

Abstract
Chromatin regulatory mechanisms play a major role in the control of gene expression programs during normal development and are disrupted in specific disease states, particularly in cancer. Important mediators of chromatin regulatory processes can broadly be classified into writers, erasers, and readers of covalent chromatin modifications that modulate eukaryotic gene transcription and maintain the integrity of the genome. The reversibility and disease-specific nature of these chromatin states make these regulators attractive therapeutic targets. As such, there is an ever-increasing number of candidate therapies aimed at targeting cancer-associated chromatin states that are in various stages of preclinical and clinical development. In this review, we discuss recent advances that have been made in the rational therapeutic targeting of chromatin regulatory mechanisms and highlight certain cancers where there is a specific rationale to assess these therapeutic approaches.
Introduction
Epigenetics refers to the description of phenotypic outcomes that are not attributable to changes in underlying DNA sequence. At the resolution of a single gene, the chromatin modifications associated with a particular locus and its distant regulatory elements play a major role in determining the final state of gene activation or repression during organismal or lineage-specific development and are thus often referred to as epigenetic modifications or epigenetic states. These states are conferred via several central epigenetic processes including posttranslational histone modification, DNA methylation, and expression of non-coding RNAs (Goldberg et al., 2007). Over the past two decades, much has been revealed about the mechanisms governing epigenetic regulation on a genome-wide scale, particularly in the wake of the development of several major technological advances, including but not limited to whole genome sequencing, chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq), RNA-seq, bisulfite sequencing, and chromosome conformation capture. Concerted epigenomic efforts like the ENCODE Project and the NIH Roadmap Epigenomics Mapping Consortium have employed these techniques on a wide variety of cell types to provide an important framework for cataloguing the myriad epigenetic modifications that modulate lineage- and disease-specific gene expression programs (Bernstein et al., 2010; Consortium, 2004).
It is now well established that chromatin or epigenetic regulation plays a nonredundant role not only in normal development but also in the pathogenesis of a number of disease states, including cancer where the role in hematologic malignancies is most well developed. In the case of acute myeloid leukemia (AML), whole-genome sequencing of 200 AML patients demonstrated that a significant proportion of these cases harbored nonsynonymous mutations in epigenetic regulators, with 44% of these samples found to have DNA-methylation-related mutations, and 43% having mutations in other chromatin modifiers or cohesin-complex genes (Cancer Genome Atlas Research Network, 2013). The relative frequency with which recurrent mutations are now known to occur in epigenetic regulators, together with the tissue and disease specificity of the epigenetic program, make the processes that control the epigenome an attractive therapeutic target for cancer and other disease states. Thus, ever since the FDA approved the DNA methyltransferase 1 (DNMT1) inhibitor 5-azacitidine in 2004, and the histone deacetylase (HDAC) inhibitor vorinostat in 2006, two of the earliest therapies to target epigenetic mechanisms, for the treatment of specific hematologic malignancies, the number of candidate drugs targeting the epigenome has grown significantly (see Table 1). In this review, we will highlight recent developments, novel strategies, and potential pitfalls in targeting epigenetic drivers of disease (see Figure 1). As the preponderance of these therapies were discovered within the context of and directed at treating hematologic malignancies, the discussion will be focused primarily on this disease subset, but selected relevant discoveries made within the context of other disease states will be discussed as well.
Table 1. Classes of Epigenetic Therapeutic Targets in Preclinical and Clinical Development.
| Class | Epigenetic Regulator |
Function | Relevant Diseases | Stage of Drug Development |
ClinicaiTrials.gov Identifier |
References |
|---|---|---|---|---|---|---|
| Histone methyltransferases | DOTlL | H3K79 methylation | AML, ALL | Phase I trial | NCT01684150, NCT02141828 | Stein et al., 2014 |
| EZH2 | H3K27 methylation | B cell lymphomas, solid tumors | Phase I trial | NCT02395601, NCT01897571, NCT02082977 | Fillmore et al., 2015; Kim et al., 2013 | |
| Histone demethylases | LSD1 | H3K4 demethylation | AML, SCLC | Phase I trial | NCT02177812, NCT02034123, NCT02273102, NCT02261779 | Harris et al., 2012; Mohammad et al., 2015; Schenk et al., 2012 |
| Histone acetyltransferases | p300/CBP | H3K27 acetylation | AML, prostate cancer | Preclinical development | Gao et al., 2013; Santer et al., 2011 | |
| Histone deacetylases | HDAC 1, 2, etc. | Histone deacetylation | Multiple myeloma, cutaneous T cell lymphomas | FDA-approved (panobinostat, vorinostat romidepsin) | Mann et al., 2007b; Piekarz et al., 2011; San-Miguel et al., 2014 | |
| DNA methyltransferases | DNMTl | Maintenance DNA methylation | MDS, CMML | FDA-approved (decitabine, azacitidine) | Kaminskas et al., 2005 | |
| DNMT3A | de novo DNA methylation | AML, MDS | Preclinical development | Aidawsari et al., 2015 | ||
| Chromatin readers | BET | Binding acetylated lysines | AML, MDS, lymphoma, multiple myeloma, glioblastoma multiforme, solid tumors, NMC advanced | Phase I trial | NCT02296476, NCT01713582, NCT02259114, NCT01949883, NCT02157636, NCT02158858, NCT02303782 | Delmore et al., 2011; Filippakopoulos and Knapp, 2014; Mertz et al., 2011; Zuber et al., 2011 |
Figure 1. Strategies for Targeting Epigenetic Regulators.
