

Abstract
One challenge in studying chronic infectious and inflammatory disorders is understanding how host pattern recognition receptors (PRRs), specifically toll-like receptors (TLRs), sense and respond to pathogen- or damage-associated molecular patterns, their communication with each other and different components of the immune system, and their role in propagating inflammatory stages of disease. The discovery of innate immune activation through nucleic acid recognition by intracellular PRRs such as endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) and cytoplasmic proteins (absent in melanoma 2 and DNA-dependent activator of interferon regulatory factor) opened a new paradigm: Nucleic acid sensing is now implicated in multiple immune and inflammatory conditions (e.g., atherosclerosis, cancer), viral (e.g., human papillomavirus, herpes virus) and bacterial (e.g., Helicobacter pylori, pneumonia) diseases, and autoimmune disorders (e.g., systemic lupus erythematosus, rheumatoid arthritis). Clinical investigations reveal the overexpression of specific nucleic acid sensors in diseased tissues. In vivo animal models show enhanced disease progression associated with receptor activation. The involvement of nucleic acid sensors in various systemic conditions is further supported by studies reporting receptor knockout mice being either protected from or prone to disease. TLR9-mediated inflammation is also implicated in periodontal diseases. Considering that persistent inflammation in the oral cavity is associated with systemic diseases and that oral microbial DNA is isolated at distal sites, nucleic acid sensing may potentially be a link between oral and systemic diseases. In this review, we discuss recent advances in how intracellular PRRs respond to microbial nucleic acids and emerging views on the role of nucleic acid sensors in various systemic diseases. We also highlight new information on the role of intracellular PRRs in the pathogenesis of oral diseases including periodontitis and oral cavity cancer, which might offer future possibilities for disease prevention and therapy.
Keywords: inflammation, toll-like receptor, AIM2, DAI, periodontal disease, infection
Introduction
The human body is composed of over 100 trillion microbial cells living in symbiosis with the host. It is now well established that many chronic inflammatory conditions are a consequence of imbalance between host-microbiota interactions, giving rise to a dysbiotic community, deregulated immune responses, and eventually disease outcomes. Innate immunity has historically been regarded as temporary, nonspecific, and the first line of defense against microorganisms until the adaptive immune response is fully activated. However, recent advances in understanding how innate immune cells detect and respond to the microbiome have demonstrated specificity in the response. This specificity includes germline-encoded receptors on innate immune cells known as pattern recognition receptors (PRRs) (Pandey et al. 2015). These families of receptors include membrane-associated, endosomal, and cytoplasmic PRRs that sense and bind pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide, lipoteichoic acid, flagellin, fimbriae, and nucleic acids. The binding of PAMPs to PRRs initiates a signal transduction cascade, culminating in the release of inflammatory molecules that coordinate the activation of innate immunity and eventually the development of adaptive immune responses. While innate immunity is critical to combat pathogens, deregulated inflammation results in tissue destruction and disease progression. Therefore, understanding how innate immune cells integrate signals in a dysbiotic ecosystem is at the forefront of comprehending disease pathogenesis and etiology in many diseases including oral diseases such as periodontitis.
The immunogenicity of bacterial DNA (bDNA) has been well documented in early pioneering studies (Yamamoto et al. 1992). The discovery of nucleic acid recognition by specific intracellular PRRs (Table) opened a new paradigm: Nucleic acid sensors are now implicated in multiple diseases of infectious and immunological origins including those that are associated with severe periodontitis. Oral tissues are constantly exposed to microbes, and continuous cell turnover provides a platform for nucleic acid abundance within the oral cavity either as a result of phagocytosis or the release of extracellular DNA that forms part of the biofilm structure. In fact, emerging evidence reveals microbial nucleic acid sensing, specifically toll-like receptor 9 (TLR9)–mediated inflammation, as one of the novel inflammatory pathways in periodontal disease (PD) pathogenesis (Hajishengallis and Sahingur 2014). Therefore, a better understanding of nucleic acid recognition pathways can provide new insights into PD pathogenesis, prevention, and therapy.
Table.
Nucleic Acid Sensors and Signaling Molecules.
| Nucleic Acid Sensor | Agonist | Signaling Molecules |
|---|---|---|
| Endosomal | ||
| TLR3 | dsRNA | AP-1, NF-κB, IRF3, IRF7 |
| TLR7 | ssRNA | AP-1, NF-κB, IRF5, IRF7 |
| TLR8 | ssRNA | AP-1, NF-κB, IRF5, IRF7 |
| TLR9 | Unmethylated CpG DNA, dsDNA | AP-1, NF-κB, IRF5, IRF7 |
| Cytosolic | ||
| AIM2 | dsDNA | Caspase-1 |
| DAI | dsDNA | NF-κB, IRF3, IRF7 |
| RIG-I | Short ssRNA, dsRNA | AP-1, NF-κB, IRF3, IRF7 |
| MDA5 | Long dsRNA | AP-1, NF-κB, IRF3, IRF7 |
| LGP2 | dsRNA | AP-1, NF-κB, IRF3, IRF7 |
| IFI16 | dsDNA | NF-κB, caspase-1, IRF3 |
| DHX9 | dsRNA | IRF7 |
| DHX36 | dsDNA | IRF7 |
| DDX3 | dsRNA | IRF3, IRF7 |
| DDX41 | dsDNA | IRF3, IRF7 |
| DDX60 | ssRNA, dsRNA, dsDNA | IRF3, IRF7 |
| STING | Cyclic dinucleotides | NF-κB, IRF3, IRF7 |
| cGAS | dsDNA | IRF3, IRF7 |
| RNA Pol III | AT-rich dsDNA | IPS-1, IRF3, IRF7 |
| Ku70 | dsDNA | IRF1, IRF7 |
| LRRFIP1 | dsDNA, dsRNA | β-catenin, IRF3, IRF7 |
AIM2, absent in melanoma 2; AP-1, activator protein 1; AT, adenine-thymine; cGAS, cyclic GMP-AMP synthase; DAI, DNA-dependent activator of interferon regulatory factor; DDX, DEAD box protein; DHX, DEAH box protein; ds, double-stranded; IFI16, interferon γ–inducible protein 16; IPS-1, interferon β promoter stimulator 1; IRF, interferon regulatory factor; LGP2, Laboratory of Genetics and Physiology 2; LRRFIP1, leucine-rich repeat (in FLII) interacting protein 1; MDA5, melanoma differentiation–associated protein 5; NF-κB, nuclear factor κ light-chain enhancer of activated B cells; RIG-I, retinoic acid–inducible gene 1; ss, single-stranded; STING, stimulator of interferon genes; TLR, toll-like receptor.
