

Abstract
Introduction
This study determined the pharmacokinetics and pharmacodynamics of (R)- and (S)-ketamine and (R)- and (S)-norketamine following a 5-day moderate dose, as a continuous (R,S)-ketamine infusion in complex regional pain syndrome (CRPS) patients.
Materials and methods
Ketamine was titrated to 10–40 mg/h and maintained for 5 days. (R)-and (S)-Ketamine and (R)- and (S)-norketamine pharmacokinetic and pharmacodynamic studies were performed. Blood samples were obtained on Day 1 preinfusion, and at 60–90, 120–150, 180–210, and 240–300 min after the start of the infusion, on Days 2, 3, 4, 5, and on Day 5 at 60 min after the end of infusion. The plasma concentrations of (R)- and (S)-ketamine and (R)- and (S)-norketamine were determined using enantioselective liquid chromatography-mass spectrometry.
Results
Ketamine and norketamine levels stabilized 5 h after the start of the infusion. (R)-Ketamine clearance was significantly lower resulting in higher steady-state plasma concentrations than (S)-ketamine. The first-order elimination for (S)-norketamine was significantly greater than that of (R)-enantiomer. When comparing the pharmacokinetic parameters of the patients who responded to ketamine treatment with those who did not, no differences were observed in ketamine clearance and the first-order elimination of norketamine.
Conclusion
The results indicate that (R)- and (S)-ketamine and (R)- and (S)-norketamine plasma concentrations do not explain the antinociceptive activity of the drug in patients suffering from CRPS.
Keywords: ketamine, norketamine, CRPS, pharmacokinetics, pharmacodynamics, enantioselective, liquid chromatography, mass spectrometry
INTRODUCTION
Complex regional pain syndrome (CRPS) is associated with pain that is out of proportion to the inciting injury, which is neuropathic in nature and is regional in distribution.1 A significant percentage of CRPS patients who do not respond to conventional treatment have disease recurrence2 along with the spread of illness from the area of original injury.3 A critical factor in the initiation of central sensitization is the release of the magnesium block of the N-methyl-D-aspartic acid (NMDA) receptor that results in calcium influx and the initiation of intracellular enzymatic cascades, thus increasing the excitability of pain transmission neurons.4,5 This observation led to the use of the NMDA antagonist, ketamine to block the receptor in neuropathic pain states. Indeed, recent studies have demonstrated that an extended infusion of subanesthetic doses of ketamine produced significant levels of pain relief in CRPS patients.6,7
Ketamine is a chiral compound that is administered in USA as a racemic (50:50) mixture of its enantiomers, (R)-and (S)-ketamine. It has been previously demonstrated that (R)- and (S)-ketamine have significantly different pharmacodynamic activities as (S)-ketamine is the more potent analgesic agent, while the posthypnotic stimulatory properties and agitated behavior associated with ketamine have been attributed to (R)-ketamine.8,9 In addition, ketamine is extensively metabolized by N-demethylation producing norketamine, which is also a NMDA receptor antagonist, that may also exhibit enantioselective pharmacological activity, e.g. (S)-norketamine has an eightfold higher affinity than (R)-norketamine in a rat cortical wedge preparation.10
The use of (R,S)-ketamine in clinical anesthesia and analgesia has resulted in a number of studies regarding the enantioselective pharmacokinetics and pharmacodynamics of ketamine in humans.11–13 The previous studies determined the fate of ketamine after a single dose of the racemate administered as a single i.v. dose to surgical patients11, a short (<20 min) continuous infusion to healthy volunteers12,13 and as oral, sublingual, suppository, and nasal formulations.13 The pharmacokinetics and pharamcodynamics of (S)-ketamine in volunteers have also been studied after the short infusion of the single enantiomer.12,14 However, to date, no studies have reported the pharmacokinetic and pharmacodynamic parameters of (R)- and (S)-ketamine and (R)- and (S)-norketamine following an extended infusion. Thus, the primary objective of this study was to 2develop a pharmacokinetic model that reflects the plasma profiles of (R)- and (S)-ketamine and (R)-and (S)-norketamine in patients administered a moderate dose of (R,S)-ketamine as an inpatient 5-day continuous infusion. During the study, the clinical effect of the treatment was also assessed. A secondary objective was to construct preliminary pharmacodynamic models of ketamine and norketamine as a guide to the development of future clinical studies.