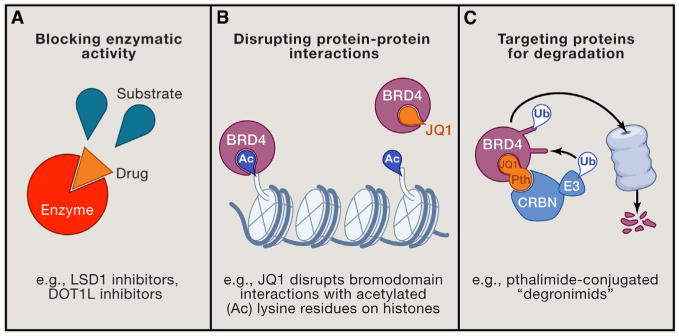
(A) Inhibiting enzymatic activity with competitive small molecule compounds, (B) disrupting protein-protein interactions, and (C) using phthalimide-conjugated small molecules to redirect difficult-to-target proteins for proteasomal degradation via the cereblon (CRBN) E3 ubiquitin (Ub) ligase complex.
Targeting DNA Methylation and Histone Deacetylation
Histone Deacetylases
Histone deacetylase (HDAC) inhibitors, together with hypomethylating agents, represent the only two classes of epigenetic therapies currently approved by the FDA. Suberoylanilide hydroxamic acid (SAHA), otherwise known as vorinostat, was developed as a differentiation agent in the 1990s and shortly afterward was found to be a potent HDAC inhibitor with antitumor activity (Richon et al., 2001). Vorinostat was the first HDAC inhibitor to be FDA-approved in 2006 for the treatment of advanced cutaneous T cell lymphoma (CTCL) (Mann et al., 2007). Since then, three additional HDAC inhibitors are now approved for the treatment of hematologic malignancies including CTCL and multiple myeloma (Tanaka et al., 2015).
Given the proven clinical efficacy of HDAC inhibitors, these compounds have now been extensively studied in combinations. HDAC inhibitors in conjunction with other epigenetic targeted therapies including EZH2 and LSD1 inhibitors have preclinical activity in AML (Fiskus et al., 2009, 2014), and HDAC inhibitors combined with imatinib has the added benefit of targeting quiescent CML progenitors that otherwise would be resistant to imatinib therapy alone (Zhang et al., 2010). The detailed history of the development and mechanisms of HDAC inhibitors has recently been reviewed elsewhere (Falkenberg and Johnstone, 2014).
DNA Methyltransferases
DNA methylation is an epigenetic modification that is relevant to a number of processes, including normal development, X chromosome inactivation, maintenance of stem cells, and disease. DNA methylation occurs on CpG dinucleotides via the addition of a methyl group to the C5 position of cytosines, which generates 5-methylcytosine (5mC); DNA hypermethylation is generally, but not always, associated with gene silencing (Galm et al., 2006). This modification is mediated by DNA methyltransferases (DNMTs), with DNMT1 functioning to maintain pre-existing DNA methylation and DNMT3A and DNMT3B acting as de novo methyltransferases (You and Jones, 2012).
The hypomethylating agents decitabine and azacitidine are DNA methyltransferase 1 inhibitors that have proven clinical efficacy in the treatment of MDS and to a somewhat lesser extent, AML. Recently, these agents have been shown to induce antitumor “memory” at very low doses and after transient exposure (Tsai et al., 2012). This occurs in the absence of cytotoxicity that can be seen with much higher doses, which causes cell cycle arrest, DNA damage, and apoptosis. Notably, this phenomenon was observed in both solid and liquid tumors. This raises the hypothesis that hypomethylating agents can be used to sensitize tumors to other therapies, which was subsequently validated in NSCLC patients (Juergens et al., 2011). In another study, azacitidine treatment resulted in immune activation characterized by gene signatures associated with interferon signaling, antigen presentation, and cytokine production, which raises the hypothesis that hypomethylating agents may perhaps sensitize tumors to immunotherapies such as nivolumab, ipilimumab, and lenalidomide (Li et al., 2014a). The development of inhibitors of DNA methylation remains an area of significant drug development and has been reviewed extensively (Ahuja et al., 2014).
Targeting Histone Methylation
The roles of histone methyltransferases in disease pathogenesis have been studied most extensively within the context of leukemias harboring rearrangements in the mixed lineage leukemia (MLL1) gene since this was the first identified cancer-associated mutation of a histone methyltransferase. As a consequence of these chromosomal translocations, a fusion oncoprotein is generated whereby the amino-terminal portion of MLL1 is joined in frame to the carboxyl terminal portion of one of over 70 partner genes, the most common of which include AF4, AF9, AF10, ENL, and ELL (Meyer et al., 2013). This fusion results in the loss of the native SET domain located near the carboxyl terminus of MLL1, which harbors the catalytic methyltransferase activity to methylate lysine 4 of histone H3 (H3K4). Via interactions with a subset of MLL fusion partners, two nuclear protein complexes, DOT1L and the super elongation complex (SEC), can be aberrantly recruited to MLL fusion target genes to drive leukemogenic gene expression (Deshpande et al., 2012; Lin et al., 2010; Mohan et al., 2010; Smith et al., 2011). The ongoing detailed mechanistic characterization of these mechanisms has led to a number of potential points for therapeutic intervention.
