

Abstract
This study investigates the molecular mechanisms by which minocycline, a second generation tetracycline, prevents cardiac myocyte death induced by in utero cocaine exposure. Timed mated pregnant Sprague-Dawley (SD) rats received one of the following treatments twice daily from embryonic (E) day 15–21 (E15–E21): (i) intraperitoneal (IP) injections of saline (control); (ii) IP injections of cocaine (15 mg/kg BW); and (iii) IP injections of cocaine + oral administration of 25 mg/kg BW of minocycline. Pups were killed on postnatal day 15 (P15). Additional pregnant dams received twice daily IP injections of cocaine (from E15–E21) + oral administration of a relatively higher (37.5 mg/kg BW) dose of minocycline. Minocycline treatment continued from E15 until the pups were sacrificed on P15. In utero cocaine exposure resulted in an increase in oxidative stress and fetal cardiac myocyte apoptosis through activation of c-Jun-NH2-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK)-mediated mitochondria-dependent apoptotic pathway. Continued minocycline treatment from E15 through P15 significantly prevented oxidative stress, kinase activation, perturbation of BAX/BCL-2 ratio, cytochrome c release, caspase activation, and attenuated fetal cardiac myocyte apoptosis after prenatal cocaine exposure. These results demonstrate in vivo cardioprotective effects of minocycline in preventing fetal cardiac myocyte death after prenatal cocaine exposure. Given its proven clinical safety and ability to cross the placental barrier and enter into the fetal circulation, minocycline may be an effective therapy for preventing cardiac consequences of in utero cocaine exposure.
Keywords: Cocaine, Cardiac myocyte apoptosis, Minocycline, Cytochrome c release, Mice
Introduction
Substance abuse has increased substantially in the past decade and cocaine is the most frequently reported drug of abuse in emergency room visits [1, 2]. Cocaine is common in pregnant women with a history of substance abuse, which readily crosses the placenta into the fetal circulation [3] affecting the placenta and fetus with detrimental consequences. Offspring born to mothers with a history of cocaine abuse have a high incidence of congenital cardiovascular malformations, including ischemia, myocarditis and development of cardiomyopathy, and arrhythmias [4–6]. There is accumulating evidence that prenatal cocaine exposure increases cardiac myocyte apoptosis in neonatal rats and its susceptibility to ischemia–reperfusion injury in both adolescent and adult males [7, 8]. Earlier in vitro studies also suggest the involvement of the mitochondria-dependent apoptotic pathway in cardiac myocyte apoptosis in response to cocaine exposure [9]. Cardiac myocyte apoptosis plays an important role in the morphogenesis and remodeling of mammalian heart during the first 2 postnatal weeks [10, 11] and has been implicated as a mechanism in the development of cardiac failure of both ischemic and non-ischemic origins [12–15]. Despite the recognition that inhibition of cardiac myocyte apoptosis is a logical target for novel therapies for preventing heart failure [14, 15], little is known about the mechanisms by which in utero cocaine exposure triggers the mitochondria-dependent death pathway and cardiac myocyte apoptosis in the offspring and its therapeutic intervention. An understanding of these mechanisms is indispensable for the rational design of antiapoptotic therapies.
Mitogen-activated protein kinases (MAPKs) comprise a family of serine/threonine kinases that function as critical mediators of a variety of extracellular signals [16, 17]. Members of the MAPK super-family include the extracellular signal-regulated kinase (ERK), the c-Jun NH2-terminal kinase (JNK), also known as stress-activated protein kinase (SAPK), and the p38 MAP kinase (p38 MAPK). ERK1 and ERK 2 are activated in response to growth stimuli and promote cell growth, whereas both JNK and p38 MAPK are activated in response to a variety of environmental stresses and inflammatory signals and promote apoptosis and growth inhibition [16, 17]. The role of these MAPKs in the regulation of fetal cardiac myocyte apoptosis is not well known and the conclusions of several studies indicate that the regulation of apoptosis by MAPKs varies depending on tissue type, nature of the apoptotic stimulus, and duration of their activation [16, 17]. Additionally, in cardiac myocytes, the role of these kinases has been reported to even vary between in vitro and in vivo settings [18]. Li et al. [9] have provided evidence indicating that cocaine induces apoptosis in fetal rat myocardial cells (FRMCs) through activation of p38 MAPK together with the inhibition of the ERK. However, we do not know whether these pathways are also the key pathways for induction of apoptosis in fetal cardiac myocytes after prenatal cocaine exposure.
Minocycline, a second generation tetracycline, has been shown to effectively inhibit cytosolic translocation of cytochrome c and activation of the mitochondria-dependent death pathway in various cell systems [19–22], which is also the key pathway for cardiac myocyte and skeletal muscle cell apoptosis [23–25]. Minocycline is orally bioavailable and has a proven safety record in human [26]. This compound is well tolerated in prolonged administration protocol and has robust neuroprotective effects in rodent models of neurodegeneration, cerebral ischemia, and traumatic brain injury [26, 27]. Like neuronal systems, minocycline is also effective in protecting male germ cells from undergoing apoptosis triggered by deprivation of gonadotropins [21] or by heat stress [22]. We therefore hypothesize that minocycline, through inhibition of p38 MAPK and JNK-mediated mitochondria-dependent death pathway would prevent fetal cardiac myocyte death induced by prenatal cocaine exposure. Thus, in the present study, we examined the minocycline-mediated signal transduction pathways that attenuate cardiac myocyte apoptosis triggered by prenatal cocaine exposure.
Materials and methods
Animals and experimental protocol
Time-mated pregnant Sprague-Dawley (SD) rats were purchased from Charles River Laboratories (Wilmington, MA) and housed in a standard animal facility under controlled temperature (22°C) and photoperiod (12 h of light, 12 h of darkness) with food and water ad libitum.
In experiment 1, pregnant dams received one of the following treatments twice daily from embryonic (E) day 15 to 21 (E15 to E21): (i) intraperitoneal (IP) injections of saline (control); (ii) IP injections of 15 mg/kg BW of cocaine [7, 28]; and (iii) IP injections of cocaine + oral administration of 25 mg/kg BW of minocycline [21]. Pups were killed on postnatal (P) day 15 (P15), a critical time point, in which cardiac myocyte apoptosis plays a pivotal role in the morphogenesis and remodeling of the heart [10, 11]. Given the gender specific effects of in utero cocaine exposure with male pups being more susceptible than female pups [8, 28], we elected to use only the male pups.
