

Abstract
Understanding the genetic basis of adaptation to climate is of paramount importance for preserving and managing genetic diversity in plants in a context of climate change. Yet, this objective has been addressed mainly in short-lived model species. Thus, expanding knowledge to nonmodel species with contrasting life histories, such as forest trees, appears necessary. To uncover the genetic basis of adaptation to climate in the widely distributed boreal conifer white spruce (Picea glauca), an environmental association study was conducted using 11,085 single nucleotide polymorphisms representing 7,819 genes, that is, approximately a quarter of the transcriptome.
Linear and quadratic regressions controlling for isolation-by-distance, and the Random Forest algorithm, identified several dozen genes putatively under selection, among which 43 showed strongest signals along temperature and precipitation gradients. Most of them were related to temperature. Small to moderate shifts in allele frequencies were observed. Genes involved encompassed a wide variety of functions and processes, some of them being likely important for plant survival under biotic and abiotic environmental stresses according to expression data. Literature mining and sequence comparison also highlighted conserved sequences and functions with angiosperm homologs.
Our results are consistent with theoretical predictions that local adaptation involves genes with small frequency shifts when selection is recent and gene flow among populations is high. Accordingly, genetic adaptation to climate in P. glauca appears to be complex, involving many independent and interacting gene functions, biochemical pathways, and processes. From an applied perspective, these results shall lead to specific functional/association studies in conifers and to the development of markers useful for the conservation of genetic resources.
Keywords: environmental gradient, genetic basis of adaptation, genomics of adaptation, population genomics, white spruce
Introduction
One of the major aims of evolutionary biology is to unravel the genetic basis of important biological processes such as adaptation. However, although evidence for local adaptation in plants and animals continues to accumulate (Linhart and Grant 1996; Leimu and Fischer 2008; Hereford 2009; Weigel 2012; Luquet et al. 2015), its genetic basis remains poorly understood. In the context of climate change, deciphering the genetic architecture of adaptive traits becomes increasingly critical as it controls the ability of populations to respond to natural selection (Etterson and Shaw 2001), which may allow them to avoid extirpation. Genomics helps answer fundamental questions related to the genetic architecture of adaptation, such as determining the identity and the number of genes involved, their effects, their interactions, their function, as well as the origin and fate of adaptive alleles, at the genomic scale (Hendry 2013; Wray 2013). This is well illustrated by studies focusing on the model species Arabidopsis thaliana (Fournier-Level et al. 2011; Hancock et al. 2011), where genome-wide data were used to identify a set of ecologically relevant genes across the entire species native range. They showed that most of these genes differed across environments, which supports the view that local adaptation involves alleles that are beneficial in one environment but neutral elsewhere (conditional neutrality), rather than alleles that are beneficial in one environment and deleterious in another (antagonistic pleiotropy) (Colautti et al. 2012). They also identified several molecular functions and biological processes that may be important for adaptation in A. thaliana. Thus, such empirical studies in model and nonmodel species (Prunier et al. 2011; Evans et al. 2014; Huber et al. 2014; Yoder et al. 2014; see also Ellegren 2014) can lead to a better understanding of the adaptation process, with regard to genetic architecture, dynamics of selection at adaptive genes, and biological processes involved.
Theoretical studies have also addressed the effects of the complex interplay between selection and other evolutionary forces, such as gene flow, on the genetic architecture of adaptation (reviewed in Olson-Manning et al. 2012; Savolainen et al. 2013). Specifically, Kremer and Le Corre (2012) and Le Corre and Kremer (2012) have shown that under high gene flow, adaptation should start to build up mainly from covariance of alleles between populations, before allele frequency shifts occur. Consequently, when selection is recent, little genetic differentiation among populations should be expected at most adaptive loci. However, selection acting in prolonged periods under high gene flow should favor genetic architectures characterized by fewer alleles with large effects, and more tightly linked (Yeaman and Whitlock 2011). Empirical studies in plants, including trees, have found a variety of genetic architectures, from few large-effect alleles to many small-effect alleles and combinations of both, depending on the species and the trait considered (Bradshaw et al. 1998; Beaulieu et al. 2011; Pelgas et al. 2011; Li et al. 2013; Prunier et al. 2013). Despite the recent progress made on the theoretical ground, more empirical studies are needed to better link theoretical predictions with empirical evidence.
Another key issue is the functional characterization of genes involved in adaptation (Wray 2013). Theory predicts that the selective constraints on genes will differ according to their level of connectivity with other genes or their position in cellular response pathways or gene networks, be they regulatory or metabolic (Olson-Manning et al. 2012). Empirical studies on environmental response and genetic adaptation in plants have found a great diversity of putative functions, from enzymes to transcription factors, related to stress response, development, phenology, and other adaptive traits (Seki et al. 2002; Namroud et al. 2008; Eckert et al. 2010; Prunier et al. 2011; Chen et al. 2012; McKown et al. 2014). This pattern is consistent with the frequent cross-talks that occur between different pathways and networks (Fujita et al. 2006).
Studies of adaptive traits in perennial plants such as forest trees often revealed the presence of clinal variation in phenotypes, as well as genetic differentiation among populations, suggesting that local adaptation is pervasive in trees (reviewed in Alberto et al. 2013; Kremer et al. 2014) despite high gene flow, and despite that many species from temperate and boreal biomes have relocated only recently in the Holocene. White spruce (Picea glauca [Moench] Voss) is a coniferous tree of the North American boreal forest with a large latitudinal distribution. The species has colonized its current range during the first half of the Holocene (Payette 1993). It is also an anemophilous species characterized by extensive gene flow (Jaramillo-Correa et al. 2001; Namroud et al. 2008). Evidence for local adaptation in relation to climate was previously found in white spruce populations, both at the phenotypic and molecular levels (Li et al. 1997; Jaramillo-Correa et al. 2001; Lesser and Parker 2004; Namroud et al. 2008), but using a limited number of DNA markers.
In this study, we examined the genetic basis of adaptation to climate for a large part of the white spruce transcriptome by investigating associations between environmental variables and frequencies of 11,085 single nucleotide polymorphisms (SNPs) representative of 7,819 expressed genes. Based on theoretical grounds, we expect to find dozens to hundreds of genes putatively involved in genetic adaptation to climate with moderate differentiation, because gene flow is extensive in white spruce and local climatic selection would be relatively recent, given the postglacial recolonization of white spruce and its long generation time (Bouillé and Bousquet 2005). We also expect to find mostly small allele frequency shifts, if adaptation occurs mainly through conditional neutrality, and if adaptation builds up mainly from covariance, given that local selection would be quite recent. Finally, because climate (and environmental factors correlated to climate) may select several traits linked to growth, phenology, and physiology (Alberto et al. 2013; Franks et al. 2014), we expect to find a great diversity of functions and processes involved in genetic adaptation to climate. In order to detect genes under selection along temperature and precipitation gradients, we conducted an environmental association study in P. glauca populations from eastern Canada following a large latitudinal transect and climatic gradient. Covariation between allele frequencies and climatic variables, as well as decision trees obtained with the Random Forest (RF) algorithm, were used to identify a limited set of loci putatively under selection. Functional and ecological annotations were used to characterize the functions and processes encompassed by the statistically most robust adaptive genes.
Materials and Methods
Sampling and Environmental Variables
Twigs were collected on 198 white spruce individuals sampled in 41 populations from eastern Canada (1–13 individuals per population; fig. 1 and supplementary table S1, Supplementary Material online).
Fig. 1.—

Location of the 41 populations of Picea glauca sampled in eastern Canada. Numbers identify each population (supplementary table S1, Supplementary Material online). Bold numbers indicate the 27 populations containing at least 4 sampled individuals (see text). Circle color represents mean annual temperature, from warmer (red) to cooler (dark blue). Circle size represents total annual precipitation, from drier (small size) to moister (large size). The green area represents the distribution range of P. glauca in the region.
Mean annual temperature and total annual precipitation were the two climatic factors retained for this study. Indeed, temperature and precipitation regimes are expected to change drastically with anticipated global warming and are likely to affect adaptation of white spruce populations (Andalo et al. 2005). Moreover, white spruce traits related to growth and phenology were shown to be well correlated with latitude (Li et al. 1997; Jaramillo-Correa et al. 2001), which strongly covaries with mean annual temperature in the studied region (r = −0.89, N = 41, P < 0.001). Some of these traits are also correlated with longitude (Li et al. 1997; Jaramillo-Correa et al. 2001), which is correlated to total annual precipitation (r = −0.52, N = 41, P < 0.001) and mean annual temperature (r = −0.47, N = 41, P = 0.002) in the studied region. Finally, many other climatic variables are correlated to mean annual temperature or total annual precipitation in the studied region (Prunier et al. 2012), so that investigating these two variables could give insights into adaptation to climate in general. Data averaged over 30 years (1981–2010) were obtained for each population by extrapolating data from nearby weather stations using the simulation model of Régnière (1996) implemented in BioSIM. Across the populations sampled, mean annual temperature and total annual precipitation varied from −3.8°C to 6.8 °C and from 705 to 1587 mm, respectively (fig. 1 and supplementary table S1, Supplementary Material online). The two climatic variables were moderately correlated across populations (r = 0.45, P < 0.01).
DNA Extraction and Genotyping
DNA was extracted from 100 mg of needles and buds using the NucleoSpin 96 Plant II kit (Macherey-Nagel, Duren, Germany) and the DNeasy 96 Plant Kit (Qiagen, Mississauga, Canada). A minimum of 80 ng of template genomic DNA per sample was used for genotyping.
