

Abstract
Myxobacteria, phylogenetically located in the delta division of the Proteobacteria, are well known for characterized social behaviors and large genomes of more than 9 Mb in size. Myxococcus fulvus is a typical species of the genus Myxococcus in the family Myxococcaceae. M. fulvus 124B02, originally isolated from a soil sample collected in Northeast China, is the one and only presently known myxobacterial strain that harbors an endogenous autonomously replicating plasmid, named pMF1. The endogenous plasmid is of importance for understanding the genome evolution of myxobacteria, as well as for the development of genetic engineering tools in myxobacteria. Here we describe the complete genome sequence of this organism. M. fulvus 124B02 consists of a circular chromosome with a total length of 11,048,835 bp and a circular plasmid of 18,634 bp. Comparative genomic analyses suggest that pMF1 has a longstanding sustention within myxobacteria, and probably contributes to the genome expansion of myxobacteria.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0121-y) contains supplementary material, which is available to authorized users.
Keywords: Myxococcus fulvus 124B02, Complete genome sequence, Endogenous plasmid, Autonomously replicate, Genome expansion
Introduction
The gliding Gram-negative myxobacteria are characterized by complex social behaviors, e.g. cells moving on solid surfaces in swarms, preying on other microorganisms in a ‘wolf-like’ pattern, and, when nutrients are depleted, developing into myxospores embodied in fruiting bodies [1, 2]. In addition, myxobacteria are able to produce various secondary metabolites and macromolecule degradation enzymes, not only having potential in applications but also probably working as ecological weapons against other living microorganisms [3–5]. Myxobacteria possess large genomes. For instance, the genomes of Myxococcus xanthus DK1622 and the halotolerant M. fulvus HW-1 are 9.14 Mb [6] and 9.03 Mb [7] in size, while the genomes of Sorangium cellulosum even reach to 13.03 Mb in strain So ce56 [8] and 14.78 Mb in strain So0157-2 [9], respectively. The So0157-2 genome is still the largest one reported in prokaryotes.
Extrachromosomal autonomously replicating genetic materials are normally absent from myxobacterial cells. Up to now, pMF1, originally discovered from M. fulvus 124B02 [10], is still the one and only endogenous plasmid that is able to replicate autonomously in myxobacterial cells. Genome sequencing of M. fulvus 124B02 is thus meaningful for understanding the evolution of myxobacterial genomes, and providing clues for the presence of pMF1 in strain 124B02. Here we report the complete genome sequence and analyses of M. fulvus 124B02.
Organism information
Classification and features
Strain 124B02 was isolated from a soil sample collected in Northeast China [11]. Vegetative cells of the strain are slender rods with tapering ends, 0.6-0.8 × 4–8 μm. The fruiting bodies are spherical or slightly pear-shaped with a diameter of 50–250 μm and a yellow red color. The strain did not grow expansively, but into membranaceous clumps on CYE solid plates. When grown in liquid CYE medium, the cells grew into spherical clumps. Figure 1 shows morphological characteristics of M. fulvus 124B02. The optimal growth pH for strain 124B02 is in the range of 6.8–7.6, and the optimal growth temperature ranges between 26 °C and 32 °C. The predominant fatty acids of M.fulvus 124B02 cells were determined as iso-C15:0 (33.18 %), C16:1 ω5c (20.19 %), iso-C14:0 3-OH (6.27 %), C16:0 (5.79 %) and C14:0 (5.65 %). 2-hydroxy and 3-hydroxy fatty acids are the major hydroxyl fatty acid components of strain 124B02.
Fig. 1.
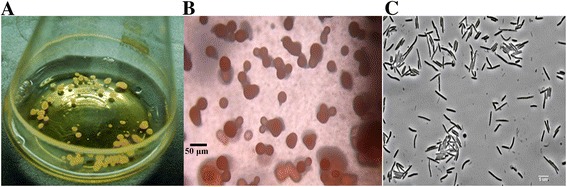
Morphological characteristics of M. fulvus 124B02. a Spherical clumps of strain 124B02 cells cultivated in liquid CYE medium. b Fruiting bodies formed on the TPM development medium. The pictures were taken after six days of incubation under a stereoscopic microscope, Bar = 50 μm. c Vegetative cells, Bar=5 μm
Figure 2 is a phylogenetic tree of the 16S rRNA gene sequences showing the location of M. fulvus 124B02 in the Cystobacterineae suborder of myxobacteria (the GenBank accession number of the 16S rRNA gene sequence of strain 124B02 is EU137665). All three 16S rRNA gene copies in the genome of strain 124B02 are identical, but differ by two nucleotides from the previously published 16S rRNA sequence generated from M.fulvus 124B02 (EU137665). According to the morphological and phylogenetic characteristics, M. fulvus 124B02 was determined as a typical strain of Myxococcus fulvus (Table 1 shows the classification and general features of the strain).