In experiment 2 (designed after the results of the first experiment were known), additional pregnant dams received twice daily IP injections of cocaine (E15 to E21) + oral administration of a relatively higher dose (37.5 mg/kg BW) of minocycline. Minocycline treatment continued from E15 until the pups were killed on P15. Pups were kept with their mothers and allowed to be breast-fed ad libitum. Moms were not given cocaine after delivery.
At autopsy, ventricles from 10 male pups in each group were quickly removed and frozen in liquid nitrogen for subsequent measurements of oxidative stress, kinase activation, and changes in protein expression. Ventricles form five additional pups in each group were fixed with 4% formalin for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL), immunohistochemistry, and immunofluorescence studies. Ventricles from three additional pups were fixed with 2.5% glutaraldehyde, post-fixed into 1% osmium tetroxide, and processed for routine electron microscopy [22, 24]. Animal handling and experimentation were in accordance with the recommendation of the American Veterinary Medical Association and were approved by the Charles R. Drew University School of Medicine and Science animal care and use review committee.
Histology and TUNEL
Cardiac pathology was evaluated using conventional histological analysis on haematoxylin and eosin or Masson’s trichrome staining. In situ detection of cells with DNA strand breaks was performed in formalin-fixed, paraffinembedded ventricular sections by the TUNEL technique [24, 25, 29, 30] using an ApopTag-peroxidase kit (Chemicon International, Inc., San Francisco, CA). Enumeration of TUNEL-positive nuclei was carried out in ventriclular sections stained with ApopTag peroxidase kit using an American Optical Microscope with an X40 objective and a pair of X10 eyepieces. Methyl green was used as a counterstain to detect non apoptotic nuclei. A square grid fitted within one eyepiece provided a reference of 62,500 μm2. The rate of cardiac myocyte apoptosis was expressed as the percentage of the TUNEL-positive apoptotic nuclei per total nuclei (apoptotic plus non apoptotic) present within the reference area [24, 25, 30].
Measurements of reduced (GSH) and oxidized (GSSG) glutathione and 4-hydroxy-trans-2-nonenal (4-HNE) levels
The GSH/GSSG ratio in the ventricle was measured using a commercial kit (BIOXYTECH® GSH/GSSG-412™ assay kit (OXISResearch™, A division of Oxis Health Product, Inc., Portland, OR), as described previously [31]. This assay, using different sample preparations, measures both reduced (GSH) and oxidized (GSSG) concentrations and the GSH/ GSSG ratio. The omission or addition of 1-methyl-2-vinlylpyridinium trifluromethanesulfonate allows the measurement of GSH and GSSG, respectively. The GSH/GSSG ratio is inversely related to reactive oxygen species levels (ROS) levels.
Further evaluation of oxidative stress was achieved by measuring 4-HNE levels, a biomarker of oxidative stress [32, 33], using an ELISA kit (Cell Biolabs, San Diego, CA), as described previously [31]. In brief, 4-HNE protein adducts present in the sample or standard are probed with the primary 4-HNE antibody, followed by a HRP-conjugated secondary antibody. The 4-HNE protein adduct content in unknown sample is determined by comparing with the standard curve that is prepared from predetermined HNE-BSA standards.
Measurements of kinase activation
Activation of p38 MAPK and JNK in ventricular lysates were measured by TiterZyme EIA kit (Assay Designs Inc., Ann Arbor, MI), as described previously [30, 34, 35].
Immunofluorescence analysis
Activation of p38 MAPK and JNK in fetal cardiac myocytes undergoing apoptosis was also detected by confocal microscopy using double immunostaining as previously described [24, 29, 30, 34]. In brief, after deparaffinization and rehydration, tissue sections were incubated with proteinase K for 45 min at 37°C, washed in distilled water, and then incubated with a mixture containing digoxigenin-conjugated nucleotide and TdT in a humidified chamber at 37°C for 1 h and subsequently treated with antidigoxige-nin–fluorescein for 30 min in the dark. After fluorescein staining, slides were washed in PBS and incubated with blocking serum for 20 min to reduce nonspecific antibody binding. Slides were then incubated in a humidified chamber for overnight at 4°C with a rabbit polyclonal phospho-p38 MAPK, which detects p38 MAPK only when phosphorylated at tyrosine 182 (1:100) or phospho-JNK, which detects JNK only when phosphorylated at threonine 183 and tyrosine 185 antibody (1:100) from Santa Cruz Biotechnology, Santa Cruz, CA, followed by goat anti-rabbit Texas Red-labeled secondary antibody (1:100; Vector Laboratories, Burlingame, CA) for 45 min at room temperature, washed, and mounted in ProLong Antifade (Molecular Probes, Eugene, OR). Identification of apoptotic cardiac myocytes was achieved by confocal microscopy using double immunostaining with active caspase 3 (1:50; Cell Signaling Technology, Beverly, MA) and α-actinin (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), a marker of cardiac myocyte [36, 37]. For negative controls, sections were either treated only with secondary antibody or primary antibody that was pre-incubated with blocking peptide, and no signals were detected.
Western blotting
Western blotting was performed using ventricular lysates as described previously [24, 25, 29, 30, 34]. In brief, proteins (50–80 lg) were separated on a 4–12% SDS-poly-acrylamide gel with MES or MOPS buffer purchased from Invitrogen (Carlsbad, CA, USA) at 200 V. Gel was transferred on a Immuno-blot PVDF Membrane (Bio-Rad, Hercules, CA) overnight at 4°C. Membranes were blocked in blocking solution (0.3% Tween 20 in Tris-buffered saline and 10% nonfat dry milk) for 1 h at room temperature then probed using rabbit polyclonal phospho-JNK (1:200), phospho-p38 MAPK (1:200), and BAX (1:200) from Santa Cruz Biotechnology; BCL-2 (1:200), phospho-44/42 MAPK, which detects endogenous levels of ERK1 and ERK 2 only when phosphorylated at threonine 202 and tyrosine 204 (1:300), cleaved caspase 9 (1:200) and cleaved caspase 3 from Cell Signaling Technology, Inc, Beverly, MA, antibodies for 1 h at room temperature or overnight at 4°C with constant shaking. Following 3 9 10-min washes in TBS-T buffer, membranes were then incubated in anti-rabbit IgG-HRP (Amersham Biosciences, Piscataway, NJ) secondary antibody at a 1:2000 dilution. All antibodies were diluted in blocking buffer. For immunodetection, membranes were washed three times in TBS-T wash buffer, incubated with ECL solutions per the manufacturer’s specifications (Amersham Biosciences), and exposed to Hyper film ECL. The membranes were stripped and reprobed with a rabbit polyclonal glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:2000) for normalization of the loading. Band intensities were determined using Quantity One software from Bio-Rad (Hercules, CA). All measurements were replicated at least three times.