In this review article, we attempt to introduce new perspectives to study the role of microbial nucleic acid sensing in the pathogenesis of diseases related to the oral cavity. We present information on how intracellular TLRs sense and respond to microbial nucleic acids and the association of these sensors with systemic diseases, and we highlight new information on the emerging role of nucleic acid sensing in PD pathogenesis.
Microbial Nucleic Acid Receptors
Nucleic acids are recognized by innate receptors that reside intracellularly either in the cytoplasm or in the endolysosomal compartment (Table). The intracellular localization of these sensors is thought to be evolutionarily conserved, minimizing the likelihood of triggering autoimmune reactions. We will focus on the nucleic acid sensors that have been studied in the course of periodontal inflammation, which primarily includes endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) and cytoplasmic sensors (absent in melanoma 2 [AIM2] and DNA-dependent activator of interferon regulatory factor [DAI]), and systemic conditions related to these receptors that are also associated with periodontitis.
Endosomal TLRs
While each endosomal TLR shows specificity in ligand recognition, the initial trafficking pathway to encounter the ligand and the subsequent downstream signaling pathways show some redundancy. Prior to ligand interaction, all intracellular TLRs reside in the endoplasmic reticulum (ER), and they undergo essential processing steps in order to achieve correct folding, positioning, and activation. While in the ER and during trafficking to endosomes through the Golgi apparatus, they remain in an uncleaved state and only undergo proteolytic cleavage to attain functional activation after the arrival to endolysosomes due to the acidic environment (Lee and Barton 2014). The mechanisms that determine the selective export of TLRs from the ER are still not fully elucidated. Some studies reported the presence of a small number of receptors in the endosomal compartment in the absence of activating stimuli, suggesting the existence of a self-perpetuating, low-frequency trafficking process. A number of chaperone proteins are required for shuttling from the ER to endosomes, which include gp96, PRAT4, UNC93B, and adaptor protein complex 2 and 4 (Lee and Barton 2014) (Fig. 1). Each chaperone protein is involved with its specific receptor, assisting in trafficking, folding, and final activation. Before entering endosomes, TLR9 and TLR3 are thought to shuttle to the plasma membrane first, where they still remain folded by the chaperone proteins and are inactive. After reaching the endosomes, TLRs are proteolytically cleaved by cathepsin and asparagine endopeptidase to yield an active receptor (Pandey et al. 2015). An acidic environment within the endosome is particularly important for the function of the receptors, as studies have determined that agents such as chloroquine and bafilomycin, which prevent acidification of the endolysosomal compartment, can inhibit TLR activation. Together, these processing steps produce a functional endosomal receptor that recognizes nucleic acids (Lee and Barton 2014; Pandey et al. 2015).
Figure 1.

Cellular trafficking pathways of the nucleic acid sensing toll-like receptors (TLRs). The chaperone protein UNC93B associates with TLR3, TLR7, and TLR9 and facilitates their exit from the endoplasmic reticulum (ER) to the Golgi apparatus via coat protein complex II vesicles. With the exception of TLR3, TLR7 and TLR9 require the additional folding chaperone proteins gp96 and PRAT4 for exiting the ER. TLRs are sorted into endosomes through the Golgi apparatus. Prior to endosomal localization, TLR3 and TLR9 translocate to the cell membrane, where they remain functionally inactive. TLR7 and TLR9 shuttling into endosomes require adaptor protein complex 4 and 2, respectively. After the arrival to the endosome, the TLRs are cleaved by cathepsin and asparagine endopeptidase (AEP) to yield an active, functional receptor.
Following nucleic acid engagement within the endolysosomal compartment, all sensors, except TLR3, signal through MYD88 and activate nuclear factor κ light-chain enhancer of activated B cells (NF-κB), activator protein 1 (AP-1), and mitogen-activated protein kinase signaling pathways, which stimulate proinflammatory activities, but also the interferon regulatory factor pathway, which can induce type I interferon and anti-inflammatory activities (Pandey et al. 2015) (Fig. 2). TLR3 signals through TRIF, culminating in the expression of type I interferons and proinflammatory cytokines (Pandey et al. 2015).
Figure 2.

Signaling pathways of nucleic acid sensors. Toll-like receptor 3 (TLR3) recognizes dsRNA to initiate a signaling cascade with the recruitment of toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon β (TRIF), signaling through TANK-binding kinase (TBK), resulting in the nuclear translocation of interferon regulatory factor 3 (IRF3) to induce type I interferon production. Additionally, TRIF can signal via tumor necrosis factor receptor–associated factor 6 (TRAF6), the transforming growth factor β–activated kinase 1 (TAK1)/TAK-binding protein (TAB) complex, and the IκB kinase (IKK) complex to activate nuclear factor κ light-chain enhancer of activated B cells (NF-κB) for the induction of proinflammatory cytokines. The recognition of viral ssRNA and CpG DNA activates TLR7 and TLR9, respectively, to recruit myeloid differentiation primary response gene 88 (MYD88). MYD88 recruitment triggers a signaling cascade via interleukin-1 receptor–associated kinase 4 (IRAK4) to activate activator protein 1 (AP-1), NF-κB, and IRF5 for the transcription of proinflammatory cytokines and type I interferons. Concurrently, MYD88 signals via TRAF6 and TRAF3 for the IRF7-mediated transcription of type I interferons. Absent in melanoma 2 (AIM2) recognizes dsDNA for the recruitment of caspase-1 via apoptosis-associated speck-like protein containing a CARD (ASC) to activate interleukin 1β (IL-1β) and IL-18. dsDNA recognition by DNA-dependent activator of interferon regulatory factor (DAI) triggers TBK1, signaling through the IKKε complex for the IRF3-mediated transcription of type I interferons. Additionally, DAI-mediated dsDNA recognition activates receptor-interacting protein 1 (RIP1)/RIP3 signaling through the IKK complex to activate NF-κB for the transcription of proinflammatory cytokines.
