

Abstract
Background
Isepamicin is a weakly toxic but highly active aminoglycoside antibiotic derivative of gentamicin B. Gentamicin B is a naturally occurring minor component isolated from Micromonospora echinospora. 2ʹ-NH2-containing gentamicin C complex is a dominant compound produced by wild-type M. echinospora; by contrast, 2ʹ-OH-containing gentamicin B is produced as a minor component. However, the biosynthetic pathway of gentamicin B remains unclear. Considering that gentamicin B shares a unique C2ʹ hydroxyl group with kanamycin A, we aimed to design a new biosynthetic pathway of gentamicin B by combining twelve steps of gentamicin biosynthesis and two steps of kanamycin biosynthesis.
Results
We blocked the biosynthetic pathway of byproducts and generated a strain overproducing JI-20A, which was used as a precursor of gentamicin B biosynthesis, by disrupting genK and genP. The amount of JI-20A produced in M. echinospora ∆K∆P reached 911 μg/ml, which was 14-fold higher than that of M. echinospora ∆P. The enzymes KanJ and KanK necessary to convert 2ʹ-NH2 into 2ʹ-OH from the kanamycin biosynthetic pathway were heterologously expressed in M. echinospora ΔKΔP to transform JI-20A into gentamicin B. The strain with kanJK under PermE* could produce 80 μg/ml of gentamicin B, which was tenfold higher than that of the wild-type strain. To enhance gentamicin B production, we employed different promoters and gene integration combinations. When a PhrdB promoter was used and kanJ and kanK were integrated in the genome through gene replacement, gentamicin B was generated as the major product with a maximum yield of 880 μg/ml.
Conclusion
We constructed a new biosynthetic pathway of high-level gentamicin B production; in this pathway, most byproducts were removed. This method also provided novel insights into the biosynthesis of secondary metabolites via the combinatorial biosynthesis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-015-0402-6) contains supplementary material, which is available to authorized users.
Keywords: Micromonospora echinospora, Gentamicin B, Metabolic engineering, Artificial biosynthetic pathway
Background
Aminoglycoside antibiotics have been widely used to treat severe bacterial infections; for instance, streptomycin was administered as the first effective anti-tuberculosis agent. Other aminoglycoside antibiotics include 2-deoxystreptamine (2-DOS) containing gentamicin, kanamycin, neomycin, and butirosin. However, the critical resistance mechanism of aminoglycoside antibiotics in pathogens is enzymatic inactivation. As such, semi-synthetic aminoglycosides have been created to overcome pathogen enzymatic inactivation. For example, isepamicin is developed by introducing (S)-3-amino-2-hydroxypropionyl side chains to the 1-amino group of gentamicin B. This side chain can block the modification by various aminoglycoside-modifying enzymes. Thus, isepamicin is highly stable against aminoglycoside-inactivating enzymes [1].
Isepamicin is manufactured from gentamicin B, which is co-produced in Micromonospora echinospora. Gentamicin C complex is produced by M. echinospora as its major product. By contrast, gentamicin A, B, and X are yielded by M. echinospora as minor components [2]. Gentamicin A and X are intermediates of gentamicin C biosynthesis. However, gentamicin B is not an intermediate of the gentamicin C biosynthetic pathway because gentamicin B cannot be biotransformed by M. echinospora into any gentamicin C component [2]. Thus far, the biosynthetic pathway of gentamicin B remains unclear.
The biosynthetic pathway of gentamicin has been elucidated (Fig. 1). The gene cluster of gentamicin has also been cloned [3, 4]. The genes involved in the biosynthesis of pseudodisaccharide paromamine and pseudotrisaccharide gentamicin A2 have been identified [5]. The production of gentamicin X2 from A2 involves four enzymes: oxidoreductase, transaminase, and two methyltransferases [6]. X2 can also be transformed into JI-20A by C-6ʹ dehydrogenase and transaminase [7–9]. Moreover, GenK is the methyltransferase of C-6ʹ in gentamicin, and genK disruption generates a gentamicin C1a-overproducing strain [10–12]. GenP is a phosphotransferase participating in 3ʹ,4ʹ-deoxygenation in the gentamicin biosynthetic pathway [13]. If genP is disrupted, JI-20A,JI-20B, and JI-20Ba accumulate in mutant strains [9]. In this study, genK and genP were disrupted simultaneously to generate strains producing JI-20A, which was designed as a precursor of gentamicin B biosynthesis (Fig. 1).
Fig. 1.