Disruptor of Telomeric Silencing 1-like
Disruptor of Telomeric Silencing 1-like (DOT1L) is the only known mammalian enzyme that catalyzes methylation of lysine 79 on histone H3 (H3K79), a chromatin modification that is generally associated with actively transcribed genes (Steger et al., 2008). In 2005, Zhang and colleagues first implicated DOT1L as a potential therapeutic target in leukemia based on the finding that it interacts with the MLL fusion partner AF10 and that MLL-AF10 target genes were decorated with H3K79 methylation (Okada et al., 2005). Subsequent studies demonstrated that aberrant H3K79 methylation profiles were found in other more common subtypes of MLL-rearranged leukemias (Guenther et al., 2008; Krivtsov et al., 2008). Furthermore, suppression of DOT1L led to downregulation of critical leukemia genes driven by the MLL-AF4 fusion, thereby highlighting a potentially broad requirement for DOT1L in MLL-rearranged leukemias (Krivtsov et al., 2008). The requirement of DOT1L for MLL-rearranged leukemia development was convincingly demonstrated by a series of studies using conditional mouse models (Bernt et al., 2011; Jo et al., 2011; Nguyen et al., 2011). In several independent mouse models, loss of DOT1L profoundly hindered the leukemogenicity of MLL fusion-transformed cells. Epigenomic studies revealed that gene targets directly bound by MLL fusion products are associated with abnormally high levels of H3K79 dimethylation, particularly at the loci of the leukemogenic oncogenes MEIS1 and HOXA-cluster genes whose upregulation constitute a hallmark feature of MLL-rearranged leukemias (Guenther et al., 2008; Krivtsov et al., 2008).
The role of the DOT1L complex member AF10 was defined in multiple subtypes of AML vis-à-vis its ability to potentiate DOT1L-mediated conversion of H3K79 mono-methylation to di-methylatedstates and thereby maintain aberrant expression of HOXA9 and MEIS1 (Deshpande et al., 2014). Genetic loss of AF10 caused a significant reduction of H3K79 di- and trimethylation, which corresponded to a reduction in HOXA gene expression and impairmentin the transforming potential of both MLL and non-MLL fusions. These findings were supported by elegant structure-function studies demonstrating that the stoichiometry of DOT1L binding to the MLL fusion complex can be manipulated to titrate the level and state of H3K79 methylation at MLL fusion target genes and thus leukemia development (Kuntimaddi et al., 2015). Furthermore, the critical transforming ability of AF10 as a component of the DOT1L complex suggests that the interface of the DOT1L-AF10 interaction may also be a therapeutic target.
Intriguingly, the observation that HOXA-cluster gene expression is dampened when conversion of H3K79 to higher order states is blocked raised the hypothesis that DOT1L inhibition may be a viable therapeutic option in leukemias with high HOXA gene expression but otherwise without MLL rearrangements. This was tested and validated in AML cells transformed by NUP98-NSD1 as well as in primary AML cells cultured in vitro (Deshpande et al., 2014; Sarkaria et al., 2014).
The dependency of MLL-rearranged leukemias on DOT1L enzymatic activity spurred the development of small molecule DOT1L inhibitors, which are actively under investigation in clinical trials (NCT02141828 and NCT01684150 at https://clinicaltrials.gov/). Early findings show that some patients do achieve morphologic and cytogenetic remissions (Stein et al., 2014). Although these positive responses were achieved with DOT1L inhibitor monotherapy, there is a significant precedent for resistance to arise in the setting of single-agent targeted therapies, similar to observations made early during the nascence of cytotoxic chemotherapy regimens. Examples include tyrosine kinase inhibitor (TKI) resistance in chronic myelogenous leukemia (CML) and EGFR mutant non-small cell lung cancers (NSCLC). In these cases, patients exhibit an initial response to therapy but eventually develop disease progression as secondary mutations ultimately arise. As such, targeted therapies administered in rational combinations provide a strategy to achieve deeper responses by potentially derailing acquired resistance mechanisms or by enhancing the sensitivity of tumor cells to the primary therapy. Furthermore, the fact that DOT1L inhibitors and many other small molecule inhibitors that target chromatin regulatory mechanisms often induce cell cycle arrest and/or cellular differentiation supports the concept that combinations of therapeutics will be necessary to fully eradicate malignant clones.
A number of groups have used shRNA or CRISPR-Cas9 mediated screens in combination with a small molecule of interest to identify proteins/pathways that modulate sensitivity to specific small molecules (Shalem et al., 2015). This approach has recently been used to identify mechanisms that could be targeted to enhance sensitivity to DOT1L inhibition. Chen et al. employed a genome-wide shRNA screen to identify genes that, when knocked down, allowed for cell growth to occur in murine MLL-AF9-driven leukemia cells upon DOT1L deletion (Chen et al., 2015). The NAD-dependent lysine deacetylase SIRT1 was the top hit identified in this screen, and adding a pharmacologic activator of SIRT1 resulted in an increased susceptibility of leukemia cells to DOT1L inhibition. Such approaches highlight how vulnerabilities in the combinatorial effects of chromatin modifiers can be unveiled and thus be exploited via rational combinations of targeted epigenetic therapies. We expect that similar experiments with multiple different small molecules will help guide future combination approaches.
MLL Complex
MLL1 and MLL2 function as large macromolecular complexes comprised of multiple subunits. Several of the core components of these complexes act as allosteric regulators of histone methyltransferase activity, including WDR5, RBBP5, and ASH2L (Hughes et al., 2004). Given the role of wild-type MLL1 in leukemia initiation and maintenance (Thiel et al., 2010), disrupting components of the MLL1 complex has become an attractive therapeutic strategy. For example, small molecule inhibitors aimed at disrupting the interaction between MLL1 and WDR5, such as the cyclic compound MM-401, have shown activity in inhibiting leukemia cell proliferation (Cao et al., 2014). The tumor suppressor menin is another essential component of the MLL complex. Menin binds MLL1, MLL2, and MLL1-fusion proteins and is necessary for transcription of MLL target genes. Disruption of the MLL-menin interaction blocks the development of acute leukemia in mice (Yokoyama et al., 2005). Structural studies of the MLL-menin interface revealed bivalent binding of menin at two sites on MLL1, a high-affinity motif on MLL1 (MBM1) and a low-affinity motif (MBM2) (Grembecka et al., 2010). A number of menin-MLL inhibitors targeting MBM1 have been developed, including peptidomimetics such as MCP-1 and thienopyrimidines such as MI-2, a derivatized version MI-2-2, MI-136, MI-463, and MI-503 (Borkin et al., 2015; Grembecka et al., 2012; Shi et al., 2012; Zhou et al., 2013). These inhibitors demonstrated activity in blocking MLL fusion-driven leukemic transformation, and given the potentially broad role for MLL1/MLL2 in the regulation of gene expression it is likely that further indications for menin inhibitors will be identified.