The 14,842 SNPs successfully genotyped were distributed across 9,938 expressed genes. They were genotyped with two Illumina iSelect Infinium arrays (Pavy, Gagnon, et al. 2013) at the McGill University Genome Quebec Innovation Center (Montreal, Canada). SNPs that did not match all the following criteria were discarded: call rate ≥90%, minor allele frequency ≥5%, and absolute fixation index FIS ≤ 0.5. A subset of 11,085 SNPs located in 7,819 genes was retained, representing 24% of the estimated number of expressed genes in white spruce (Rigault et al. 2011). Overall, there was less than 1% missing genotypes. The average number of SNPs per gene was 1.4 (ranging from 1 to 11), with 6,218 genes (79.5%) harboring only one SNP. Mean total expected heterozygosity (HE) estimated over 7,819 SNPs (one random SNP per gene) was 0.329, while mean observed heterozygosity (Ho) was 0.324.
Some of the studied genes were putatively involved in wood formation, growth, and adaptation to biotic and abiotic factors, and were selected based on previous studies (Pavy, Deschênes, et al. 2013). An overview of biological processes, molecular functions, and cellular components represented in the 7,819 genes used here is provided in supplementary figure S1, Supplementary Material online.
Data Analysis
Spatial Genetic Structure
Neutral population structure was investigated because it can lead to the detection of false positives when identifying loci under selection. Population structure was first tested using the Bayesian algorithms implemented in STRUCTURE (Pritchard et al. 2000; Falush et al. 2003) and BAPS (Corander et al. 2006) on a subset of 4,000 SNPs (one SNP per gene in 4,000 random genes) for computational reasons. This large SNP subset is assumed to be largely representative of neutral variation. STRUCTURE was run using the admixture model and correlated allele frequencies for 500,000 iterations after a burn-in period of 100,000 iterations. For each value of K ranging between 1 and 10, 10 independent runs were performed and population structure was assessed graphically using the maximum-likelihood method and the ΔK method of Evanno et al. (2005). For BAPS, 5 runs were performed for several values of K between 1 and 41 (the actual number of populations sampled) and the partition yielding the highest estimated probability was considered optimal. In both methods, the spatial information on the origin of populations or individuals was not taken into account.
Population structure was further assessed by estimating FST, by using a Mantel test, and by performing principal component analyses (PCAs). FST among the 27 populations carrying at least 4 individuals was estimated in GenAlEx 6.5 (Peakall and Smouse 2012), using the same subset of 4,000 SNPs previously mentioned. A matrix of pairwise FST among populations was generated in GenAlEx with the same data set. Next, a matrix of pairwise geographic distances between populations was computed from geographic coordinates and the correlation between genetic and geographic matrices was estimated using 999 permutations in GenAlEx. PCA was applied separately to individuals (198 individuals) and to populations (27 populations carrying at least 4 individuals), using the prcomp function in R (R Development Core Team 2010), in order to 1) summarize neutral genetic variation in the studied region and 2) produce principal components useful to correct for population structure in regression (which uses genetic data at the population level) and RF (which uses individual genotypes) analyses. Because population scores and individual scores on the first components produced were further used in regression and RF analyses, respectively (see below), all 11,085 SNPs were retained to perform the PCA. Input genetic data were allele frequencies within populations, and genotypes within individuals coded as 0 (homozygote), 0.5 (heterozygote), or 1 (alternate homozygote).
Detection of Loci Under Selection
Random Forest Analysis
The RF algorithm (Breiman 2001) produces decision trees that recursively split observations according to several predictor variables, resulting in a model that may achieve a good predictive accuracy. Its particularity is that it introduces two layers of randomness to improve model accuracy: 1) each tree is grown from a bootstrap sample of the observations and 2) at each node in a tree the best split is selected based on a random subset of predictors. The construction of one tree thus follows these steps: 1) take a bootstrap sample of the observations (∼2/3); 2) at each node, select a random subset of size mtry of predictor variables and split the data with the one that provides the best split of the observations (based on residual mean square error [MSE] in the dependent variable); and 3) recursively split the data until each terminal node is pure or contains a minimum number of observations (default value of 5 in this study). For each tree, the ∼1/3 of data that was not used to train the model (out-of-bag data, OOB) was used to compute several estimates. First, the constructed model was used to compare the actual value of the dependent variable of the OOB samples with the one predicted by the model. Across the forest of decision trees, this procedure computes the variance in the dependent variable that is explained by the model. Second, the values of each predictor used in a given tree were permuted. The resulting change in global MSE enables estimating the importance of the predictor in the model: if permuting the values of a predictor does alter model accuracy and increases error, then this predictor is important, and vice versa. Across all the decision trees, this permutation procedure leads to an importance measure (increase in % MSE) for each predictor. Interaction between predictors is implicitly taken into account during model training. For instance, if a SNP does not have a good splitting power by itself, but splits the samples very well after another SNP had split them, its importance will be high and this could reveal an interaction between these two SNPs (fig. 2).
Fig. 2.—
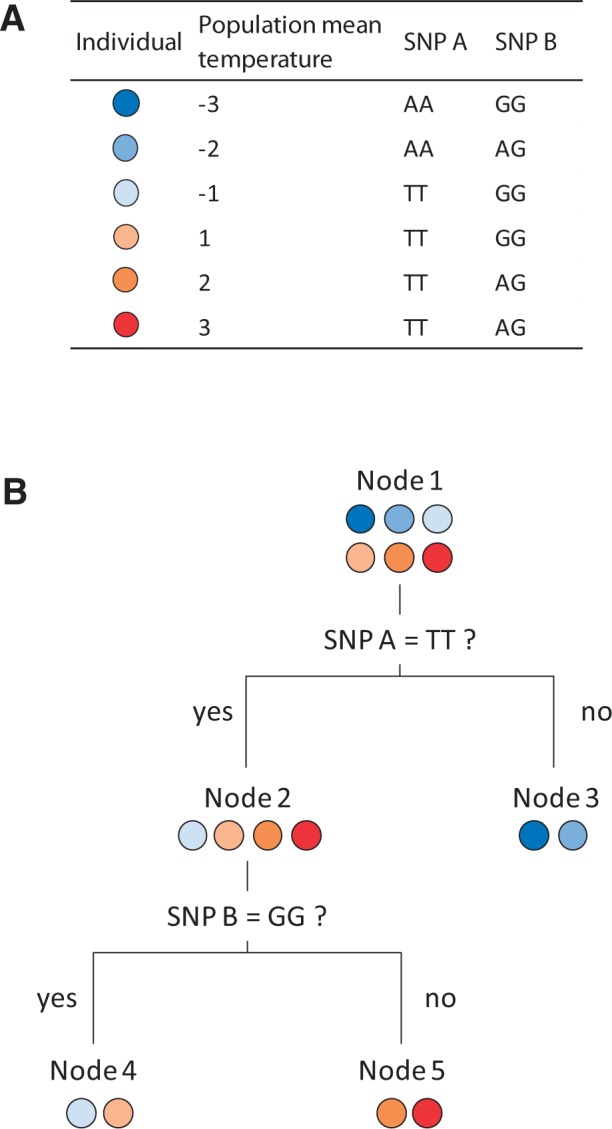
Toy example of the detection of important SNPs in RF. (A) Toy data set representing six individuals located in six populations with different mean annual temperatures, genotyped at two loci. (B) RF splits the individuals at the first node according to their genotype at SNP A, which leads to two rather homogeneous child nodes in terms of temperature. Splitting individuals at the first node according to their genotype at SNP B would have led to more heterogeneous child nodes, so RF would rather split the individuals using SNP A at the first node. Although SNP B had a poor splitting power at the first node, it has a good splitting power at the second node. Permuting the values of SNP A or SNP B would increase heterogeneity (variance) in the child nodes, meaning that they are important in the model. In this example, SNP A would thus have a main effect, while the effect of SNP B would depend on genotype at SNP A, suggesting an interaction. In regression, we would find SNP A as associated to temperature, contrary to SNP B, whereas it might still be important in adaptation to temperature through an interaction with another SNP. Note that in the actual RF analysis using many more individuals, nodes are not split (and thus considered terminal) when containing five individuals or less.
Regression trees (instead of classification trees) were used because the dependent variable (temperature or precipitation) was continuous. The objective of the analysis was to identify a restricted set of SNPs that together explain most of the variance in temperature or precipitation among individuals, indicating that they may be under selection for these variables. The RF analysis was performed in R with the randomForest package (Liaw and Wiener 2002). The data set contained less than 1% missing genotypes, which were imputed using the rfImpute function. The default mtry value was used (number of SNPs/3) and the number of decision trees ntree was set to 10,000. For the RF analysis with precipitation, one population (population 25 in fig. 1 and supplementary table S1, Supplementary Material online) was discarded because it had an outlying precipitation value and led to many likely false positive relationships. First, a full model including all SNPs was computed to estimate the importance of each SNP. Because few of them had a high importance in the full model (see Results and Discussion), we considered the 100 most important SNPs in RF for each climatic variable, to which we applied a backward elimination procedure aiming at finding the most important ones (Diaz-Uriarte et al. 2006; Holliday et al. 2012). The first step of this procedure was to build a model with the 100 top SNPs, and discard the least important one based on at least 5 independent runs of the model. This procedure was iterated until the model contained only two SNPs (the minimal model possible). For each model, the proportion of explained variance was estimated. Null models with 2, 5, 10, 15, 20, and 50 random SNPs were run several times to estimate whether the backward elimination approach was efficient in selecting important SNPs (Holliday et al. 2012).
A weak population structure related to isolation-by-distance (IBD) was detected in the studied region, as indicated by significant Mantel test (see Results and Discussion). Because no option is implemented in RF to directly account for population structure, the effect of genetic differences among individuals due to neutral processes was subtracted from the dependent variable (i.e., temperature or precipitation) before performing the analysis (Holliday et al. 2012). This was done by regressing temperature and precipitation values on scores of individuals on the first axis of a PCA performed on individuals (see Results and Discussion), and then using residuals as the dependent variable in RF.