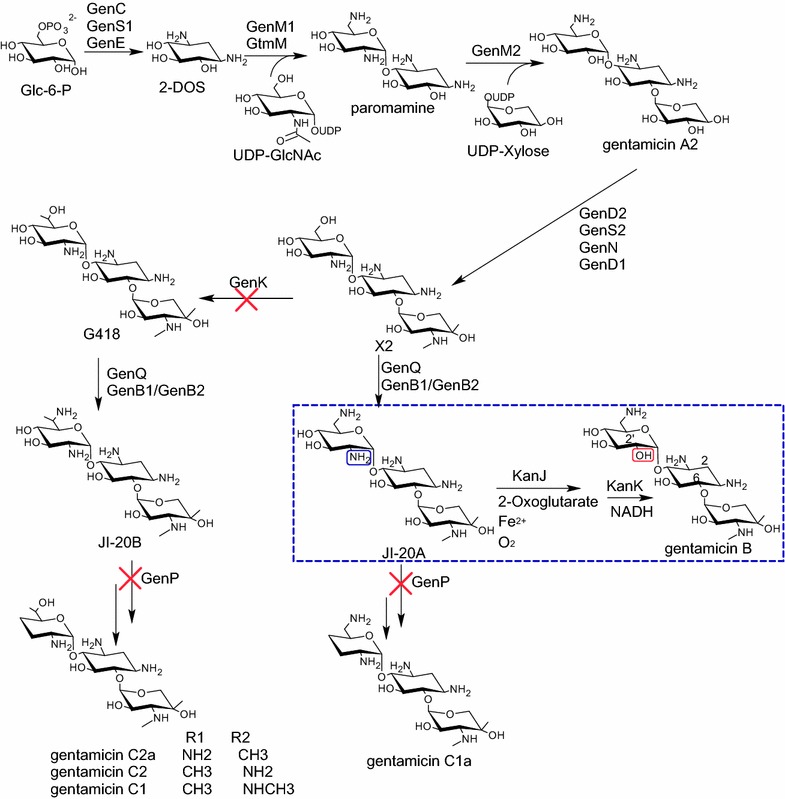
Biosynthetic pathway for gentamicin and the proposed strategy for gentamicin B biosynthetic pathway (inside the dashed line)
The structure of gentamicin B resembles that of both gentamicin C and kanamycin A. Unlike most 2-DOS-containing aminoglycosides, kanamycin A contains a hydroxy group at C2ʹ; gentamicin B shares this unique structure with kanamycin A. The methylated pentose ring of gentamicin B is also similar to that of gentamicin C and is designated as garaosamine. Gentamicin B can be synthesized via a biosynthetic pathway similar to that of kanamycin A on the basis of molecular structure. In the kanamycin A biosynthetic pathway in Streptomyces kanamyceticus, 2ʹ-NH2 is converted into 2ʹ-OH by using KanJ and KanK; in the process, 2-oxoglutarate, Fe2+, O2, and NADH are utilized [14]. We hypothesized that gentamicin B could be biosynthesized from JI-20A, a compound structurally similar to gentamicin B, except NH2 group at C2ʹ (Fig. 1). Thus, JI-20A could be converted into gentamicin B in JI-20A-overproducing strain via two steps catalyzed by KanJ and KanK. Using the assembly and metabolically engineered biosynthesis pathway of gentamicin B, we can eliminate byproducts and improve gentamicin B production.
Results and discussion
Construction of JI-20A-overproducing strain by disrupting genP and genK genes
The original strain can potentially generate gentamicin B; as such, we determined whether gentamicin B is synthesized from the 2ʹ-amino-containing precursor by the KanJ and KanK homologs. However, the genes homologous to kanJ and kanK have yet to be detected in M. echinospora. Considering that the biosynthetic pathway of gentamicin B remains unclear, we aimed to construct an artificial pathway of gentamicin B biosynthesis. JI-20A and gentamicin B are similar in terms of chemical structure except at C2ʹ. JI-20A contains an amino group at C2ʹ, whereas gentamicin B comprises a hydroxyl group at the same position. The structural difference between JI-20A and gentamicin B is similar to the difference between kanamycin A and B. Considering that kanamycin B is converted into kanamycin A by KanJ and KanK, we determined whether KanJ and KanK can also transform JI-20A into gentamicin B.
To generate a JI-20A-producing strain, we simultaneously disrupted the 6ʹ-methyltransferase gene genK and the 3ʹ-phosphotransferase gene genP in this study (Fig. 2). We separately generated genP- and genK-disrupting strains in our previous work [6, 7]. The genP-disrupting strain does not produce gentamicin C complex, which are the main products of the wild-type strain but are the main byproducts in gentamicin B production. In this study, the genK-disrupting plasmid was introduced to M. echinospora ∆P to obtain the double-gene-disrupting strain M. echinospora ∆K∆P (Additional file 1: Figure S1A). The disrupting strain was fermented and its products were analyzed through high-performance liquid chromatography with evaporative light scattering detector (HPLC-ELSD). JI-20B and JI-20Ba were the main products of M. echinospora ∆P but were undetectable in M. echinospora ∆K∆P (Fig. 3). JI-20A was produced up to 911 μg/ml, which was 14-fold higher than that produced by M. echinospora ∆P. We blocked the biosynthesis of gentamicin C1, C1a, C2, C2a, JI-20B, and JI-20Ba and generated a JI-20A-overproducing strain by disrupting genP and genK.
