

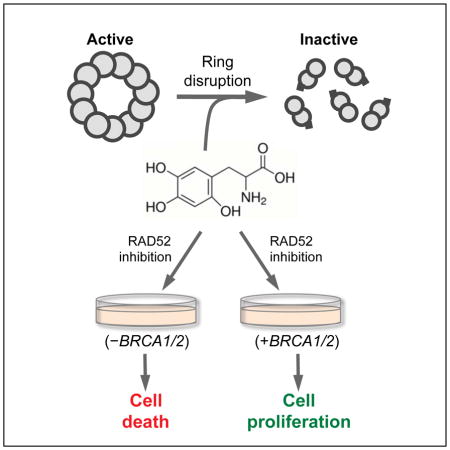
SUMMARY
Suppression of RAD52 causes synthetic lethality in BRCA deficient cells. Yet pharmacological inhibition of RAD52, which binds single-strand DNA (ssDNA) and lacks enzymatic activity, has not been demonstrated. Here, we identify the small molecule 6-hydroxy-dopa (6-OH-dopa) as a major allosteric inhibitor of the RAD52 ssDNA binding domain. For example, we find that multiple small molecules bind to and completely transform RAD52 undecamer rings into dimers, which abolishes the ssDNA binding channel observed in crystal structures. 6-OH-dopa also disrupts RAD52 heptamer and undecamer ring superstructures, and suppresses RAD52 recruitment and recombination activity in cells with negligible effects on other double-strand break repair pathways. Importantly, we show that 6-OH-dopa selectively inhibits the proliferation of BRCA deficient cancer cells, including those obtained from leukemia patients. Taken together, these data demonstrate small molecule disruption of RAD52 rings as a promising mechanism for precision medicine in BRCA deficient cancers.
Keywords: Homologous Recombination, DNA Repair, Synthetic Lethality, Single-Strand Annealing, Genome Instability, Cancer, High-Throughput Screening, Cancer Therapeutics
Graphical abstract
INTRODUCTION
Homologous recombination (HR) is an essential DNA repair pathway due to its accurate and predominant role in DNA break repair during S-phase and G2(Li and Heyer, 2008; Moynahan and Jasin, 2010; Sung and Klein, 2006). Heritable mutations in HR factors, most notably BRCA1 and BRCA2, cause genome instability and predispose to breast and ovarian cancer(Lok and Powell, 2012; Moynahan and Jasin, 2010). BRCA deficiencies are also observed in a variety of other cancers including lung, prostate and pancreatic cancers, melanomas, and leukemias(Cramer-Morales et al., 2013; Francis et al., 2010; Mai et al., 2009; Wang et al., 2014). BRCA deficient-like phenotypes due to spontaneous mutations or defects in HR that are independent of BRCA1/2 are also commonly observed in cancer cells(Ceccaldi et al., 2015; McCabe et al., 2006; Turner et al., 2004). Because HR defective cells are impaired in their ability to repair DNA breaks during S-phase and G2, DNA damage caused during replication causes severe growth defects in these cells, with little or no effect in normal cells. Thus, drugs that induce DNA damage or further inhibit DNA repair during replication can cause synthetic lethality in BRCA deficient cells while sparing normal cells(Farmer et al., 2005; McCabe et al., 2006).
The ability to target BRCA deficient cells for killing has received wide attention due to the potential development of non-toxic drugs for personalized medicine. A notable example includes Poly (ADP-ribose) polymerase 1 (PARP-1) inhibitors, which cause replication dependent DNA breaks and thus preferentially kill BRCA deficient cells(Farmer et al., 2005; McCabe et al., 2006). In so far, PARP-1 inhibitors, such as the recently approved drug olaparib, have shown promise in the clinic, barring some side effects(Kaufman et al., 2015; Lord and Ashworth, 2012). However, considering that PARP-1 has wide ranging roles in transcription, translation, telomere maintenance, chromatin and cellular stress response, in addition to DNA repair, its inhibition inevitably causes a large number of short-term, and possibly long-term, side effects in normal cells(Farmer et al., 2005; Gibson and Kraus, 2012; Ji and Tulin, 2010; Lord and Ashworth, 2012; Thomas and Tulin, 2013). Identifying and characterizing new drug targets that exclusively perform DNA repair as a backup to HR during S-phase and G2 will lead to the development of personalized medicine for BRCA deficient cancer patients with a significantly lower risk of side effects.
Previous studies demonstrate that cells deficient in BRCA1/2 or associated proteins in this pathway (PALB2, RAD51B/C/D, XRCC2/3) combined with a deficiency in recombination factor RAD52 are synthetic lethal(Chun et al., 2013; Feng et al., 2011; Lok et al., 2012; Lok and Powell, 2012). Cells and mice deficient in only RAD52, however, are viable with no apparent phenotypes(Feng et al., 2011; Lok and Powell, 2012; Rijkers et al., 1998). Thus, these studies have revealed a new vulnerability in BRCA deficient cells which may be exploited to target these cells for killing. For example, drugs that inhibit RAD52 activity are likely to cause synthetic lethality in BRCA deficient cells in a similar manner to PARP-1 inhibitors, but potentially have no side effects(Lok and Powell, 2012).