Fig. 2.
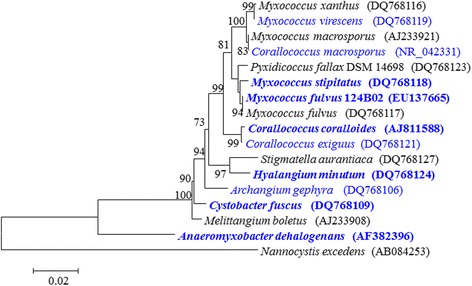
Phylogenetic tree showing the position of M. fulvus 124B02 within the Cystobacterineae. The tree was inferred from 1,445 aligned bases [35, 36] of the 16S rRNA gene sequence under the maximum likelihood criterion [37] and rooted with Nannocystis excedens. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are the supporting values from 1,000 bootstrap replicates. Lineages with type strain genome sequencing projects registered in GOLD [38] are shown in blue and published genomes in bold
Table 1.
Classification and general features of M. fulvus 124B02 according to the MIGS recommendations [39]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS[40] | |
| Phylum Proteobacteria | TAS[41] | ||
| Class Deltaproteobacteria | TAS[42] | ||
| Order Myxococcales | TAS[43] | ||
| Suborder Cystobacterineae | TAS[44] | ||
| Family Myxococcaceae | TAS[45] | ||
| Genus Myxococcus | TAS[46] | ||
| Species Myxococcus fulvus | TAS[47, 48] | ||
| Strain 124B02 | TAS[10] | ||
| Gram strain | Negative | TAS[44] | |
| Cell shape | Rod | TAS[1] | |
| Motility | Motile | TAS[1] | |
| Sporulation | Myxospore | TAS[1] | |
| Temperature range | Mesophile, 25–35 °C | TAS[44] | |
| Optimum temperature | 26–32 °C | TAS[44] | |
| pH range; Optimum | 6.4–8.8; 6.8–7.6 | TAS[47] | |
| Carbon source | Macromolecules such as proteins | TAS[44] | |
| MIGS-6 | Habitat | Soil | IDA |
| MIGS-6.3 | Salinity | Non-halophile | NAS |
| MIGS-22 | Oxygen requirement | Aerobic | TAS[1] |
| MIGS-15 | Biotic relation | Free-living | NAS |
| MIGS-14 | Pathogenicity | Non-pathogen | NAS |
| Biosafety level | 1 | TAS[49] | |
| Isolation | Soil | IDA | |
| MIGS-4 | Geographic location | Changchun, China | IDA |
| MIGS-5 | Sample Collection | 1999 | IDA |
| MIGS-4.1 | Latitude | not reported | |
| MIGS-4.2 | Longitude | not reported | |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | not reported |
aEvidence codes – TAS traceable author statement, i.e. the direct report in the literature, NAS non-traceable author statement, i.e. not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence. These evidence codes are from the Gene Ontology project [19]
Genome sequencing information
Genome project history
This organism was selected for sequencing because of its evolutionary significance as the only presently known myxobacterial strain bearing an endogenous plasmid. The genome project of M. fulvus 124B02 was deposited in the Genome Online Database and the complete genome sequence of strain 124B02 was deposited in GenBank under the accession number of CP006003. A summary of the project information is shown in Table 2.
Table 2.
Genome sequencing project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic libraries: one 454 single read library,one 454 pair end library (3 kb), one Illumina pair end library (350) |
| MIGS-29 | Sequencing platforms | Roche 454 GS FLX, Illumina GAII |
| MIGS-31.2 | Fold coverage | 25.8 × pyrosequence; 86.7 × Illumina |
| MIGS-30 | Assemblers | Newbler assembler V2.3, Phrap, Velvet |
| MIGS-32 | Gene calling method | GeneBank |
| Locus Tag | MFUL124B02 | |
| GenBank ID | CP006003 | |
| GenBank Date of Release | May 6, 2015 | |
| GOLD ID | Gp0043396 | |
| BIOPROJECT | PRJNA203240 | |
| MIGS-13 | Source material identifier | M 206081 |
| Project relevance | The host of plasmid pMF1 |
Growth conditions and genomic DNA preparation
M. fulvus 124B02 was cultivated in the CTT growth medium containing 1 % casitone, 10 mM Tris–HCl, 1 mM KH2PO4-K2HPO4, 8 mM MgSO4, pH 7.6. The cells were harvested by centrifugation after five days of incubation at 30 °C. DNA was extracted from the cell mass using the methods described previously [12] with slight modifications. Briefly, approximately 50–100 mg cell pellets were suspended in 500 μl TE buffer, containing 25 mM Tris–HCl (pH 8.0), 25 mM EDTA, and 2 mg/ml lysozyme. The mixture was incubated at 37 °C for 1 h with periodic gentle inversion for cell lysis. Then, 2.5 μl proteinase K was added to a final concentration of 100 μg/ml, and the mixture was incubated at 37 °C for additional 1 h. The total protein was removed with Tris-saturated phenol-chloroform-isoamyl alcohol (25:24:1, pH 8.0). To precipitate DNA, 0.1 volume of 3 M sodium acetate (pH 5.3) and the same volume of isopropyl alcohol were added to the final supernatant. The DNA pellet was washed with 70 % ethanol twice, air-dried, and dissolved in 50 μl ddH2O.