Fig. 2.
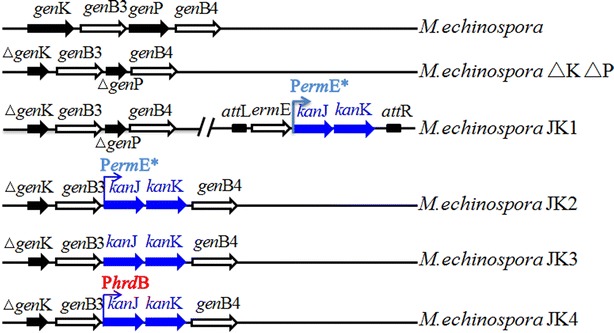
Genotypes of M. echinospora and its recombinant strains
Fig. 3.
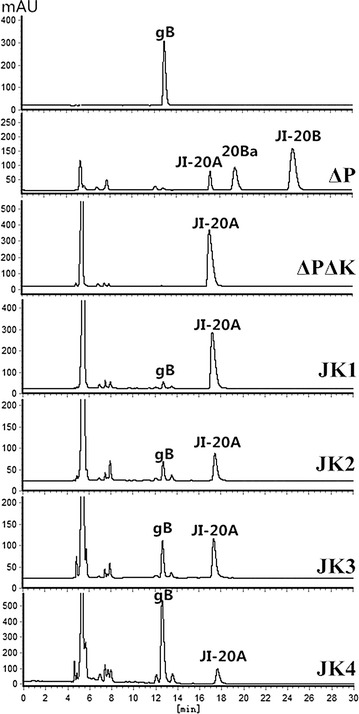
HPLC-ELSD analysis of gentamicin production in fermentations. gB gentamicin B, 20Ba JI-20Ba
Construction of a new gentamicin B biosynthetic pathway through the heterologous expression of kanJ and kanK
The kanJ and kanK genes under the control of the strong promoter PermE* were cloned into the site-specific integration plasmid pEAP1 to construct pSPUJK1. pSPUJK1 was introduced to M. echinospora ∆K∆P through conjugation and exconjugants were selected through erythromycin resistance; as a result, M. echinospora JK1 was generated (Additional file 1: Figure S1B). M. echinospora JK1 was fermented under the same condition used to ferment the wild-type strain. The fermentation products of M. echinospora JK1 were also analyzed through HPLC-ELSD. Compared with the wild-type strain, M. echinospora JK1 produced a new product exhibiting a retention time similar to that of gentamicin B (Fig. 3).
To determine the structure of the new product of M. echinospora JK1, we analyzed the purified product through mass spectrometry. The exact mass of the product was 482.26 (m/z 483.26), which corresponds to gentamicin B (Fig. 4). Furthermore, the protonated fragments were the same as those of the reported mass spectra of gentamicin B [15], that is, the glycoside bond of gentamicin B cleavage formed the fragment b + c (m/z 324.17). The glycoside found at the C6–O of 2-DOS decomposed; thus, the fragments b + c + x (m/z 366.18) were produced. Nevertheless, gentamicin B yields the same molecular weight as that of gentamicin X2, which is another minor component of gentamicin. To verify the structure of the new product, we recorded the corresponding 1H and 13C NMR data. The 1H spectrum of the new product was similar to that of gentamicin B [16] (Additional file 2: Figure S2). Moreover, the 13C NMR data of the new product (Table 1) were identical with the reported NMR data of gentamicin B [17]. MS and NMR data demonstrated that the product from M. echinospora JK1 is gentamicin B.
Fig. 4.
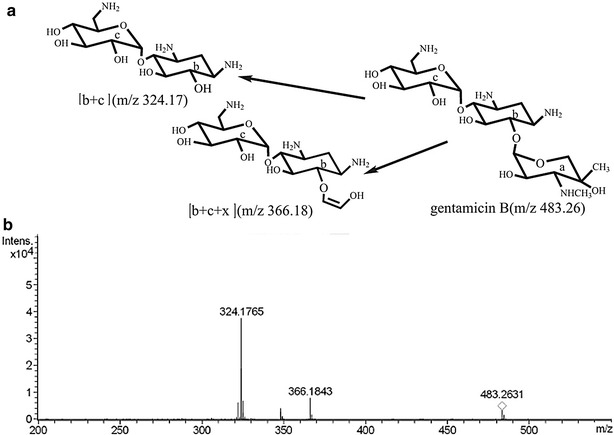
Structural determination of gentamicin B by MS/MS. a Structure of gentamicin B. b Mass spectra of the new product from M. echinosporaJK1
Table 1.