Measurements of mitochondrial cytochrome c release into cytosol
Measurements of cytosolic translocation of mitochondrial cytochrome c were achieved by subcellular fractionation and Western blotting as described previously [21, 22, 24, 29, 34]. The purity of the mitochondrial and cytosolic fractions were validated by Western blotting using anti-bodies to cytochrome c oxidase subunit IV (COX IV; 1:500, Molecular Probes, Eugene, OR) and actin (1:2000; Sigma-Aldrich), respectively.
Statistical analysis
Statistical analyses were performed using the SigmaStat 2.0 Program (Jandel Cooperation, San Rafael, CA). Data are presented as mean ± SE unless otherwise indicated. We used one-way ANOVA to compare group differences. If overall ANOVA revealed significant differences, post hoc comparisons were performed using Tukey’s test or Student–Newman–Keuls test. Differences were considered significant if P < 0.05.
Results
Concomitant administration of minocycline (25 mg/kg BW) from E15 to E21 fails to prevent in utero cocaine-induced activation of p38 MAPK, JNK, caspases, and fetal cardiac myocyte apoptosis (experiment 1)
We first assessed whether concomitant administration of minocycline (25 mg/kg BW) from E15 to E21 can prevent fetal cardiac myocyte apoptosis induced by in utero cocaine exposure. Apoptosis was detected by TUNEL. Compared with controls, where no apoptosis was detected (Fig. 1a), prenatal cocaine exposure resulted in a marked increase in the incidence of cardiac myocyte apoptosis in the ventricle at P15 (Fig. 1b). Incidence of apoptosis was essentially similar between cocaine and cocaine plus minocycline treated groups (Fig. 1b). We also quantitated the incidence of apoptosis, expressed as the percentage of TUNEL-positive nuclei per total nuclei (apoptotic plus non-apoptotic nuclei) counted in a unit reference area, in various treatment groups. The incidence of fetal cardiac myocyte apoptosis was very low in controls (1.68 ± 0.22) but exhibited a significant (P < 0.001) increase at P15 (7.23 ± 0.52) after prenatal cocaine exposure. No significant changes in the number apoptotic nuclei were noted between cocaine and cocaine plus minocycline treated ± 0.68) groups. The identity of apoptotic cardiac myocytes was characterized by double immunofluorescence staining for α-actinin, a cardiac myocyte marker [36, 37], and caspase 3 (Fig. 1c, d). Electron microscopic observation further confirmed the apoptotic nature and the identity of dying cells as cardiac myocytes (Fig. 1e, f). Consisting with the findings of a recent study [38], we found no perivascular or interstitial fibrosis in ventricles of neonates after short-term (from E15 to E21) prenatal cocaine exposure (data not shown).
Fig. 1.

In situ detection of cardiac myocyte apoptosis detected by TUNEL assay. At P15, compared with controls (a), in which little or no apoptosis is detected, a marked increase in the incidence of cardiac myocyte apoptosis is evident in the ventricles after prenatal cocaine exposure (b). Concomitant administration of minocycline (25 mg/kg BW) from E15 to E21 fails to prevent in utero cocaine exposure-induced activation of cardiac myocyte apoptosis in fetal hearts. Scale bar × 50 μm. c Representative examples of cardiac myocytes stained with a-actinin. Chromatin was stained with DAPI. Scale bar × 15 μm. d Co-staining for caspase 3 (green) and a-actinin (red) shows activation of caspase 3 in cardiac myocytes. Scale bar × 10 μm. e, f The overall morphology of apoptotic cardiac myocytes. Note varying degrees of chromatin condensation and fragmentation typical of apoptosis. Scale bar × 2 μm (e) and 1 μm (f). g Western blot analysis of ventricular lysates shows increased levels of phospho-p38 MAPK (p-p38 MAPK) and phospho-JNK (p-JNK) and active caspase 9 and caspase 3 in neonatal hearts after prenatal cocaine exposure compared with controls. Concomitant administration of minocycline fails to prevent in utero cocaine exposure-induced activation of p38 MAPK, JNK, and caspases. The gels are representative of three pups in each group from one of three separate experiments (n × 10 pups per group). GAPDH in the immunoblot is shown as a loading control. Con, Control; Coc, Cocaine; and Coc + M, Cocaine plus minocycline (Color figure online)
In utero cocaine exposure also resulted in increased expression of phospho-p38 MAPK, phospho-JNK, active caspase 9, and active caspase 3 in ventricular lysates as evidenced by immunoblotting (Fig. 1g). However, prenatal cocaine exposure had no effect on ERK activation (Fig. 1g). Consistent with its failure to prevent fetal cardiac myocyte apoptosis, minocycline treatment, within the study paradigm, had no discernible effect on activation of p38 MAPK, JNK, and caspases 9 and 3 (Fig. 1g). These findings were substantiated by densitometric analysis (data not shown).
Continued minocycline (37.5 mg/kg BW) treatment from E15 to P15 attenuates fetal cardiac myocyte apoptosis (experiment 2)
In this study, as a first step, we examined whether continued administration of a relatively higher dose (37.5 mg/kg BW) of minocycline from E15 until the pups were killed on P15 would confer resistance to in utero cocaine exposure-induced fetal cardiac myocyte apoptosis. Minimal cardiac myocyte apoptosis was detected in controls (Fig. 2a). As in the first experiment, in utero cocaine exposure resulted in a marked increase in the incidence of cardiac myocyte apoptosis at P15 (Fig. 2b). Minocycline treatment, within the study paradigm, markedly suppressed fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure (Fig. 2c). Morphometric analysis further confirmed histological findings and revealed that minocycline treatment significantly (P < 0.001) prevented cardiac myocyte apoptosis after prenatal cocaine exposure to levels compared to controls (Fig. 2d).