Linear and Quadratic Regression Analyses
Covariation between allele frequencies and climatic variables was estimated through linear and quadratic regressions taking into account the weak population structure related to IBD. Populations with less than four sampled individuals were discarded to increase the reliability of regression models estimation. The analysis was thus performed using data from 27 populations, represented by 171 individuals. The population with an outlying precipitation value was also discarded in regression analysis for precipitation (see above). In addition to linear regression models, quadratic associations were assessed because the relationship between allele frequency and environmental variables may not necessarily be linear, for instance when an allele is only selected on a restricted part of the gradient (Manel et al. 2010; Prunier et al. 2012). Associations between allele frequencies and climatic variables were tested with logistic regression using the glm function in R. The weak population structure was accounted for by adding a covariate in the regression model. Values of this covariate were population scores on the first axis of a PCA performed on population allele frequencies (see Results and Discussion). The original variables were standardized to avoid collinearity between the linear and the quadratic terms. First, the best model (linear or quadratic) was selected using a likelihood-ratio test because the linear model is nested within the quadratic model. Then, the significance of the linear and the quadratic terms (when present in the model) were assessed using a Wald test. To account for multiple testing, a false discovery rate (FDR; Benjamini and Hochberg 1995) was applied to P values of linear terms and P values of quadratic terms. A SNP was considered associated with a given climatic variable if at least one term of the model (linear and/or quadratic) was significant after the correction. Both FDR = 0.10 (permissive threshold) and FDR = 0.05 (strict threshold) were considered.
Gene Annotations
Although linkage disequilibrium (LD) is generally weak in P. glauca, it is heterogeneous among genes and along the genome (Namroud et al. 2010; Pavy, Namroud, et al. 2012; Pavy, Pelgas, et al. 2012). Thus, we cannot firmly exclude that the gene bearing a SNP showing a signature of selection is the actual gene selected (as opposed to a nearby gene, if both are located in a genomic region with high LD). In the following, we assume that each gene harboring a SNP showing a selection signal is actually under selection. Given that we mainly discuss classes (e.g., gene families, functional classes, biological processes) rather than individual genes, and that LD is generally weak in P. glauca, we trust that our conclusions are not sensitive to this issue.
Several functional annotations were retrieved from public databases to describe the studied genes: keywords from Uniprot KB (Uniprot Consortium 2014), metabolic pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2014), and A. thaliana homologs from The Arabidopsis Information Resource (TAIR) (Lamesch et al. 2012). PFAM annotations, including protein families and domains, were retrieved from the P. glauca gene catalog (Rigault et al. 2011).
BLASTP was run against the protein database Uniprot-SwissProt. Keywords associated with the best hit with an e-value < 1 × 10−10 were retrieved with the seqret program from EMBOSS (Rice et al. 2000). Specifically, the keywords corresponding to the widely used categories “biological process,” “molecular function,” and “cellular component” were used. Uniprot keywords could be assigned to 5,090 genes (∼65% of the 7,819 genes), and represented diverse biological processes (144 terms), molecular functions (35 terms), and cellular components (38 terms; supplementary figure S1, Supplementary Material online).
Putative metabolic pathways were assigned to genes based on annotations from the manually curated KEGG database. First, the best KEGG Orthology number (KO) was assigned to each gene using the KEGG Automatic Annotation Server based on nucleotide sequence and the single-directional best hit method against a database including A. thaliana and poplar (Populus trichocarpa) genes. Next, the Search Pathway tool was used against poplar (rather than A. thaliana because poplar is a tree) to find the pathways that included the retrieved KO. Along with specific pathway terms (e.g., “galactose metabolism”), the subcategory associated with each pathway (e.g., “carbohydrate metabolism”), and the corresponding broader category (e.g., “metabolism”) were also retrieved. Among the 7,819 studied genes, 2,871 (∼37%) were annotated with a KO. They encompassed 1,728 different KO terms, of which 853 corresponded to a KEGG pathway term, representing a total of 121 poplar KEGG pathways. In total, pathway annotations were available for 1,462 of the 7,819 genes (∼19%). These pathways represented the major KEGG pathway terms categories: “metabolism,” “genetic information processing,” “cellular processes,” “environmental information processing,” and “organismal systems.” Within these categories, the major subcategories represented in our data set were (excluding the noninformative “Global and overview maps” term) as follows: “carbohydrate metabolism,” “translation,” and “folding, sorting, and degradation.”
Putative homologs between P. glauca and A. thaliana genes was assessed using BLASTP (e-value < 1 × 10−10) against the TAIR database. A literature mining tool (EVEX) (Van Landeghem et al. 2011) was then used to retrieve information about gene regulation and interacting networks, for each homologous gene described in public databases.
To search for ecologically relevant annotations, gene sequences were compared with genes whose expression under environmental stress or seasonal transitions has been studied in angiosperms and in spruces: in rice in response to drought (Zhang et al. 2012), in A. thaliana in response to cold, drought, salt, or UV-B (Kilian et al. 2007) (at 1 and 24 h for each stress, pooled between roots and shoots), in Eucalyptus camaldulensis in response to water stress (Thumma et al. 2012), and during bud set in Sitka spruce, Picea sitchensis (Holliday et al. 2008) (between any time point and day 0). Sequence data were retrieved from the supplementary materials of the cited articles, from online databases, or directly from the authors. First, P. glauca genes that did not match any sequence on the gene chip used by these authors in their expression experiment were discarded. Then, the remaining P. glauca genes were compared with the differentially expressed genes (DEGs). When only raw data were available, the DEG status of genes was determined using the same criteria as those reported in the original papers (see supplementary table S2, Supplementary Material online, for more information regarding DEG datasets). For angiosperms, only sequence pairs with at least 50% identity over at least 100 amino acids, and BLASTP e-value < 1 × 10−10 were considered. For P. sitchensis sequences, the identity threshold was set to 95% identity at the nucleic level.
Picea glauca transcriptome was analyzed during the growth-to-dormancy transition by El Kayal et al. (2011) and Galindo-Gonzalez et al. (2012). These studies relied on an early version of the Arborea transcriptomic array (Pavy et al. 2008) and therefore did not include all the genes studied herein. Thus, transcriptomic profiles were only available for 4,186 genes (∼54%). Out of them, 2,258 (∼54%) were DEGs in forming buds (El Kayal et al. 2011), 1,415 (∼34%) were DEGs in whole stems (Galindo-Gonzalez et al. 2012), and 1,085 (∼26%) genes were differentially expressed in both studies. Of the 5,832 genes matching a P. sitchensis gene studied by Holliday et al. (2008), 1,098 (∼19%) matched a DEG with the above criteria. For the sake of simplicity, we use “bud set” to refer to the transition from growth to dormancy studied by Holliday et al. (2008), El Kayal et al. (2011), and Galindo-Gonzalez et al. (2012), although many other physiological states are related to this transition. Indeed, the transition from growth to dormancy is characterized by the cessation of growth, bud set, dormancy, and acquisition of cold hardiness. The timing of these stages is of paramount importance for fitness because early bud set might limit tree growth while late bud set increases susceptibility to cold injuries. All the above-mentioned transcriptomic studies used biological replicates, but used various combinations of fold change thresholds and/or statistical thresholds (corrected or uncorrected P values).
All these annotations were used to provide a description of genes carrying SNPs likely under selection and to infer their putative functional importance. Fisher’s exact tests were performed to determine whether genes significantly matching a given annotation were enriched within the set of genes under selection compared with the remaining genes. Finding significant enrichments would mean that the set of genes putatively under selection is different from a random set of genes of the same size. Enrichment tests were only performed on terms represented by at least five members in the whole data set.
Results and Discussion
Spatial Genetic Structure
According to BAPS, all 198 individuals belonged to a single genetic cluster (log(likelihood) = −798,380; P = 1). STRUCTURE revealed a similar trend, although the optimal partition was K = 2 (supplementary fig. S2, Supplementary Material online). The detected population structure was weak and had little biological meaning, with only four individuals being assigned to the second genetic cluster identified (supplementary fig. S3, Supplementary Material online). These individuals were not misidentified given that they had similar rate of missing data than for the rest of the trees (less than 1.5%) and given that their heterozygosity rate was similar to that of other trees (0.18–0.34 vs. 0.17–0.35, respectively). A drastic decrease in heterozygosity would have been expected if these individuals belonged to the sympatric but nonhybridizing and phylogenetically distantly related black spruce (Picea mariana) (Bouillé et al. 2011), given the low level of SNP sharing between white spruce and black spruce (Pavy, Gagnon, et al. 2013). Three of these four individuals belonged to the same population, located at the western end of the studied region (population 8 on fig. 1), while the fourth individual belonged to another population (population 15 on fig. 1). The overall pattern is thus consistent with the fact that all populations were sampled across a single white spruce glacial lineage.
Consistently, overall genetic population differentiation was weak, although significantly different from zero (FST = 0.021, P = 0.001, 999 permutations). Mantel test revealed that genetic and geographic distances between populations were weakly correlated (r = 0.27; P = 0.02, 999 permutations): pairs of populations farther away tended to be more genetically differentiated, likely due to slight IBD.
Although the first axis of the PCA analysis on individuals only explained 1.3% of the total genetic variation among individuals (supplementary fig. S4A, Supplementary Material online), it summarized well the genetic structure detected with the population structure analyses. Indeed, it clearly separated the four individuals detected in STRUCTURE from the rest of the samples, as their score was greater than 48, while all remaining individuals had scores comprised between −13 and 12. The first principal component primarily separated individuals according to longitude: the correlation between individuals scores on the first principal component and longitude was strong and significant (r = −0.42, P < 0.0001). This correlation was even stronger when removing the four outlying individuals (r = −0.68, P < 0.0001). The first axis of the PCA analysis on populations explained 6.8% of the genetic variation among populations (supplementary fig. S4B, Supplementary Material online) and effectively summed up the IBD pattern observed. Indeed, the pairwise differences in population scores on this component were correlated to geographic distances between populations (Mantel test, r = 0.27; P = 0.02, 999 permutations). This component also separated populations according to longitude: the correlation between populations scores on the first principal component and longitude was strong and significant (r = −0.52, P = 0.005). Populations having the highest scores on the first principal component were the westernmost populations, which included population 8 (which carried most individuals belonging to the STRUCTURE small second cluster) (supplementary table S1, Supplementary Material online).