Cytoplasmic Nucleic Acid Sensors
The signaling pathways activated by 2 cytoplasmic nucleic acid sensors, AIM2 and DAI, are distinct from endosomal TLRs (Fig. 2). AIM2 belongs to the hematopoietic interferon–inducible nuclear protein with 200 amino acid repeats (HIN-200) family and consists of a ligand-binding domain called HIN and another domain called pyrin (PYD) that interacts with the adaptor protein, apoptosis-associated speck-like protein containing a CARD (ASC) (caspase recruitment domain), of the inflammasome (Wu and Chen 2014). In the absence of the ligand, the HIN and PYD domains engage in an intramolecular complex, preventing inflammasome activation. Upon ligand binding, the HIN domain of AIM2 releases the PYD domain to interact with ASC of the inflammasome, activating subsequent signaling cascades that generate active caspase-1. In turn, catalytically active caspase-1 cleaves the inactive pro–interleukin 1β (IL-1β) and pro–IL-18 cytokines (Fig. 2).
Following engagement with its ligand, DAI interacts with receptor-interacting protein 1 (RIP1) via the RIP homotypic interaction motif (RHIM). An additional kinase, RIP3, also binds to the RHIM domain of DAI. Together, these kinases enhance NF-κB activation. DAI can also form a complex with the serine/threonine kinase TBK1 and IRF3 (Wu and Chen 2014) (Fig. 2).
Microbial Nucleic Acid Sensing in Periodontitis
Periodontitis is a chronic inflammatory disease that leads to the local destruction of periodontal tissues and also impacts systemic health. It is now well characterized that periodontal tissue destruction is a result of deregulated inflammation in response to microbial dysbiosis (Lamont and Hajishengallis 2015).
Oral tissues are composed of various cells of myeloid and nonmyeloid origins that possess cytosolic, membrane-associated, and secreted PRRs such as TLRs, NOD-like receptors, RIG-I–like receptors, and C-type lectin receptors. The engagement of PRRs with oral PAMPs activates inflammatory signaling cascades, triggering increased cytokine/chemokine production and cell trafficking to the subgingival crevice. In relation to periodontitis, one particular challenge is understanding how the host PRRs, specifically TLRs, sense and respond to PAMPs in a transitioning dysbiotic environment and their role in propagating the inflammatory stages of disease. Therefore, current research efforts focus on understanding the interplay between keystone pathogens, host immunity, and the cellular and molecular mechanisms by which the two lead to excessive, detrimental inflammation and periodontal bone loss.
Data linking innate immune sensors, particularly TLRs, with periodontitis pathology continue to accumulate as they relate to inflammatory responses both in local tissues and at distant sites. The involvement of membrane-bound receptors (TLR2 and TLR4) that can respond to cell surface–associated periodontal PAMPs has been well studied, and TLR2 has now been implicated in inflammation induced by Porphyromonas gingivalis (a keystone periodontal pathogen) (Lamont and Hajishengallis 2015). Most recent studies have also highlighted the participation of endosomal TLRs in periodontal inflammation. Among these, TLR9-triggered immune responses emerge as a novel inflammatory pathway in periodontitis pathogenesis (Appendix Table). Clinical studies have demonstrated increased TLR9 gene and protein expression in gingival tissues associated with chronic periodontitis (Rojo-Botello et al. 2012; Sahingur et al. 2013) (Fig. 3A, B). It is also well documented that the host genetic background affects the susceptibility to periodontitis. Consistent with this, the presence of specific polymorphisms (SNPs) in the promoter region of the TLR9 gene has been reported in individuals with chronic periodontitis (Holla et al. 2010; Sahingur et al. 2011). In silico analyses revealed that TLR9 SNPs are located in the promoter region corresponding to the transcriptional activator–binding site (NF-κB and Sp-1) (Ng et al. 2010), presumably having a functional role in gene expression. Complementing clinical studies, in vitro investigations demonstrated that periodontitis-associated bDNA up-regulates several genes of the innate immune response and induces the production of proinflammatory cytokines through TLR9 (Sahingur et al. 2010; Kim et al. 2012; Sahingur et al. 2012). Furthermore, utilizing an integrative gene prioritization method (Zhan et al. 2014), TLR9 has also been identified as one of the most promising candidate genes involved in the pathogenesis of periodontitis. Recently, using TLR9 knockout (TLR9–/–) mice and wild-type (WT) controls in a murine model of P. gingivalis–induced periodontitis, our group reported the first proof-of-concept evidence that TLR9 signaling mediates the induction of periodontal bone loss (Kim et al. 2015) (Fig. 3C, D). This pivotal in vivo study identified TLR9 as one of the key immune sensors in periodontal inflammation, thereby revealing a new therapeutic target for periodontitis. As small molecule inhibitors of TLR9 are already available, it will be important to determine their therapeutic effect in PD (Connolly and O’Neill 2012). It was also revealed that P. gingivalis DNA injections in rats could induce antibody production against fimbriae and prevent periodontal bone loss (Han et al. 2014). This finding also warrants further studies to explore the potential of bDNA and nucleic acid sensing in vaccine development.
Figure 3.

Microbial nucleic acid sensors in periodontitis. (A) Toll-like receptor 8 (TLR8), TLR9, and DNA-dependent activator of interferon regulatory factor (DAI) gene expression were significantly elevated in periodontitis lesions compared to healthy sites, and TLR9 expression was the highest of all the other innate sensors. (B) Immunohistochemistry analyses revealed increased TLR9 and DAI expression in periodontitis lesions compared to healthy tissues. Adapted from Sahingur et al. (2013). (C) Following the induction of periodontitis using oral gavage with Porphyromonas gingivalis, bacteria-infected TLR9–/– mice did not exhibit significant alveolar bone loss compared to sham-infected TLR9–/– mice. There was significant bone loss in P. gingivalis–infected wild-type (WT) mice compared to sham-infected WT mice and P. gingivalis–infected TLR9–/– mice. (D) Micro–computed tomography images of P. gingivalis–infected TLR9–/– and WT mice versus sham-infected controls. Adapted from Kim et al. (2015). *P < 0.05, **P < 0.01.
Still, the host response to periodontal microbiota is the sum of several immune and inflammatory pathways induced by the recognition of different PAMPs by different PRRs. Intriguingly, it was also revealed that TLR9 deficiency can affect the extent of inflammatory responses to TLR2 and TLR4 ligands, despite similar TLR2 and TLR4 expression in WT versus TLR9–/– cells (Kim et al. 2015), possibly through crosstalk through downstream signaling pathways. Another study also demonstrated the synergistic induction of antimicrobial factors including peptidoglycan recognition proteins and β-defensins in human oral epithelial cells when activated with a combination of synthetic TLR9, NOD1, or NOD2 agonists, suggesting crosstalk among these sensors as well (Uehara and Takada 2008). Thus, further investigations are required to fully characterize the extent of communication between TLR9, other innate sensors, and the oral microbiome to identify effective therapeutic targets to halt periodontal inflammation.