13C NMR spectral data for gentamicin B [16] and the new compound from M. echinospora JK1 (for atom numbering see Fig. 1)
| Atom | Gentamicin B (δ) | New compound (δ) |
|---|---|---|
| 1 | 50.6 | 49.7 |
| 2 | 28.3 | 27.5 |
| 3 | 48.4 | 47.5 |
| 4 | 79.0 | 78.3 |
| 5 | 73.1 | 72.3 |
| 6 | 84.6 | 83.8 |
| 1′ | 96.6 | 95.9 |
| 2′ | 71.7 | 70.6 |
| 3′ | 73.0 | 72.0 |
| 4′ | 71.6 | 69.8 |
| 5′ | 69.5 | 68.6 |
| 6′ | 41.1 | 40.1 |
| 1′′ | 102.0 | 101.2 |
| 2′′ | 67.2 | 67.6 |
| 3′′ | 64.2 | 63.3 |
| 4′′ | 70.8 | 70.6 |
| 5′′ | 68.6 | 66.2 |
| 3′′–N–CH3 | 35.3 | 34.3 |
| 4′′–CH3 | 21.7 | 20.8 |
Although gentamicin B production is greatly improved in M. echinospora JK1 compared with that of the wild-type strain, the yield is only 85 μg/ml in M. echinospora JK1. A large quantity of JI-20A in M. echinospora JK1 was also found in the fermentation broth. Therefore, the expression levels of kanJ and kanK were genetically manipulated to improve gentamicin B production.
Enhancing gentamicin B production through the genetic manipulation of kanJK
The genetic stability of the strains applied in industrial fermentation is very important. However, gentamicin B production in M. echinospora JK1 decreased from 162 μg/ml to 80 μg/ml in five generations of unselected passages (Fig. 5a). We proposed that the instability of M. echinospora JK1 is caused by the homologous recombination between the PermE* upstream of kanJK and the native promoter of ermE, which was used as a selection marker gene in pSPUJK1. In addition, the instability may be caused by chromosomal rearrangements or plasmid elimination from the chromosome, which can occur when a φC31-derived plasmid is used in strains containing multiple pseudo attB sites [18].
Fig. 5.
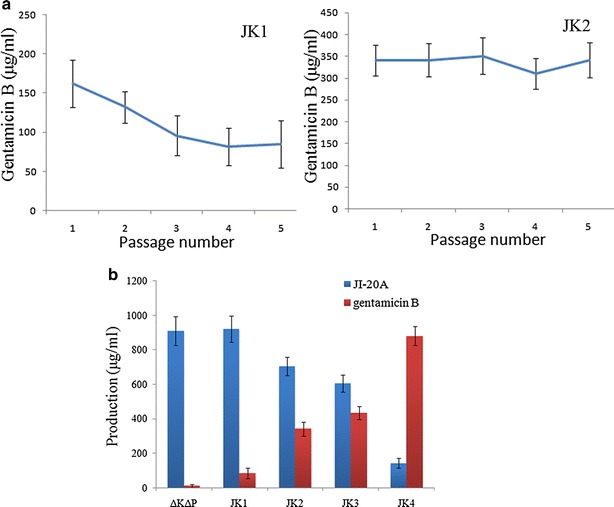
Production and stability analysis of gentamicin B-producing strains. a Stability of gentamicin B production in M. echinosporaJK1 and JK2 strains. b Production analysis of gentamicin B in M. echinospora ΔKΔP and kanJK heterologous expression strains. Error bars represent standard deviations. Samples were analyzed through HPLC and quantified on the basis of the peak areas
To avoid genetic instability, we integrated kanJ and kanK into the chromosome through homologous recombination instead of site-specific insertion. Considering that genP has been disrupted in M. echinospora ∆K∆P, we designed kanJ and kanK to replace genP (Additional file 1: Figure S1C). To reduce JI-20A production and improve gentamicin B production, we placed kanJ and kanK under the control of the strong promoter PermE* (Fig. 2). Moreover, the fermentation broth of the gene replacement strain M. echinospora JK2 was analyzed through HPLC-ELSD. Figure 5a shows that the gentamicin B production by M. echinospora JK2 was more stable than that by M. echinospora JK1. Gentamicin B production increased to 342 μg/ml. However, M. echinospora JK2 produced a considerable quantity of JI-20A.
The native promoter of genP was used to improve gentamicin B production. To eliminate any possibility of polar effects on other genes, we designed kanJK as a replacement of the open reading frame of genP, and no other elements were introduced (Additional file 1: Figure S1D). As such, the native ribosome binding site (RBS) and promoter of genP (or promoter of its operon) were employed by kanJK. Although the regulatory elements of genP have yet to be determined, we proposed that all regulatory elements of genP can be used. In the fermentation of M. echinospora JK3, kanJK was expressed as gentamicin B was produced in JK3. Gentamicin B production also reached 436 μg/ml. PermE*, which exhibits a strong promoter activity, is widely used for the gene expression in Actinomyces. However, this work found that PermE* is weaker than the native promoter of genP.This phenomenon occurred possibly because PermE* originates from Saccharopolyspora erythraea; as such, PermE* cannot be as effective in M. echinospora as in S. erythraea. Another possible cause is the ineffectiveness of the RBS of the construction because the translation initiation of PermE*-kanJ-kanK is weak when analyzed with the RBS Calculator [19].
