconjugates targeting cancer-associated or even tumour-specific targets on malignant cells that can be readily identified owing to the advances in genomic medicine.
Clinical testing of several additional anticancer therapies is underway that expands and applies the immunoconjugate concept in novel ways. Marks et al.123 developed a method to select optimal antibodies for internalization by cancer cells. They then used this method to create highly specific antibodies coupled to liposomes containing doxorubicin, which demonstrated significant antitumour effects in mice.124 Exploration of this method in human subjects has potential merit. The use of antibody-loaded liposomes to deliver a therapeutic entity has also been explored as a novel method of immunoconjugate development. In animal models, these immunoliposomes are stable, and have demonstrated delivery of the cytotoxic chemotherapeutic agents to the tumour—some example include the delivery of vinca-alkaloids using anti-HER2 targeting antibodies and doxorubicin using the anticancer antibody 2C5.125,126 Immunoliposomes also have been employed as delivery vehicles targeting the p53 tumour suppressor.127 The therapeutic potential of immunoliposomes have also been explored in some phase I studies. For example, an anti-EGFR antibody has been used to deliver doxorubicin to solid tumours via a liposome carrier.128 In a separate study, p53 nanoparticles have been delivered to solid tumours, restoring p53 tumour suppression function.129 In both studies, treatment was well tolerated and clinical activity was observed, warranting further investigation.
A different approach for achieving highly specific tumour cell targeting is represented by the development of antibody binding domain-modified oncolytic viruses that target tumours, where viral tropisms are regulated in such a way that they only target and are toxic to the tumour cells.130,131 These antibody-retargeted viruses have the potential to serve as alternative payload delivery systems. Adenoviral-based vector delivery systems have also demonstrated similar potential. For example, an adenoviral vector conjugated to etoposide has been designed to target pancreatic cancer that overexpress c-erb-B2, and was found to effectively inhibit tumour growth in a mouse model.132
Recently, efforts to develop better immunoconjugates for cancer therapy have focused on bispecific antibodies, in which the antibody binds both a tumour antigen and the therapeutic entity. The antibody is administered first, allowed time to saturate its binding of the tumour antigen and the excess unbound antibody is cleared from the bloodstream; the therapeutic entity (radioactive immunoconjugates have been explored in preclinical models) is then introduced and binds to the antibody in a high concentration.133 Fournier and Schirrmacher have extended this approach by employing the Newcastle Disease Virus as a tumour cell vaccine to serve as the therapeutic entity. The vaccine consists of patient-derived tumour cells infected by the virus which, when anchored to the tumour via the bispecific antibody, redirects the host immune response against the tumour.134 The dual functionality of bispecific antibodies has been considered analogous to immunocytokines, which essentially function as immunoconjugates in cancer treatment.135 Unconjugated cytokine therapy may be limited because cytokines are unable to localize to the tumour itself.136 However, if delivered specifically to a tumour in an immunoconjugate construct, the cytokine serves as the therapeutic entity in these molecules, recruiting immune-mediated tumour cells that are targeted by the host immune system. The antitumour activity of cytokine immunoconjugates has been demonstrated with IL-2, TNF-α, and IL-12.137–139 Cytokine immunoconjugates remain in phase I and II clinical trial development.140–146
Many highly effective anticancer drugs, radioisotopes, and toxins are limited in their use or are not used at all due to the comorbid collateral damage that they cause. Current data with immunoconjugates clearly demonstrate that such agents can be repurposed when modified and targeted by antibodies. In contrast to current versions of targeted therapy, the resulting immunoconjugate–drug toxicity properties can be focused on malignant cells, limiting comorbidities and overcoming many of the drug resistance mechanisms that nowadays limit the durability of targeted agents, such as BRAF inhibitors.32
Conclusions
Several important therapeutic immunoconjugates have been developed to date, employing both pharmacological and radioisotope effector agents, with toxin conjugates under development as well. In some cases, the antibodies used in the development of these immunoconjugates have involved the repurposing of existing antibodies. The feasibility and utility of this repurposing suggests that an important next step might be the linkage of various cytotoxins to existing molecules, both antibody and effector, whose clinical development was aborted.