Fig. 2.
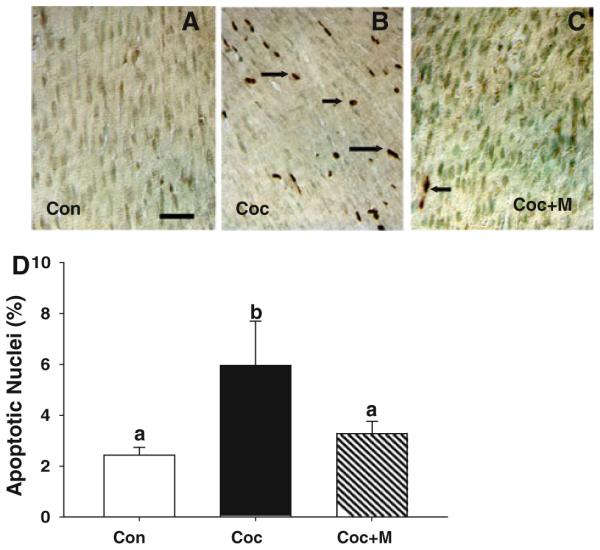
Continued minocycline (37.5 mg/kg BW) treatment from E15 up to P15 attenuates fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure. a Compared with control neonates, where no apoptosis is detected, prenatal cocaine administration results in a marked increase in cardiac myocyte apoptosis (b) and that can be effectively prevented by minocycline treatment (c). Scale bar × 50 μm. d Quantitative changes in the incidence of cardiac myocyte apoptosis among various treatment groups. Apoptotic rate was expressed as the percentage of TUNEL positive nuclei per total nuclei (apoptotic plus nonapoptotic nuclei) counted in a unit reference area. Values are the mean ± SEM of five pups per group. Means with unlike superscripts are significantly (P < 0.001) different
Continued minocycline treatment from E15 to P15 prevents in utero cocaine exposure-induced oxidative stress and activation of p38 MAPK, JNK, and caspases in fetal hearts
We next investigated the mechanisms by which minocycline protects fetal cardiac myocyte apoptosis induced by prenatal cocaine exposure. To determine whether in utero cocaine exposure induces oxidative stress in fetal hearts, we measured the GSH/GSSG ratio, which is inversely related to ROS levels, in ventricular samples. Compared to controls, there was a significant (P < 0.01) increase in oxidative stress as evidenced by low GSH/GSSG ratio in the ventricles at P15 after prenatal cocaine exposure (Fig. 3a). We also examined another biomarker of oxidative stress, the lipid peroxidation marker 4-HNE [32, 33] in the ventricles of neonates. As shown in Fig. 3b, in utero cocaine exposure also led to a significant (P < 0.001) increase in the levels of 4-HNE protein adducts in fetal hearts compared to controls. Notably, continued minocycline (37.5 mg/kg BW) treatment from E15 up to P15 significantly prevented in utero cocaine exposure-induced oxidative stress in fetal hearts (Fig. 3a, b).
Fig. 3.

Continued minocycline treatment from E15 up to P15 prevents in utero cocaine exposure-induced oxidative stress and activation of p38 MAPK and JNK. Ventricles from pups born to cocaine-treated pregnant dams exhibit significantly greater oxidative stress, as evidenced by low (P < 0.01) GSH/GSSG ratio (a) and increased (P < 0.001) 4-HNE levels (b) compared to those of control neonates. Minocycline treatment significantly prevented in utero cocaine exposure-induced oxidative stress in fetal hearts. Values are the mean ± SEM of ten pups per group. Means with unlike superscripts are significantly different. EIA reveals a significant (P < 0.005) increase in both phospho-JNK (c) and phopsho-p38 MAPK (d) levels in fetal hearts after prenatal cocaine exposure when compared with control neonates, which could be significantly (P < 0.005) prevented by minocycline treatment. Values are the mean ± SEM of ten pups per group. Means with unlike superscripts are significantly different
Since oxidative stress can promote activation of both p38 MAPK and JNK, which through mitochondria-dependent intrinsic pathway signaling promotes apoptosis in various cell types [reviewed in 39], we examined the contribution of these kinases in fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure by EIA as well as by immunoblotting. EIA revealed a significant (P < 0.005) increase in both phospho-JNK (Fig. 3c) and phospho-p38 MAPK (Fig. 3d) levels in fetal ventricles after prenatal cocaine exposure when compared with controls, which could be significantly (P < 0.005) prevented by minocycline treatment. As shown in Fig. 4a, immunoblot analysis also revealed significantly (P < 0.05) increased levels of both phospho-p38 MAPK (by 1.5-fold) and phospho-JNK (by 1.7-fold) in ventricles from pups at P15 after prenatal cocaine exposure compared with that of control pups. Given the opposing effects of ERK and JNK-p38 MAPK on apoptosis [16, 17], we next investigated the contribution of ERK in fetal cardiac myocyte death after prenatal cocaine exposure. Unlike p38 MAPK or JNK, in utero cocaine exposure had no effect on ERK activation. Continued minocycline (37.5 mg/kg BW) treatment from E15 up to P15 significantly (P < 0.05) prevented such in utero cocaine exposure-induced activation of p38 MAPK and JNK in fetal ventricles (Fig. 4a). Double immunofluorescence staining of phospho-p38 MAPK or phospho-JNK and TUNEL in ventricular sections of pups after prenatal cocaine exposure further confirmed activation of p38 MAPK or JNK (data not shown) in fetal cardiac myocytes undergoing apoptosis (Fig. 4b).
Fig. 4.