JI-20A accumulation in the fermentation broths of M. echinospora JK2 and JK3 indicated the inefficiency of KanJ and KanK. Thus, an increase in KanJK expression may enhance their total activity and gentamicin B production. The PhrdB promoter of Streptomyces coelicoloris stronger than PermE* [20]. Therefore, a kanJK under the control of PhrdB was constructed and introduced to M. echinospora ∆K∆P; as a result, a mutant strain M. echinospora JK4 was generated (Additional file 1: Figure S1E).
The HPLC analysis of the fermentation broth of M. echinospora JK4 revealed that gentamicin B production reached 880 μg/ml; by contrast, JI-20A production decreased to 143 μg/ml. Figure 5b shows that gentamicin B production increased by 2.5-fold when the PhrdB promoter was used compared with that when PermE* was used. This result confirmed that the expression level of KanJK was enhanced by replacing the promoter PermE* with PhrdB.
The genome analysis of Actinomyces revealed the presence of numerous gene clusters encoding secondary metabolites [21]. Actinomyces can be metabolically engineered to overproduce native metabolites or analogs. With the development of synthetic biological tools, improved bioinformatics tools of metabolic engineering, and enhanced sensitivity and sophistication of analytical methods, secondary metabolite production is feasible in various cellular factories [22].
Conclusions
We successfully established an artificial biosynthetic pathway to achieve a high-level production of gentamicin B. The genes genK and genP were disrupted in M. echinospora to producethe JI-20A, which is the precursor of gentamicin B. JI-20Aproduction in the gene-disrupting strain M. echinospora ΔKΔP reached 911 μg/ml, which was 14-fold higher than that of M. echinospora ∆P. We disrupted the biosynthesis of gentamicin C1, C1a, C2, C2a, JI-20B, and JI-20Ba by disrupting genP and genK. The removal of byproducts in fermentation broth will be beneficial to purification because antibiotic purification is costly and time consuming.
An artificial pathway for the conversion of JI-20A to gentamicin B was constructed through the heterologous overexpression of kanJ and kanK in M. echinospora ΔKΔP. The kanJK-overexpressing strain under the control of PermE* can produce 80 μg/ml gentamicin B, which was tenfold higher than that of the wild-type strain. Different promoters and gene integration combinations were investigated to improve gentamicin B production. When the PhrdB promoter was used and kanJ and kanK were integrated in the genome through gene replacement, gentamicin B was produced as a major product with a maximum yield of 880 μg/ml.These results confirmed that microbiological strains can be engineered through the metabolic engineering of an intrinsic biosynthetic pathway and the introduction of exogenous genes to produce high yields of target products.
Methods
Bacterial strains, plasmids, media, and culture conditions
The strains and plasmids used in this work are listed in Table 2. Escherichia coli Top10 was used as the cloning host grown on Luria–Bertani (LB) liquid or solid medium. Liquid ATCC172 was used for the vegetative growth of M. echinospora. The conjugal transfer was performed on MS agar. Solid slanting medium was used for M. echinospora sporulation. The previously described media and culture conditions were used for gentamicin production [8].
Table 2.