Genome sequencing and assembly
Genome sequencing and assembly were performed in Shanghai Majorbio Bio-Pharm Technology Co., Ltd. The genome was sequenced with a combination of the Roche 454 GS FLX and Illumina GAII sequencing platforms. The 454 pyrosequencing reads, containing 285.8 Mb draft data, were firstly assembled using the Newbler assembler V2.3, producing 51 contigs in 23 scaffolds. This initial assembly was converted into a phrap assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. The clean data from Illumina GAII sequencing were assembled with Velvet assembler and the consensus sequences were shredded into 800-bp overlapped fake reads, which were assembled with the 454 draft data. In total, the combination of the Illumina and 454 sequencing platforms produced 112.5× coverage of the genome. The final assembly contained 738,315 pyro sequences and 12,776,900 Illumina reads. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Then the Phred/Phrap/Consed software package [13–15] was used for quality assessment. Possible misassembles were corrected by sequencing the cloned bridging PCR fragments. We designed primers for the amplification of 76 gap regions to close gaps and to improve the quality of the finished genome. Gaps between contigs were closed by editing in Consed, PCR amplification and 3730 sequencing. The wrong bases were corrected by comparing with Illumina GAII data after the genome cyclization, using BWA (0.7.3a) [16] and samtools (0.1.19) [17]. The error rate of the completed genome sequence is less than 1 bp in 100,000 bp.
Genome annotation
The genome was annotated automatically in GenBank. In addition, we predicted Cluster Regularly Interspaced Short Palindromic Repeats (CRISPRS) with PILER-CR [18]. We analyzed the predicted protein sequences against the National Center for Biotechnology Information (NCBI) non-redundant database, Gene Ontology [19], KEGG [20], and COG [21] databases for functional annotation. The results were summarized with the InterProScan [22] software. To analyze the COG annotation, hits with an E-value < = 1e-5 were first retained. Then, only the best hit was selected for each protein. Signal peptides and transmembrane helices of all annotated proteins were predicted using SignalP 4.1 Sever [23] and TMHMM Sever v. 2.0 respectively.
Genome properties
The genome statistics are provided in Table 3, Table 4 and Fig. 3.M. fulvus 124B02 consists of a circular chromosome with a total length of 11,048,835 bp and a circular plasmid of 18,634 bp. The G+C contents of the chromosome and the plasmid are 69.96 % and 68.7 %, respectively. There were 8,515 predicted coding sequences (CDSs) in the genome, including 9 rRNAs and 80 tRNAs. The protein coding sequences occupied 86.13 % of the whole genome sequence. The majority of the protein-coding genes (5,042, 58.24 % of the total) were assigned putative functions in categories of orthologous group (COG), while the remaining ones were annotated as hypothetical proteins. The distribution of genes in COGs functional categories is presented in Table 5.
Table 3.
Information of M. fulvus 124B02 genome and the endogenous plasmid
| Label | Size (bp) | Topology | INSDC identifier | RefSeq ID |
|---|---|---|---|---|
| Chromosome | 11,048,835 | Circular | CP006003 | NZ_CP006003.1 |
| Plasmid pMF1 | 18,634 | Circular | EU137666.1 | NC_010372.1 |
Table 4.