a Western blot analysis of ventricular lysates shows significantly (P < 0.05) increased levels of phospho-p38 MAPK and phospho-JNK in neonates after prenatal cocaine exposure compared with control neonates. Minocycline treatment significantly (P < 0.05) prevents such in utero cocaine exposure-induced activation of these kinases. However, in utero cocaine exposure had no effect on ERK activation. The gels are representative of two neonates in each group from one of three separate experiments (n × 10 pups per group). GAPDH in the immunoblot is shown as a loading control. b Double-immunofluorescence staining for TUNEL (green) and phospho-p38 MAPK (red) from pups born to cocaine-treated mothers show activation of p38 MAPK in fetal cardiac myocyte undergoing apoptosis (shown as yellow). Scale bar × 25 μm (Color figure online)
Because the ratio of anti-apoptotic and pro-apoptotic BCL-2 family members such as BCL-2/BAX constitutes a rheostat that sets the thresholds for susceptibility to apoptosis in the intrinsic pathway signaling [40], we examined the expression of profiles of BAX and BCL-2 and activation of caspases by immunoblotting (Fig. 5). Compared to controls, we found a significant (P < 0.05) increase in BAX by 2.1 fold with no apparent changes in BCL-2 levels in ventricular lysates from 15-day-old neonates after prenatal cocaine exposure, suggesting perturbation of the BAX/BCL-2 ratio. Continued minocycline (37.5 mg/kg BW) treatment significantly (P < 0.05) prevented in utero cocaine exposure-induced up regulation of BAX in fetal hearts. Interestingly, we found significantly (P < 0.05) elevated levels of BCL-2 over the values measured in controls or in neonates after in utero cocaine exposure in the ventricles of neonates after minocycline treatment. Compared with control neonates, where no expression was detected, we found significantly (P < 0.001) increased expression of both active caspase 9 (by 4.0-fold) and caspase 3 (by 9.4-fold) levels in fetal hearts after in utero cocaine exposure. Such activation of caspases in fetal hearts was significantly (P < 0.001) prevented by minocycline.
Fig. 5.
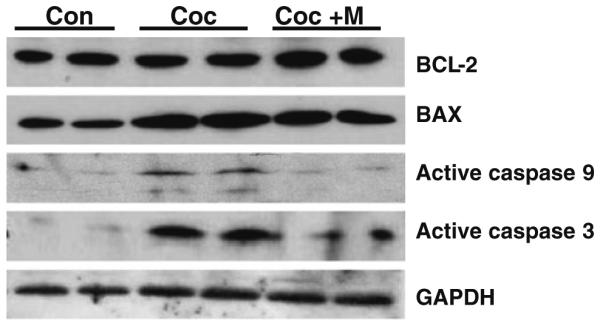
In utero cocaine exposure-induced activation of p38-MAPK and JNK is further associated with a significant (P < 0.05) increase in BAX expression (BCL-2 levels remain unaltered) and active caspases 9 and 3 levels (P < 0.001) as detected by immunoblotting. Minocycline treatment significantly (P < 0.05) up-regulates BCL-2 levels, suppresses prenatal cocaine exposure-induced changes in the BAX levels and prevents (P < 0.001) activation of caspases in fetal hearts. The gels are representative of two neonates in each group from one of three separate experiments (n × 10 pups per group). GAPDH in the immunoblot is shown as a loading control
Continued minocycline (37.5 mg/kg BW) treatment from E15 up to P15 prevents in utero cocaine exposure-induced cytochrome c release from mitochondria in fetal hearts
Given that inhibition of cytochrome c release is essential for minocycline-mediated protection of neurons [19, 20, 27] or male germ cells [21, 22], we next examined the cytochrome c release from mitochondria in various treatment groups (Fig. 6). Ventricular lysates were fractionated into cytosolic and mitochondrial fractions and analyzed by Western blotting. COX IV was used to determine the purity of the mitochondrial fraction. The absence of COX IV in the cytosolic fractions (Fig. 6a) confirmed that the cytosolic preparations were free of mitochondrial contamination. As shown in Fig. 6b, no cytochrome c was detected in cytosol from the ventricles of control neonates. In contrast, cytosolic accumulation of cytochrome c was readily detected in fetal ventricles after in utero cocaine exposure. Minocycline significantly (P < 0.05) prevented such cocaine-induced release of cytochrome c from mitochon-dria into the cytosol (Fig. 6b). These findings were further substantiated by densitometric analysis (Fig. 6c).
Fig. 6.

Minocycline treatment from E15 up to P15 suppresses in utero cocaine exposure-induced cytochrome c release from mitochondria. Ventricular lysates from neonates were fractionated into cytosolic and mitochondrial fractions and analyzed by Western blotting. a Representative Western blots of mitochondrial (MITO) and cytosolic fractions of neonates show the presence of COX IV only in mitochondrial fractions but not in cytosolic fractions. The absence of COX IV in the cytosolic fractions confirmed that the cytosolic preparations were free of mitochondrial contamination. b Representative Western blots of cytosolic fractions from various treatment groups show accumulation of cytochrome c in ventricles of neonates born to cocaine-treated moms and that can be significantly (P < 0.05) suppressed by minocycline treatment. No cytochrome c is detected in the cytosol of ventricles from control neonates. The gels are representative of two neonates in each group from one of three separate experiments. Actin in the immunoblot is shown as a loading control. c Densitometric analysis shows significant (P < 0.05) inhibition of cocaine-induced cytochrome c release after minocycline treatment
Discussion
Cardiac myocyte apoptosis plays an important role in the morphogenesis and remodeling of mammalian heart during the first 2 postnatal weeks [10, 11] and has been implicated as a mechanism in the development of cardiac failure of either ischemic or non-ischemic origin [12–15]. In fact, Wencker et al. [12], using transgenic mice with inducible cardiac myocyte apoptosis, have demonstrated that low levels of apoptosis (levels that are 4.0 to 10-fold lower than those seen in human heart failure cases) are sufficient to cause a lethal, dilated cardiomyopathy. Conversely, inhibition of cardiac myocyte apoptosis in this murine model largely prevents the development of this syndrome [12]. Thus preventing fetal cardiac myocyte apoptosis is an attractive therapeutic strategy to prevent cardiovascular diseases associated with the loss of terminally differentiated cardiac myocytes. A growing body of evidence demonstrates that minocycline has a robust neuroprotective effect in rodent models of neurodegeneration, cerebral ischemia, and traumatic brain injury [19, 20, 27, 41]. This compound is equally effective in protecting testicular germ cells from apoptosis triggered by hormone deprivation [21] or by heat stress [22]. In congruence with these findings, here we show that minocycline is also effective in pro-tecting fetal cardiac myocytes from apoptosis induced by in utero cocaine exposure. Minocycline-mediated protection of fetal cardiac myocyte death occurs through suppression of oxidative stress and p38 MAPK and JNK-mediated intrinsic (mitochondria-dependent) pathway signaling. Our results further demonstrate that suppression of cardiac myocyte apoptosis is only possible through continued treatment of minocycline to pregnant dams from E15 until the pups were sacrificed at P15.