METHODS
Subjects and Treatment Protocol
After approval from the Cooper University Institutional Review Board, 16 patients with a primary diagnosis of CRPS gave written informed consent to participate in this prospective study. Exclusion criteria included allergies to ketamine, clonidine, midazolam, or known contraindications to ketamine use which included severe arterial hypertension, hyperthyroidism, ischemic heart disease, or heart failure. Patients who had a history of substance or drug abuse or suspected somatoform pain disorder were excluded. Patients were admitted to a monitored telemetry unit and maintained on their current pain medications during the infusion period. Ketamine was mixed in a 500 ml bag of normal saline and started at an infusion rate of 10 mg/h and titrated to a maximum of 40 mg/h to achieve comfort without evidence of significant side effects or oxygen desaturation (<92%). By the end of Day 1, the infusion was stabilized to 30.4 ± 4.8 mg/h and maintained for 5 days with 24 h monitoring of the subject During the titration period, an advanced practice nurse and a research assistant collected the study data and blood samples.
Pharmacokinetic and Pharmacodynamic Studies
Blood sampling
Blood samples (7 ml) were obtained on Day 1 before starting the infusion, and at 60–90, 120–150, 180–210, and 240–300 min after the initiation of the infusion, on Days 2, 3, 4, 5 (morning collection), and on Day 5 at 60 min after the conclusion of the infusion. The samples were centrifuged and the plasma was collected and frozen at −80°C until analysis.
Plasma analysis
The plasma concentrations of (R)- and (S)-ketamine and (R)- and (S)-norketamine were determined using a previously reported enantioselective liquid chromatography-mass spectrometry method.11 In brief, the target analytes were extracted from the plasma using solid-phase extraction and analyzed using a liquid chromatographic stationary phase containing immobilized α1-acid glycoprotein (Chiral-AGP). The mobile phase consisted of 2-propranol-ammonium acetate buffer [10 mM, pH 7.6] (6:94, v/v) delivered at 0.5 ml/min at 25°C. Single ion monitoring was used to quantify the target compounds at m/z 238.1 (ketamine) and m/z 224.1 (norketamine).
Pharmacokinetics
All ketamine and norketamine concentration-time data were modeled simultaneously using a nonlinear mixed effects approach. The rate of change of each ketamine enantiomer plasma concentrations was described using a single-compartment model:
| (1) |
where A represents the amount of ketamine in the central compartment, K0 is the zero-order infusion rate, and kel is the first-order elimination rate constant The initial condition to eq. 1 is zero, and plasma ketamine concentrations, Cp(R,S), were defined as: Cp(R,S) = A(R,S)/V(R,S), where V is the apparent volume of distribution. Individual enantiomer infusions were set equal to 50% of the total infusion to account for the racemic mixture. The rate of change of norketamine concentrations, Cm(R,S), was also defined by a single compartment:
| (2) |
where kf and km are first-order rate constants of metabolite formation and elimination, and the initial condition is zero.
Pharmacokinetic parameters were estimated using the maximum likelihood expectation maximization algorithm as implemented in ADAPT (v5beta, Biomedical Simulations Resource, University of Southern California, Los Angeles, CA). Parameters were assumed to be log-normally distributed within the population and the error variance model was defined as:
| (3) |
where Vari is the variance of the ith data point, σ is a variance parameter, and Y is the ith model predicted value. Selection criteria during the model development process were based on the goodness-of-fit plots, stability of log-likelihood values, and distribution of residuals. Final estimated parameters were reported as population mean estimates and inter-individual variability (IIV, CV%). Differences between selected enantiomer pharmacokinetic parameters within individuals and between groups (i.e., responders and nonresponders) were tested using paired or two-sample independent t-tests. Statistical comparisons were conducted using Minitab (v15), and P-values less than or equal to 0.05 were considered significant.
Pharmacodynamics
Individual pain scores (PSs) were used to calculate the probability of achieving a PS of less than or equal to 3 as a function of time. The time course of this probability indicated a slow progressive increase with time despite drug and metabolite concentrations reaching steady-state within 24 h (Results section). Although a biophase distribution delay has been used to characterize such hysteresis in modeling the probability of specific PSs,12 a relatively long delay in drug distribution equilibration did not seem physiologically plausible. Therefore, the probability of a PS less than or equal to 3, pr(s ≤ 3), was assumed to be modified by a latent variable (m) which was described by an indirect response model13 driven by mean S-ketamine concentrations:
| (4) |
where kin is a zero-order production rate constant, EC50 is the S-enantiomer drug concentration producing 50% of inhibition, and kout is a first-order removal rate constant Mean estimated pharmacokinetic parameters and infusion rates were used in eq. 1 to fix the driving function for eq. 4. The initial or baseline condition of eq. 4 was fixed to 1, and thus may be simplified to:
| (5) |
The response variable was defined as:
| (6) |
where β is the initial or baseline probability. This model assumes that drug can completely inhibit production, and m → 0 and pr(s ≤ 3) → 1 when Cp(S) is much greater than the EC50. The EC50 and kout parameters were estimated using maximum likelihood nonlinear regression in ADAPT, and the variance model was defined according to eq. 3.