In addition to this repurposing of antibodies already in clinical use, the future of immunoconjugate therapy will rely on the development of new techniques in other areas of cancer medicine that can drive the identification of new targets and the consequent de novo production of new antibodies. These techniques involve the selection of antigens to serve as cancer-specific antibody targets as well as optimization of the internalization of the antibodies by the tumour cells. Some tumour-specific antigens that could be targeted by immunoconjugates might emerge from intense genomic analysis of tumours; for example, from the identification of new driver mutations.147 The tumour-specific protein products of these mutations would represent attractive targets for the construction of an immunoconjugate, as toxicity to normal cells would be minimized. Other approaches also include the use novel radiotracers, for example linked to a HER2 receptor antibody for positron emission tomography imaging. This approach has enabled the identification of antibody structural features that influence cellular internalization and subsequent tumour catabolism, and are associated with variability in antibody affinity for the antigen.148,149 Finally, antibody therapy can be incorporated into vaccination strategies. Vaccine-induced immune responses can be enhanced by trastuzumab therapy in breast cancer patients.150,151 This approach might have additional utility in augmenting immunoconjugate therapy that uses trastuzumab or other monoclonal antibodies as the delivery vehicle of the therapeutic entity to the tumour site.
The therapeutic potential for immunoconjugates represents a ‘call to arms’ for cancer researchers. As the appreciation for clonal heterogeneity increases, it will be important to develop new therapies to overcome the drug resistance that could arise as a consequence of the variability in antigen expression as well as other resistance mechanisms. Immunoconjugates have the potential to target this variability, both with the repurposing of available resources and with the development of new agents.
Key points.
■ Immunoconjugates represent an efficient strategy to exploit the tumor-targeting properties of monoclonal antibodies and the tumor-killing properties of various types of toxins
■ Immunoconjugates are composed of an antibody, a linker and a therapeutic entity
■ Pharmacologics, radioisotopes, and toxins can all be used as therapeutic entity in an immunoconjugate; all three types of therapeutic entities are actively being investigated
■ Future immunoconjugate development will entail the repurposing of existing tumor-targeting antibodies as immunoconjugate delivery vehicles, as well as the design of antibodies with novel tumor-targeting specificities
Review criteria.
We searched for original articles focusing on immunoconjugate therapy in MEDLINE and PubMed that were published between 1985 and 2014. We also searched for ongoing clinical trials in Clinicaltrials.gov. The search terms we used were “immunoconjugate”, “monoclonal antibody”, and “cancer”. All papers identified were English-language full-text papers. We also searched the reference lists of identified articles for further papers.
Acknowledgements
L.M.W. is supported by National Institute of Health Awards CA51880 and CA50633.
L.M.W. is an advisor and a consultant for Abbvie, Celldex Pharmaceuticals, Jounce Pharmaceuticals, and Merrimack Pharmaceuticals; he has stock options in Cellde Pharmaceuticals, Jounce Pharmaceuticals and Merrimack Pharmaceuticals and his research has been funded by Symphogen AG.
Footnotes
Competing interests
The other authors declare no competing interests.
Author contributions
B.G.S., D.A. and L.M.W. researched data for this article, reviewed and edited the manuscript before submission, provided substantial contribution to discussion of content and wrote the manuscript.