It is also likely that interaction among nucleic acid sensors and other innate sensing molecules may be important for PD effects on systemic conditions. For example, a study showed that periodontal pathogens can induce HIV-1 reactivation in monocytes/macrophages by the enhanced activation of TLR2 and TLR9 (González et al. 2010). Another study reported the enhanced expression of TLR2 and TLR9 in gingival tissues of periodontitis patients with type 2 diabetes compared to those without diabetes (Rojo-Botello et al. 2012). Thus, delineating the cooperation among different receptors and signaling pathways in the course of periodontal inflammation may also help to characterize the link between periodontitis and certain systemic diseases. The involvement of microbial nucleic acid sensors with systemic diseases that are associated with periodontitis will be discussed in the next section.
While TLR9 is the most studied nucleic acid sensor in periodontal inflammation (Appendix Table), the participation of other sensors in periodontitis has also been investigated. The increased expression of TLR8 and DAI were reported in chronic periodontitis lesions compared to healthy sites (Sahingur et al. 2013) (Fig. 3A, B). The same study also reported no significant difference in AIM2 expression in chronic periodontitis lesions compared to healthy tissues (Sahingur et al. 2013). In contrast, a recent study (Xue et al. 2015) reported significantly increased AIM2 expression in chronic periodontitis lesions. The increased expression of AIM2 was also demonstrated in gingival fibroblasts in response to oral biofilm ex vivo (Bostanci et al. 2011). Another study showed that P. gingivalis could induce IL-1β secretion and inflammatory cell death via NLRP3 and AIM2 inflammasome activation (Park et al. 2014). Additionally, it has been reported that TLR3 signaling can induce cytokine secretion in human oral keratinocytes and fibroblasts (Fukui et al. 2013). Damage-associated molecular patterns (DAMPs) are endogenous molecules released from damaged or necrotic cells. It was demonstrated that TLR3 activation by necrotic cell supernatants can induce cytokine production and TLR2 expression in gingival cells possibly through nucleic acids released upon cell death, acting as DAMPs (Mori et al. 2015). Thus, it is likely that besides TLR9, other endosomal and cytoplasmic nucleic acid sensors might also be involved in periodontal inflammation either through PAMP or DAMP recognition. However, it is still too early to draw any conclusions due to the limited number of investigations.
It is noteworthy to point out that one common feature of nucleic acid receptors is that they are also implicated in the recognition of viral nucleic acids. While the bacterial etiology of periodontitis is well accepted, the contribution of viruses in PD pathology has also been supported by a number of studies (Slots 2005). In fact, the association of bacteria and viruses has been suggested to potentially create a favorable environment for pathogen survival, persistence, and enhanced inflammatory responses. Since there is still insufficient evidence to define such interactions, it can only be speculated that nucleic acid sensing may potentially interfere and modulate the pathological outcomes related to viral and bacterial infections within the oral cavity.
Microbial Nucleic Acid Sensing in Systemic Diseases Associated with Periodontitis
While the activation of innate immune sensors is certainly important to control infections, deregulated immune responses can promote unfavorable clinical outcomes, leading to chronic diseases. In relation to nucleic acid receptors, their aberrant activation is thought to contribute to the pathogenesis of several systemic conditions (Fig. 4), some of which are also associated with periodontitis. Given the fact that periodontal bDNA has been isolated in tissues related to some of these conditions, it is plausible to speculate that microbial DNA sensing may be a link between periodontitis and the systemic diseases that are reviewed below.
Figure 4.
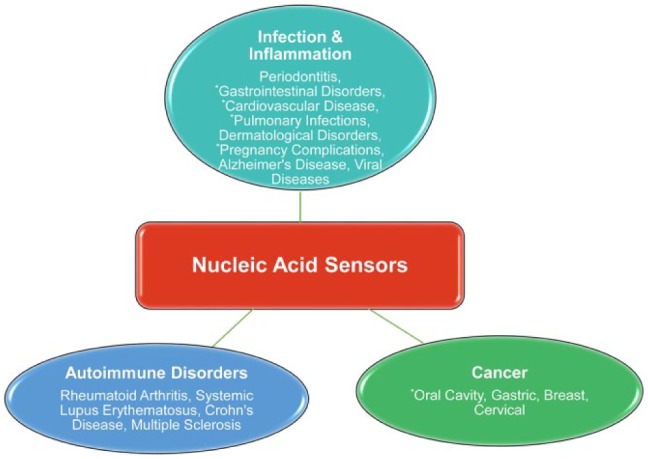
Diseases related to nucleic acid sensors. Nucleic acid sensing has been implicated in the pathogenesis of several diseases including infectious and inflammatory conditions, autoimmune disorders, and cancer. *Systemic disorders associated with periodontal disease and highlighted in this review.
Nucleic Acid Sensors and Cardiovascular Disease
Cardiovascular disease (CVD) is one of the leading causes of morbidity and mortality in the industrialized world and driven by atherosclerosis. Chronic bacterial and viral infections are considered to play a role in the development of atherosclerosis, and now, there is an increasing body of evidence supporting the role of nucleic acid sensing and the activation of TLRs in the initiation and progression of the different stages of atherosclerotic lesions. Primary human arterial and venous endothelial and smooth muscle cells isolated from carotid atheroma samples were found to be responsive to bDNA (Erridge et al. 2008). It has also been reported that compared to TLR9–/– mice, WT mice displayed increased proinflammatory cytokine production and NF-κB activation in response to bDNA and that bDNA caused myocardial dysfunction by reducing cardiomyocyte contractility (Knuefermann et al. 2008). Endothelial cells also express TLR9, and bDNA activation of endothelial cells via TLR9 promotes neutrophil chemotaxis (El Kebir et al. 2009). Furthermore, heart failure associated with polymicrobial sepsis was ameliorated in TLR9–/– mice compared to their WT counterparts (Lohner et al. 2013). Mitochondria evolved from symbiogenesis with prokaryotes and contain hypomethylated DNA similar to bacteria. Recently, heart failure due to inflammation has been associated with TLR9 activation from endogenous mitochondrial DNA released from cardiomyocytes during external hemodynamic stress (Oka et al. 2012). Opposing outcomes were reported for the role of TLR3 in atherosclerosis, with some studies suggesting a protective role and others reporting detrimental effects (Cole et al. 2011; Lundberg et al. 2013). A recent study revealed that TLR7 activation is protective for atherosclerosis by constraining macrophage activation (Salagianni et al. 2012). Additionally, there is also evidence for the contribution of AIM2 to CVD, with a recent study identifying increased AIM2 expression in the necrotic core of atherosclerotic carotid lesions and in the vasa vasorum neovasculature of aortic aneurysms (Hakimi et al. 2014).