Statistics of M. fulvus 124B02 genome
| Attribute | Value | % of totala |
|---|---|---|
| Genome size (bp) | 11,048,835 | 100.00 |
| DNA coding (bp) | 9,516,537 | 86.13 |
| DNA G + C content (bp) | 7,730,063 | 69.96 |
| DNA scaffolds | 0 | |
| Total genes | 8,658 | 100.00 |
| Protein-coding genes | 8,515 | 98.58 |
| RNA genes | 89 | 1.03 |
| Pseudogenes | 54 | 0.62 |
| Genes in internal clusters | 505 | 5.83 |
| Genes with function prediction | 4,749 | 54.85 |
| Genes assigned to COGs | 5,042 | 58.24 |
| Genes with Pfam domain | 5,929 | 68.48 |
| Genes with signal peptides | 984 | 11.37 |
| Genes with transmembrances | 1,605 | |
| CRISPR repeats | 1 |
aThe total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome
Fig. 3.
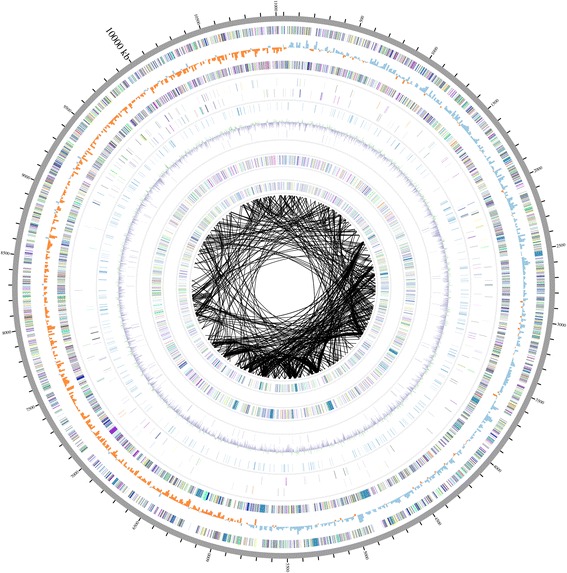
Schematic map of the genome. From outside: Circle 1, genome positions in kb (started from dnaA); Circles 2 and 4, predicted protein coding sequences (CDSs) on the forward (outer wheel) and the reverse (inner wheel) strands, colored according to COG classification; Circle 3, GC skew plot; The positive GC skew value (blue) corresponds to leading strand and negative GC value (orange) correspond to lagging strand. Circles 5 and 6, expanded genes in M. fulvus 124B02, compared with M. stipitatus DSM 14675; Circles 7, putative virus and prophage-derived CDSs; Circle 8, GC content showing deviations from the average (69.96 %); Circle 9 and 10, putative plasmid-derived CDSs (leading strand, 1,879 CDSs; lagging strand, 2,046 CDSs); Core circle, paralogous CDSs
Table 5.
The genes of M. fulvus 124B02 genome in COG functional categories
| Code | Value | %a | Description |
|---|---|---|---|
| J | 203 | 2.38 | Translation, ribosomal structure and biogenesis |
| A | 5 | 0.06 | RNA processing and modification |
| K | 416 | 4.89 | Transcription |
| L | 205 | 2.41 | Replication, recombination and repair |
| B | 3 | 0.04 | Chromatin structure and dynamics |
| D | 44 | 0.52 | Cell cycle control, cell division, chromosome partitioning |
| Y | - | - | Nuclear structure |
| V | 129 | 1.51 | Defense mechanisms |
| T | 415 | 4.87 | Signal transduction mechanisms |
| M | 329 | 3.86 | Cell wall/membrane/envelope biogenesis |
| N | 117 | 1.37 | Cell motility |
| Z | - | - | Cytoskeleton |
| W | - | - | Extracellular structures |
| U | 65 | 0.76 | Intracellular trafficking, secretion, and vesicular transport |
| O | 235 | 2.76 | Posttranslational modification, protein turnover, chaperones |
| C | 227 | 2.67 | Energy production and conversion |
| G | 205 | 2.41 | Carbohydrate transport and metabolism |
| E | 342 | 4.02 | Amino acid transport and metabolism |
| F | 94 | 1.10 | Nucleotide transport and metabolism |
| H | 148 | 1.74 | Coenzyme transport and metabolism |
| I | 283 | 3.32 | Lipid transport and metabolism |
| P | 191 | 2.24 | Inorganic ion transport and metabolism |
| Q | 177 | 2.08 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 767 | 9.01 | General function prediction only |
| S | 442 | 5.19 | Function unknown |
| - | 3,473 | 40.79 | Not in COGs |
aThe percentage is based on the total number of protein coding genes in M. fulvus 124B02 genome
Insights from the genome sequence