Altogether, these results suggest that there is a weak spatial genetic structure in the studied region, likely driven by IBD. From a phylogeographical perspective, these results indicate that only one glacial lineage occurs in the studied region, which would have colonized eastern Canada from an Appalachian refugium following the last glacial maximum (de Lafontaine et al. 2010). From a methodological point of view, these results show how genotyping thousands of SNPs might reveal cryptic population structure, even in species with high gene flow. Because selection could result in small genotype or allele frequency shifts among populations in such species, it is necessary to control for neutral genetic structure to minimize the detection of false positives. In this respect, principal component scores proved to be good proxies for population structure.
Detection of Genes Under Selection
Figure 3 illustrates examples of significant linear and quadratic relationships between allelic frequencies and climatic variables. The results of the two approaches (regressions and RF controlling for population structure using PCA scores) are summarized in table 1. Out of the 11,085 SNPs tested, regression analyses identified 33 polymorphisms (0.3% of tested SNPs) significantly correlated to variations in mean annual temperature and/or total annual precipitation at FDR = 0.05, with only two of them being associated to precipitation. Several of these SNPs showed a quadratic relationship with no significant linear term, which means that they would not have been detected by testing a linear regression model alone. Using the less stringent FDR of 0.10 led to the detection of roughly four times more SNPs (1.2% of tested SNPs), mainly for temperature (table 1).
Fig. 3.—
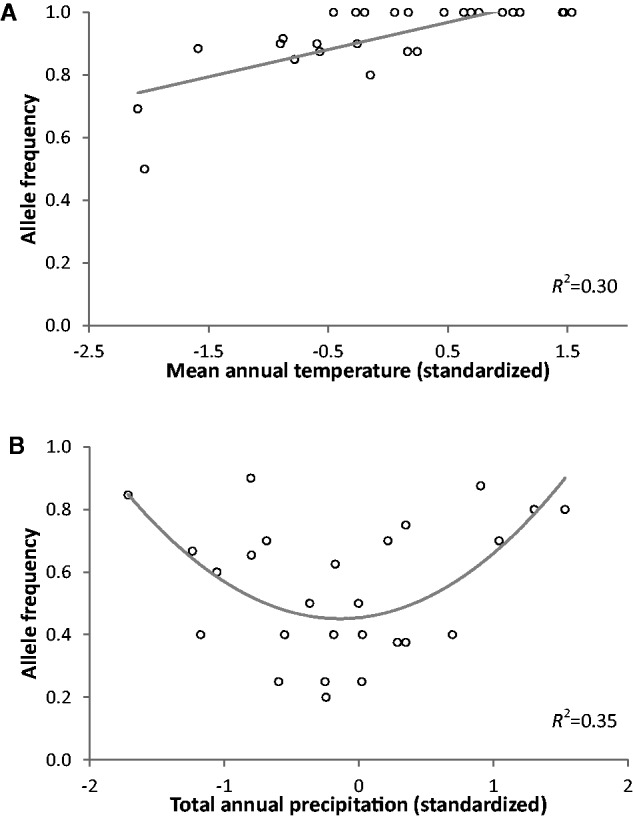
Examples of significant relationships between climatic variables and SNP allele frequencies. (A) Linear relationship between temperature and SNP allele frequency (in a β-tubulin gene). (B) Quadratic relationship between precipitation and SNP allele frequency (in an ATP-binding cassette transporter gene). Note the typically low R2 values observed.
Table 1.
Overview of the Number of SNPs and Genes Showing Strong Signals of Climate Selection in Picea glauca from the Analyzing 11,085 SNPs in 7,819 Genes
| Number of Significant SNPs (Genes) |
|||
|---|---|---|---|
| Mean Annual Temperature | Total Annual Precipitation | Union | |
| FDR = 0.05 | |||
| Linear effect | 24 (22) | 2 (2) | 24 (22) |
| Quadratic effect | 10 (10) | 0 (0) | 10 (10) |
| Linear and quadratic effects | 33 (31) | 2 (2) | 33 (31) |
| FDR = 0.10 | |||
| Linear effect | 95 (84) | 4 (4) | 96 (85) |
| Quadratic effect | 43 (40) | 2 (2) | 44 (41) |
| Linear and quadratic effects | 134 (121) | 5 (5) | 136 (123) |
| Random Forest | |||
| Top 10 | 10 (10) | 10 (9) | 19 (18) |
| Union of regression and RF | |||
| FDR = 0.05 and RF top 10 | 39 (36) | 11 (10) | 46 (43) |
| FDR = 0.10 and RF top 10 | 138 (124) | 13 (12) | 146 (132) |
In the RF analysis, relatively few SNPs had a high importance value in the full models (i.e., models including all 11,085 SNPs): only four and five SNPs had an importance value greater than 10% (percentage increase in MSE), for temperature and precipitation, respectively. The full models explained 19% and 9% of temperature and precipitation variance, respectively. To identify the most significant SNPs, a backward elimination procedure was performed (see Materials and Methods). For both climatic variables, the proportion of explained variance increased when adding SNPs into the minimal RF model (i.e., containing only the two best SNPs), reaching a plateau at around 10 SNPs (∼50–55% variance explained; fig. 4). In both cases, null models including between 2 and 50 random SNPs explained near zero variance (fig. 4), indicating that the procedure was efficient in selecting SNPs related to temperature and precipitation variations. Thus, the 10 most important SNPs identified by RF (representing 10 genes for temperature and 9 genes for precipitation, with one gene common to both variables) were considered highly significant.
Fig. 4.—
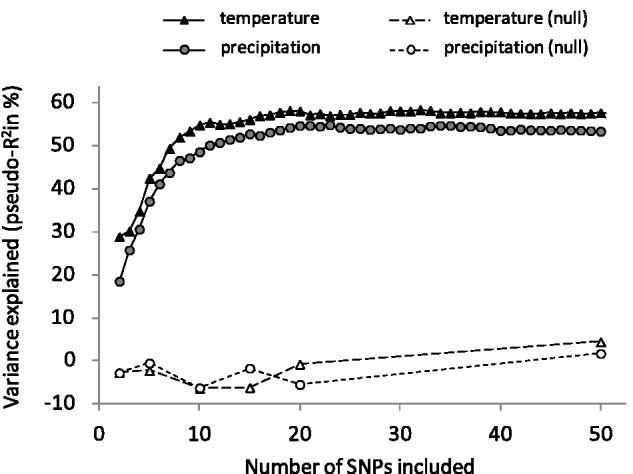
Variance explained by Random Forest models with various numbers of SNPs included in the backward elimination procedure (see Materials and Methods). Models containing between 50 and 100 SNPs are not displayed because the plateau shown here extended until 100 SNPs. Variance explained by randomly selected SNPs (null models) could be slightly negative, meaning that these SNPs were not important. Each point represents the variance explained by the corresponding model, averaged over at least five independent runs of this model.
In order to minimize the impact of false positives on subsequent analyses, we retained only the most significant SNPs detected with each approach. This subset included the 33 SNPs associated to temperature and/or precipitation in regressions at FDR = 0.05, and the 19 SNPs identified as the top 10 in RF for temperature and precipitation (table 1). Altogether, this subset contained 46 SNPs located in 43 genes. It should be noted that discarding the four individuals which were found to be genetically divergent according to the STRUCTURE analysis had little impact on the detection of significant SNPs and their genes. In this scenario, 36 of the 43 genes detected were still significant. The remaining 7 genes carried SNPs strongly associated to climatic variables, although their significance was just above the thresholds (2 were significant in regression at FDR = 0.10, 5 were in the top 15 in RF, and 1 was in the top 30 in RF). This is likely a result of slightly reduced statistical power. Thus, most of the results discussed in the following sections apply to this strict set of 43 genes detected with all trees considered. This set is assumed to include true adaptive genes with a very small rate of false positives given the correction for population structure and the statistically stringent criteria used. Although it is very likely that these genes are under climatic selection, we cannot exclude that some of them are selected by other environmental factors (e.g., biotic or edaphic factors) that may be correlated to mean annual temperature or total annual precipitation. The lists of genes significantly associated to climate are provided in supplementary table S3, Supplementary Material online (for both FDR = 0.05 and FDR = 0.10).
Number of Gene Loci Involved in Adaptation to Climate in Picea glauca
Depending on the statistical stringency of the FDR criterion used, the total number of putatively adaptive genes varied from 43 (carrying 46 SNPs associated to climate) to 132 (carrying 146 SNPs associated to climate; table 1), representing 0.55% or 1.69% of the total number of genes tested, respectively. The proportion of genes showing signals of selection in this study is comparable, although at the lower end, with that reported in previous genome scan studies in plants, which typically reported between 1% and 20% of loci to be under selection following variable levels of statistical stringency (Holderegger et al. 2008; Zulliger et al. 2013; Cullingham et al. 2014). Out of the 43 genes most significantly associated to climate, 32 were mapped on the genome of P. glauca and were located on 9 of the 12 linkage groups (Pavy, Pelgas, et al. 2012; Pavy et al., in preparation), with no obvious clustering according to recombination distance.