Emerging studies in the basic understanding of the roles of intracellular PRRs in atherosclerosis are promising and may open up new therapeutic avenues. While the link between periodontitis and CVD is also well documented, the exact mechanism is poorly understood. The presence of periodontal bDNA has been reported in atherosclerotic plaques (Armingohar et al. 2014), and it is likely that nucleic acid sensing may be a possible link between PD and CVD. Yet, more definitive studies are needed to identify these interactions.
Nucleic Acid Sensors and AdversePregnancy Outcomes
Numerous environmental and genetic factors can predispose women to adverse pregnancy outcomes (APOs) such as preterm labor, preeclampsia, preterm premature ruptures of membranes, low birth weight, and intrauterine growth retardation. The oral cavity can act as a reservoir to deliver microbial species and their by-products to the placenta, amniotic fluid, and fetal circulation, triggering immune responses via PRRs. Persistent inflammatory responses at the maternal-fetal interface may eventually have an impact on the outcome of the pregnancy. Oral bDNA has been isolated in the intrauterine environment (Fardini et al. 2010). The expression of endosomal TLRs has been demonstrated at the maternal-fetal interface including placental tissue, decidua, and amnion both by immune and nonimmune cells (Koga et al. 2014). Preterm labor and preeclampsia have been associated with TLR9 activity to hypomethylated fetal DNA and placenta-derived mitochondrial DNA, which were prevented by chloroquine, a TLR9 antagonist (Goulopoulou et al. 2012; Scharfe-Nugent et al. 2012). Increased basal levels of TLR9 and proinflammatory cytokines were determined in plasmacytoid dendritic cells of women with preterm labor and preeclampsia, implicating a possible role of TLR9 in contributing to the overall increased inflammatory state of these patients (Panda et al. 2012). There is also evidence for the contribution of TLR3 to APOs. TLR3 activation by poly I:C injections resulted in preterm labor in a murine model, and the effect was ameliorated in TLR3–/– mice (Koga et al. 2009). While studies on the role of viruses and APOs are scarce in humans due to the lack of a global marker for detecting viral genomes, murine studies suggest that future work is warranted to further characterize the role of viral infections and TLR3 in APOs.
Nucleic Acid Sensors and Pulmonary Diseases
Lower respiratory tract infections and pulmonary diseases are major causes of mortality worldwide, affecting individuals regardless of age, sex, and race. Microbes play a role in the initiation of these diseases via their spread from the oropharyngeal tract by mucus aspirations or devices such as endotracheal tubes. An oral route for pulmonary infections has been suggested following the isolation of periodontitis-associated bacteria from human pneumonia aspirations. Improved oral hygiene and decreased oral microbiota burdens have also been associated with the reduction of pulmonary diseases (Linden et al. 2013). Recent studies have also implicated innate immune cell activation via endosomal TLRs in the development of pulmonary infections. In Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus mouse models, TLR9–/– was protective with increased proinflammatory cytokines, IL-1β, and nitric oxide production in alveolar macrophages (Benmohamed et al. 2014) and decreased levels of TNF-α, respectively (Parker and Prince 2012). However, the exact role of TLR9 in pulmonary infections may be more complicated, with some studies suggesting a protective role (Bhan et al. 2007). Evidence also exists that TLR3 contributes to viral infections associated with pulmonary injuries. TLR3 is highly expressed in alveolar epithelial cells of patients with acute respiratory distress syndrome, an acute lung injury disorder characterized by trauma-induced edema and collagen accumulation. These patients are usually plagued by recurring viral infections, and TLR3–/– mice have been shown to be protected from lung injury–induced viral infections (Murray et al. 2008). Additionally, evidence also exists for the role of AIM2 to Streptococcus pneumoniae infections. A recent study using both in vivo and in vitro pneumococcal models suggested that type I interferons regulate AIM2 expression and subsequent IL-18 production in response to S. pneumoniae (Fang et al. 2014).
Nucleic Acid Sensors and Gastrointestinal Diseases
Inflammatory bowel disease (IBD), including colitis and Crohn disease, is driven by chronic gastrointestinal tract inflammation, resulting in severe weight loss, diarrhea, pain, and fatigue. An association between IBD and oral lesions such as periodontitis and dental caries has been suggested through oral microbiome–driven changes in the gut (Lankarani et al. 2013). Changes in the gut microbiota are also associated with IBD, and studies have shown that the expression of TLR9 in intestinal epithelial cells plays an important role in discriminating between commensal and pathogenic bacteria. The exposure of epithelial cells to pathogenic Escherichia coli DNA induces IL-8 production, while exposure to probiotic lactobacillus DNA prevented the induction of IL-8 (de Kivit et al. 2014). It has been shown that the gut microbiota expresses both immunostimulatory and immunosuppressive DNA motifs, with the latter limiting colitis (Bouladoux et al. 2012). Microbial DNA from different oral bacteria also shows differences in immunostimulatory activity (Sahingur et al. 2012). Thus, it will be important to elucidate whether the suppressive DNA motifs in the oral microbial genome can modify immunopathology and affect disease progression. Studies using a murine colitis model have shown that disease severity is increased upon CpG DNA (TLR9 agonist) co-administration and that adverse outcomes were alleviated in TLR9–/– mice (Obermeier et al. 2002). It has been also suggested that the spatial distribution of TLR9 within the cell governs the inflammatory response. TLR9 expression has been demonstrated both at the apical and basal epithelium membranes, where activation elicits differential responses, with the apical epithelium promoting an anti-inflammatory, tolerant response marked by increased barrier functions and the basolateral epithelium producing a proinflammatory response (de Kivit et al. 2014).