Until now, 22 myxobacterial genomes have been released in NCBI database. Except for the anaerobic myxobacteria, whose genomes are approximately 5 Mb, all the aerobic myxobacteria have rather large genomes, ranging from 9.03 Mb of M. fulvus HW-1 to 14.78 Mb of S. cellulosum So0157-2. Compared with the other sequenced Myxococcus genomes, i.e. 9.14 Mb of M. xanthus DK1622 [6], 9.03 Mb of M. fulvus HW-1 [7], and 10.35 Mb of M. stipitatusDSM 14675 [24], the genome of M. fulvus 124B02 is rather large. It is known that horizontal gene transfer (HGT) [25, 26] and intra-chromosomal gene duplication (IGD) [27, 28] are two major contributors for the expansion of most prokaryotic genomes. BLASTP searching against the other three sequenced Myxococcus genomes revealed 576 strain-specific duplications in the strain 124B02 genome (the core circle in Fig. 3), accounting for 6.7 % of the total CDSs. The exogenous genetic materials may be introduced into bacterial genomes via plasmids, prophages, virus, integrative conjugative elements, insertion sequence elements or other unclassified elements [29]. Of the total 8,492 CDSs in M. fulvus 124B02 genome, 3926 (46.2 %) were probably derived from plasmids (circles 9 & 10 in Fig. 3), which is similar to that in other myxobacteria [6, 9]. We conducted an all-blast-all analysis using BLASTP program with an E-value cutoff of 1e-5, and the results were transferred into OrthoMCL package to extract the paralogous and orthologous proteins. Interestingly, the phylogenomic analysis indicated that M.fulvus 124B02 is closer to M. stipitatusDSM 14675, rather than M. xanthus DK1622 or M. fulvus HW-1 (Fig. 4a), which was also supported by the genome synteny analysis (Fig. 4b). We found that the major differences between M. fulvus 124B02 and the other three Myxococcus strains were those protein sequences for the metabolism and environment adaption processes [Additional file 1: Table S1, Additional file 2: Table S2, Additional file 3: Table S3] and of those strain-specific genes. For example, according to the COG catalog, the major differences between M. fulvus 124B02 and M. stipitatusDSM 14675 were in the families of lipid transport and metabolism (p-value is 0.0076, Fisher’s exact test, two-tailed test), transcription (p-value is 0.0097, Fisher’s exact test, two-tailed test), secondary metabolites biosynthesis, transport and catabolism (p-value is 0.0015, Fisher’s exact test, two-tailed test) and replication, recombination and repair (p-value is 0.0251, Fisher’s exact test, two-tailed test). M. fulvus 124B02 had approximately 1,230 kb strain-specific fragments, which scattered throughout the whole genome (circles 5 & 6 in Fig. 3). Additional file 4: Table S4 lists the strain specific genes of replication, recombination and repair family, of which the number of M.fulvus 124B02 is less than M.stipitatusDSM 14675.
Fig. 4.
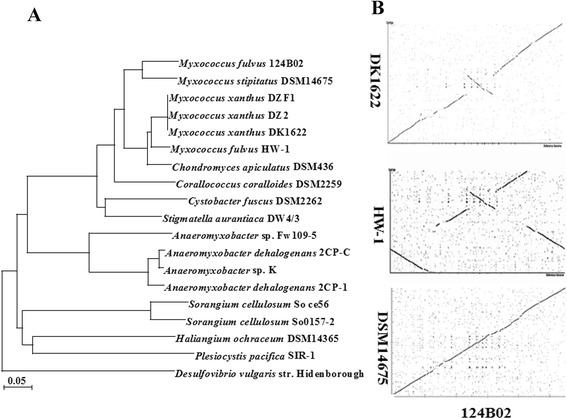
Phylogenomic analyses of M. fulvus 124B02. a A whole-genome phylogenomic tree of M. fulvus 124B02 and other sequenced myxobacterial strains using Co-phylog [50]. b The schematic maps of whole-genome synteny using blastn in NCBI between M. fulvus 124B02 and the other three sequenced Myxococcus strains, i.e. M. xanthus DK1622, M. fulvus HW-1 and M. stipitatus DSM 14675. Genomic inversions were observed in M. fulvus 124B02 vs. M. xanthus DK1622 and M. fulvus 124B02 vs. M. fulvus HW-1