References
- 1.Paul Ehrlich—Biographical. Nobelprize.org. 2014 [online], http://www.nobelprize.org/nobel_prizes/medicine/laureates/1908/ehrlich-bio.html. [Google Scholar]
- 2.Anderson CS, Quinones R, Olson MR. Determining efficacy and side effects from the concurrent use of chemotherapy and radiation therapy for the management of adult solid tumors [abstract] J. Clin. Oncol. 2014;32(Suppl.):e17640. [Google Scholar]
- 3.Van Cutsem E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs CS, et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: results from the BICC-C study. J. Clin. Oncol. 2007;25:4779–4786. doi: 10.1200/JCO.2007.11.3357. [DOI] [PubMed] [Google Scholar]
- 5.Hochster HS, et al. Safety and efficacy of oxaliplatin-fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer (mCRC): results of the TREE-Study. J. Clin. Oncol. 2008;26:3523–3529. doi: 10.1200/JCO.2007.15.4138. [DOI] [PubMed] [Google Scholar]
- 6.Reichert JM, et al. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 7.Pro B, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J. Clin. Oncol. 2012;30:2190–2196. doi: 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- 8.Younes A, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012;30:2183–2189. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verma S, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 11.Teicher BA, Chari RVJ. Antibody conjugate therapeutics: challenges and potential. Clin. Cancer Res. 2011;17:6389–6397. doi: 10.1158/1078-0432.CCR-11-1417. [DOI] [PubMed] [Google Scholar]
- 12.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008;60:1421–1434. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chari RVJ. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 14.Kapoor P, et al. Anti-CD20 monoclonal antibody therapy in multiple myeloma. Br. J. Haematol. 2008;141:135–148. doi: 10.1111/j.1365-2141.2008.07024.x. [DOI] [PubMed] [Google Scholar]
- 15.Yarden Y. The EGFR family and its ligands in human cancer: signaling mechanisms and therapeutic opportunities. Eur. J. Cancer. 2001;37:S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 16.Colcher D, et al. Pharmacokinetics and biodistribution of genetically-engineered antibodies. Q. J. Nucl. Med. 1998;42:225–241. [PubMed] [Google Scholar]
- 17.Wells A. EGF receptor. Int. J. Biochem. Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 18.Colby DW, et al. Development of a human light chain variable domain (V(L)) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J. Mol. Biol. 2004;342:901–912. doi: 10.1016/j.jmb.2004.07.054. [DOI] [PubMed] [Google Scholar]
- 19.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 20.Cao Y, et al. Single-chain antibody-based immunotoxins targeting Her2/neu: design optimization and impact of affinity on antitumor efficacy and off-target toxicity. Mol. Cancer Ther. 2012;11:143–153. doi: 10.1158/1535-7163.MCT-11-0519. [DOI] [PubMed] [Google Scholar]
- 21.Ducancel F, Muller BH. Molecular engineering of antibodies for therapeutic and diagnostic purposes. mAbs. 2012;4:445–457. doi: 10.4161/mabs.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiner LM, et al. I. A human tumor xenograft model of therapy with a bispecific monoclonal antibody targeting c-erbB-2 and CD16. Cancer Res. 1993;53:94–110. [PubMed] [Google Scholar]
- 23.Yokota T, et al. Microautoradiographic analysis of the normal organ distribution of radioiodinated single-chain Fv and other immunoglobulin forms. Cancer Res. 1993;53:3776–3783. [PubMed] [Google Scholar]
- 24.Adams GP, et al. Optimization of in vivo tumor targeting in SCID mice with divalent forms of 741F8 anti-c-erbB-2 single-chain Fv: effects of dose escalation and repeated i.v. administration. Cancer Immunol. Immunother. 1995;40:299–306. doi: 10.1007/BF01519629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams GP, et al. Enhanced tumor specificity of 741F8–1 (sFv’)2, an anti-c-erbB-2 single-chain Fv dimer, mediated by stable radioiodine conjugation. J. Nucl. Med. 1995;36:2276–2281. [PubMed] [Google Scholar]
- 26.Saga T, et al. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. Proc. Natl Acad. Sci. USA. 1995;92:8999–9003. doi: 10.1073/pnas.92.19.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams GP, et al. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998;58:485–490. [PubMed] [Google Scholar]