Menin, in addition to its role in leukemogenesis, has been implicated in other malignancies. Recently, MLL1 was identified as a coactivator associated with the androgen receptor (AR) in castration-resistant prostate cancer (Malik et al., 2015). Pharmacologic disruption of the MLL1-menin interaction with MI-136 blocked androgen-stimulated proliferation of prostate cancer cells. Menin inhibition has also been identified as a potential therapeutic opportunity in diffuse intrinsic pontine glioma (DIPG), a rare but extremely aggressive brainstem tumor that frequently harbors somatic mutations in the H3F3A gene encoding H3.3 (Funato et al., 2014). Further studies aimed at interrogating the mechanisms by which menin and the MLL complex cooperate in these diseases are necessary, but these initial studies are encouraging and suggest that MLL-menin inhibition may be an interesting approach in multiple cancers. An important next step in all cases will be to perform thorough structure-function studies of complex components in order to determine the specific mechanisms at play in various cancers.
Enhancer of Zeste Homolog 2
Enhancer of zeste homolog 2EZH2 is a member of the Polycomb repressive complex 2 (PRC2), together with embryonic ectoderm development (EED), suppressor of zeste 12 (SUZ12), Jumonji/ARID domain-containing protein 2 (JARID2), and RBBP4. EZH2 has H3K27 methyltransferase activity that marks target loci and is associated with gene repression and chromatin silencing. The context dependence of epigenetic modifiers in tumorigenesis is highlighted by the opposing roles of EZH2 in different malignancies. EZH2 is amplified in solid tumors (Bachmann et al., 2006; Wagener et al., 2008) and gain-of-function Y641 mutations that cause H3K27 hypermethylation are found in B cell lymphomas (Morin et al., 2010). The PRC2 complex is also important in a mouse model of AML (Neff et al., 2012). In contrast, inactivating mutations or deletions of EZH2 have been identified in MDS, myeloproliferative neoplasms (MPN), and T-ALL, which implicates EZH2 as a tumor suppressor in these diseases (Ernst et al., 2010; Khan et al., 2013; Ntziachristos et al., 2012). This illustrates the biological complexity of H3K27 methylation and suggests that differential target gene silencing may be what ultimately defines whether EZH2 functions as an oncogene or a tumor suppressor.
In the case of tumors harboring gain-of-function mutations, EZH2 is an obvious therapeutic target. A number of small molecule inhibitors targeting EZH2 catalytic activity have demonstrated antiproliferative activity in human lymphoma cell lines and xenograft models (Campbell et al., 2015; Knutson et al., 2014a). Some of these compounds have entered early phase clinical trials for patients with relapsed or refractory B cell lymphomas or with advanced solid tumors (NCT02395601, NCT01897571, and NCT02082977 at clinicaltrials.gov). Additionally, similar to menin inhibitors and other compounds that disrupt the MLL complex, Orkin and colleagues engineered a stabilized alpha helix of EZH2 peptides that block H3K27 trimethylation by disrupting the interaction between EZH2 and EED (Kim et al., 2013). Treatment of MLL-AF9 AML cells with these peptides caused growth arrest and differentiation. Thus, disruption of the PRC2 complex represents another interesting potential therapeutic approach.
In addition to the focus on EZH2 as a target in mutant lymphomas, a unique susceptibility to EZH2 inhibition has been demonstrated in tumors harboring loss-of-function mutations in the SWI/SNF chromatin remodeling complex. SWI/SNF complexes play an important role in regulating gene expression via ATP-dependent mobilization of nucleosomes at promoters and enhancers, and recurrent mutations in subunits of these complexes have been identified in at least 20% of all human cancers (Kadoch et al., 2013; Shain and Pollack, 2013). Loss-of-function mutations of SNF5 (SMARCB1), a core member of the SWI/SNF complex, have been found in malignant rhabdoid tumors, and conditional loss of function leads to cancer in mice, thus confirming the role of SNF5 as a tumor suppressor (Roberts et al., 2000; Versteege et al., 1998). More recently SNF5 mutations have been identified in a number of other tumors including epithelioid sarcomas and melanomas (Stockman et al., 2015; Sullivan et al., 2013). Important insight into a potential therapeutic opportunity in this disease was gained when Roberts and colleagues demonstrated epigenetic antagonism between EZH2 and SNF5 at Polycomb targets in SNF5-deficient tumors (Wilson et al., 2010). Conditional inactivation of EZH2 prevented tumor formation driven by loss of SNF5, suggesting that SNF5 deficiency confers PRC2 dependence and thus potential sensitivity toEZH2 inhibition. Sensitivity to EZH2 inhibition was indeed observed in a xenograft mouse model of SMARCB1 mutant malignant rhabdoid tumors, providing further support for the potential use of EZH2 inhibitors in treating SNF5 mutant cancers (Knutson et al., 2013).