Nucleic Acid Sensors and Oral Cavity Cancer
Squamous cell carcinoma (SCC) is the most frequently occurring malignancy of the oral cavity and adjacent sites, representing over 90% of all cancers. While smoking, alcohol consumption, and exposure to human papillomavirus are the most widely accepted etiological factors for oral cavity cancer, the molecular mechanisms triggering tumorigenesis are poorly understood. The tumor microenvironment plays an important role in the initiation and progression of malignant disease, and individuals who are prone to chronic inflammatory disorders have an increased risk of cancer development. An association between chronic periodontitis and cancer both in the oral cavity and at distant sites has been reported, and plausible biological mechanisms linking the 2 conditions have been reviewed (Sahingur and Yeudall 2015). The modulation of inflammation by TLRs plays a crucial role and acts as a double-edged sword in tumor development and progression, inducing both tumor-promoting and antitumor responses. Among these, TLR9 has been implicated in oral SCC. TLR9 expression was reported to be significantly elevated in the tissues of oral SCC, and increased receptor expression was correlated with increased tumor size and clinical stage (Kotrashetti et al. 2013). In vitro studies revealed that the activation of TLR9 can mediate oral cancer cell migration by up-regulating MMP-2, and tumor cell proliferation by up-regulating cyclin D1 expression, both in an AP-1–dependent manner (Ruan, Thorn et al. 2014; Ruan, Zhang et al. 2014). Increased IL-1α and IL-6 production in oral SCC cells treated with a TLR9 agonist was also reported (Ruan, Thorn et al. 2014). Another clinical investigation reported a strong correlation between TLR2, TLR4, and TLR9 expression and increased tumor invasion in tongue SCC (Mäkinen et al. 2015). This study also revealed that increased TLR9 expression correlated with advanced tumor size. The expression of TLR3 and TLR7 has also been detected both in oral SCC tissues and oral SCC cell lines, and the activation of these receptors resulted in increased cytokine expression and apoptosis (Ahn et al. 2012; He et al. 2014). It is plausible that prolonged inflammation in periodontal tissues due to the activation of innate sensors by periodontal microbial products may promote a tumor-favorable environment and warrants further investigation.
Concluding Remarks and Future Perspectives
Rapid progress has been made in recent years in understanding the nucleic acid recognition by the host immune system and its relevance in the pathogenesis of several diseases. Therapeutics targeting nucleic acid sensors are already available. The identification of the immunostimulatory effects of periodontal bDNA and TLR9-mediated periodontal inflammation opens a new avenue in dental research, revealing a novel therapeutic target for PD. Since nucleic acid sensors are also implicated in the recognition of viral nucleic acids and DAMPs, it will be crucial to characterize whether they play a role in virus-related disorders and modulate bacterial-viral interactions within the oral cavity and how the interaction of DAMPs with host sensors contribute to oral inflammation. Moreover, given that microbial nucleic acid sensors have been implicated in systemic diseases that are associated with periodontitis, and periodontal bDNA has been isolated from sites distant from the oral cavity, further studies are warranted to delineate if nucleic acid sensing pathways can be a link between oral and systemic diseases. Areas that require future investigations to delineate the relation between intracellular PRRs and oral diseases are modeled in Figure 5. An improved understanding of the specific cells and molecular pathways involved in microbial DNA sensing and their interaction with other components of the immune system and oral microbiome in the course of oral disease and other pathological conditions will ultimately lead to better oral and systemic health.
Figure 5.
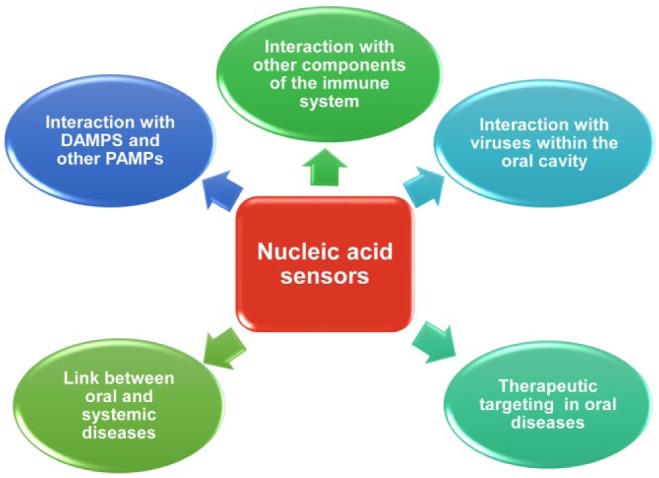
Potential areas of investigation in relation to nucleic acid sensing within the oral cavity.
Author Contributions
K.E. Crump, S.E. Sahingur, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. Both authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
The study was supported by U.S. Public Health Service grants DE025037, DE022836, and KL2TR000057-03 to S.E.S. and K12GM093857 to support K.E.C. from the National Institutes of Health.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Ahn MY, Kwon SM, Cheong HH, Park JH, Lee J, Min SK, Ahn SG, Yoon JH. 2012. Toll-like receptor 7 agonist, imiquimod, inhibits oral squamous carcinoma cells through apoptosis and necrosis. J Oral Pathol Med. 41(7):540–546. [DOI] [PubMed] [Google Scholar]
- Armingohar Z, Jorgensen JJ, Kristoffersen AK, Abesha-Belay E, Olsen I. 2014. Bacteria and bacterial DNA in atherosclerotic plaque and aneurysmal wall biopsies from patients with and without periodontitis. J Oral Microbiol. [epub ahead of print 2014. May 15]. doi: 10.3402/jom.v6.23408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmohamed F, Medina M, Wu YZ, Maschalidi S, Jouvion G, Guillemot L, Chignard M, Manoury B, Touqui L. 2014. Toll-like receptor 9 deficiency protects mice against Pseudomonas aeruginosa lung infection. PLoS One. 9(3):e90466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhan U, Lukacs NW, Osterholzer JJ, Newstead MW, Zeng X, Moore TA, McMillan TR, Krieg AM, Akira S, Standiford TJ. 2007. TLR9 is required for protective innate immunity in gram-negative bacterial pneumonia: role of dendritic cells. J Immunol. 179(6):3937–3946. [DOI] [PubMed] [Google Scholar]
- Bostanci N, Meier A, Guggenheim B, Belibasakis GN. 2011. Regulation of NLRP3 and AIM2 inflammasome gene expression levels in gingival fibroblasts by oral biofilms. Cell Immunol. 270(1):88–93. [DOI] [PubMed] [Google Scholar]