In this study, we detected a much greater number of putatively adaptive genes for temperature than for precipitation (table 1). This suggests that more genes are involved in adaptation along the temperature gradient than along the precipitation gradient across the studied region. This could indicate that, compared with the precipitation gradient, more environmental factors impose selective pressures on P. glauca populations along the temperature gradient, and/or more traits are involved in adaptation along this gradient, and/or traits selected along this gradient are controlled by more genes. These explanations are not mutually exclusive, but they are difficult to disentangle with the data at hand. It is still worth noting that, in P. glauca, genetic variation in traits related to growth and phenology is mainly structured by latitude (which is the main driver of the temperature gradient) in the studied region (Li et al. 1997), and that several quantitative trait loci, each explaining a small percentage of variance, have been identified for these traits (Pelgas et al. 2011). These results are consistent with the finding that more genes may be involved in adaptation to temperature. Another possible explanation for the low number of genes involved in adaptation to precipitation may relate to the fact that white spruce is mainly found in mesic soil moisture conditions across the boreal forest. Climax species occurring on xeric sites are preferably jack pine or black spruce (Bonan and Shugart 1989; Desponts and Payette 1992), with low soil moisture being recurrently reported as a growth limiting factor in white spruce (Wang and Klinka 1996; Barber et al. 2000). Similarly, hydric sites are generally occupied by eastern larch or black spruce at the final stage of boreal forest succession (Ritchie 1957; Viereck 1970; Bonan and Shugart 1989). Hence, white spruce may lack the adaptive genetic background necessary to develop on a wide array of moisture conditions. Finally, the low number of significant associations with precipitation may indicate that mostly small-effect genes are at play, which would require greater statistical power to be detected.
We found that several SNPs ranked as important in the multilocus RF analysis were not significant according to the univariate linear or quadratic regression analyses. This trend suggests that these SNPs may be involved in adaptation through interactions with other SNPs (Boulesteix et al. 2012). It should be noted that genes detected both by RF and regression analyses may also be in interaction with other genes. This is consistent with the view that epistatic interactions contribute to the genetic architecture of traits and their response to selection, along with additive genetic effects (Le Corre and Kremer 2012; Hendry 2013; Wray 2013).
From a methodological perspective, our results showed the clear benefit of extending linear univariate methods for the detection of loci under selection by assessing nonlinear responses to selection (e.g., quadratic regression) (Manel et al. 2010; Prunier et al. 2012; this study). For instance, if a substitution is only beneficial in populations with intermediate temperatures, its frequency would increase in this part of the temperature gradient due to selection. This would result in a bell-shape relationship between allele frequency and temperature rather than a linear one. Thus, testing for quadratic relationships may allow detecting those loci that may be selected on a limited part of the gradient (Prunier et al. 2012). The results obtained with RF highlight the need to implement multivariate methods taking into account interactions between loci. Combining such methods should provide a more realistic picture of the genetic basis of adaptation (Sork et al. 2013; Bourret et al. 2014).
It will be insightful to compare all the above results with results from association studies in white spruce (ongoing project), in order to better understand which phenotypic traits are controlled by the genes identified herein (as well as their interactions). Integrating such results with those of studies on quantitative trait genetic variation across populations (such as those of Li et al. 1997; Jaramillo-Correa et al. 2001) will allow to disentangle which environmental factor selects which trait, which genes and gene interactions control these traits, and how these relationships lead to the observed patterns of phenotypic and adaptive genetic variation.
Small to Moderate Allele Frequency Shifts
We estimated changes in allele frequency along the climatic gradients using the equation of the logistic regression model for each of the 46 SNPs most strongly associated to climate. Mean differences between extreme allele frequencies were 0.31 ± 0.08 (range 0.04–0.46, N = 39) and 0.32 ± 0.08 (range 0.21–0.47, N = 11) for the temperature and the precipitation gradients, respectively. Mean differences for temperature and precipitation were similar for SNPs identified with linear and quadratic regressions.
These results suggest that although moderate to strong differentiation in quantitative adaptive traits along geographical gradients related to precipitation and temperature is observed in white spruce (Li et al. 1997), selection along the climatic gradients only led to small or moderate allele frequency shifts at individual gene loci. A similar trend for smaller sets of tested and significant gene SNPs was also observed in other largely distributed boreal spruces such as the North American P. mariana, shown to harbor significant differentiation in quantitative adaptive traits (Prunier et al. 2011), and along a temperature gradient in Picea abies in Scandinavia (Chen et al. 2012). Such a trend may be explained by several factors which cannot be disentangled with the current data set but are worth examining. First, though significant, selection intensity at individual loci is likely weak in P. glauca along the climatic gradients investigated, as previously suggested for other largely distributed tree species (Alberto et al. 2013). Second, the homogenizing effect of extensive pollen gene flow may counteract selection-driven divergence. Furthermore, it is possible that local adaptation involves only weak antagonistic pleiotropy or conditional neutrality, where a beneficial allele in one environment has only small deleterious or neutral effect in other environments. In such a case, differences in allele frequencies are expected to remain limited or to disappear over time at the selected locus, due to gene flow (Anderson et al. 2012). Finally, assuming that current P. glauca populations in the south of the studied region established about 12,000 cal. yr BP (calibrated years before past) and that the northernmost populations studied herein established about 7,000 cal. yr BP (Payette 1993; Richard 1993; see also de Lafontaine et al. 2010), and assuming a conservative generation time of at least 50 years (Bouillé and Bousquet 2005), modern populations would have been under climatic selection for no more than 140–240 generations. This means that local climatic selection in extant populations is rather recent. All these factors might also act in combination to impede divergence in allele frequencies at selected loci along environmental gradients. For instance, Kremer and Le Corre (2012) and Le Corre and Kremer (2012) showed theoretically that even under high divergent selection, small or no differentiation is expected at loci implicated in quantitative trait variation when their number is large and gene flow is high. This is in agreement with the trends observed here, including the small percentage of significant gene SNPs.
Utility and Limitations of Random Forest in Genome-Wide Environmental Association Studies
Our results indicate that RF provides valuable insights in environmental association studies, due to several desirable properties (see Materials and Methods). Besides, without a multilocus approach such as RF, several loci putatively interacting with other loci along the climatic gradients would not have been detected, despite being among the most important ones.
We also identified two main limitations to the current RF approach in environmental association studies. First, RF did not give high importance values to SNPs that had a strong quadratic effect (and thus were significant in quadratic regression). Although RF performs nonlinear regressions, this is possibly due to the fact that contrary to linear relationships, the three genotypic classes at a given locus may have similar temperature or precipitation mean values when the relationship is strongly bell shaped. Second, although RF takes into account interactions between SNPs, the detection of interactions might not be efficient in high-dimension data sets where main effects are more easily detected than interactions (Winham et al. 2012). This means that many more potentially interacting SNPs are likely to occur in the data set. Despite these drawbacks, our study shows how multilocus methods, such as RF, can provide new insights compared with single-locus methods (Sork et al. 2013; Bourret et al. 2014).
Functional Enrichments in Genes Related to Local Adaptation to Climate in Picea glauca
The strict set of 43 genes carrying the most significant SNPs included 33 (∼77%) genes similar to protein families from the PFAM database, representing 44 different families or domains (25 with at least 5 members in the whole data set and then used for enrichment tests). They included 28 genes (∼65%) associated to an Uniprot keyword, with 20 biological processes, 19 molecular functions, and 16 cellular components (20, 18, and 16 terms, respectively, with at least 5 members in the whole data set). In total, 22 (∼51%) had a KEGG Orthology (KO) annotation, of which 18 were assigned to a pathway in poplar, representing 24 different pathway terms (23 with at least 5 members in the whole data set).
Statistical power was quite low in tests for enrichments in functional annotations: except for PFAM terms, less than 50% of the 43 genes were annotated. Still, we detected some significant enrichments. At FDR = 0.05, the 43 genes associated to climate were enriched in the pathway “Cellular processes” (2 genes vs. 0.8 expected by chance). At FDR = 0.10, the 43 genes associated to climate were enriched in the PFAM term “Leucine rich repeat” [PFAM: PF13516.1] (4 genes vs. 0.06 expected by chance) and the pathway “Transport and catabolism” (4 genes vs. 0.8 expected by chance). At P < 0.05 (uncorrected P value), we detected enrichments in the “mRNA surveillance” pathway (2 genes vs. 0.6 expected by chance), the “Plant–pathogen interaction” pathway (2 genes vs. 0.4 expected by chance), and the Uniprot keyword “Rotamase” (2 genes vs. 0.1 expected by chance). These results show that the strict set of 43 adaptive genes differed from the distribution of the other genes and that the enriched functional categories may be especially important for climate adaptation in P. glauca. Enrichment tests performed on the less stringent set of 132 genes associated to climate (table 1) revealed more functional enrichments that may also be especially important for adaptation in P. glauca (e.g., CBS and NAF domains, “environmental information processing,” “signal transduction,” “nucleus,” “activator”; supplementary fig. S5, Supplementary Material online).
Genetic Adaptation to Climate Encompasses a Great Diversity of Molecular Functions and Biological Processes
Overall, the 43 adaptive genes carrying the most significant SNPs were well characterized, with 88% of them having a putative molecular function assigned after database and literature information on putative homologs. A diversity of molecular functions was found. Among enzymes, six classes were represented (fig. 5). Along with hydrolases, transferases were the most represented enzymes and included several kinases and methyltransferases. Through their role in phosphorylation, kinases are involved in the signal transduction needed to convey stress signals into cellular responses (Hirt 1997). Several methyltransferases belonged to the biosynthetic pathway of secondary metabolites. Most of the transcriptional regulators (fig. 5) were transcription factors, which belonged to several families (e.g., IAA, AP2). The other most populated classes were ubiquitin-related proteins and actors of the photosynthesis. All the other molecular functions found were so diverse that they could hardly be grouped (fig. 5). Moreover, except for most of the above-mentioned families, gene families were represented only by a few members or even by a single gene in the strict set. This large functional diversity observed was consistent with that reported in environmental and phenotypic association studies, as well as transcriptomic studies conducted in trees (Holliday et al. 2008; Eckert et al. 2010; Prunier et al. 2011; McKown et al. 2014), and in plants in general (Seki et al. 2002; Kilian et al. 2007; Yoder et al. 2014). The biological processes in which the 43 adaptive genes carrying highly significant SNPs are likely involved were also very diverse (fig. 6). In the following sections, we discuss this diversity of functions and processes involved in climate adaptation in P. glauca, along with some of the most remarkable of the 43 adaptive genes identified.