Blood level data measured across time were analyzed using ANOVA with repeated measures. Post hoc analysis was used to compare differences between means after confirming significant main effects, and a P < 0.05 was considered statistically significant Demographic data are presented as means ± standard deviation whereas drug blood level data as means ± standard error of the mean (SEM). The analyses were performed using Systat Software version 11.00.01 (Systat Software, Chicago, IL).
RESULTS
Patient Characteristics
The subjects in this study included 15 females and one male whose age ranged from 17 to 47 yr (mean 33 ± 10.2 yr), height from 156.2 to 182.9 cm (mean 166.1 ± 7.7 cm), and weight from 41.5 to 118.2 kg (mean 67.9 ± 19.3 kg). The 5-day moderate dose ketamine therapy produced variable analgesic effects in these CRPS patients. Ten out of 16 (10/16) subjects had significant reduction (≥30%) in pain levels compared with baseline preinfusion PSs. Six out of 16 (6/16) subjects reported no significant pain reduction (≤15%; Table 1).
TABLE 1.
The data represent the baseline (column 2) and end of study pain scores (column 3) for all 16 CRPS study subjects receiving (R,S)-ketamine as a continuous infusion for 5 days
| Subject number | Pain level predosing (Day 1) | Pain level, Day 5 | ΔPS |
|---|---|---|---|
| 1 | 4.0 | 1.0 | 3.0 |
| 2 | 8.5 | 7.5 | 0.5 |
| 3 | 8.0 | 0.0 | 8.0 |
| 4 | 8.0 | 8.0 | 0.0 |
| 5 | 10.0 | 5.0 | 5.0 |
| 6 | 8.0 | 7.0 | 1.0 |
| 7 | 8.0 | 10.0 | −2.0 |
| 8 | 7.0 | 4.0 | 3.0 |
| 9 | 7.0 | 2.0 | 5.0 |
| 10 | 10.0 | 6.0 | 4.0 |
| 11 | 10.0 | 10.0 | 0.0 |
| 12 | 10.0 | 10.0 | 0.0 |
| 13 | 10.0 | 2.0 | 8.0 |
| 14 | 10.0 | 4.0 | 6.0 |
| 15 | 10.0 | 7.0 | 3.0 |
| 16 | 8.0 | 3.0 | 5.0 |
|
| |||
| Means | 8.5 | 5.4 | 3.1 |
| SD | 0.4 | 0.8 | 3.0 |
The ΔPS (column 4) is the difference in the reported pain score (PS) from Day 1 to Day 5 calculated as PS(Dayl) – PS(Day 5).
Pharmacokinetics
Serial blood samples were obtained from all of the subjects in the study and complete profiles were obtained for 13/16 subjects. The samples were analyzed using the validated assay and (R)-ketamine, (S)-ketamine, and their respective N-demethylated metabolites, i.e. norketamine, were detected and quantified in all of the samples obtained after the initiation of the study. The average (R)- and (S)-ketamine plasma concentrations peaked at 240–300 min after the start of the infusion and were significantly (P < 0.05) increased from baseline at all time points (Fig. 1). A similar trend was noted for the average (R)- and (S)-norketamine plasma concentrations, although the average peak levels were not obtained until Day 2 of the infusion (Fig. 1). It is interesting to note that the plasma concentrations of ketamine remained greater than those of norketamine throughout the sampling period. This is different than the profiles observed with single i.v. administrations in which norketamine levels rapidly surpass those of ketamine11,13 but is consistent with the data from the recent study of (S)-ketamine, in which the levels of (S)-ketamine remained higher than (S)-norketamine until the end of the 2-h infusion.14
Fig. 1.