- Bouladoux N, Hall JA, Grainger JR, dos Santos LM, Kann MG, Nagarajan V, Verthelyi D, Belkaid Y. 2012. Regulatory role of suppressive motifs from commensal DNA. Mucosal Immunol. 5(6):623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, Davies AH, Flavell RA, Feldmann M, Monaco C. 2011. Unexpected protective role for toll-like receptor 3 in the arterial wall. Proc Natl Acad Sci U S A. 108(6):2372–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly DJ, O’Neill LA. 2012. New developments in toll-like receptor targeted therapeutics. Curr Opin Pharmacol. 12(4):510–518. [DOI] [PubMed] [Google Scholar]
- de Kivit S, Tobin MC, Forsyth CB, Keshavarzian A, Landay AL. 2014. Regulation of intestinal immune responses through TLR activation: implications for pro- and prebiotics. Front Immunol. 5:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kebir D, Jozsef L, Pan W, Wang LL, Filep JG. 2009. Bacterial DNA activates endothelial cells and promotes neutrophil adherence through TLR9 signaling. J Immunol. 182(7):4386–4394. [DOI] [PubMed] [Google Scholar]
- Erridge C, Burdess A, Jackson AJ, Murray C, Riggio M, Lappin D, Milligan S, Spickett CM, Webb DJ. 2008. Vascular cell responsiveness to toll-like receptor ligands in carotid atheroma. Eur J Clin Invest. 38(10):713–720. [DOI] [PubMed] [Google Scholar]
- Fang R, Hara H, Sakai S, Hernandez-Cuellar E, Mitsuyama M, Kawamura I, Tsuchiya K. 2014. Type I interferon signaling regulates activation of the absent in melanoma 2 inflammasome during Streptococcus pneumoniae infection. Infect Immun. 82(6):2310–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardini Y, Chung P, Dumm R, Joshi N, Han YW. 2010. Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect Immun. 78(4):1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui A, Ohta K, Nishi H, Shigeishi H, Tobiume K, Takechi M, Kamata N. 2013. Interleukin-8 and CXCL10 expression in oral keratinocytes and fibroblasts via toll-like receptors. Microbiol Immunol. 57(3):198–206. [DOI] [PubMed] [Google Scholar]
- González OA, Li MT, Ebersole JL, Huang CB. 2010. HIV-1 reactivation induced by the periodontal pathogens Fusobacterium nucleatum and Porphyromonas gingivalis involves toll-like receptor 4 and 9 activation in monocytes/macrophages. Clin Vaccine Immunol. 17(9):1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulopoulou S, Matsumoto T, Bomfim GF, Webb RC. 2012. Toll-like receptor 9 activation: a novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin Sci (Lond). 123(7):429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Sahingur SE. 2014. Novel inflammatory pathways in periodontitis. Adv Dent Res. 26(1):23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi M, Peters A, Becker A, Bockler D, Dihlmann S. 2014. Inflammation-related induction of absent in melanoma 2 (AIM2) in vascular cells and atherosclerotic lesions suggests a role in vascular pathogenesis. J Vasc Surg. 59(3):794–803. [DOI] [PubMed] [Google Scholar]
- Han X, LaRosa KB, Kawai T, Taubman MA. 2014. DNA-based adaptive immunity protect host from infection-associated periodontal bone resorption via recognition of Porphyromonas gingivalis virulence component. Vaccine. 32(2):297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Huang X, Ni Y, Shi P, Wang Z, Han W, Hu Q. 2014. Functional toll-like receptor 3 expressed by oral squamous cell carcinoma induced cell apoptosis and decreased migration. Oral Surg Oral Med Oral Pathol Oral Radiol. 118(1):92–100. [DOI] [PubMed] [Google Scholar]
- Holla LI, Vokurka J, Hrdlickova B, Augustin P, Fassmann A. 2010. Association of toll-like receptor 9 haplotypes with chronic periodontitis in Czech population. J Clin Periodontol. 37(2):152–159. [DOI] [PubMed] [Google Scholar]
- Kim PD, Xia-Juan X, Crump KE, Abe T, Hajishengallis G, Sahingur SE. 2015. Toll-like receptor 9-mediated inflammation triggers alveolar bone loss in experimental murine periodontitis. Infect Immun. 83(7):2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Jo AR, Jang da H, Cho YJ, Chun J, Min BM, Choi Y. 2012. Toll-like receptor 9 mediates oral bacteria-induced IL-8 expression in gingival epithelial cells. Immunol Cell Biol. 90(6):655–663. [DOI] [PubMed] [Google Scholar]
- Knuefermann P, Schwederski M, Velten M, Krings P, Ehrentraut H, Rüdiger M, Boehm O, Fink K, Dreiner U, Grohé C, et al. 2008. Bacterial DNA induces myocardial inflammation and reduces cardiomyocyte contractility: role of toll-like receptor 9. Cardiovasc Res. 78(1):26–35. [DOI] [PubMed] [Google Scholar]
- Koga K, Cardenas I, Aldo P, Abrahams VM, Peng B, Fill S, Romero R, Mor G. 2009. Activation of TLR3 in the trophoblast is associated with preterm delivery. Am J Reprod Immunol. 61(3):196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Izumi G, Mor G, Fujii T, Osuga Y. 2014. Toll-like receptors at the maternal-fetal interface in normal pregnancy and pregnancy complications. Am J Reprod Immunol. 72(2):192–205. [DOI] [PubMed] [Google Scholar]
- Kotrashetti VS, Nayak R, Bhat K, Hosmani J, Somannavar P. 2013. Immunohistochemical expression of TLR4 and TLR9 in various grades of oral epithelial dysplasia and squamous cell carcinoma, and their roles in tumor progression: a pilot study. Biotech Histochem. 88(6):311–322. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Hajishengallis G. 2015. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. 21(3):172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankarani KB, Sivandzadeh GR, Hassanpour S. 2013. Oral manifestation in inflammatory bowel disease: a review. World J Gastroenterol. 19(46):8571–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, Barton GM. 2014. Trafficking of endosomal toll-like receptors. Trends Cell Biol. 24(6):360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden GJ, Lyons A, Scannapieco FA. 2013. Periodontal systemic associations: review of the evidence. J Clin Periodontol. 40(Suppl 14):S8–S19. [DOI] [PubMed] [Google Scholar]
- Lohner R, Schwederski M, Narath C, Klein J, Duerr GD, Torno A, Knuefermann P, Hoeft A, Baumgarten G, Meyer R, et al. 2013. Toll-like receptor 9 promotes cardiac inflammation and heart failure during polymicrobial sepsis. Mediators Inflamm. 2013:261049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg AM, Ketelhuth DF, Johansson ME, Gerdes N, Liu S, Yamamoto M, Akira S, Hansson GK. 2013. Toll-like receptor 3 and 4 signalling through the TRIF and TRAM adaptors in haematopoietic cells promotes atherosclerosis. Cardiovasc Res. 99(2):364–373. [DOI] [PubMed] [Google Scholar]