Fig. 5.—

Putative molecular functions of the 43 Picea glauca genes carrying highly significant SNPs associated with climate (table 1 and supplementary table S3A, Supplementary Material online). The numbers on top of the histogram indicate the percentage of these genes found in each class. Genes with homologs of known function were split into several functional categories. Enzymes were distributed across six of the seven categories of the enzyme classification, but ubiquitin ligases were included in the «ubiquitin related proteins» category. Some included such a high diversity of proteins that they were grouped in the category named “other.” Within bars, the left number indicates how many of the 43 adaptive genes matched differentially expressed genes (DEGs) in spruces only (Holliday et al. 2008; El Kayal et al. 2011; Galindo-Gonzalez et al. 2012); the right number indicates the number of adaptive genes matching DEGs in angiosperms only (rice, Zhang et al. 2012; Arabidopsis thaliana, Kilian et al. 2007; Eucalyptus camaldulensis, Thumma et al. 2012); the number in parentheses indicates the number of adaptive genes matching DEGs both in spruces and angiosperms (for angiosperms, identity ≥50% over at least 100 nucleotides, and BLASTP e-value < 1 × 10−10; for Picea sitchensis, identity ≥95% over at least 100 nucleotides, and BLASTN e-value < 1 × 10−10). The isomerase gene did not match any DEG.
Fig. 6.—

Functional classification of the 43 Picea glauca genes carrying highly significant SNPs associated with climate. Each line corresponds to one of the 43 adaptive genes. The categorization into functional classes was determined after transcriptome analyses and literature search about the functions of homologs (see Materials and Methods, Supplementary Table S3A). 1Adaptive genes detected by regression (FDR = 0.05) or Random Forest (top 10) alone (pale blue), or by both methods (dark blue). 2Adaptive genes differentially expressed (or matching a Picea sitchensis DEG) during bud set in P. glauca or P. sitchensis alone (yellow), or in both species (orange). The experiments were published by El Kayal et al. (2011) and Galindo-Gonzalez et al. (2012) for P. glauca, and by Holliday et al. (2008) for P. sitchensis. 3Picea glauca adaptive genes matching an Arabidopsis gene transcriptionally regulated by cold and/or drought and/or salt and/or UV-B stresses (Kilian et al. 2007). 4Genes with homologs involved in defense mechanisms against several pathogens (e.g., bacteria, nematodes, herbivory). 5Genes involved in development, growth, or seed development. 6Genes involved in secondary metabolism, lignin accumulation. Letters and arrows on the right indicate the main genes discussed in the text. (a) CIPK20, (b) β-tubulin, (c) PP2A catalytic subunit, (d) PP2A regulatory subunit.
Genetic Adaptation to Climate Involves Genes Related to Development, Metabolism, and Stress Response
In total, 11 (∼26%) sequences were similar to proteins involved in stress response (fig. 6) in addition to two sequences involved in proteolysis, a crucial mechanism underlying protein plasticity and therefore plant survival under abiotic stress (Stone 2014). Moreover, 9 (∼21%) sequences were involved in plant development, including seed development, the auxin pathway, ABA signaling, or phenology (fig. 6). One of the two top genes for temperature in RF belonged to the β-tubulin family (>90% identity over 426 amino acids with A. thaliana β-tubulin sequences [TAIR: AT1G75780.1, AT5G62690.1, AT5G62700.1, AT5G44340.1, AT1G20010.1, AT2G29550.1, AT5G23860.1, AT4G20890.1]). β-tubulins, which are components of microtubules, are involved in important processes such as cell division and expansion, and cell wall formation (Nick 1998). Some β-tubulins have been shown to be differentially expressed under biotic and abiotic stresses in Arabidopsis (Chu et al. 1993; Kreps et al. 2002; Ascencio-Ibanez et al. 2008). Other highly significant genes detected in regression and/or RF included three genes putatively coding for two oxidoreductases and one isomerase whose homologs are involved in photosynthesis, one gene putatively coding for a transferase whose homolog is involved in the accumulation of lipids in seeds, and one gene putatively coding for a superoxide dismutase whose homolog is involved in tolerance to oxidative stress (fig. 6).
One adaptive gene [GenBank: BT111298] belonged to the sucrose non-fermenting-1 (SNF1) related kinase 3 (SnRK3 or CIPK, CBL-interacting protein kinase) family (Hrabak et al. 2003). This gene was differentially expressed during bud set in P. glauca, and matched DEGs during bud set in P. sitchensis (Holliday et al. 2008), DEGs in E. camaldulensis under water stress (Thumma et al. 2012), and DEGs in A. thaliana submitted to all four studied stresses (Kilian et al. 2007) (see Materials and Methods). This gene was downregulated in stems and in forming buds during bud set in P. glauca (El Kayal et al. 2011; Galindo-Gonzalez et al. 2012). It likely encodes a kinase: the P. glauca protein sequence was 65% identical over 425 amino acids to A. thaliana CIPK20 [TAIR: AT5G45820.1]. Its closest homolog was CIPK16 of Vitis vinifera [RefSeq: NP_001267894] (72% identity over 434 amino acids). CIPKs are activated by CBL (calcineurin B-like) proteins that bind to the regulatory NAF domain. CBL proteins are sensors of the second messenger Ca2+, and the CIPK/CBL network is thus an important mediator of different signaling pathways (Luan 2009; Yu et al. 2014). SnRK3 proteins are involved in the response to abiotic stresses (Coello et al. 2011). For instance, SnRK3 mutations are responsible for the “salt overly sensitive” phenotypes of SOS Arabidopsis mutants (Liu and Zhu 1998; Liu et al. 2000). Several CIPKs are also involved in ABA responses (Gong et al. 2002; Guo et al. 2002; Kim et al. 2003). For example, activation of CIPK20 is required to activate the ABA signaling pathway involved in seed germination and growth elongation inhibition in A. thaliana (Gong et al. 2002).
The many processes and pathways likely involved in climate adaptation in P. glauca were well reflected by such adaptive genes, and by the enriched functional categories. These findings were consistent with the known cross-talks between responses to different stresses (Cheong et al. 2002; Fraire-Velazquez et al. 2011), and between development and stress response (Krasensky and Jonak 2012). Altogether, our results showed that adaptation to climate in P. glauca involves many mechanisms playing at several cellular and organismal levels.
Genetic Adaptation to Climate Involves Genes Related to Bud Set and Phenology
Among the 43 adaptive genes carrying highly significant SNPs, 29 had a known expression profile, of which 18 and 14 were differentially expressed during bud set in buds (El Kayal et al. 2011) and whole stems (Galindo-Gonzalez et al. 2012), respectively. Expression and sequence data were also compared with DEGs during bud set in the closely related species P. sitchensis (Holliday et al. 2008) (identity ≥ 95% over at least 100 bp), although some paralogy between annotated genes in both spruces cannot be excluded. Among the 43 adaptive genes, 34 matched a P. sitchensis gene studied by these authors, including 9 that were differentially expressed. No significant enrichment in DEGs in P. glauca or P. sitchensis was found (P > 0.10 in all cases).
Altogether, six genes (fig. 6; six first rows) were declared as involved in climate adaptation by our study and the three transcriptomic studies of Holliday et al. (2008), El Kayal et al. (2011), Galindo-Gonzalez et al. (2012). They belonged to different protein families: a universal stress protein [PFAM: PF00582.21], a dirigent-like protein [PFAM: PF03018.9], a putative cyclase [PFAM:PF04199.8], a protein tyrosine kinase [PFAM: PF07714.12] with a NAF domain [PF03822.9] (the CIPK20 homolog discussed above), a protein from the linker histone H1 and H5 family [PFAM: PF00538.14], and one orphan. Furthermore, some of the 43 adaptive genes identified herein were homologs of key components of flowering regulation in angiosperms. Indeed, two of them encoded two of the three subunits of the PP2A (phosphatase 2A; the regulatory and the catalytic subunits), which is involved in the regulation of flowering time in Arabidopsis through its interaction with the flowering locus C gene (Heidari et al. 2013). These genes may be adaptive through their role in phenological changes, especially those linked to bud set, and through other processes discussed above, such as development and stress response.
Genetic Adaptation to Climate Involves Genetic and Epigenetic Control of Transcription
The 43 adaptive genes carrying highly significant SNPs included several transcription factors (fig. 5), histones, and a putative SWI/SNF chromatin remodeling ATPase, suggesting that the control of transcription is one important mechanism for adaptation, involving both transcription factors and also likely epigenetic modifications. Both processes are contribute to acclimation and response to stress in plants (Singh et al. 2002; Kim et al. 2010; Bräutigam et al. 2013; Baulcombe and Dean 2014; Han and Wagner 2014). Finally, some of the 43 adaptive genes were also likely involved in the preservation of genome integrity and DNA repair (fig. 6), two processes critical for plant survival.
Conclusions
In this study, the use of linear and quadratic regressions, as well as the RF algorithm, allowed detecting dozens of loci likely involved, directly or indirectly, into genetic adaptation to climate in P. glauca. Most of these genes had homologs likely involved in responses to various biotic and abiotic stresses, as well as in development or phenological changes. Some of these genes are reported as integrators of the stress response from signal reception to transduction to activation of mechanisms for protecting cells and genomes. Our results support the view that several adaptive genes identified in this study are involved in cross-talks between processes, in part through the regulation of transcription or by phosphorylation. Altogether, our results are consistent with theoretical and empirical studies suggesting that adaptation involves functionally diverse loci with small allele frequency shifts when selection is recent in species with high gene flow.