Strains and plasmids used in this study
| Strains or plasmids | Relevant characteristic | Reference or source |
|---|---|---|
| Strains | ||
| E. coli TOP10 | F-mcrAΔ(mrr-hsdRMS-mcrBC), φ80lacZΔM15,ΔlacX74, deoR, recA1, araD139Δ(ara-leu)7697, galU, galK, rpsL(StrR), endA1, nupG | Invitrogen |
| E. coli ET12567/pUZ8002 | Methylation defective, strain used in E. coli-streptomyces intergeneric conjugation | [25] |
| S. kanamyceticus | Kanamycin producing strain | CGMCC4.1441 |
| M. echinospora | Wild-type strain, gentamicin C1a, C2, C2a, and C1 producer | ATCC 15835 |
| M. echinosporaΔP | M. echinospora with disrupted genP | [7] |
| M. echinosporaΔKΔP | M. echinospora with disrupted genK and genP | This study |
| M. echinospora JK1 | Heterologous, genome-based expression of kanJ and kanK, with replacement of the native promoter by PermE* in M. echinosporaΔKΔP. | This study |
| M. echinospora JK2 | M. echinosporaΔKΔP + heterologous expression of kanJ and kanK under the promoter PermE*, insertion at genP locus in M. echinospora ΔKΔP | This study |
| M. echinospora JK3 | Heterologous expression of kanJ and kanK under promoter PgenP, insertion at genP locus in M. echinospora ΔKΔP | This study |
| M. echinospora JK4 | Heterologous expression of kanJ and kanK under promoter PhrdB, insertion at genP locus in M. echinospora ΔKΔP | This study |
| Plasmids | ||
| pKC1139 | E. coli-streptomyces shuttle vector, AmR | [26] |
| pSPU241 | pIJ2925 derivative carrying the Streptomyces constitutive promoter PermE* and To terminator, AmpR | [8] |
| pEAP1 | pSET152 carrying ermE, the apramycin resistance-conferring gene aac(3)IV was replaced by the ampicillin resistance-conferring gene bla, AmpR, ErmR | [8] |
| pSPU503 | pKC1139 carrying homologous arms of genK (gacD), used in genK disruption | [12] |
| pJK1 | pEAP1 carrying PermE*-kanJ-kanK, used in generating M. echinospora JK1 | This study |
| pJK2 | pKC1139 carrying homologous arms and kanJK, used in generating M. echinospora JK2 | This study |
| pJK3 | pKC1139 carrying homologous arms and PermE*-kanJ-kanK, used in generating M. echinospora JK3 | This study |
| pJK4 | pKC1139 carrying homologous arms andPhrdB-kanJ-kanK, used in generating M. echinospora JK4 | This study |
Amp R ampicillin resistance, Am R ampramycin resistance, Erm R erythromycin resistance
Construction of kanJ and kanK expression plasmids
DNA isolation and manipulation were performed as described by Sambrook [23]. Additional file 3: Table S1 lists the primers used in this work. The kanJ and kanK genes were amplified from the genomic DNA of S. kanamyceticus. The primers were designed using the biosynthetic gene sequence of kanamycin (GenBank accession number: AJ628422.2) and gentamicin (GenBank accession number:AJ628149.4). The primers Pkanjk-up1 and Pkanjk-down1 were used to amplify a 1.95 kb fragment containing intact kanJ and kanK. The PCR product was digested with HindIII and BamHI and then ligated to pSPU241; thus, pJK241 was generated. The 2.6 kb insert containing PermE*-kanJK-To was recovered as a BglII fragment and then inserted into the same site of pEAP1 to generate pSPUJK1.
The primers Ph1 and Ph2 were used to amplify the downstream homologous arm and to generate a gene replacement vector with kanJK genes under PermE*. The primers Ph3 and Ph4 were utilized to amplify the upstream homologous arm. Pkanjk-up2 and Pkanjk-down2 were also employed to amplify the intact kanJK fragment. Perm-up and Perm-down were used to amplify PermE*. PermE*, kanJK, and the upstream homologous arm fragment were fused through overlap extension PCR in accordance with a previously described procedure [24]. The overlapping PCR product was ligated with pMD18-T (Takara) and digested with XbaI and SmaI; the downstream homologous arm was digested with XbaI and SmaI. Afterward, the digested fragments were ligated to pKC1139; thus, pSPUJK2 was generated.
The primers Ph1 and Ph2 were used to amplify the downstream homologous arm and the primers Ph33 and Ph4 were utilized to amplify the upstream homologous arm to generate a vector for gene replacement. Pkanjk-up3 and Pkanjk-down2 were also used to amplify kanJK. The overlap extension PCR was employed using the primers Ph4 and Pkanjk-down2 to fuse the kanJK fragment and the upstream homologous arm. The overlap PCR product was ligated using pMD18-T(Takara) and then digested with XbaI and SmaI. The downstream homologous arm was digested with XbaI and SmaI. The digested fragments were then ligated to pKC1139; thus, pSPUJK3 was generated.
The primers Ph34 and Ph4 were used to amplify the upstream homologous arm to generate a gene replacement vector with kanJK genes under the strong promoter PhrdB. Pkanjk-up4 and Pkanjk-down2 were utilized to amplify the intact kanJK fragment. Phrd-up and Phrd-down were also used to amplify PhrdB. The upstream homologous arm fragment, PhrdB, and kanJK were then fused through overlap extension PCR. The overlap PCR product was then ligated with pMD18-T and then digested with XbaI and SmaI. The downstream homologous arm was digested with XbaI and SmaI. The digested fragments were ligated to pKC1139. Thus, pSPUJK4 was generated.
Construction of genK-disrupting and kanJK-expressing strains
genK-disrupting and kanJK-expressing plasmids were introduced to M. echinospora through conjugation on MS medium at 28 °C for 24 h. After the medium was spread with 50 mg of apramycin (erythromycin was used for pSPUJK1) and 100 mg of pipemidic acid per liter, incubation was performed at 28 °C for 7 days. Exconjugants were initially selected to determine the apramycin-resistance (the first crossover event) phenotype because pSPU503, pSPUJK2, pSPUJK3, and pSPUJK4 plasmids contain an apramycin-resistance gene; the apramycin-sensitive (the second crossover event) phenotype was then used to isolate the strains via the desired double-crossover homologous recombination event. Genomic DNA was extracted and used as a template DNA in PCR. The PCR products were subjected to DNA sequencing to demonstrate that kanJ and kanK exist in the kanJK-expressing strains.