- 28.Zhang Y, Pastan I. High shed antigen levels within tumours: an additional barrier to immunoconjugate therapy. Clin. Cancer Res. 2008;14:7981–7986. doi: 10.1158/1078-0432.CCR-08-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams GP, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61:4750–4755. [PubMed] [Google Scholar]
- 30.Ackerman ME, Pawlowski D, Wittrup KD. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Mol. Cancer Ther. 2008;7:2233–2240. doi: 10.1158/1535-7163.MCT-08-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer A, et al. Radioimmunoguided surgery in colorectal cancer using a genetically engineered anti-CEA single-chain Fv antibody. Clin. Cancer Res. 2000;5:1711–1719. [PubMed] [Google Scholar]
- 32.Singh R, Zhang Y, Pastan I, Kreitman RJ. Synergistic antitumor activity of anti-CD25 recombinant immunotoxin LMB-2 with chemotherapy. Clin. Cancer Res. 2012;18:152–160. doi: 10.1158/1078-0432.CCR-11-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasquetto MV, Vecchia L, Covini D, Digilio R, Scotti C. Targeted drug delivery using immunoconjugates: principles and applications. J. Immunother. 2011;34:611–628. doi: 10.1097/CJI.0b013e318234ecf5. [DOI] [PubMed] [Google Scholar]
- 34.Marks JD, et al. Human antibody fragments specific for human blood group antigens from a phage display library. Biotechnology. 1993;11:1145–1149. doi: 10.1038/nbt1093-1145. [DOI] [PubMed] [Google Scholar]
- 35.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 36.Griffiths AD, Duncan AR. Strategies for selection of antibodies by phage display. Curr. Opin. Biotechnol. 1998;1:102–108. doi: 10.1016/s0958-1669(98)80092-x. [DOI] [PubMed] [Google Scholar]
- 37.Feldhaus MJ, Siegel RW. Yeast display of antibody fragments: a discovery and characterization platform. J. Immunol. Methods. 2004;290:69–80. doi: 10.1016/j.jim.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Graff CP, Chester K, Begent R, Wittrop KD. Directed evolution of an anti-carcinoembrynoic antigen sc Fv with a 4-day monovalent dissociation half-time at 37°C. Protein Eng. Des. Sel. 2004;17:293–304. doi: 10.1093/protein/gzh038. [DOI] [PubMed] [Google Scholar]
- 39.Dubreuil O, et al. Fine tuning of specificity of an anti-progesterone antibody by first and second sphere residue engineering. J. Biol. Chem. 2005;280:24880–24887. doi: 10.1074/jbc.M500048200. [DOI] [PubMed] [Google Scholar]
- 40.Clark LA, et al. Affinity enhancement of an in vivo matured therapeutic antibody using structure-based computational design. Protein Sci. 2006;15:949–960. doi: 10.1110/ps.052030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat. Biotechnol. 2007;25:1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int. Immunol. 2003;15:187–195. doi: 10.1093/intimm/dxg018. [DOI] [PubMed] [Google Scholar]
- 43.Covell DG, et al. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab')2, and Fab' in mice. Cancer Res. 1986;46:3969–3978. [PubMed] [Google Scholar]
- 44.Adams GP, et al. Prolonged in vivo tumour retention of a human diabody targeting the extracellular domain of human HER2/neu. Br. J. Cancer. 1998;77:1405–1412. doi: 10.1038/bjc.1998.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu AM. Engineered antibodies for molecular imaging of cancer. Methods. 2014;65:139–147. doi: 10.1016/j.ymeth.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olafsen T, Sirk SJ, Olma S, Shen CK, Wu AM. ImmunoPET using engineered antibody fragments: fluorine-18 labeled diabodies for same-day imaging. Tumour Biol. 2012;33:669–677. doi: 10.1007/s13277-012-0365-8. [DOI] [PubMed] [Google Scholar]
- 47.Sanderson RJ. In vivo drug-linker stability of an anti-CD30 dipeptide-liked auristatin immunoconjugate. Clin. Cancer Res. 2005;11:843–852. [PubMed] [Google Scholar]
- 48.Ducry L, Stump B. Antibody-drug congjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 49.Junutula JR, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 50.Koblinski JE, Ahran M, Sloane BF. Unraveling the role of proteases in cancer. Clin. Chim. Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 51.Sjogren HO, et al. Human carcinomas in athymic mice and rats and syngeneic rat carcinomas in immunocompetant rates. Cancer Res. 1997;57:4530–4536. [PubMed] [Google Scholar]
- 52.Saleh MN, et al. Phase I trial of the anti-lewis Y drug immunoconjugate BR96-doxorubicin in patients with lewis Y-expressing epithelial tumors. J. Clin. Oncol. 2000;18:2282–2292. doi: 10.1200/JCO.2000.18.11.2282. [DOI] [PubMed] [Google Scholar]