Two independent studies in different cancer models also highlight the possible utility of EZH2 inhibitors as sensitizers to standard cytotoxic chemotherapy. In one study, treatment of human lymphoma in a xenograft model with an EZH2 inhibitor plus standard CHOP chemotherapy demonstrated synergistic effects (Knutson et al., 2014b). In another report, Fillmore et al. observed a stark contrast in responsiveness of distinct genetic subsets of NSCLC tumor cells to the topoisomerase II inhibitor etoposide after EZH2 inhibition. While BRG1- and EGFR mutant NSCLC cells treated with EZH2 inhibitors exhibited an enhanced susceptibility to etoposide therapy, BRG1- and EGFR-wild-type tumors displayed resistance to etoposide by virtue of their ability to upregulate BRG1 to counter EZH2 inhibition (Fillmore et al., 2015). Given the potential for single-agent targeted therapies to fail due to resistance, adding epigenetic therapies to standard cytotoxic chemotherapy regimens that have proven clinical efficacy is a promising strategy. However, as demonstrated in the case of NSCLC, it will be important to understand the genetic and epigenetic determinants of enhanced sensitivity to combination regimens in order to help identify those patients that will benefit most from these therapies.
Lysine-Specific Demethylase 1
Lysine-specific demethylase 1 (LSD1) is a member of the FAD-dependent amine oxidase family of enzymes and can demethylate mono- and di-methylated lysines, specifically lysines 4 and 9 on histone H3 (H3K4me1/2 and H3K9me1/2, respectively) (Shi et al., 2004). When LSD1 is targeted to specific loci via repressive complexes, it demethylates H3K4me1/2 thus promoting gene silencing. In addition, LSD1 has since been identified in a number of activating complexes. When recruited by these complexes to target genes, LSD1 demethylates the repressive H3K9me2 mark leading to activation of gene expression (Metzger et al., 2005).
Recent studies have identified a functional link between LSD1 and leukemia maintenance (Harris et al., 2012). Pharmacologic inhibition of LSD1 induced differentiation of mouse AML cells and impaired the ability of these cells to cause leukemia in recipient mice. Simultaneously, another group found that LSD1 inhibition, when coupled with all-trans retinoic acid (ATRA) therapy, could induce differentiation and suppress leukemia engraftment in AML cell lines that otherwise were insensitive to ATRA (Schenk et al., 2012). These studies provided the initial preclinical rationale to develop LSD1 inhibitors, either as monotherapy or in combination with ATRA, as a potential approach for patients with AML. These and other preclinical data have prompted multiple phase I trials using either irreversible LSD1 inhibitors such as GSK2879552 as a single agent or tranylcypromine in combination with ATRA (NCT02177812 and NCT02261779 at https://clinicaltrials.gov/, respectively).
LSD1 is also overexpressed in solid tumors (Lv et al., 2012). A recent cell line screen demonstrated that the LSD1 inhibitor GSK2879552 has activity against small cell lung carcinomas (SCLC) (Mohammad et al., 2015). Furthermore, a DNA hypomethylation signature correlated with sensitivity to LSD1 inhibition in SCLC cells. This is a potentially important finding, because a reproducible biomarker that can predict response to LSD1 inhibitor therapy is currently lacking. Given that the mechanisms by which LSD1 inhibition slows cancer cell proliferation remain unclear, a genetic or epigenetic signature that correlates with response would be extremely useful to help inform ongoing and future clinical trials.
Jumonji D3
Jumonji D3 (JMJD3) and UTX are the only known members of the Jumonji family of deoxygenases that have H3K27 demethylase activity and have recently been investigated as therapeutic targets in T-ALL. The majority of T-ALL cases have activating NOTCH1 mutations, and roughly one quarter of T-ALL cases have loss-of-function mutations or deletions in members of PRC2, such as EZH2 and SUZ12, implicating a tumor suppressor role of PRC2 in T-ALL (Ntziachristos et al., 2012). Recent work from Aifantis and colleagues demonstrated that NOTCH1 activation antagonizes PRC2 by inducing loss of H3K27 trimethylation. If transcriptional repression mediated by the H3K27me3 mark has a tumor suppressive function, then inhibition of the enzymes that mediate removal of that chromatin modification, namely H3K27 demethylases, may confer therapeutic benefit.
This hypothesis was tested by Ntziachristos et al. (2014). Strikingly, UTX and JMJD3 had divergent roles in T-ALL pathogenesis. While UTX functions as a tumor suppressor and is inactivated in T-ALL, JMJD3 binds important NOTCH1 targets and is required for T-ALL initiation and maintenance. Treatment of T-ALL cell lines with the histone demethylase inhibitor GSKJ4 impaired proliferation and induced apoptosis. Although this small molecule compound targets both JMD3 and UTX, it appears more potent against JMJD3, which was consistent with the finding that expression profiles of T-ALL cell lines in the setting of JMJD3 knockdown and GSKJ4 treatment were similar to each other but not to UTX knockdown (Ntziachristos et al., 2014). These studies demonstrate that targeting JMJD3 may be a potential treatment for T-ALL by virtue of the functional antagonism between the leukemogenic NOTCH1 pathway and tumor suppressor role of PRC2, and more broadly provide support for the idea that loss-of-function mutations in histone methyltransferases might be targeted by inhibition of the corresponding demethylase.
Targeting Histone Acetylation
Acetyl-Lysine Readers
In recent years, major progress has been made in characterizing epigenetic “reader” domains that are important for recognizing posttranslational chromatin modifications and for recruiting downstream effector proteins to specific loci to activate lineage- and disease-specific gene expression programs. Included among these “readers” are bromodomains; these domains are highly conserved and recognize acetylated lysine residues. In humans, there are 61 bromodomains identified within 46 bromodomain-containing proteins (Filippakopoulos and Knapp, 2014). BRD4 is one of the best characterized among these members, and its multiple roles in regulating transcription intersect a number of distinct pathways ranging from transcriptional initiation and elongation to super-enhancer assembly. These functions have recently been reviewed by Basheer and Huntly (2015).