Emerging evidence suggests that cardiac oxidative stress is an early event of cocaine administration, which severely compromises the cardiac antioxidative system and causes myocyte damage [42–44]. Consistent with the role of oxidative stress, in the present study, we found pups those were born to cocaine treated pregnant dams had greater oxidative stress, as evidenced by low GSH/GSSG ratio and increased levels of 4-HNE. Minocycline is indeed effective in mitigating in utero cocaine exposure-induced oxidative stress in fetal hearts. Thus, it is conceivable that minocycline may protect cardiac consequences of prenatal cocaine exposure by suppressing oxidative stress.
Oxidative stress has been implicated in apoptotic signaling in various cell types, including neonatal rat cardiac myocytes [23, 45]. One possible mechanism by which oxidative stress can induce fetal cardiac myocyte apoptosis after prenatal cocaine exposure is through stimulation of p38 MAPK and JNK signaling, resulting in the activation of the mitochondria-dependent intrinsic pathway signaling [35, 39, 46]. Indeed, in the present study, we found activation of both of these kinases in ventricles of the neonates after in utero cocaine exposure compared to that of control neonates. Earlier in vitro studies have shown activation of both p38 MAPK and JNK in cultured rat neonatal cardiac myocytes after ischemia and reperfusion and their inhibition results in attenuation of cardiac myocyte apoptosis [23]. Li et al. [9] have also provided in vitro evidence for a role of p38 MAPK in cocaine-induced neonatal cardiac myocyte apoptosis. Collectively, the data reported herein provide for the first time an in vivo evidence for involvement of p38 MAPK and JNK in fetal cardiac myocyte apoptosis following in utero cocaine exposure. The signal for activation of p38 MAPK and JNK most likely emanates from in utero cocaine exposure-induced increase in oxidative stress in fetal hearts. Importantly, we further show that continued minocycline treatment significantly suppressed cocaine-induced activation of p38 MAPK and JNK in fetal hearts. Inhibition of p38 MAPK has been known to be associated with minocycline-mediated neuroprotection [19, 47, 48]. In this study, we have provided evidence for involvement of JNK signaling in minocycline-mediated cardioprotection. Thus, minocycline-mediated in vivo cardioprotection in neonates born to pregnant dams receiving cocaine involves inhibition of both JNK and p38 MAPK activation.
The downstream signaling events that couple activation of p38 MAPK and JNK with fetal cardiac myocyte death after in utero cocaine exposure have not been previously identified. Several lines of evidence indicate that these kinases can induce apoptosis in various cell systems through perturbation of the BAX/BCL-2 rheostat, cytochrome c release from mitochondria, and subsequent activation of the initiator caspase 9 and the executioner caspase 3 [24, 29, 34, 49, 50]. Indeed, in the present study, we found that activation of JNK and p38 MAPK is associated with stimulation of the mitochondria-dependent apoptotic pathway characterized by the perturbation of the BAX/ BCL-2 rheostat, cytosolic translocation of mitochondrial cytochrome c and activation of the initiator caspase 9. Intriguingly, we further show that minocycline treatment significantly prevented in utero cocaine exposure-induced cytochrome c release from mitochondria and activation of caspases 9 and 3. This is consistent with earlier works on various cell systems indicating that minocycline-mediated protection of apoptosis occurs through suppression of cytochrome c release and subsequent activation of caspase 9 and caspase 3 [19–22, 27, 48].
In summary, we have extended the protective role of minocycline to neonatal cardiac myocyte apoptosis and provided new insights into the molecular mechanisms by which minocycline prevents in utero cocaine exposure-induced fetal cardiac myocyte apoptosis. Minocycline appears to work at various steps of the signal transduction pathways involving suppression of oxidative stress, activation of JNK and p38 MAPK, perturbation of the BAX/ BCL-2 ratio, cytochrome c release, and activation of caspases 9 and 3 (Fig. 7), which may explain why minocycline has a protective effect in a wide range of cells. Minocycline with its proven clinical safety may become an effective therapy for preventing cardiac consequences of prenatal cocaine exposure.
Fig. 7.
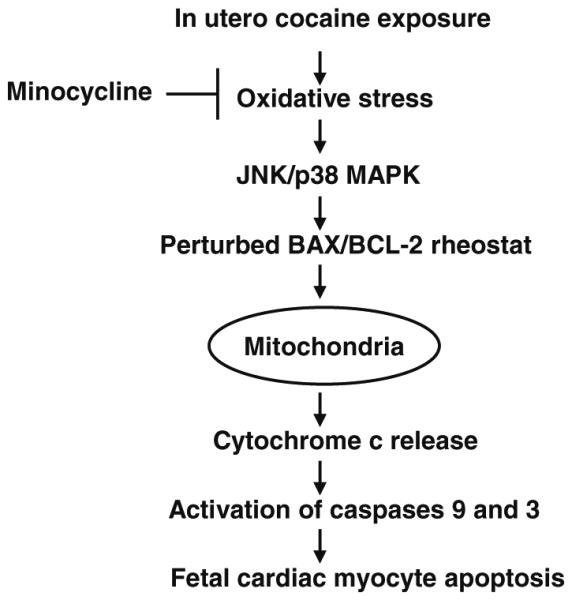
Key signaling pathways involved in minocycline-mediated protection of cardiac myocytes from apoptosis trigged by in utero cocaine exposure. Increased oxidative stress generated in response to prenatal cocaine exposure can result in activation of both JNK and p38 MAPK in fetal hearts. It is likely that activation of these signaling cascades promote fetal cardiac myocyte apoptosis through perturbation of the BAX/BCL-2 rheostat and subsequent activation of cytochrome c-mediated death pathway. Minocycline seems to be working at various steps of the signal transduction pathways involving suppression of oxidative stress, activation of JNK and p38 MAPK, perturbation of the BAX/BCL-2 ratio, cytochrome c release, and activation of caspases 9 and 3
Acknowledgments
This work was supported by MIDARP grant from NIH (to I.S-H and A.P.S-H). Additional support was provided by the NIH/NIGMS Program (S06 GM068510; A.P.S-H) and by the NIH-NCCR Accelerating Excellence in Translation Science Grant (U54 RR026138; Norris, KC). S.K.M. is supported by grants form the Department of Veteran Affairs and the National Institutes of Health (RO1 DA011311 and PO HL58120).