- 53.Wong JYC, et al. Pilot trial evaluating an 123I-labeled 80-kilodalton engineered anticarcinoembryonic antigen antibody fragment in patients with colorectal cancer. Clin. Cancer Res. 2004;10:5014–5021. doi: 10.1158/1078-0432.CCR-03-0576. [DOI] [PubMed] [Google Scholar]
- 54.Kreitman RJ, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox in patients with hairy cell leukemia. J. Clin. Oncol. 2012;30:1822–1828. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hassan R, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci. Transl. Med. 2013;5:208ra147. doi: 10.1126/scitranslmed.3006941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dosio F, Brusa P, Cattel L. Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components. Toxins. 2011;3:848–883. doi: 10.3390/toxins3070848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sutherland MS, et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem. 2006;281:10540–10547. doi: 10.1074/jbc.M510026200. [DOI] [PubMed] [Google Scholar]
- 58.US Department of Health and Human Services Brentuximab vedotin information. US Food and Drug Administration. 2012 [online], http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm287672.htm. [Google Scholar]
- 59.Ujjani C, Cheson BD. The current status and future impact of targeted therapies in non-Hodgkin lymphoma. Expert Rev. Hematol. 2013;6:191–203. doi: 10.1586/ehm.13.6. [DOI] [PubMed] [Google Scholar]
- 60.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01851200?term=antibody+drug+conjugate+solid+tumors&rank=6. [Google Scholar]
- 61.Ansell SM, et al. Phase I/II study of an anti-CD30 monoclonal antibody in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J. Clin. Oncol. 2007;25:2764–2769. doi: 10.1200/JCO.2006.07.8972. [DOI] [PubMed] [Google Scholar]
- 62.LoRusso PM, Weiss D, Guardino E, Grish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011;17:6437–6447. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 63.Lewis Phillips GD, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 64.Barok M, Tanner M, Koninki K, Isola J. Trasuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011;13:R46. doi: 10.1186/bcr2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burris HA, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011;29:398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 66.Krop IE, et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trasuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J. Clin. Oncol. 2012;30:3234–3241. doi: 10.1200/JCO.2011.40.5902. [DOI] [PubMed] [Google Scholar]
- 67.US Department of Health and Human Services Ado-trastuzumab emtansine. US Food and Drug Administration. 2013 [online], http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm340913.htm. [Google Scholar]
- 68.Bross PF, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001;7:1490–1496. [PubMed] [Google Scholar]
- 69.Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011;17:6417–6427. doi: 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- 70.Petersdorf S, et al. Preliminary results of Southwest Oncology Group Study S0106: an international intergroup phase 3 randomized trial comparing the addition of gemtuzumab ozogamicin to standard induction therapy versus standard induction therapy followed by a second randomization to post-consolidation gemtuzumab ozogamicin versus no additional therapy for previously untreated acute myeloid leukemia [abstract] Blood. 2009;114:a790. [Google Scholar]
- 71.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01564784?term=inotuzumab+ozogamicin&recr=Open&rank=5. [Google Scholar]
- 72.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01535989?term=inotuzumab+ozogamicin&recr=Open&rank=3. [Google Scholar]
- 73.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01562990?term=inotuzumab+ozogamicin&recr=Open&rank=4. [Google Scholar]
- 74.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01664910?term=inotuzumab+ozogamicin&recr=Open&rank=1. [Google Scholar]
- 75.Ribrag V, et al. A dose-escalation study of SAR3419, an anti-CD19 antibody maytansinoid conjugate, administered by intravenous infusion once weekly in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2014;20:213–220. doi: 10.1158/1078-0432.CCR-13-0580. [DOI] [PubMed] [Google Scholar]
- 76.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01440179?term=SAR3419&rank=3. [Google Scholar]
- 77.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01472887?term=SAR3419&rank=1. [Google Scholar]