Our results also highlight the importance of extending current methods for the detection of loci under selection with complementary methods accounting for more realistic patterns of adaptive genetic variation in natural populations such as nonlinear patterns and interaction effects. Doing so will provide a more rigorous foundation to compare the results of empirical environmental association studies with theoretical predictions on the dynamics and the genetic architecture of adaptation. Ultimately, it should result in a better understanding and monitoring of ecologically relevant genetic variation in an era of global environmental change, which is particularly crucial for perennial plant species with long generation times such as conifers.
Supplementary Material
Supplementary figures S1–S5 and tables S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors acknowledge S. Blais and F. Gagnon for their help with SNP data quality control, A. Deschênes, J. Prunier, J. Laroche, and G. de Lafontaine (Institute for Systems and Integrative Biology and Canada Research Chair in Forest and Environmental Genomics, Université Laval) for helpful discussions, R. Saint-Amant (Natural Resources Canada) for help with BioSIM, M. Mazerolle (UQAT) for statistical advices, J. Holliday (Virginia Tech) and A. Liaw (Merck Research Labs) for their advices regarding Random Forest. We also thank A. Montpetit and his team at the McGill University Genome Quebec Innovation Centre for genotyping the trees. This work was supported by the SMarTForests project with funding from Genome Canada and Génome Québec, and a Natural Sciences and Engineering Research Council of Canada grant to J.B.
Literature Cited
- Alberto FJ, et al. 2013. Potential for evolutionary responses to climate change - evidence from tree populations. Glob Change Biol. 19:1645–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andalo C, Beaulieu J, Bousquet J. 2005. The impact of climate change on growth of local white spruce populations in Québec, Canada. Forest Ecol Manag. 205:169–182. [Google Scholar]
- Anderson JT, Willis JH, Mitchell-Olds T. 2012. Evolutionary genetics of plant adaptation. Trends Genet. 27:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascencio-Ibanez JT, et al. 2008. Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during Geminivirus infection. Plant Physiol. 148:436–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber VA, Juday GP, Finney BP. 2000. Reduced growth of Alaskan white spruce in the twentieth century from temperature-induced drought stress. Nature 405:668–673. [DOI] [PubMed] [Google Scholar]
- Baulcombe DC, Dean C. 2014. Epigenetic regulation in plant responses to the environment. Cold Spring Harb Perspect Biol. 6:a019471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J, et al. 2011. Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics 188:197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Met. 57:289–300. [Google Scholar]
- Bonan GB, Shugart HH. 1989. Environmental factors and ecological processes in boreal forests. Annu Rev Ecol Syst. 20:1–20. [Google Scholar]
- Bouillé M, Bousquet J. 2005. Trans-species shared polymorphisms at orthologous nuclear gene loci among distant species in the conifer Picea (Pinaceae): implications for the long-term maintenance of genetic diversity in trees. Am J Bot. 92:63–73. [DOI] [PubMed] [Google Scholar]
- Bouillé M, Senneville S, Bousquet J. 2011. Discordant mtDNA and cpDNA phylogenies indicate geographic speciation and reticulation as driving factors for the diversification of the genus Picea. Tree Genet Genomes. 7:469–484. [Google Scholar]
- Boulesteix AL, Janitza S, Kruppa J, König IR. 2012. Overview of Random Forest methodology and practical guidance with emphasis on computational biology and bioinformatics.Technical Report Number 129. Department of Statistics, University of Munich. [Google Scholar]
- Bourret V, Dionne M, Bernatchez L. 2014. Detecting genotypic changes associated with selective mortality at sea in Atlantic salmon: polygenic multilocus analysis surpasses genome scan. Mol Ecol. 23:4444–4457. [DOI] [PubMed] [Google Scholar]
- Bradshaw HD, Otto KG, Frewen BE, McKay JK, Schemske DW. 1998. Quantitative trait loci affecting differences in floral morphology between two species of Monkeyflower (Mimulus). Genetics 149:367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam K, et al. 2013. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol Evol. 3:399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. 2001. Random forests. Mach Learn. 45:5–32. [Google Scholar]
- Chen J, et al. 2012. Disentangling the roles of history and local selection in shaping clinal variation of allele frequencies and gene expression in Norway spruce (Picea abies). Genetics 191:865–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong YH, et al. 2002. Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis. Plant Physiol. 129:661–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B, Snustad DP, Carter JV. 1993. Alteration of beta-tubulin gene expression during low-temperature exposure in leaves of Arabidopsis thaliana. Plant Physiol. 103:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coello P, Hey SJ, Halford NG. 2011. The sucrose non-fermenting-1-related (SnRK) family of protein kinases: potential for manipulation to improve stress tolerance and increase yield. J Exp Bot. 62:883–893. [DOI] [PubMed] [Google Scholar]
- Colautti RI, Lee CR, Mitchell-Olds T. 2012. Origin, fate, and architecture of ecologically relevant genetic variation. Curr Opin Plant Biol. 15:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Marttinen P. 2006. Bayesian identification of admixture events using multi-locus molecular markers. Mol Ecol. 15:2833–2843. [DOI] [PubMed] [Google Scholar]
- Cullingham CI, Cooke JEK, Coltman DW. 2014. Cross-species outlier detection reveals different evolutionary pressures between sister species. New Phytol. 204:215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lafontaine G, Turgeon J, Payette S. 2010. Phylogeography of white spruce (Picea glauca) in eastern North America reveals contrasting ecological trajectories. J Biogeogr. 37:741–751. [Google Scholar]
- Desponts M, Payette S. 1992. Recent dynamics of jack pine and its northern distribution limit in northern Quebec. Can J Bot. 70:1157–1167. [Google Scholar]
- Diaz-Uriarte R, Alvarez de Andres S. 2006. Gene selection and classification of microarray data using random forest. BMC Bioinformatics 7:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert AJ, et al. 2010. Back to nature: ecological genomics of loblolly pine (Pinus taeda, Pinaceae). Mol Ecol. 19:3789–3805. [DOI] [PubMed] [Google Scholar]
- El Kayal W, et al. 2011. Molecular events of apical bud formation in white spruce, Picea glauca. Plant Cell Environ. 34:480–500. [DOI] [PubMed] [Google Scholar]
- Ellegren H. 2014. Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 29:51–63. [DOI] [PubMed] [Google Scholar]
- Etterson JR, Shaw RG. 2001. Constraint to adaptive evolution in response to global warming. Science 294:151–154. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 14:2611–2620. [DOI] [PubMed] [Google Scholar]
- Evans LM, et al. 2014. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat Genet. 46:1089–1096. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier-Level A, et al. 2011. A map of local adaptation in Arabidopsis thaliana. Science 333:86–89. [DOI] [PubMed] [Google Scholar]
- Fraire-Velazquez S, Rodriguez-Guerra R, Sanchez-Calderon L. 2011. Abiotic and biotic stress response crosstalk in plants. In: Shanker A, Venkateswarlu B, editors. Abiotic stress response in plants—physiological, biochemical and genetic perspectives. Rijeka (Croatia): InTech; p. 3–26. [Google Scholar]
- Franks SJ, Weber JJ, Aitken S. 2014. Evolutionary and plastic responses to climate change in terrestrial plant populations. Evol Appl. 7:123–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, et al. 2006. Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol. 9:1–7. [DOI] [PubMed] [Google Scholar]
- Galindo-Gonzalez LM, et al. 2012. Integrated transcriptomic and proteomic profiling of white spruce stems during the transition from active growth to dormancy. Plant Cell Environ. 35:682–701. [DOI] [PubMed] [Google Scholar]
- Gong D, Gong Z, Guo Y, Chen X, Zhu JK. 2002. Biochemical and functional characterization of PKS11, a novel Arabidopsis protein kinase. J Biol Chem. 277:28340–28350. [DOI] [PubMed] [Google Scholar]
- Guo Y, et al. 2002. A calcium sensor and its interacting protein kinase are global regulators of abscisic acid signalling in Arabidopsis. Dev Cell. 3: 233–244. [DOI] [PubMed] [Google Scholar]
- Han SK, Wagner D. 2014. Role of chromatin in water stress responses in plants. J Exp Bot. 65:2785–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, et al. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 333:83–86. [DOI] [PubMed] [Google Scholar]
- Heidari B, Nemie-Feyissa D, Kangasjärvi S, Lillo C. 2013. Antagonistic regulation of flowering time through distinct regulatory subunits of protein phosphatase 2A. PLoS One 8:e67987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry AP. 2013. Key questions in the genetics and genomics of eco-evolutionary dynamics. Heredity 111:456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hereford J. 2009. A quantitative survey of local adaptation and fitness trade-offs. Am Nat. 173:579–588. [DOI] [PubMed] [Google Scholar]
- Hirt H. 1997. Multiple roles of MAP kinases in plant signal transduction. Trends Plant Sci. 2:11–15. [Google Scholar]
- Holderegger R, et al. 2008. Land ahead: using genome scans to identify molecular markers of adaptive relevance. Plant Ecol Divers. 1:273–283. [Google Scholar]
- Holliday JA, Ralph SG, White R, Bohlmann J, Aitken S. 2008. Global monitoring of autumn gene expression within and among phenotypically divergent populations of Sitka spruce (Picea sitchensis). New Phytol. 178:103–122. [DOI] [PubMed] [Google Scholar]
- Holliday JA, Wang T, Aitken S. 2012. Predicting adaptive phenotypes from multilocus genotypes in Sitka spruce (Picea sitchensis) using Random Forest. G3 (Bethesda) 2:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabak EM, et al. 2003. The Arabidopsis CDPK-SnRK superfamily of protein kinases. Plant Physiol. 132:666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber CD, Nordborg M, Hermisson J, Hellmann I. 2014. Keeping it local: evidence for positive selection in Swedish Arabidopsis thaliana. Mol Biol Evol. 31:3026–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Correa JP, Beaulieu J, Bousquet J. 2001. Contrasting evolutionary forces driving population structure at expressed sequence tag polymorphisms, allozymes and quantitative traits in white spruce. Mol Ecol. 10:2729–2740. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, et al. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42:D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian J, et al. 2007. The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 50:347–363. [DOI] [PubMed] [Google Scholar]