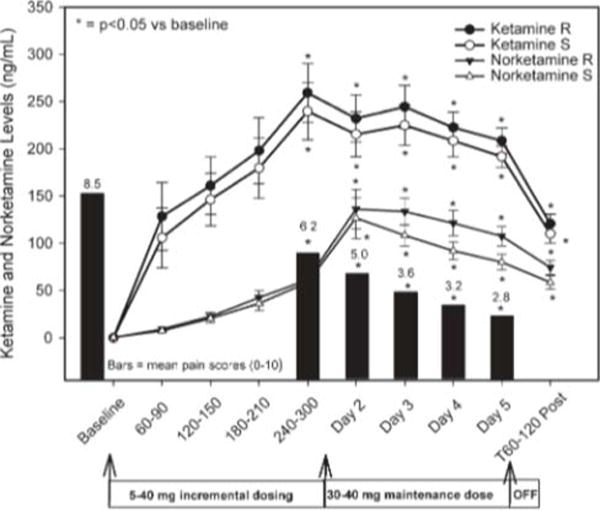
The mean ± SEM ketamine levels for the R (closed circles) and S enantiomers (open circles). Norketamine levels are similarly illustrated with closed and open triangles. The X axis shows the baseline and infusion period with the corresponding ketamine and norketamine blood levels during the 4–5 h incremental dosing period. The subjects’ pain scores are illustrated along the X axis to provide a reference to the achieved level of analgesia Blood levels for both ketamine and norketamine were significantly (P < 0.05) increased from baseline at all time points with a corresponding decrease in pain scores.
The pharmacokinetic profiles of (R)- and (S)-ketamine and (R)- and (S)-norketamine enantiomers were reasonably described using simple linear one-compartment models (eqs. 1 and 2). Representative concentration-time profiles for two of the study subjects are shown in Figure 2, and diagnostic plots of observed versus model predicted values are shown in Figure 3. Although there was a slight bias at high ketamine concentrations in some select subjects, there was generally good agreement between ketamine and norketamine fitted profiles. Final estimated pharmacokinetic model parameters are listed in Table 2.
Fig. 2.
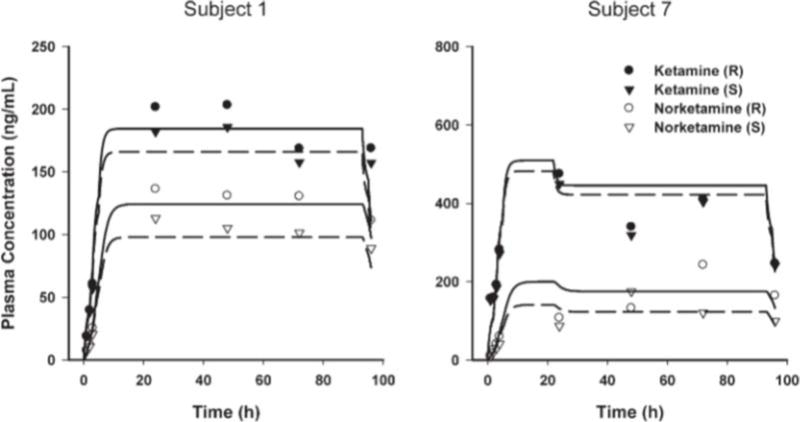
Time course of (R,S)-ketamine and (R,S)-norketamine plasma concentrations in two representative subjects. Symbols show measured concentrations. Lines are model-predicted profiles using individual pharmacokinetic parameters, where solid and dashed lines represent the R- and S-enantiomers, respectively.
Fig. 3.
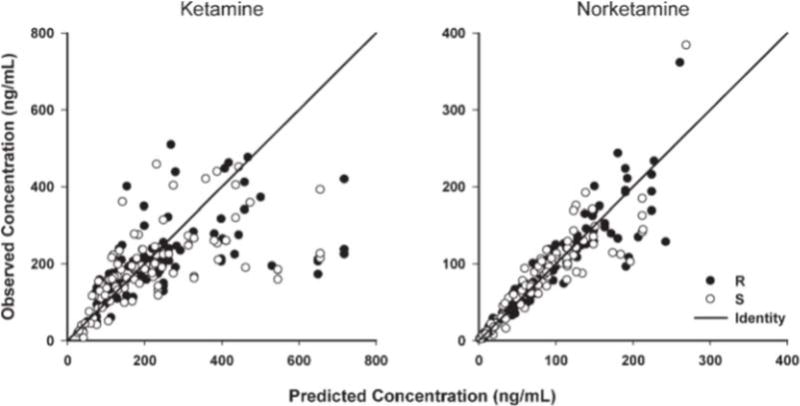
Diagnostic plots for me final population pharmacokinetic model showing the agreement between observed and individual predicted concentrations.
TABLE 2.