Contributor Information
Indrani Sinha-Hikim, Division of Endocrinology, Metabolism, and Molecular Medicine, Charles R. Drew University of Medicine and Science, Los Angeles 90059, CA, USA.
Ruoqing Shen, Division of Endocrinology, Metabolism, and Molecular Medicine, Charles R. Drew University of Medicine and Science, Los Angeles 90059, CA, USA.
Ify Nzenwa, Division of Endocrinology, Metabolism, and Molecular Medicine, Charles R. Drew University of Medicine and Science, Los Angeles 90059, CA, USA.
Robert Gelfand, Division of Endocrinology, Metabolism, and Molecular Medicine, Charles R. Drew University of Medicine and Science, Los Angeles 90059, CA, USA.
Sushil K. Mahata, Department of Medicine, UCSD, San Diego, CA, USA VA San Diego Healthcare System, San Diego, CA, USA.
Amiya P. Sinha-Hikim, Division of Endocrinology, Metabolism, and Molecular Medicine, Charles R. Drew University of Medicine and Science, Los Angeles 90059, CA, USA indranisinhahikim@cdrewu.edu
References
- 1.DAWN Report: Drug Abuse Warning Network October 2002. http://www.samhsa.gov/oas/major DAWN.
- 2.DAWN Report: Drug Abuse Network 2005. major DAWN http://www.drug abuse.gov/PDF/CEWG/AdvReport606.pdf.
- 3.Feng Q. Postnatal consequences of prenatal cocaine exposure and myocardial apoptosis: does cocaine in utero imperil the adult heart? Br J Pharmacol. 2005;144:887–888. doi: 10.1038/sj.bjp.0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCord J, Jneid H, Hollander JE, de Lemos JA, Cercek B, Hsue P, Gibler WB, Ohman EM, Drew B, Philippides G, Newby LK. Management of cocaine associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897–1907. doi: 10.1161/CIRCULATIONAHA.107.188950. [DOI] [PubMed] [Google Scholar]
- 5.Mone SM, Gillman MW, Miller TL, Herman EH, Lipshultz SE. Effects of environmental exposures on the cardiovascular system: prenatal period through adolescence. Pediatrics. 2004;113:1058–1069. [PubMed] [Google Scholar]
- 6.Gaithner K. Cocaine abuse in pregnancy: an evolution from placenta to pandemonium. Southern Med J. 2008;101:783–784. doi: 10.1097/SMJ.0b013e31817f1f5e. [DOI] [PubMed] [Google Scholar]
- 7.Bae S, Zhang L. Prenatal cocaine exposure increases apoptosis of neonatal rat heart and heart susceptibility to ischemia-reperfusion injury in 1-month-old rat. Br J Pharmacol. 2005;144:900–907. doi: 10.1038/sj.bjp.0706129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer KD, Zhang L. Short- and ling-term adverse effects of cocaine abuse during pregnancy on the heart development. Ther Adv Cardiovasc Dis. 2009;3:7–16. doi: 10.1177/1753944708099877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li G, Xiao Y, Zhang L. Cocaine-induces apoptosis in fetal rat myocardial cells through p38 mitogen-activated protein kinase and mitochondrial/cytochrome c pathways. J Pharmacol Exp Ther. 2005;312:112–119. doi: 10.1124/jpet.104.073494. [DOI] [PubMed] [Google Scholar]
- 10.Kajstura J, Mansukhani M, Cheng W, Reiss K, Krajewski S, Reed JC, Qiaini F, Sonnenblick EH, Anversa P. Programmed cell death and expression of the protooncogene bcl-2 in myocytes during postnatal maturation of the heart. Exp Cell Res. 1995;219:110–121. doi: 10.1006/excr.1995.1211. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez E, Siddiquee Z, Shohet RV. Apoptosis and proliferation in the neonatal murine heart. Dev Dyn. 2001;221:302–310. doi: 10.1002/dvdy.1139. [DOI] [PubMed] [Google Scholar]
- 12.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role of cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scarabelli TM, Gottlieb RA. Functional and clinical repercussions pf myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: old and new concepts after 10 years of contributions. Cell Death Differ. 2004;11:S144–S152. doi: 10.1038/sj.cdd.4401544. [DOI] [PubMed] [Google Scholar]
- 14.Foo RS-Y, Mani K, Kitsis RN. Death begins failure in the heart. J Clin Invest. 2005;115:565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee Y, Gustafsson AB. Role of apoptosis in cardiovascular disease. Apoptosis. 2009;14:536–548. doi: 10.1007/s10495-008-0302-x. [DOI] [PubMed] [Google Scholar]
- 16.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 17.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 18.Liang Q, Mollkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li M, Sarang S, Liu AS, Hartely DM, Chu Wu D, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM. Minocycline inhibits caspase-independent and-dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci USA. 2003;100:10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castanares M, Vera Y, Erkkila K, Kyttanen S, Lue Y, Dunkel L, Wang C, Swerdloff RS, Sinha Hikim AP. Minocycline upregulates BCL-2 levels in mitochondria and attenuates male germ cell apoptosis. Biochem Biophys Res Commun. 2005;337:663–669. doi: 10.1016/j.bbrc.2005.09.101. [DOI] [PubMed] [Google Scholar]
- 22.Vera Y, Rodriguez S, Castanares M, Lue Y, Atienza V, Wang C, Swerdloff RS, Sinha Hikim AP. Functional role of caspases in heat-induced testicular germ cell apoptosis. Biol Reprod. 2005;72:516–522. doi: 10.1095/biolreprod.104.034520. [DOI] [PubMed] [Google Scholar]
- 23.Sun H-Y, Wang N-P, Halkos M, Kerendi F, Kin H, Guyton RA, Vinten-Johansen J, Zhao Z-Q. Postconditioning attenuates cardiomyocyte apoptosis by inhibiting JNK and p38 mitogen-activated protein kinase signaling pathways. Apoptosis. 2006;11:1583–1593. doi: 10.1007/s10495-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 24.Sinha-Hikim I, Braga M, Shen R, Sinha Hikim AP. Involvement of c-Jun NH2-terminal kinase and nitric oxide-mediated mitochondria-dependent intrinsic pathway signaling in cardiotoxin-induced muscle cell death: role of testosterone. Apoptosis. 2007;12:1965–1978. doi: 10.1007/s10495-007-0120-6. [DOI] [PubMed] [Google Scholar]