Antibiotic isolation and analysis
The pH of the culture broth was adjusted to 2.0 by using H2SO4. The acidified broth was agitated for 30 min and then centrifuged at 11,378×g 10 min. The pH of the supernatant was readjusted to 7.0 with NaOH. The pretreated supernatant was centrifuged again at 11,378×g for 10 min. The supernatant was then applied to strongly acidic resin 001 × 7 (Shandong Lukang Record Pharmaceutical Co., Ltd).The bound substances were eluted with 2 mol/L NH4OH. Second cation-exchange chromatography was performed on weakly acidic resin D152 (Shandong Lukang Record Pharmaceutical Co., Ltd). The bound substances were eluted with the gradient elution of NH4OH (from 0.1 to 1.0 mol/L).
The elution from the acidic resin was used as the sample for the reversed-phase HPLC-ELSD analysis in a reverse C18 column at an evaporation temperature of 45 °C, nitrogen pressure of 3.5 bar, and a mobile phase of 0.2 mol/L trifluoroacetatic acid–methanol (97:3) at a flow rate of 0.6 ml/min. Authentic gentamicin B was used as standard. The purified products were analyzed using an LC/MS/MS instrument (Bruker micrOTOF-Q). The mass spectrometer was set in a positive mode. 1H and 13C NMR data were recorded on Bruker AV600 at 600 MHz frequency, and D2O was used as a solvent.
Authors’ contributions
XN performed the experiments, analyzed the primary data, and prepared the manuscript. ZS constructed M. echinospora JK3 and JK4 strains and assisted in fermentation experiments. YG assisted in the MS and NMR data analysis and revised the manuscript. HC assisted in product purification. HX supervised the whole research and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (8127341), Specialized Research Fund for the Doctoral Program of Higher Education (20122134130001), and the General Project of Scientific Research of the Education Department of Liaoning Province (L2014389).
Competing interests
The authors declare that they have no competing interests.
Additional file
10.1186/s12934-015-0402-6 Schematic representation and confirmation of recombination strains. (A) Gene disruption of genK and genP. (B) Expression of kanJ and kanK by site specific intergration. (C) Expression of kanJ and kanK with the promoter PermE*. (D) Expression of kanJ and kanK with the native promoter of genP. (E) Expression of kanJ and kanK with promoter PhrdB.
10.1186/s12934-015-0402-6 1H NMR spectrum of the new compound from kanJK expression strains.
10.1186/s12934-015-0402-6 Sequence of primers used in this study.
Contributor Information
Xianpu Ni, Email: nixianpu126@126.com.
Zhenpeng Sun, Email: 1026625249@qq.com.
Yawen Gu, Email: gyw0636@163.com.
Hao Cui, Email: cuihao102@163.com.
Huanzhang Xia, Email: xiahz612@sina.com.
References
- 1.Petrikkos G, Giamarellou H, Tsagaraki C, Pefanis A. Evaluation of the efficacy and safety of isepamicin in the treatment of various bacterial infections. J Chemother. 1995;7(Suppl 2):161–164. [PubMed] [Google Scholar]
- 2.Testa RT, Tilley BC. Biotransformation, a new approach to aminoglycoside biosynthesis. II. Gentamicin. J Antibiot. 1976;29(2):140–146. doi: 10.7164/antibiotics.29.140. [DOI] [PubMed] [Google Scholar]
- 3.Kharel MK, Basnet DB, Lee HC, Liou K, Moon YH, Kim JJ, Woo JS, Sohng JK. Molecular cloning and characterization of a 2-deoxystreptamine biosynthetic gene cluster in gentamicin-producing Micromonospora echinospora ATCC15835. Mol Cells. 2004;18(1):71–78. [PubMed] [Google Scholar]
- 4.Unwin J, Standage S, Alexander D, Hosted T, Jr, Horan AC, Wellington EM. Gene cluster in Micromonospora echinospora ATCC15835 for the biosynthesis of the gentamicin C complex. J Antibiot. 2004;57(7):436–445. doi: 10.7164/antibiotics.57.436. [DOI] [PubMed] [Google Scholar]