pMF1 is a low copy number plasmid, containing 23 predicted ORFs [10]. The plasmid has no obvious beneficial genes for persistence in host, such as the genes encoding for antibiotic resistance, virulence, or growth phenotypes. All the predicted genes in pMF1 are of unknown functions, except the replication system (pMF1.13-pMF1.16) and the partitioning system (pMF1.21-pMF1.23), both of which were determined by narrowing-down of sequence fragments [10, 30, 31]. While the pMF1.19 and pMF1.20 genes were on the lagging strand, the others were located on the leading strand (Fig. 5). Interestingly, BLASTP searching against the GenBank database showed that pMF1.19 and pMF1.20 had multiple homologues, mostly in pairs, in different myxobacterial genomes, except that of Anaeromyxobacter. For example, there were at least ten homologues of pMF1.19 and nine of pMF1.20 in M. xanthus DK1622. The predicted protein products of these two genes contained the conserved Pfam09535 and Pfam09533 domains, but lack significant sequence similarities to any known protein families. These function-unknown homologues exist in myxobacteria only. The identities of these homologues ranged from 31 % (WP_011550397, M. xanthus DK1622) to 74 % (MFUL124B02_18095 of M. fulvus 124B02) for pMF1.19 and from 30 % (WP_011550387, M.xanthus DK1622) to 88.0 % (MFUL124B02_18100 of M. fulvus 124B02) for pMF1.20 (Fig. 6a and b are phylogenetic trees of the pMF1.19 and pMF1.20 homologues, respectively). The homologues with highest similarities to pMF1.19 and pMF1.20 are both in M. fulvus 124B02 host genome, which suggested that pMF1 was more closely related with this strain than other myxobacteria. The homologues of pMF1.19 and pMF1.20 in the genome of M. fulvus 124B02 are summarized in an additional file [Additional file 5: Table S5]. In addition to pMF1.19 and pMF1.20, the pMF1.1- pMF1.5 proteins each had a unique homologue in M. stipitatusDSM 14675, from MYSTI_04154 to MYSTI_04158 (YP 007361138.1-YP 007361142.1) (Fig. 6c). It is also noted that, although there is no gene coding for mobility systems [32], and we have not yet observed conjugative transfer of the plasmid between Myxococcus strains, the pMF1.2 and its homologue MYSTI_04155 had an AAA_10 and TraC-F-type motifs, both were reported to relate to conjugative transfer [33, 34].
Fig. 5.
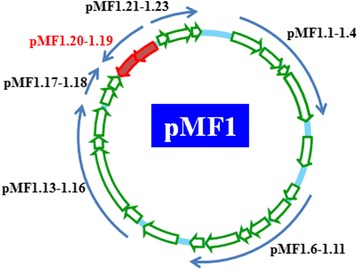
The organization of genes in pMF1. Except for pMF1.19 and pMF1.20 on the lagging strand, the other genes were located on the leading strand. A blue arrow means an operon
Fig. 6.
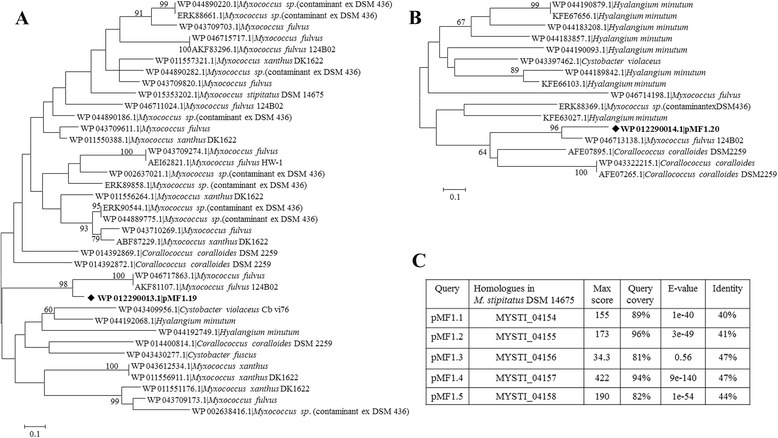
Homologues of pMF1 ORFs in myxobacterial genomes. a & b are the unrooted phylogenetic trees of the pMF1.19 and pMF1.20 proteins and their homologues, respectively. c is the summary of the homologues of pMF1.1-1.5 in M. stipitatus DSM 14675. The homologous proteins that have identity >= 40 %, E. value < 1×10−5 for pMF1.19, and identity >= 40 %, E. value < 1×10−5 for pMF1.20. The phylogenetic trees were constructed using maximum likelihood program with the Poisson correction distance model of MEGA5 [51]. The bootstrapping supports for the interior branch length of the trees were from 1,000 replicates
Conclusions
M. fulvus 124B02 is a typical strain of Myxococcus fulvus. The complete genome sequence of M. fulvus 124B02 is much larger than the other sequenced genomes of Myxococcus strains. The phylogenomic analysis of total genome sequence indicates that M. fulvus 124B02 is closer to M. stipitatusDSM 14675, rather than M. xanthus DK1622 or M. fulvus HW-1. Multiple copies of the pMF1.19 and pMF1.20 homologues in different myxobacterial strains suggest that myxobacterial genomes are open, not only to being subject to integrate foreign DNA sequences but also to being duplicated by self, in which the pMF1 plasmid played important roles. The bioinformatics analyses, together with the similar G+C contents of pMF1 and myxobacterial genomes, suggested that pMF1 had a longstanding co-adaption with myxobacteria, probably involving in the expansion of myxobacterial genomes.