- 78.Maric G, Rose AA, Annis MG, Siegel PM. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. Onco. Targets Ther. 2013;6:839–852. doi: 10.2147/OTT.S44906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Celdex Therapeutics Final Data from Celldex Therapeutic’s CDX-011 phase 2 study in metastatic breast cancer supports overall survival benefit in patients with high GPNMB expression. 2012 [online], http://ir.celldex.com/releasedetail.cfm?ReleaseID=725688. [Google Scholar]
- 80.Naumovski L, Junutula JR. Glembatumumab vedotin, a conjugate of an anti-glycoprotein non-metastatic melanoma protein B mAb and monomethyl auristatin E for the treatment of melanoma and breast cancer. Curr. Opin. Mol. Ther. 2010;12:248–257. [PubMed] [Google Scholar]
- 81.Hamid O, et al. Frequent dosing and GPNMB expression with CDX-011, an antibody-drug conjugate, in patients with advanced melanoma. J. Clin. Oncol. 2010;28:15s. [Google Scholar]
- 82.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01997333?term=glembatumumab&rank=1. [Google Scholar]
- 83.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT00346385?term=antibody+drug+conjugate+solid+tumors&rank=11. [Google Scholar]
- 84.Woll P. Abstract B237: Clinical experience of IMGN901 (BB-10901, huN901-DM1) in patients with Merkel cell carcinoma (MCC) Mol. Cancer Ther. 2009;8:B237. [Google Scholar]
- 85.Gan HK, et al. A phase I study evaluating ABT-414 in combination with temozolamide for subjects with recurrent or unresectable glioblastoma. J. Clin. Oncol. 2014;32:5s. [Google Scholar]
- 86.Burris HA, et al. A phase I study of DNIB0600A, an antibody-drug conjugate targeting NaPi2b, in patients with non-small cell lung cancer or platinum-resistant ovarian cancer. J. Clin. Oncol. 2014;32:5s. [Google Scholar]
- 87.Marx JL. In: A Revolulation in Biotechnology. Marx JL, editor. The Press Syndicate of the University of Cambridge; 1989. pp. 145–158. [Google Scholar]
- 88.Witzig TE, et al. Treatment with ibritumomab tiuxetan radioimmunotherapy in patients with rituximab-refractory follicular non-Hodgkin’s lymphoma. J. Clin. Oncol. 2002;20:3262–3269. doi: 10.1200/JCO.2002.11.017. [DOI] [PubMed] [Google Scholar]
- 89.US Department of Health and Human Services Ibritumomab tiuxetan product approval information. US Food and Drug Administration. 2009 [online], http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm093379.htm. [Google Scholar]
- 90.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01434472?term=Ibritumomab+tiuxetan&recr=Open&rank=3. [Google Scholar]
- 91.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01686165?term=Ibritumomab+Tiuxetan&recr=Open&rank=7. [Google Scholar]
- 92.Kaminski MS, et al. Pivotal study of iodine I 131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin’s lymphomas. J. Clin. Oncol. 2001;19:3918–3928. doi: 10.1200/JCO.2001.19.19.3918. [DOI] [PubMed] [Google Scholar]
- 93.Timmerman L. Why good drugs sometimes fail: the Bexxar story. Xconomy. 2013 [online], http://www.xconomy.com/national/2013/08/26/why-good-drugs-sometimes-fail-in-the-market-the-bexxar-story/ [Google Scholar]
- 94.Steiner M, Neri D. Antibody-radionucleotide conjugates for cancer therapy: historical considerations and new trends. Clin. Cancer Res. 2011;17:6406–6416. doi: 10.1158/1078-0432.CCR-11-0483. [DOI] [PubMed] [Google Scholar]
- 95.Song H, Sgouros G. Radioimmunotherapy of solid tumors: searching for the right target. Current Drug Deliv. 2011;8:26–44. doi: 10.2174/156720111793663651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sharkey RM, Karacay H, Govindan SV, Goldenberg DM. Combination radioimmunotherapy and chemoimmunotherapy involving different or the same targets improves therapy of human pancreatic carcinoma xenograft models. Mol. Cancer Ther. 2011;10:1072–1081. doi: 10.1158/1535-7163.MCT-11-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01956812?term=Clivatuzumab&rank=1. [Google Scholar]
- 98.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01125085?term=radioimmunotherapy+solid&rank=1. [Google Scholar]
- 99.Press OW, et al. A comparative evaluation of conventional and pretargeted radioimmunotherapy of CD-20 expressing lymphoma xenografts. Blood. 2001;98:2535–2543. doi: 10.1182/blood.v98.8.2535. [DOI] [PubMed] [Google Scholar]
- 100.Knox SJ, et al. Phase II trial of Yttrium-90-DOTA-biotin pretargeted by NR-LU-10 antibody/streptavidin in patients with metastatic colorectal cancer. Clin. Cancer Res. 2000;6:406–414. [PubMed] [Google Scholar]