- 25.Braga M, Sinha Hikim AP, Datta S, Ferrini M, Brown D, Kovacheva EL, Gonzalez-Cadavid NF, Sinha-Hikim I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–832. doi: 10.1007/s10495-008-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 27.Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci USA. 2004;101:3071–3076. doi: 10.1073/pnas.0306239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae S, Gilbert RD, Ducsay CA, Zhang L. Prenatal cocaine exposure increases heart susceptibility to ischaemia-reperfusion injury in adult male but not female rats. J Physiol. 2005;565:149–158. doi: 10.1113/jphysiol.2005.082701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vera Y, Erkkila K, Wang C, Nunez C, Kyttanen S, Lue Y, Dunkel L, Swerdloff RS, Sinha Hikim AP. Involvement of p38 mitogen-activated protein kinase and inducible nitric oxide synthase in apoptotic signaling of murine and human male germ cells after hormone deprivation. Mol Endocrinol. 2006;20:1597–1609. doi: 10.1210/me.2005-0395. [DOI] [PubMed] [Google Scholar]
- 30.Kovacheva EL, Sinha Hikim AP, Shen R, Sinha I, Sinha-Hikim I. Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology. 2010;151:628–638. doi: 10.1210/en.2009-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha-Hikim I, Shen R, Lee W-NP, Crum A, Vaziri ND, Norris KC. Effect of a novel cystine based glutathione precursor on oxidative stress in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2010;299:C638–C642. doi: 10.1152/ajpcell.00434.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohen R, Nysks A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reaction, and methods for their quantification. Toxicol Pathol. 2002;30:620–650. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- 33.Tam NNC, Gao Y, Leung Y-K, Ho S-M. Androgenic regulation of oxidative stress in the rat prostate: involvement of NAD(P)H oxidases and antioxidant defense machinery during prostatic involution and regrowth. Am J Pathol. 2003;163:2513–2522. doi: 10.1016/S0002-9440(10)63606-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia Y, Castellanos J, Wang C, Sinha-Hikim I, Lue Y, Swerdloff RS, Sinha Hikim AP. Mitogen-activated protein kinase signaling in male germ cell apoptosis. Biol Reprod. 2009;80:771–780. doi: 10.1095/biolreprod.108.072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinha-Hikim I, Shen R, Kovacheva E, Crum A, Vaziri ND, Norris KC. Inhibition of apoptotic signaling in spermine-treated vascular smooth muscle cells by a novel glutathione precursor. Cell Biol Int. 2010;34:503–511. doi: 10.1042/CBI20090349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuzmenkin A, Liang H, Xu G, Pfannkuche K, Eichhorn H, Fatima A, Luo H, Saric T, Werning M, Jaenisch R, Hescheler J. Functional characterization of cardiomyocytes derived from murine induced pluripotent cells in vitro. FASEB J. 2009;23:4168–4180. doi: 10.1096/fj.08-128546. [DOI] [PubMed] [Google Scholar]
- 37.Jin M-S, Shi S, Zhang Y, Yan Y, Sun X-D, Liu W, Liu H-W. Icariin-mediated differentiation of mouse adipose-derived stem cells into cardiomyocytes. Mol Cell Biochem. 2010;344:1–9. doi: 10.1007/s11010-010-0523-5. [DOI] [PubMed] [Google Scholar]
- 38.Duysen EG, Li B, Carlson M, Li Y-F, Wieseler S, Hinrichs SH, Lockridge O. Increased hepatotoxicity and cardiac fibrosis in cocaine-treated butyrulchokinesterase knockout mice. Basic Clin Pharmacol Toxicol. 2008;103:514–521. doi: 10.1111/j.1742-7843.2008.00259.x. [DOI] [PubMed] [Google Scholar]
- 39.Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an oxidant. Cell Death Differ. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 40.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 41.Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist. 2005;11:308–322. doi: 10.1177/1073858405275175. [DOI] [PubMed] [Google Scholar]
- 42.Boess F, Ndikum-Moffor FM, Boelsterli UA, Roberts SM. Effects of cocaine and its oxidative metabolites on mitochondrial respiration and generation of reactive oxygen species. Biochem Pharamacol. 2000;60:615–623. doi: 10.1016/s0006-2952(00)00355-5. [DOI] [PubMed] [Google Scholar]
- 43.Kovacic P. Role of oxidative metabolites of cocaine in toxicity and addiction: oxidative stress and electron transfer. Med Hypo. 2005;64:350–356. doi: 10.1016/j.mehy.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 44.Fan L, Sawbridge D, George V, Teng L, Bailey A, Kitchen I, Li J-M. Chronic cocaine-induced cardiac oxidative stress and mitogen activated protein kinase activation: the role of Nox2 oxidase. J Phramacol Exp Ther. 2009;328:99–106. doi: 10.1124/jpet.108.145201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JK, Pedram A, Razandi M, Levin ER. Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J Biol Chem. 2006;281:6760–6767. doi: 10.1074/jbc.M511024200. [DOI] [PubMed] [Google Scholar]
- 46.Circu ML, Aw TY. Glutathione and apoptosis. Free Radic Res. 2008;42:689–706. doi: 10.1080/10715760802317663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excytotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei X, Zhao L, Liu J, Dodel RC, Farlow MR, Du Y. Minocycline prevents gentamicin-induced ototoxicity by inhibiting p38 MAP kinase phosphorylation and caspase 3 activation. Neuroscience. 2005;131:513–521. doi: 10.1016/j.neuroscience.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 49.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavel RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 50.Lei K, Nimnual A, Zong W-X, Kennedy NJ, Flavel RA, Thompson CB, Bar-Sagi D, Davis RJ. The Bax subfamily of Bcl-2 related proteins is essential for apoptotic signal transduction by c-Jun NH2-terminal kinase. Mol Cell Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]