- 5.Park JW, Hong JSJ, Parajuli N, Jung WS, Park SR, Lim SK, Sohng JK, Yoon YJ. Genetic dissection of the biosynthetic route to gentamicin A2 by heterologous expression of its minimal gene set. Proc Natl Acad Sci USA. 2008;105(24):8399–8404. doi: 10.1073/pnas.0803164105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang C, Huang F, Moison E, Guo J, Jian X, Duan X, Deng Z, Leadlay PF, Sun Y. Delineating the biosynthesis of gentamicin X2, the common precursor of the gentamicin C antibiotic complex. Chem Biol. 2015;22(2):251–261. doi: 10.1016/j.chembiol.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo J, Huang F, Huang C, Duan X, Jian X, Leeper F, Deng Z, Leadlay PF, Sun Y. Specificity and promiscuity at the branch point in gentamicin biosynthesis. Chem Biol. 2014;21(5):608–618. doi: 10.1016/j.chembiol.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni X, Sun Z, Zhang H, He H, Ji Z, Xia H. Genetic engineering combined with random mutagenesis to enhance G418 production in Micromonospora echinospora. J Ind Microbiol Biotechnol. 2014;41(9):1383–1390. doi: 10.1007/s10295-014-1479-3. [DOI] [PubMed] [Google Scholar]
- 9.Gu Y, Ni X, Ren J, Gao H, Wang D, Xia H. Biosynthesis of epimers C2 and C2a in the gentamicin C complex. ChemBioChem. 2015;16(13):1933–1942. doi: 10.1002/cbic.201500258. [DOI] [PubMed] [Google Scholar]
- 10.Hong W, Yan L. Identification of gntK, a gene required for the methylation of purpurosamine C-6′ in gentamicin biosynthesis. J Gen Appl Microbiol. 2012;58(5):349–356. doi: 10.2323/jgam.58.349. [DOI] [PubMed] [Google Scholar]
- 11.Karki S, Kim JY, Park SH, Kwon HJ. Gene inactivation study on gntK, a putative C-methyltransferase gene in gentamicin biosynthesis from Micromonospora echinospora. J Korean Soc Appl Biol Chem. 2012;55(3):1–4. doi: 10.1007/s13765-012-2041-5. [DOI] [Google Scholar]
- 12.Li D, Li H, Ni X, Zhang H, Xia H. Construction of a gentamicin C1a-overproducing strain of Micromonospora purpurea by inactivation of the gacD gene. Microbiol Res. 2013;168(3):263–267. doi: 10.1016/j.micres.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Shao L, Chen J, Wang C, Li J, Tang Y, Chen D, Liu W. Characterization of a key aminoglycoside phosphotransferase in gentamicin biosynthesis. Bioorg Med Chem Lett. 2013;23(5):1438–1441. doi: 10.1016/j.bmcl.2012.12.064. [DOI] [PubMed] [Google Scholar]
- 14.Sucipto H, Kudo F, Eguchi T. The last step of kanamycin biosynthesis: unique deamination reaction catalyzed by the alpha-ketoglutarate-dependent nonheme iron dioxygenase KanJ and the NADPH-dependent reductase KanK. Angew Chem Int Ed Engl. 2012;51(14):3428–3431. doi: 10.1002/anie.201108122. [DOI] [PubMed] [Google Scholar]
- 15.Zupančič-Kralj Lucija. Identification of gentamicin impurities by liquid chromatography tandem mass spectrometry. JPharm Biomed Anal. 2009;50(5):1037–1043. doi: 10.1016/j.jpba.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Bérdy J, Pauncz JK, Vajna ZM, Horváth G, Gyimesi J, Koczka I. Metabolites of gentamicin-producing Micromonospora species. I. Isolation and identification of metabolites. J Antibiot. 1977;30(11):945–954. doi: 10.7164/antibiotics.30.945. [DOI] [PubMed] [Google Scholar]
- 17.Nagabhushan TL, Cooper AB, Tsai H, Daniels PJL, Miller GH. The syntheses and biological properties of 1-N-(S-4-amino-2-hydroxybutyryl)-gentamicin B and 1-N-(S-3-amino-2-hydroxypropionyl)-gentamicin B. J Antibiot. 1978;31(7):681–687. doi: 10.7164/antibiotics.31.681. [DOI] [PubMed] [Google Scholar]
- 18.Combes P, Till R, Bee S, Smith MC. The streptomyces genome contains multiple pseudo-attB sites for the (phi)C31-encoded site-specific recombination system. J Bacteriol. 2002;184(20):5746–5752. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27(10):946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du D, Zhu Y, Wei J, Tian Y, Niu G, Tan H. Improvement of gougerotin and nikkomycin production by engineering their biosynthetic gene clusters. Appl Microbiol Biotechnol. 2013;97(14):6383–6396. doi: 10.1007/s00253-013-4836-7. [DOI] [PubMed] [Google Scholar]
- 21.Ōmura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci USA. 2001;98(21):12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab Eng. 2012;14(3):189–195. doi: 10.1016/j.ymben.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3. New York: Cold spring harbor laboratory press; 2001. [Google Scholar]
- 24.Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 2004;32(2):e19–19. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene. 1992;111(1):61–68. doi: 10.1016/0378-1119(92)90603-M. [DOI] [PubMed] [Google Scholar]
- 26.Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene. 1992;116(1):43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]