- 101.Park SI, et al. Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: a preclinical model toward optimal consolidation therapy to eradicate minimal residual disease. Blood. 2010;116:4231–4239. doi: 10.1182/blood-2010-05-282327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baidoo KE, Yong K, Brechbiel MW. Molecular pathoways: targeted α-particle radiation therapy. Clin. Cancer Res. 2013;19:530–537. doi: 10.1158/1078-0432.CCR-12-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim YS, Brechbiel MW. An overview of targeted alpha therapy. Tumour Biol. 2012;33:573–590. doi: 10.1007/s13277-011-0286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gould BJ, et al. Phase I study of an anti-breast cancer immunotoxin by continuous infusion: report of a targeted toxic effect not predicted by animal studies. J. Natl Cancer Inst. 1989;81:775–781. doi: 10.1093/jnci/81.10.775. [DOI] [PubMed] [Google Scholar]
- 105.Weiner LM, et al. Phase I evaluation of an anti-breast cancer monoclonal antibody 260F9-recombinant ricin A chain immunoconjugate. Cancer Res. 1989;49:4062–4067. [PubMed] [Google Scholar]
- 106.Fitzgerald DJ, Wayne AS, Kreitman RJ, Pastan I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res. 2011;71:6300–6309. doi: 10.1158/0008-5472.CAN-11-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chaudhary VK, et al. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature. 1989;339:394–397. doi: 10.1038/339394a0. [DOI] [PubMed] [Google Scholar]
- 108.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01829711?term=moxetumomab+pasudotox&rank=1. [Google Scholar]
- 109.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01891981?term=moxetumomab+pasudotox&rank=2. [Google Scholar]
- 110.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT00659425?term=moxetumomab+pasudotox&rank=4. [Google Scholar]
- 111.Bogner C, et al. Immunotoxin BL22 induces apoptosis in mantle cell lymphoma (MCL) cells dependent on Bcl-2 expression. Br. J. Haematol. 2009;148:99–109. doi: 10.1111/j.1365-2141.2009.07939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Traini R, et al. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of Pseudomonas exotoxin-based proteins to the cell cytosol. Mol. Cancer Ther. 2010;9:2007–2015. doi: 10.1158/1535-7163.MCT-10-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Amundson SA, et al. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000;60:6101–6110. [PubMed] [Google Scholar]
- 114.Mattoo AR, Pastan I, Fitzgerald D. Combination treatmens with the PKC inhibitor, enzastaurin, enhance the cytotoxicity of the anti-mesothelin immunotoxin, SS1P. PLoS ONE. 2013;8:e75576. doi: 10.1371/journal.pone.0075576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu W, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl Acad. Sci. USA. 2012;109:11782–11787. doi: 10.1073/pnas.1209292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Onda M, et al. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl Acad. Sci. USA. 2008;105:11311–11316. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pai LH, et al. Inhibition of antibody response to Pseudomonas exotoxin by 15-deoxyspergualin in mice. Cancer Res. 1990;50:7750–7753. [PubMed] [Google Scholar]
- 118.Gelber EE, et al. Effect of immunosuppressive agents on the immunogenicity and efficacy of an immunotoxin in mice. Clin. Cancer Res. 1998;4:1297–1304. [PubMed] [Google Scholar]
- 119.Hassan R, Williams-Gould J, Watson T, Pai-Scherf L, Pastan I. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB-1. Clin. Cancer Res. 2004;10:16–18. doi: 10.1158/1078-0432.ccr-1160-3. [DOI] [PubMed] [Google Scholar]
- 120.Mazor R, et al. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc. Natl Acad. Sci. USA. 2012;109:E3597–E3603. doi: 10.1073/pnas.1218138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Basu A, et al. Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjugate Chem. 2006;17:618–630. doi: 10.1021/bc050322y. [DOI] [PubMed] [Google Scholar]
- 122.Kreitman RJ. Immunotoxins for targeted cancer therapy. AAPS J. 2006;8:E532–E551. doi: 10.1208/aapsj080363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Becerril B, Poul MA, Marks JD. Towards selection of internalizing antibodies from phage libraries. Biochem. Biophys. Res. Commun. 1999;255:386–393. doi: 10.1006/bbrc.1999.0177. [DOI] [PubMed] [Google Scholar]
- 124.Nielsen UB, et al. Therapeutic efficacy of anti-ErbB2 immunoliposomes targeted by phage antibody selected for cellular endocytosis. Biochim. Biophys. Acta. 2002;1591:109–118. doi: 10.1016/s0167-4889(02)00256-2. [DOI] [PubMed] [Google Scholar]