- Mäkinen LK, Atula T, Häyry V, Jouhi L, Datta N, Lehtonen S, Ahmed A, Mäkitie AA, Haglund C, Hagström J. 2015. Predictive role of toll-like receptors 2, 4, and 9 in oral tongue squamous cell carcinoma. Oral Oncol. 51(1):96–102. [DOI] [PubMed] [Google Scholar]
- Mori K, Yanagita M, Hasegawa S, Kubota M, Yamashita M, Yamada S, Kitamura M, Murakami S. 2015. Necrosis-induced TLR3 activation promotes TLR2 expression in gingival cells. J Dent Res. 94(8):1149–1157. [DOI] [PubMed] [Google Scholar]
- Murray LA, Knight DA, McAlonan L, Argentieri R, Joshi A, Shaheen F, Cunningham M, Alexopolou L, Flavell RA, Sarisky RT, et al. 2008. Deleterious role of TLR3 during hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 178(12):1227–1237. [DOI] [PubMed] [Google Scholar]
- Ng MT, Van’t Hof R, Crockett JC, Hope ME, Berry S, Thomson J, McLean MH, McColl KE, El-Omar EM, Hold GL. 2010. Increase in NF-kappaB binding affinity of the variant C allele of the toll-like receptor 9 -1237T/C polymorphism is associated with Helicobacter pylori-induced gastric disease. Infect Immun. 78(3):1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier F, Dunger N, Deml L, Herfarth H, Scholmerich J, Falk W. 2002. CpG motifs of bacterial DNA exacerbate colitis of dextran sulfate sodium-treated mice. Eur J Immunol. 32(7):2084–2092. [DOI] [PubMed] [Google Scholar]
- Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al. 2012. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 485(7397):251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda B, Panda A, Ueda I, Abrahams VM, Norwitz ER, Stanic AK, Young BC, Ecker JL, Altfeld M, Shaw AC, et al. 2012. Dendritic cells in the circulation of women with preeclampsia demonstrate a pro-inflammatory bias secondary to dysregulation of TLR receptors. J Reprod Immunol. 94(2):210–215. [DOI] [PubMed] [Google Scholar]
- Pandey S, Kawai T, Akira S. 2015. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Med. 5(1):a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Na HS, Song YR, Shin SY, Kim YM, Chung J. 2014. Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect Immun. 82(1):112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker D, Prince A. 2012. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol. 189(8):4040–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo-Botello NR, Garcia-Hernandez AL, Moreno-Fierros L. 2012. Expression of toll-like receptors 2, 4 and 9 is increased in gingival tissue from patients with type 2 diabetes and chronic periodontitis. J Periodontal Res. 47(1):62–73. [DOI] [PubMed] [Google Scholar]
- Ruan M, Thorn K, Liu S, Li S, Yang W, Zhang C, Zhang C. 2014. The secretion of IL-6 by CpG-ODN-treated cancer cells promotes T-cell immune responses partly through the TLR-9/AP-1 pathway in oral squamous cell carcinoma. Int J Oncol. 44(6):2103–2110. [DOI] [PubMed] [Google Scholar]
- Ruan M, Zhang Z, Li S, Yan M, Liu S, Yang W, Wang L, Zhang C. 2014. Activation of toll-like receptor-9 promotes cellular migration via up-regulating MMP-2 expression in oral squamous cell carcinoma. PLoS One. 9(3):e92748. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sahingur SE, Xia XJ, Alamgir S, Honma K, Sharma A, Schenkein HA. 2010. DNA from Porphyromonas gingivalis and Tannerella forsythia induce cytokine production in human monocytic cell lines. Mol Oral Microbiol. 25(2):123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Gunsolley J, Schenkein HA, Genco RJ, De Nardin E. 2011. Single nucleotide polymorphisms of pattern recognition receptors and chronic periodontitis. J Periodontal Res. 46(2):184–192. [DOI] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Schifferle RE. 2012. Oral bacterial DNA differ in their ability to induce inflammatory responses in human monocytic cell lines. J Periodontol. 83(8):1069–1077. [DOI] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Voth SC, Yeudall WA, Gunsolley JC. 2013. Increased nucleic acid receptor expression in chronic periodontitis. J Periodontol. 84(10):e48–e57. [DOI] [PubMed] [Google Scholar]
- Sahingur SE, Yeudall WA. 2015. Chemokine function in periodontal disease and oral cavity cancer. Front Immunol. 6:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salagianni M, Galani IE, Lundberg AM, Davos CH, Varela A, Gavriil A, Lyytikäinen LP, Lehtimäki T, Sigala F, Folkersen L, et al. 2012. Toll-like receptor 7 protects from atherosclerosis by constraining “inflammatory” macrophage activation. Circulation. 126(8):952–962. [DOI] [PubMed] [Google Scholar]
- Scharfe-Nugent A, Corr SC, Carpenter SB, Keogh L, Doyle B, Martin C, Fitzgerald KA, Daly S, O’Leary JJ, O’Neill LA. 2012. TLR9 provokes inflammation in response to fetal DNA: mechanism for fetal loss in preterm birth and preeclampsia. J Immunol. 188(11):5706–5712. [DOI] [PubMed] [Google Scholar]
- Slots J. 2005. Herpesviruses in periodontal diseases. Periodontol 2000. 38:33–62. [DOI] [PubMed] [Google Scholar]
- Uehara A, Takada H. 2008. Synergism between TLRs and NOD1/2 in oral epithelial cells. J Dent Res. 87(7):682–686. [DOI] [PubMed] [Google Scholar]
- Wu J, Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 32:461–488. [DOI] [PubMed] [Google Scholar]
- Xue F, Shu R, Xie Y. 2015. The expression of NLRP3, NLRP1 and AIM2 in the gingival tissue of periodontitis patients: RT-PCR study and immunohistochemistry. Arch Oral Biol. 60(6):948–958. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yamamoto T, Kataoka T, Kuramoto E, Yano O, Tokunaga T. 1992. Unique palindromic sequences in synthetic oligonucleotides are required to induce INF and augment INF-mediated natural-killer activity. J Immunol. 148(12):4072–4076. [PubMed] [Google Scholar]
- Zhan YB, Zhang RM, Lv HC, Song XJ, Xu XM, Chai L, Lv WH, Shang ZW, Jiang YS, Zhang RJ. 2014. Prioritization of candidate genes for periodontitis using multiple computational tools. J Periodontol. 85(8):1059–1069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.