Acknowledgements
This work was financially supported by grants from the National Natural Science Foundation of China (grants 31471183 and 31130004).
Abbreviations
- IGD
intra-chromosomal gene duplication
Additional files
Comparison of COG assignments of the genes between M. fulvus 124B02 and M. stipitatus DSM 14675. (XLSX 11 kb)
Comparison of COG assignments of the genes between M. fulvus 124B02 and M. fulvus HW-1. (XLSX 11 kb)
Comparison of COG assignments of the genes between M. fulvus 124B02 and M. xanthus DK1622. (XLSX 11 kb)
Strain-specific genes in the family of DNA replication, recombination and repair in M. fulvus 124B02, compared with DSM 14675. (XLSX 10 kb)
Homologues of pMF1.19 and pMF1.20 in M. fulvus 124B02. (XLSX 9 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XJC, KH, LZ and YJL participated in genome sequencing analysis, bioinformatics analysis, drafted the original manuscript, and participated in the revision process. JF and XJC extracted the total genome for sequencing. XJC and YZL wrote the paper. YZL conceived the study, provided funding for the project. All the authors read and approved the final manuscript.
References
- 1.Shimkets LJ. Social and developmental biology of the myxobacteria. Microbiol Rev. 1990;54:473–501. doi: 10.1128/mr.54.4.473-501.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichenbach H. The ecology of the myxobacteria. Environ Microbiol. 1999;1:15–21. doi: 10.1046/j.1462-2920.1999.00016.x. [DOI] [PubMed] [Google Scholar]
- 3.Weissman KJ, Müller R. Myxobacterial secondary metabolites: bioactivities and modes-of-action. Nat Prod Rep. 2010;27:1276–95. doi: 10.1039/c001260m. [DOI] [PubMed] [Google Scholar]
- 4.Xiao Y, Wei X, Ebright R, Wall D. Antibiotic production by myxobacteria plays a role in predation. J Bacteriol. 2011;193:4626–33. doi: 10.1128/JB.05052-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Li X, Zhang WY, Zhou XW, Li YZ. The groEL2 gene, but not groEL1, is required for biosynthesis of the secondary metabolite myxovirescin in Myxococcus xanthus DK1622. Microbiology. 2014;160:488–95. doi: 10.1099/mic.0.065862-0. [DOI] [PubMed] [Google Scholar]
- 6.Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, et al. Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci U S A. 2006;103:15200–5. doi: 10.1073/pnas.0607335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li ZF, Li X, Liu H, Liu X, Han K, Wu ZH, et al. Genome sequence of the halotolerant marine bacterium Myxococcus fulvus HW-1. J Bacteriol. 2011;193:5015–6. doi: 10.1128/JB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO, et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol. 2007;25:1281–9. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 9.Han K, Li ZF, Peng R, Zhu LP, Zhou T, Wang LG, et al. Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci Rep. 2013;3:2101. doi: 10.1038/srep02101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao JY, Zhong L, Shen MJ, Xia ZJ, Cheng QX, Sun X, et al. Discovery of the autonomously replicating plasmid pMF1 from Myxococcus fulvus and development of a gene cloning system in Myxococcus xanthus. Appl Environ Microbiol. 2008;74:1980–7. doi: 10.1128/AEM.02143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YZ, Li J, Zhou L, Zhang Y, Hu W, Chen Q. Isolation and identification of myxobacterial sources in China. Acta Microbiol Sinica. 2000;40:652–6. [PubMed] [Google Scholar]
- 12.Kieser T. Factors affecting the isolation of CCC DNA from Streptomyces lividans and Escherichia coli. Plasmid. 1984;12:19–36. doi: 10.1016/0147-619X(84)90063-5. [DOI] [PubMed] [Google Scholar]
- 13.Ewing B, Hillier LD, Wendl MC, Green P. Base-calling of automated sequencer traces usingPhred. I. Accuracy assessment. Genome Res. 1998;8:175–85. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 14.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–94. doi: 10.1101/gr.8.3.186. [DOI] [PubMed] [Google Scholar]
- 15.Gordon D. Viewing and editing assembled sequences using Consed. Curr Protoc Bioinformatics. 2003;12:11–43. doi: 10.1002/0471250953.bi1102s02. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007;8:18. doi: 10.1186/1471-2105-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–8. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 23.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 24.Huntley S, Kneip S, Treuner-Lange A, Søgaard-Andersen L. Complete genome sequence of Myxococcus stipitatus strain DSM 14675, a fruiting myxobacterium. Genome Announc. 2013;1:e00100–13. doi: 10.1128/genomeA.00100-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gogarten JP, Townsend JP. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005;3:679–87. doi: 10.1038/nrmicro1204. [DOI] [PubMed] [Google Scholar]