Population parameter estimates of ketamine and norketamine pharmacokinetics
| Parameter | Definition | Estimate | IIVa |
|---|---|---|---|
| Structural | |||
| kel(R) (h−1) | Elimination rate constant | 0.996 | 16 |
| kel(S) (h−1) | Elimination rate constant | 0.989 | 22 |
| V(R) (L) | Apparent volume of distribution | 57.7 | 54 |
| V(S) (L) | Apparent volume of distribution | 63.5 | 55 |
| kf(R) (h−1) | Formation rate constant | 0.273 | 42 |
| kf(S) (h−1) | Formation rate constant | 0.283 | 59 |
| km(R) (h−1) | Metabolite elimination rate constant | 0.669 | 40 |
| km(S) (h−1) | Metabolite elimination rate constant | 0.819 | 19 |
| CL(R)b (L h−1) | Apparent clearance | 57.4 | 32 |
| CL(S)b (L h−1) | Apparent clearance | 62.8 | 38 |
| Variance | |||
| σ | Ketamine variance parameter | 0.46 | –c |
| σ | Norketamine variance parameter | 0.247 | –c |
Interindividual variability expressed as percent coefficient of variation.
Secondary calculated parameter.
Not applicable.
The pharmacokinetic parameters for (R)- and (S)-ketamine were similar and in good agreement with literature reported estimates.15–17 However, the clearance for R-ketamine was significantly lower (P < 0.01) resulting in higher steady-state plasma concentrations when compared with S-ketamine (Figs. 1 and 2). The difference in the clearance between (R)-and (S)-ketamine is consistent with the data from previous studies of (R,S)-ketamine after a single i.v. administration to surgical patients15 and a short (<20 min) continuous infusion to healthy volunteers.15,16 In these studies, statistically significant differences were observed in the clearance, volume of distribution and area under the curve (AUC) of (S)-ketamine and (R)-ketamine. However, a recent study by Yanagihara, et al.17 found no significant differences in clearance between (S)-ketamine and (R)-ketamine in healthy Japanese volunteers after i.v. administration of R-ketamine. The authors of the latter paper suggest that the observed differences between their data and previous studies were due to pharmacogenetic differences between the Japanese population used in their study and the European population used in the earlier studies. This assumption was based on the observation that the microsomal enzyme CYP2B6 plays a key role in the N-demethylation of ketamine18 and that this enzyme shows large interpopulation differences as CYP2B6 was undetectable in 70% of Japanese and in 15% of Caucasians.19
The estimated first-order elimination rate constant for (S)-norketamine was significantly greater (P < 0.05) than that of the (R)-enantiomer (Table 2), and (S)-norketamine steady-state concentrations were routinely lower than for (R)-norketamine (Figs. 1 and 2). The previous study by Yanagihara, et al.17 found no significant difference in the pharamcokinetic parameters of (R)- and (S)-norketamine, but the Cmax and AUC0→8 values for (R)-norketamine were greater than those of (S)-norketamine. Although the pharmacokinetic parameters of (S)-norketamine and (R)-norketamine were not determined in surgical patients after the administration of (R,S)-ketamine, the mean plasma concentrations of (R)-norketamine were greater than (S)-norketamine up to 4 h post-administration.16
Pharmacodynamics
The limited number of study subjects precludes an analysis of individual pharmacodynamic drug response. As an alternative, the relative number of subjects with a PS of less than or equal to 3 was used to calculate the probability of achieving this score level as a function of time. In this study, ketamine and norketamine plasma concentrations achieved steady-state relatively quickly (Figs. 1 and 2); however, the perceived pain relief exhibited a progressive increase over the entire duration of the study. When comparing the pharmacokinetic parameters of the patients who responded to ketamine treatment to those who did not respond, no statistically significant differences were observed in the apparent ketamine clearance and the first-order elimination rates of norketamine, P = 0.64 and 0.56, respectively. These results suggest that ketamine and norketamine exposure alone will not likely explain differences in analgesic response.
The potential lack of a relationship between ketamine and norketamine plasma levels and reduction in the reported visual analogue scale was also reflected in an indirect response model (eq. 5) linked to the probability function (eq. 6), which satisfactorily captured the time course of the drug response (Fig. 4); however, model parameters were highly correlated and estimated with poor precision (EC50 = 3.07 ng/ml, >100% CV%; kout = 0.00683 h−1, 59% CV%). One possible explanation of these results is that drug steady-state concentrations may be high relative to the actual potency (EC50), which essentially results in complete inhibition of the production of the latent variable (m). Therefore, the latent variable would decrease monoexponentially over the duration of the study, and the decrease in m, and subsequent rise in the response variable, is governed by the kout turnover parameter. The fact that the study duration was not long enough to achieve a pharmacodynamic equilibrium and the lack of washout data, may also have contributed to the correlation in pharmacodynamic parameters and problems with their identifiability. Another possibility is that downstream metabolites of ketamine and norketamine also contribute to the observed therapeutic response and are not modeled by this approach.