- Kim JM, To TK, Nishioka T, Seki M. 2010. Chromatin regulation functions in plant abiotic stress responses. Plant Cell Environ. 33:604–611. [DOI] [PubMed] [Google Scholar]
- Kim KN, Cheong YH, Grant JJ, Pandey GK, Luan S. 2003. CIPK3, a calcium sensor-associated protein kinase that regulates abscisic acid and cold signal transduction in Arabidopsis. Plant Cell 15:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasensky J, Jonak C. 2012. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J Exp Bot. 63:1593–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer A, Le Corre V. 2012. Decoupling of differentiation between traits and their underlying genes in response to divergent selection. Heredity 108:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer A, Potts BM, Delzon S. 2014. Genetic divergence in forest trees: understanding the consequences of climate change. Funct Ecol. 28:22–36. [Google Scholar]
- Kreps JA, et al. 2002. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 130:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamesch P, et al. 2012. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40:D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corre V, Kremer A. 2012. The genetic differentiation at quantitative trait loci under local adaptation. Mol Ecol. 21:1548–1566. [DOI] [PubMed] [Google Scholar]
- Leimu R, Fischer M. 2008. A meta-analysis of local adaptation in plants. PLoS One 3:e4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser MR, Parker WH. 2004. Genetic variation in Picea glauca for growth and phenological traits from provenance tests in Ontario. Silvae Genet. 53:141–148. [Google Scholar]
- Li H, et al. 2013. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet. 45:43–50. [DOI] [PubMed] [Google Scholar]
- Li P, Beaulieu J, Bousquet J. 1997. Genetic structure and patterns of genetic variation among populations in eastern white spruce (Picea glauca). Can J Forest Res. 27:189–198. [Google Scholar]
- Liaw A, Wiener M. 2002. Classification and regression by randomForest. R News. 2:18–22. [Google Scholar]
- Linhart YB, Grant MC. 1996. Evolutionary significance of local genetic differentiation in plants. Annu Rev Ecol Syst. 27:237–277. [Google Scholar]
- Liu J, Ishitani M, Halfter U, Kim CS, Zhu JK. 2000. The Arabidopsis thaliana SOS2 gene encodes a protein kinase that is required for salt tolerance. PNAS 97:3730–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhu JK. 1998. A calcium sensor homolog required for plant salt tolerance. Science 280:1943–1945. [DOI] [PubMed] [Google Scholar]
- Luan S. 2009. The CBL-CIPK network in plant calcium signaling. Trends Plant Sci. 14:37–42. [DOI] [PubMed] [Google Scholar]
- Luquet E, Léna JP, Miaud C, Plénet S. 2015. Phenotypic divergence of the common toad (Bufo bufo) along an altitudinal gradient: evidence for local adaptation. Heredity 114:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel S, Poncet B, Legendre P, Gugerli F, Holderegger R. 2010. Common factors drive adaptive genetic variation at different spatial scales in Arabis alpina. Mol Ecol. 19:3824–3835. [DOI] [PubMed] [Google Scholar]
- McKown AD, et al. 2014. Genome-wide association implicates numerous genes underlying ecological trait variation in natural populations of Populus trichocarpa. New Phytol. 203:535–553. [DOI] [PubMed] [Google Scholar]
- Namroud MC, Beaulieu J, Juge N, Laroche J, Bousquet J. 2008. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol Ecol. 17:3599–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namroud MC, Guillet-Claude C, Mackay J, Isabel N, Bousquet J. 2010. Molecular evolution of regulatory genes in spruces from different species and continents: heterogeneous patterns of linkage disequilibrium and selection but correlated recent demographic changes. J Mol Evol. 70:371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick P. 1998. Signaling to the microtubular cytoskeleton in plants. Int Rev Cytol. 184:33–80. [Google Scholar]
- Olson-Manning CF, Wagner MR, Mitchell-Olds 2012. Adaptive evolution: evaluating empirical support for theoretical predictions. Nat Rev Genet. 13:867–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, et al. 2008. Identification of conserved core xylem gene sets: conifer cDNA microarray development, transcript profiling and computational analyses. New Phytol. 180:766–786. [DOI] [PubMed] [Google Scholar]
- Pavy N, Gagnon F, Rigault P, et al. 2013. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Mol Ecol Resour. 13:324–336. [DOI] [PubMed] [Google Scholar]
- Pavy N, Deschênes A, et al. 2013. The landscape of nucleotide polymorphism among 13,500 genes of the conifer Picea glauca, relationships with functions, and comparison with Medicago truncatula. Genome Biol Evol. 5:1910–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, Namourd MC, Gagnon F, Isabel N, Bousquet J. 2012. The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity 108:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, Pelgas B, et al. 2012. A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Biol. 10:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payette S. 1993. The range limit of boreal tree species in Quebec-Labrador—an ecological and paleoecological interpretation. Rev Palaeobot Palynol. 79:7–30. [Google Scholar]
- Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelgas B, Bousquet J, Meirmans PG, Ritland K, Isabel N. 2011. QTL mapping in white spruce: gene maps and genomic regions underlying adaptive traits across pedigrees, years and environments. BMC Genomics 12:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier J, Gérardi S, Laroche J, Beaulieu J, Bousquet J. 2012. Parallel and lineage-specific molecular adaptation to climate in boreal black spruce. Mol Ecol. 21:4270–4286. [DOI] [PubMed] [Google Scholar]
- Prunier J, Laroche J, Beaulieu J, Bousquet J. 2011. Scanning the genome for gene SNPs related to climate adaptation and estimating selection at the molecular level in boreal black spruce. Mol Ecol. 20:1702–1716. [DOI] [PubMed] [Google Scholar]
- Prunier J, et al. 2013. The genomic architecture and association genetics of adaptive characters using a candidate SNP approach in boreal black spruce. BMC Genomics 14:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2010. R: A language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing. [Google Scholar]
- Régnière J. 1996. A generalized approach to landscape-wide seasonal forecasting with temperature-driven simulation models. Environ Entomol. 25:869–881. [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Richard PJH. 1993. Origine et dynamique postglaciaire de la Forêt mixte au Québec. Rev Palaeobot Palynol. 79:31–68. [Google Scholar]
- Rigault P, et al. 2011. A white spruce gene catalog for conifer genome analyses. Plant Physiol. 157:14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie JC. 1957. The vegetation of northern Manitoba II: a prisere on the Hudson Bay lowlands. Ecology 38:429–435. [Google Scholar]
- Savolainen O, Lascoux M, Merilä J. 2013. Ecological genomics of local adaptation. Nat Rev Genet. 14:807–820. [DOI] [PubMed] [Google Scholar]
- Seki M, et al. 2002. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold, and high-salinity stresses using a full-length cDNA microarray. Plant J. 31:279–292. [DOI] [PubMed] [Google Scholar]
- Singh KB, Foley RC, Onate-Sanchez L. 2002. Transcription factors in plant defense and stress response. Curr Opin Plant Biol. 5:430–436. [DOI] [PubMed] [Google Scholar]
- Sork VL, et al. 2013. Putting the landscape into the genomics of trees: approaches for understanding local adaptation and population responses to changing climate. Tree Genet Genomes. 9:901–911. [Google Scholar]
- Stone SL. 2014. The role of ubiquitin and the 26S proteasome in plant abiotic stress signaling. Front Plant Sci. 5:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumma BR, Sharma N, Southerton SG. 2012. Transcriptome sequencing of Eucalyptus camaldulensis seedlings subjected to water stress reveals functional single nucleotide polymorphisms and genes under selection. BMC Genomics 12:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uniprot Consortium. 2014. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42:D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Landeghem S, Ginter F, Van de Peer Y, Salakoski T. 2011. EVEX: a PubMed-scale resource for homology-based generalization of text mining predictions. In: Proceedings of the 2011 Workshop on Biomedical Natural Language Processing Portland, Oregon, USA: Association for Computational Linguistics p. 28–37. [Google Scholar]
- Viereck LA. 1970. Forest succession and soil development adjacent to the Chena River in interior Alaska. Arct Alp Res. 2:1–26. [Google Scholar]
- Wang GG, Klinka K. 1996. Use of synoptic variables in predicting white spruce site index. Forest Ecol Manag. 80:95–105. [Google Scholar]
- Weigel D. 2012. Natural variation in Arabidopsis: from molecular genetics to ecological genomics. Plant Physiol. 158:2–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winham SJ, et al. 2012. SNP interaction detection with Random Forests in high-dimensional genetic data. BMC Bioinformatics 13:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA. 2013. Genomics and the evolution of phenotypic traits. Annu Rev Ecol Evol Syst. 44:51–72. [Google Scholar]
- Yeaman S, Whitlock MC. 2011. The genetic architecture of adaptation under migration-selection balance. Evolution 65:1897–1911. [DOI] [PubMed] [Google Scholar]
- Yoder JB, et al. 2014. Genomic signature of adaptation to climate in Medicago trunculata. Genetics 196:1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, An L, Li W. 2014. The CGL-CIPK network mediates different signaling pathways in plants. Plant Cell Rep. 33:203–214. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu S, Zuo K, Tang K. 2012. Identification of gene modules associated with drought response in rice by network-based analysis. PLoS One 7:e33748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulliger D, Schnyder E, Gugerli F. 2013. Are adaptive loci transferable across genomes of related species? Outlier and environmental association analyses in Alpine Brassicaceae species. Mol Ecol. 22:1626–1639 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.