BRD4 was identified as a potential therapeutic target in an RNAi screen querying known chromatin regulators in an AML mouse model (Zuber et al., 2011). Treatment of AML cells with the competitive bromodomain inhibitor JQ1 resulted in potent antiproliferative effects and induced myeloid differentiation. A follow-up study by Vakoc and colleagues confirmed and extended the importance of the bromodomain in an elegant negative selection screen, this time using CRISPR-Cas9 genome editing as a means to unveil this BRD4 dependency, among others, in AML (Shi et al., 2015). This study not only validated the importance of BRD4 in AML but also precisely revealed that bromodomain 1, bromodomain 2, extra-terminal (ET) domain, and a C-terminal domain were the important determinants of BRD4 responsible for this phenotype. CRISPR-Cas9-based screens of this sort have the potential to greatly accelerate structure-function studies and provide significantly higher resolution of candidate drug targets to inform rational therapeutic design.
In addition to AML, bromodomain inhibition also has activity in a number of other malignancies, including multiple myeloma, lymphoma, and the NUT midline carcinomas (NMC), which represent a group of aggressive carcinomas characterized by the presence of NUT fusion oncogenes, the most common of which is BRD4-NUT (Delmore et al., 2011; Filippakopoulos et al., 2010; French, 2012; Mertzet al., 2011). A striking commonality among these diseases is their addiction to MYC, and the susceptibility of these malignancies to inhibitors like JQ1 can partially be attributed to their ability to suppress MYC transcription. In the case of AML, restoration of MYC expression was sufficient to rescue much of the phenotype induced by JQ1 or knockdown of BRD4 (Zuber et al., 2011).
A series of reports also showed how MYC expression is maintained in T-ALL via a BRD4-dependent manner despite inhibition of NOTCH1 (Herranz et al., 2014; Knoechel et al., 2014; Yashiro-Ohtani et al., 2014). Due to the frequency of activating NOTCH1 mutations in ALL, gamma-secretase inhibitors (GSIs) that block this pathway have been in various stages of clinical development, but their effectiveness has been hampered by resistance. These reports described MYC enhancer elements in T-ALL that were differentially occupied by NOTCH1 or BRD4. In the setting of GSI resistance, MYC expression was maintained by switching to the BRD4-occupied enhancer, thereby rendering these cells exquisitely sensitive to JQ1. Targeting of bromodomains has also been shown to overcome resistance to kinase inhibitor therapy in solid tumors. A recent report demonstrated that resistance to PI3K in a mouse model of breast cancer could be overcome with the addition of a BET inhibitor (Stratikopoulos et al., 2015). These studies demonstrate that epigenetic resistance pathways converge with genetic drivers of cancer to determine sensitivity to targeted therapies.
Despite the importance of MYC in these diseases, it is one of many genes affected by bromodomain inhibitors. Transcriptome profiling of AML cells treated with the BET inhibitor I-BET151 showed diminished expression of CDK6, CCND2, and BCL2 in addition to MYC (Dawson et al., 2011). In multiple myeloma, BRD4 was found to colocalize with the Mediator complex at thousands of active enhancers. Among these enhancers was a subset of super-enhancers characterized by broad regions of K27 acetylation and exceptionally high BRD4 and Mediator occupancy that influences transcription of major oncogenic drivers including MYC (Lovén et al., 2013).
Similar epigenomic efforts aimed at understanding where BRD4 exerts its effects were extended to AML. Roe et al. demonstrated via ChIP-seq analysis of AML cell lines that BRD4 occupancy coincided with binding of the major hematopoietic transcription factors PU.1, FLI1, ERG, C/EBPα, C/EBPβ, and MYB (Roe et al., 2015). Recruitment of the acetyl-transferases p300 and CBP to target loci by these transcription factors is the proximal event that creates a milieu of lysine acetylation that ultimately results in BRD4 occupancy at promoter and enhancer regions. Notably, bromodomain inhibition caused a reduction in the functional output of these transcription factors, suggesting that the antileukemic potency of bromodomain inhibitors is at least partially explained by their ability to perturb hematopoietic-specific transcription factor networks that cooperate to maintain an AML-specific transcriptional program.
JQ1, one of the early prototypical bromodomain inhibitors, acts by potently and selectively displacing acetylated lysines from bromodomains (Filippakopoulos et al., 2010). Its experimental utility has been highlighted in the aforementioned studies. Multiple bromodomain inhibitors have also been developed with enhanced pharmacokinetic properties, including CPI-0610, OTX015, TEN-010, and I-BET762 (GSK525762A) (Filippakopoulos and Knapp, 2014). These compounds are under investigation in early phase clinical trials for lymphoma, leukemia, MDS, and solid tumors including NMC (NCT02308761, NCT01587703, NCT02303782, NCT02158858, and NCT02157636 at https://clinicaltrials.gov/; see also Table 1).
The notion of “druggability” implies that the feasibility of targeting disease modifiers across the proteome lies on a spectrum. Drug development strategies are adept at targeting proteins with enzymatic activity but less effective at developing potent small molecules that block the functions of proteins such as transcription factors or scaffold molecules that lack intrinsic catalytic activity. Also, early studies suggest that effective targeting of chromatin regulatory processes may, in many cases, require near complete inhibition of protein function for extended periods of time. This could complicate drug development efforts. Recently, Bradner and colleagues developed a promising novel strategy that may overcome some of these issues via degradation of proteins like BRD4 by redirecting E3 ubiquitin ligase activity to these targets (Winter et al., 2015). Ubiquitination of BRD4 resulted in its rapid proteasomal degradation and treatment of mice with this compound delayed leukemia progression. Strategies to coopt this degradation pathway can likewise be extended to other proteins that have been difficult to target and represents a novel approach to target complexes that modify chromatin.