- 125.Noble CO, et al. Characterization of highly stable liposomal and immunoliposomal formulations of vincristine and vinblastine. Cancer Chemother. Pharmacol. 2009;64:741–751. doi: 10.1007/s00280-008-0923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.El-Bayoumi TA, Tochilin VP. Tumor-targeted nanomedicines: enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin. Cancer Res. 2009;15:1973–1980. doi: 10.1158/1078-0432.CCR-08-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu L, et al. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol. Cancer Ther. 2002;5:337–346. [PubMed] [Google Scholar]
- 128.Mamot C. Tolerability, safety, pharmacokinetics, and efficacy of doxorubicin-loaded anti-EGFR immunoliposomes in advanced solid tumours: a phase 1 dose-escalation study. Lancet Oncol. 2012;13:1234–1241. doi: 10.1016/S1470-2045(12)70476-X. [DOI] [PubMed] [Google Scholar]
- 129.Senzer N, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013;21:1096–1103. doi: 10.1038/mt.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kelly EJ, Hadac EM, Greiner S, Russell SJ. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008;14:1278–1283. doi: 10.1038/nm.1776. [DOI] [PubMed] [Google Scholar]
- 131.Galanis E, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu X, et al. Enhanced pancreatic cancer gene therapy by combination of adenoviral vector expressing c-erb-B2-targeted immunotoxin with a replication-competent adenovirus or etoposide. Hum. Gene Ther. 2010;21:157–170. doi: 10.1089/hum.2009.083. [DOI] [PubMed] [Google Scholar]
- 133.Sharkey RM, Goldenberg DM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J. Clin. 2006;56:226–243. doi: 10.3322/canjclin.56.4.226. [DOI] [PubMed] [Google Scholar]
- 134.Fournier P, Schirrmacher V. Bispecific antibodies and trispecific immunocytokines for targeting the immune system against cancer. BioDrugs. 2013;27:35–53. doi: 10.1007/s40259-012-0008-z. [DOI] [PubMed] [Google Scholar]
- 135.List T, Neri D. Immunocytokines: a review of molecules in clinical development for cancer therapy. Clin. Pharmacol. 2013;5:29–45. doi: 10.2147/CPAA.S49231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pasche N, Neri D. Immunocytokines: a novel class of potent armed antibodies. Drug Discov. Today. 2012;17:583–590. doi: 10.1016/j.drudis.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 137.Carnemolla B, et al. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood. 2002;99:1659–1665. doi: 10.1182/blood.v99.5.1659. [DOI] [PubMed] [Google Scholar]
- 138.Borsi L, et al. Selective targeted delivery of TNF-alpha to tumor blood vessels. Blood. 2003;102:4384–4392. doi: 10.1182/blood-2003-04-1039. [DOI] [PubMed] [Google Scholar]
- 139.Halin C, et al. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat. Biotechnol. 2002;20:264–269. doi: 10.1038/nbt0302-264. [DOI] [PubMed] [Google Scholar]
- 140.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01134250?term=F16-IL2&rank=1. [Google Scholar]
- 141.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT02054884?term=F16-IL2&rank=2. [Google Scholar]
- 142.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT02076646?term=L19-IL2&recr=Open&rank=1. [Google Scholar]
- 143.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT02076633?term=L19-IL2&recr=Open&rank=2. [Google Scholar]
- 144.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT01417546?term=NHS-IL12&rank=1. [Google Scholar]
- 145.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT02076620?term=L19-TNF&recr=Open&rank=1. [Google Scholar]
- 146.US National Library of Medicine ClinicalTrials.gov. 2014 [online], http://clinicaltrials.gov/ct2/show/NCT02076633?term=L19-IL2&recr=Open&rank=2. [Google Scholar]
- 147.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Robinson MK, et al. Quantitative immune-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005;65:1471–1478. doi: 10.1158/0008-5472.CAN-04-2008. [DOI] [PubMed] [Google Scholar]
- 149.Rudnick SI, et al. Influsence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71:2250–2259. doi: 10.1158/0008-5472.CAN-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schlom J. Therapeutic cancer vaccines: current status and moving forward. J. Natl Cancer Inst. 2012;104:599–613. doi: 10.1093/jnci/djs033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Disis ML, et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J. Clin. Oncol. 2009;27:4685–4692. doi: 10.1200/JCO.2008.20.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]