Fig. 4.
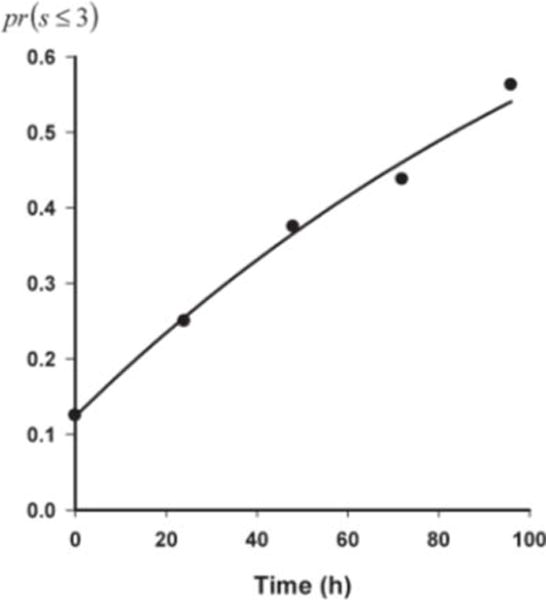
Temporal progression of the probability of achieving a pain score less than or equal to three over the duration of me study. Symbols represent calculated probability, and the line represents the model-fitted profile according to eq. 6.
DISCUSSION
This is the first study reporting the pharmacokinetic and pharmacodynamic parameters of (R)- and (S)-ketamine and (R)- and (S)-norketamine following an extended infusion over several days. The data from this study are consistent with the results of previous studies involving single administrations of the racemic drug15,16 in which (S)-ketamine had a significantly higher clearance rate and volume of distribution than (R)-ketamine and the average plasma concentrations of (R)-ketamine were consistently higher than those of (S)-ketamine (Table 2, Figs. 1 and 2). The same relative enantioselectivity was observed for norketamine as the estimated first-order elimination rate constant for (S)-norketamine was greater than that of the (R)-enantiomer (Table 2) and (S)-norketamine steady-state concentrations were routinely lower than for (R)-norketamine (Figs. 1 and 2).
At the present time, ketamine and norketamine are considered to be the active agents responsible for the antinociceptive response produced by the administration of (R,S)-ketamine, with the activity primarily residing in the (S)-enantiomers of these compounds. This assumption is based on the observations that (S)-ketamine is a more potent analgesic agent than (R)-ketamine,8 that (S)-norketamine has an eightfold higher affinity than (R)-norketamine in a rat cortical wedge preparation10 and the recent data from a study using a rat model of peripheral neuropathy which demonstrated that the antinociceptive properties of norketamine are due to (S)-norketamine.20
However, although this study involved a limited number of patients, the pharmacokinetic data and pharmacodynamic analysis suggest that the systemic exposure to ketamine and norketamine may not be responsible for all of this drug’s antinociceptive properties. The results suggest that the varied responses to treatment observed in this study, and in the study of Rabben et al.,21 may not reflect different mechanisms of pain but differences in the ability to metabolize ketamine, i.e. pharmacogenetic differences, as was observed between European and Japanese subjects17. Thus, downstream metabolites of ketamine and norketamine may play a role in its therapeutic efficacy.
Ketamine is extensively metabolized by microsomal enzymes in humans and rats producing a variety of metabolites. The primary metabolite is norketamine, which is produced by the N-demethylation of ketamine,22–25 that is primarily mediated by CYP3A4 with CYP2B6 and CYP2C9 also contributing to this transformation.18,25 Norketamine, and to a minor extent ketamine, are further transformed by ring hydroxylation into a variety of hydroxylated-metabolites and further metabolized to dehydronorketamine.22,26 Norketamine and dehydronorketamine have been reported as the primary circulating metabolites27,28 with the 24-h cumulative urinary excretion of norketamine representing 1.6% of the administered dose, dehydronorketamine 16.1% of the dose, and the remaining drug is excreted unchanged in the urine or feces or as glucuronidates of the hyroxylated metabolites.29 The enzymes responsible for the hydroxylation and dehydration of ketamine and norketamine have not been identified nor has the in vivo enantioselectivity of this process. However, in vitro studies with human liver microsomes have demonstrated that (R)- and (S)-norketamine are preferentially hydroxylated at different sites.30 The antinociceptive properties of the hydroxylated and dehydro metabolites have not been extensively studied, primarily due to an initial determination that these metabolites produced little or no anesthesia or central nervous system excitation.23
In this study, the initial analysis of the plasma samples obtained indicated that steady-state plasma concentrations of hydroxylated and dehydro metabolites of ketamine and norketamine were reached relatively early compared with the clinical response (data not shown). However, due to the lack of appropriate standards, the pharmacokinetic and pharmacodynamic profiles of these metabolites were not determined. The necessary methods are being developed and will be used in the further exploration of the application of a 5-day infusion of a moderate dose of ketamine in the treatment of CRPS.