- 26.Takeuchi N, Kaneko K, Koonin EV. Horizontal gene transfer can rescue prokaryotes from Muller’s ratchet: benefit of DNA from dead cells and population subdivision. G3 (Bethesda) 2014;4:325–39. doi: 10.1534/g3.113.009845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersson DI, Hughes D. Gene amplification and adaptive evolution in bacteria. Annu Rev Genet. 2009;43:167–95. doi: 10.1146/annurev-genet-102108-134805. [DOI] [PubMed] [Google Scholar]
- 28.Serres MH, Kerr AR, McCormack TJ, Riley M. Evolution by leaps: gene duplication in bacteria. Biol Direct. 2009;4:46. doi: 10.1186/1745-6150-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8:552–63. doi: 10.1038/nrmicro2382. [DOI] [PubMed] [Google Scholar]
- 30.Sun X, Chen XJ, Feng J, Zhao JY, Li YZ. Characterization of the partitioning system of Myxococcus plasmid pMF1. PLoS One. 2011;6:e28122. doi: 10.1371/journal.pone.0028122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng J, Chen XJ, Sun X, Wang N, Li YZ. Characterization of the replication origin of the myxobacterial self-replicative plasmid pMF1. Plasmid. 2012;68:105–12. doi: 10.1016/j.plasmid.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Smillie C, Garcillán-Barcia MP, Francia MV, Rocha EP, de la Cruz F. Mobility of plasmids. Microbiol Mol Biol R. 2010;74:434–52. doi: 10.1128/MMBR.00020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchler Bauer A, Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, et al. CDD: NCBI conserved domain database. Nucleic Acids Res. 2015;43(D):222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christie PJ, Vogel JP. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 2000;8:354–60. doi: 10.1016/S0966-842X(00)01792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18:452–64. doi: 10.1093/bioinformatics/18.3.452. [DOI] [PubMed] [Google Scholar]
- 36.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 37.Stamatakis A, Hoover P, Rougemont J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst Biol. 2008;57:758–71. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 38.Reddy TBK, Thomas A, Stamatis D, Bertsch J, Isbandi M, Jansson J, et al. The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta) genome project classification. Nucleic Acids Res. 2014;43(Database issue):D1099–106. doi: 10.1093/nar/gku950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about agenome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garrity GM, Bell JA, Lilburn T. The revised road map to the manual. In: Bergey’s Manual of Systematic Bacteriology. Springer US; 2005. pp. 159–187. [Google Scholar]
- 42.Kuever J, Rainey FA, Widdel F. Class IV. Deltaproteobacteria class nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. 2. NY: Springer; 2005. p. 922. [Google Scholar]
- 43.Tchan YT, Pochon J, Prévot AR. Etude de systématique bactérienne. VIII. Essai de classification des Cytophaga. Ann Inst Pasteur (Paris) 1948;74:394–400. [Google Scholar]
- 44.Reichenbach H, Garrity GM, Brenner DJ, Krieg NR, Staley JT. Order VIII. Myxococcales. Tchan, Pochon and Prévot 1948, 398AL. Bergey’s manual of systematic bacteriology. Springer, New York, 2005;2(Part C):1059–1072.
- 45.Jahn E. Beiträge zur botanischen Protistologie I: Die Polyangiden. Leipzig: Verlag Gebruder Borntraeger; 1924. pp. 1–107. [Google Scholar]
- 46.Lang E, Stackebrandt E. Emended descriptions of the genera Myxococcus and Corallococcus, typification of the species Myxococcus stipitatus and Myxococcus macrosporus and a proposal that they be represented by neotype strains. Request for an opinion. Int J Syst Evol Microbiol. 2009;59:2122–8. doi: 10.1099/ijs.0.003566-0. [DOI] [PubMed] [Google Scholar]
- 47.Jahn E. Myxobacteriales. Kryptogamenflora der Mark Brandenburg. 1911;5:187–206. [Google Scholar]
- 48.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30:225–30. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 49.Agents B. Technical rules for biological agents. www.baua.de TRBA 466.
- 50.Yi H, Jin L. Co-phylog: an assembly-free phylogenomic approach for closely related organisms. Nucleic Acids Res. 2013;41(7):e75. doi: 10.1093/nar/gkt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]