Additional acetyl-lysine readers beyond bromodomains are on the horizon as promising drug targets. This includes the AF9 YEATS domain, which recently was found to bind acetylated H3K9 and, to a lesser extent, acetylated H3K27 and H3K18. Recognition of these modifications leads to AF9 recruitment of DOT1L, which then mediates transcriptional regulation at target loci (Li et al., 2014b). The role of YEATS domain function in disease states awaits further characterization, but these findings raise the possibility that disrupting this reader activity may also have therapeutic implications.
Histone Acetyltransferases
While there has been much recent focus on drug development to target acetyl-lysine readers, there has been less activity to target acetyltransferases. Most work that has focused on histone acetyltransferases (HATs) has been directed at p300/CREB binding protein (CBP). P300/CBP are transcriptional coactivators that have HAT activity whose substrates include histone and non-histone proteins. H3K27 acetylation by p300/CBP marks active enhancers and promoters. Inactivating mutations in p300/CBP have been identified in lymphoma and ALL (Pasqualucci et al., 2011). In contrast, genetic inactivation or knockdown of p300 and CBP results in impaired AML initiation and maintenance, and HAT inhibitors exhibit antileukemic activity across a variety of AML subtypes (Giotopoulos et al., 2015). It seems that these are a group of enzymes that warrant further investigation as potential therapeutic targets.
Perspectives
In the early era of cytotoxic chemotherapy, chemoresistance in the setting of single-agent therapies prohibited patients from achieving durable remissions in multiple cancer subtypes. Following the example of antituberculosis therapy, in which antibiotics were administered in combination in order to block resistance mechanisms, most chemotherapy regimens given with curative intent are now administered in combinations. The postimatinib era of targeted therapy has seen similar parallels with respect to acquired resistance. Moreover, epigenetic mechanisms have recurrently been implicated in the generation of “persister” or “drug-tolerant” cells (Knoechel et al., 2014; Sharma et al., 2010), raising the interesting hypothesis that targeted epigenetic therapies have the capacity to eliminate these cells as a means to prevent acquired drug resistance. Clearly, there is a need for a deeper mechanistic understanding of disease pathogenesis in order to inform rational combinations of these targeted therapies.
As highlighted in this review, there are multiple instances in which epigenetic dysregulation is either the primary driver of disease (e.g., MLL-rearranged leukemias) or a unique dependency that is likely not to be a direct result of oncogene activation (e.g., NSCLC). In either case, preclinical studies have shown that combination epigenetic therapies appear to be effective at targeting unique vulnerabilities to achieve synergies. The utility of either shRNA- or CRISPR-Cas9-based genetic screens as a strategy to reveal these vulnerabilities has been proven in a variety of experimental settings and such approaches will continue to provide insight.
Additionally, the spatial organization of chromatin into higher order structures is clearly functionally relevant and may be important for cancer cell proliferation and survival. Portions of the genome are compartmentalized into megabase-scale units termed topologically associated domains (TADs) (Dixon et al., 2012). This organization, including subdomains within TADS, allows for selective enhancer-promoter interactions to occur within discrete regions of the genome defined by CTCF boundaries. The technology used to resolve this chromatin organization is relatively new, but it is already becoming clear that structural variants in the form of deletions, inversions, or duplications can disrupt TADs to cause diseases such as limb malformation (Lupiáñez et al., 2015). It Is likely only a matter of time before we appreciate how disruption of this domain structure is playing a role in cancer. Future studies geared toward characterizing these higher order chromatin structures in normal and disease states will be of significant interest and will likely uncover new approaches to modulate cancer associated gene expression and hopefully more therapeutic opportunities.
Epigenetic dysregulation is a common feature of many diseases, and therapeutic targeting of these regulatory pathways is beginning to be assessed in patients. However, there is still much work to be done. There are a number of likely obstacles that will influence the efficacy of these therapies against cancer, including cellular heterogeneity, resistance, and the need for deep and consistent inhibition of target function. Furthermore, we are still in the early stages of drug development for most targets that regulate chromatin and the development of new treatment strategies will require deeper understanding of pathogenic mechanisms. The complexity of effectively drugging the epigenome is significant, but there is tremendous excitement that continued focus on the biology of these processes, new small molecule development, and informed clinical trials will lead to a new class of potent anticancer agents.
Acknowledgments
S.A.A. is a consultant for Epizyme, Inc. and Imago Biosciences.