Acknowledgments
We are thankful to Ms. Jessie Dotson, RN, CNP, for her much appreciated assistance with the data collection and excellent care of every study subject.
Contract grant sponsor National Institute on Aging/NIH (Intramural Research Program)
Footnotes
This work was presented in part at the 2007 ASA Annual Meeting, October 2007, San Francisco, CA
LITERATURE CITED
- 1.Schwartzman RT, Alexander GM, Grothusen J. Pathophysiology of complex regional pain syndrome. Expert Rev Neurother. 2006;6:669–681. doi: 10.1586/14737175.6.5.669. [DOI] [PubMed] [Google Scholar]
- 2.Veldman PH, Goris RJ. Multiple reflex sympathetic dystrophy. Which patients are at risk for developing a recurrence of reflex sympathetic dystrophy in the same or another limb. Pain. 1996;64:463–466. doi: 10.1016/0304-3959(95)00160-3. [DOI] [PubMed] [Google Scholar]
- 3.Maleki J, LeBel AA, Bennett GJ, Schwartzman RJ. Patterns of spread in complex regional pain syndrome type I (reflex sympathetic dystrophy) Pain. 2000;88:259–266. doi: 10.1016/S0304-3959(00)00332-8. [DOI] [PubMed] [Google Scholar]
- 4.Correll GE, Maleki J, Gracely EJ, Muir JJ, Harbut RE. Subanesthetic ketamine infusion therapy: a retrospective analysis of a novel therapeutic approach to complex regional pain syndrome. Pain Med. 2004;5:263–275. doi: 10.1111/j.1526-4637.2004.04043.x. [DOI] [PubMed] [Google Scholar]
- 5.Kvarnstrom A, Karlsten R, Quiding H, Emanuelsson BM, Gordh T. The effectiveness of intravenous ketamine and lidocaine on peripheral neuropathic pain. Acta Anaesthesiol Scand. 2003;47:868–877. doi: 10.1034/j.1399-6576.2003.00187.x. [DOI] [PubMed] [Google Scholar]
- 6.Sigtermans MJ, van Hilten JJ, Bauer MC, Arbous MS, Marinus J, Sarton EY, Dahan A. Ketamine produces effective and long-term pain relief in patients with complex regional pain syndrome type 1. Pain. 2009;145:304–311. doi: 10.1016/j.pain.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 7.Schwartzman RJ, Alexander GM, Grothusen JR, Paylor T, Reichenberger E, Perreault M. Outpatient intravenous ketamine for the treatment of complex regional pain syndrome: a double-blind placebo controlled study. Pain. 2009;147:107–115. doi: 10.1016/j.pain.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Øye I, Paulsen O, Maurset A. Effects of ketamine on sensory perception: evidence for a role of JV-methyl-D-aspartate receptors. J Pharmacol Exp Ther. 1992;260:1209–1213. [PubMed] [Google Scholar]
- 9.White PF, Schuettler J, Shafer A, Stanski DR, Horai Y, Trevor AJ. Comparative pharmacology of the ketamine isomers. Br J Anaesthesia. 1985;57:197–203. doi: 10.1093/bja/57.2.197. [DOI] [PubMed] [Google Scholar]
- 10.Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur J Pharmacol. 1997;333:99–104. doi: 10.1016/s0014-2999(97)01116-3. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez Rosas ME, Patel S, Wainer IW. Determination of the enantiomers of ketamine and norketamine in human plasma by enantioselective liquid chromatography-mass spectrometry. J Chromatogr B. 2003;794:99–108. doi: 10.1016/s1570-0232(03)00420-3. [DOI] [PubMed] [Google Scholar]
- 12.Mandema JW, Stanski DR. Population pharmacodynamic model for ketorolac analgesia. Clin Pharmacol Ther. 1996;60:619–635. doi: 10.1016/S0009-9236(96)90210-6. [DOI] [PubMed] [Google Scholar]
- 13.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–478. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sigtermans M, Dahan A, Mooren R, Bauer M, Kest B, Sarton E, Olofsen E. S(+)-ketamine effect on experimental pain and cardiac output. Anesthesiology. 2009;111:892–903. doi: 10.1097/ALN.0b013e3181b437b1. [DOI] [PubMed] [Google Scholar]
- 15.Geisslinger G, Hering W, Thomann P, Knoll R, Kamp HD. Pharmacokinetics and pharmacodynamics of ketamine enantiomers in surgical patients using stereoselective analytical method. Br J Anaesthesia. 1993;70:666–671. doi: 10.1093/bja/70.6.666. [DOI] [PubMed] [Google Scholar]
- 16.Ihmsen H, Geisslinger G, Schuttler J. Stereoselective pharmacokinetics of ketamine: R(−)-ketamine inhibits the elimination of S(+)-ketamine. Clin Pharmacol Ther. 2001;70:431–438. doi: 10.1067/mcp.2001.119722. [DOI] [PubMed] [Google Scholar]
- 17.Yanagihara Y, Ohtani M, Kariya S, Uchino K, Hiraishi T, Ashizawa N, Aoyamo T, Yamamura Y, Yamada Y, Iga T. Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharm Drug Dispos. 2003;24:37–43. doi: 10.1002/bdd.336. [DOI] [PubMed] [Google Scholar]
- 18.Yanagihara Y, Kariya S, Ohtani M, Uchino K, Aoyama T, Yamamura Y, Iga T. Involvement of CYP2B6 in N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2001;29:887–890. [PubMed] [Google Scholar]
- 19.Shimada T, Yamazaki M, Mimura M, Inui Y, Guengerich FP. Interindividual variation in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals—studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 20.Holtman JR, Crooks PA, Johnson-Hardy JK, Hojomat M, Heven M, Wala EP. Effects of norketamine enantiomers in rodent models of persistent pain. Pharmacol Biochem Behav. 2008;90:676–685. doi: 10.1016/j.pbb.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Rabben T, Skjelbred P, Øye I. Prolonged analgesic effect of ketamine, an N-methyl-D-aspartate receptor inhibitor, in patients with chronic pain. Pharmacol Exp Ther. 1999;289:1060–1066. [PubMed] [Google Scholar]
- 22.Adams JD, Jr, Baillie TA, Trevor AJ, Castagnoli N. Studies on the biotransformation of ketamine 1—identification of metabolites produced in vitro from rat liver microsomal preparations. Biomed Mass Spectrom. 1981;8:527–538. doi: 10.1002/bms.1200081103. [DOI] [PubMed] [Google Scholar]
- 23.Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29:2396–2399. doi: 10.1021/jm00161a043. [DOI] [PubMed] [Google Scholar]
- 24.Kharasch ED, Labroo R. Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology. 1992;77:1201–1207. doi: 10.1097/00000542-199212000-00022. [DOI] [PubMed] [Google Scholar]
- 25.Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–858. doi: 10.1124/dmd.30.7.853. [DOI] [PubMed] [Google Scholar]
- 26.Turfus SC, Parkin MC, Cowan DA, Halket JM, Smith NW, Braithwaite RA, Elliot SP, Steventon GB, Kicman AT. Use of human microsomes and deuterated substrates: an alternative approach for the identification of novel metabolites of ketamine by mass spectrometry. Drug Metab Dispos. 2009;37:1769–1778. doi: 10.1124/dmd.108.026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolze S, Boulieu R. HPLC determination of ketamine, norketamine and dehydronorketamine in plasma with a high-purity reversed-phase sorbent. Clin Chem. 1998;44:560–564. [PubMed] [Google Scholar]
- 28.Williams ML, Mager DE, Parenteau H, Gudi G, Tracy TS, Mulheran M, Wainer IW. Effects of protein calorie malnutrition on the pharmacokinetics of ketamine in rats. Drug Metab Dispos. 2004;32:786–793. doi: 10.1124/dmd.32.8.786. [DOI] [PubMed] [Google Scholar]
- 29.Wieber J, Gugler R, Hengstmann JH, Dengler HJ. Pharmacokinetics of ketamine in man. Anaesthesist. 1975;24:260–263. [PubMed] [Google Scholar]
- 30.Trevor AJ, Woolf TF, Baillie TA, Adams JD. Castagnoli. Stereoselective metabolism of ketamine enantiomers. In: Kamenka JM, Domino EF, Geneste P, editors. Phencyclidine and Related Arylcyclohexylamines: Present and Future Applications. Ann Arbor, MI: NPP Books; 1983. pp. 279–289. [Google Scholar]