References
- Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest. 2014;124:56–63. doi: 10.1172/JCI69736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- Basheer F, Huntly BJ. BET bromodomain inhibitors in leukemia. Exp Hematol. 2015;43:718–731. doi: 10.1016/j.exphem.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 2015;27:589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med Chem Lett. 2015;6:491–495. doi: 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, Doench JG, Xu H, Chu SH, Qi J, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med. 2015;21:335–343. doi: 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33:563–570. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, Fazio M, Chen CW, Zhu N, Koche R, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. 2014;26:896–908. doi: 10.1016/j.ccell.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK, Kim CF. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520:239–242. doi: 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–2743. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, Iyer SP, Bearss D, Bhalla KN. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28:2155–2164. doi: 10.1038/leu.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA. Pathogenesis of NUT midline carcinoma. Annu Rev Pathol. 2012;7:247–265. doi: 10.1146/annurev-pathol-011811-132438. [DOI] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galm O, Herman JG, Baylin SB. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev. 2006;20:1–13. doi: 10.1016/j.blre.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Giotopoulos G, Chan WI, Horton SJ, Ruau D, Gallipoli P, Fowler A, Crawley C, Papaemmanuil E, Campbell PJ, Göttgens B, et al. The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene. 2015 doi: 10.1038/onc.2015.92. http://dx.doi.org/10.1038/onc.2015.92. [DOI] [PMC free article] [PubMed]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Grembecka J, Belcher AM, Hartley T, Cierpicki T. Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. J Biol Chem. 2010;285:40690–40698. doi: 10.1074/jbc.M110.172783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, Croce CM, Nakamura T, Canaani E, Young RA. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22:3403–3408. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navás D, Cordon-Cardo C, et al. A NOTCH1-driven MYC enhancer promotes T cell development transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117:4759–4768. doi: 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SN, Jankowska AM, Mahfouz R, Dunbar AJ, Sugimoto Y, Hosono N, Hu Z, Cheriyath V, Vatolin S, Przychodzen B, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013;27:1301–1309. doi: 10.1038/leu.2013.80. [DOI] [PubMed] [Google Scholar]
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–650. doi: 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–370. doi: 10.1038/ng.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014a;13:842–854. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, et al. Synergistic anti-tumor activity of EZH2 inhibitors and glucocorticoid receptor agonists in models of germinal center non-Hodgkin lymphomas. PLoS ONE. 2014b;9:e111840. doi: 10.1371/journal.pone.0111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntimaddi A, Achille NJ, Thorpe J, Lokken AA, Singh R, Hemenway CS, Adli M, Zeleznik-Le NJ, Bushweller JH. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 Di- and tri-methylation on target genes and transformation potential. Cell Rep. 2015;11:808–820. doi: 10.1016/j.celrep.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014a;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell. 2014b;159:558–571. doi: 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE. 2012;7:e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Khan AP, Asangani IA, Cieslik M, Prensner JR, Wang X, Iyer MK, Jiang X, Borkin D, Escara-Wilke J, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015;21:344–352. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ., 3rd Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Mu¨ller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schu¨le R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Meyer C, Hofmann J, Burmeister T, Gröger D, Park TS, Emerenciano M, Pombo de Oliveira M, Renneville A, Villarese P, Macintyre E, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27:2165–2176. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28:57–69. doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Mohan M, Lin C, Guest E, Shilatifard A. Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer. 2010;10:721–728. doi: 10.1038/nrc2915. [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, Xie H, Orkin SH, Armstrong SA. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci USA. 2012;109:5028–5033. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117:6912–6922. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18:298–301. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014;514:513–517. doi: 10.1038/nature13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Zhou X, Rifkind RA, Marks PA. Histone deacetylase inhibitors: development of suberoylanilide hydroxamic acid (SAHA) for the treatment of cancers. Blood Cells Mol Dis. 2001;27:260–264. doi: 10.1006/bcmd.2000.0376. [DOI] [PubMed] [Google Scholar]
- Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA. 2000;97:13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JS, Mercan F, Rivera K, Pappin DJ, Vakoc CR. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol Cell. 2015;58:1028–1039. doi: 10.1016/j.molcel.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria SM, Christopher MJ, Klco JM, Ley TJ. Primary acute myeloid leukemia cells with IDH1 or IDH2 mutations respond to a DOT1L inhibitor in vitro. Leukemia. 2014;28:2403–2406. doi: 10.1038/leu.2014.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J, Cierpicki T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EM, Garcia-Manero G, Rizzieri DA, Savona M, Tibes R, Altman JK, Jongen-Lavrencic M, Döhner H, Armstrong S, Pollock RM, et al. The DOT1L Inhibitor EPZ-5676: Safety and Activity in Relapsed/Refractory Patients with MLL-Rearranged Leukemia; 56th American Society of Hematology Annual Meeting and Exposition; San Francisco, CA. 2014. [Google Scholar]
- Stockman DL, Curry JL, Torres-Cabala CA, Watson IR, Siroy AE, Bassett RL, Zou L, Patel KP, Luthra R, Davies MA, et al. Use of clinical next-generation sequencing to identify melanomas harboring SMARCB1 mutations. J Cutan Pathol. 2015;42:308–317. doi: 10.1111/cup.12481. [DOI] [PubMed] [Google Scholar]
- Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, Parsons R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell. 2015;27:837–851. doi: 10.1016/j.ccell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Modern Pathology. 2013;26:385–392. doi: 10.1038/modpathol.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Roberts JM, Qi J, Bradner JE. Inhibitors of emerging epigenetic targets for cancer therapy: a patent review (2010–2014) Pharm Pat Anal. 2015;4:261–284. doi: 10.4155/ppa.15.16. [DOI] [PubMed] [Google Scholar]
- Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, Hua X. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Wagener N, Holland D, Bulkescher J, Crnkovic-Mertens I, Hoppe-Seyler K, Zentgraf H, Pritsch M, Buse S, Pfitzenmaier J, Haferkamp A, et al. The enhancer of zeste homolog 2 gene contributes to cell proliferation and apoptosis resistance in renal cell carcinoma cells. Int J Cancer. 2008;123:1545–1550. doi: 10.1002/ijc.23683. [DOI] [PubMed] [Google Scholar]
- Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, Knoechel B, Lanauze C, Louis L, Forsyth KS, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci USA. 2014;111:E4946–E4953. doi: 10.1073/pnas.1407079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyer-son M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Strauss AC, Chu S, Li M, Ho Y, Shiang KD, Snyder DS, Huettner CS, Shultz L, Holyoake T, Bhatia R. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17:427–442. doi: 10.1016/j.ccr.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Liu L, Huang J, Bernard D, Karatas H, Navarro A, Lei M, Wang S. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J Med Chem. 2013;56:1113–1123. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]