Much of our knowledge of how RAD52 functions has been derived from studies in the yeast model Saccharomyces cerevisiae. For example, yeast Rad52 plays a major role in HR by facilitating loading of Rad51 recombinase onto RPA coated ssDNA which is necessary for strand invasion and subsequent steps of HR(Krogh and Symington, 2004; Liu and Heyer, 2011). This function is consistent with the ability of Rad52 to bind ssDNA, RPA and Rad51. Since Rad52 plays a prominent role in Rad51 loading, yeast deficient in Rad52 are impaired in HR and highly sensitive to genotoxic agents(Krogh and Symington, 2004). Rad52 in yeast and higher eukaryotes is also essential for the repair of DSBs by single-strand annealing (SSA)(Kagawa et al., 2002; Krogh and Symington, 2004; Singleton et al., 2002). In this pathway, RAD52 uses sequence microhomology along ssDNA exposed by nuclease processing of DSBs to recombine broken DNA which results in chromosomal deletions(Krogh and Symington, 2004).
In contrast to yeast, RAD51 loading in mammalian cells is primarily performed by the BRCA pathway, whereas RAD52 appears to play a backup role(Feng et al., 2011; Lok et al., 2012; Lok and Powell, 2012). For example, previous studies have showed that suppression of RAD52 expression has little or no effect on HR in BRCA proficient cells(Feng et al., 2011; Lok et al., 2012). Yet, suppression of RAD52 in BRCA deficient cells further reduces HR and RAD51 loading, and causes synthetic lethality(Feng et al., 2011; Lok et al., 2012). Hence, these studies suggest that RAD52 might promote the survival of BRCA deficient cells by facilitating RAD51 dependent HR. Considering that RAD52 is also essential for SSA which functions during S-phase, its annealing activity probably also contributes to the survival of BRCA defective cells. Although the exact mechanism by which RAD52 promotes the survival of BRCA deficient cells remains unclear, a recent report showed that its ssDNA binding activity is essential for this role(Cramer-Morales et al., 2013). Altogether, these studies indicate that small molecule inhibitors of the RAD52 ssDNA binding domain are likely to target BRCA deficient cells for killing.
The crystal structure of the RAD52 ssDNA binding domain, for example residues 1-209, has been solved by two groups(Kagawa et al., 2002; Singleton et al., 2002). Remarkably, eleven monomers form an undecamer ring with a positively charged channel around its outer surface that likely interacts with ssDNA(Kagawa et al., 2002; Singleton et al., 2002). Although DNA binding motifs are traditionally considered to be “undruggable”, small molecule disruption of protein complexes is a promising strategy for inhibiting multi-subunit DNA binding proteins(Scheuermann et al., 2013). Here, we identify and characterize a small molecule allosteric inhibitor of RAD52 that disrupts ring structures of the protein and selectively impairs the growth of BRCA deficient cancer cells.
RESULTS
Identification of RAD52 Small Molecule Inhibitors
Since recent studies have shown that the proliferation of BRCA deficient cells depends on RAD52 ssDNA binding activity(Cramer-Morales et al., 2013), we set out to identify inhibitors of the ssDNA binding domain. A fluorescence polarization (FP) assay that detects RAD52 ssDNA binding was optimized and adapted for high-throughput screening as follows (Fig. 1a, left). A 29 nucleotide (nt) ssDNA probe conjugated with fluorescein (FAM) was incubated with increasing concentrations of wild-type RAD52 which forms a 323 kDa heptamer ring and higher-order heptamer ring superstructures(Lloyd et al., 2002; Stasiak et al., 2000; Van Dyck et al., 1998; Van Dyck et al., 2001). After 30 min FP was determined which is inversely proportional to the rotation rate of the ssDNA probe (Fig. 1a, middle). RAD52 binding caused an increase in polarization due to slowed rotation of the ssDNA probe. FP reactions, with and without RAD52, performed in 384-well high-throughput format using a liquid handler resulted in a high Z’ score of 0.82, demonstrating that the assay was consistent and suitable for high-throughput screening (Fig. 1a, right).
Figure 1. Identification of RAD52 inhibitors by high-throughput screening.

a, Schematic of fluorescence polarization (FP) assay (left). Plot showing RAD52 binding to ssDNA probe. Kd = 25 nM. Data shown as average ± s.d. from 4 independent experiments (middle). Plot showing 176 high signal (RAD52 with ssDNA probe) and 176 low signal (ssDNA probe only) FP control reactions performed in 384-well high-throughput format. Z’ factor = 0.82, mP = millipolarization. b, Structures of select identified RAD52 inhibitors. c, Plot showing IC50 for RAD52 inhibitors. d, Schematic of electrophoresis mobility shift assay (EMSA)(left). Non-denaturing gel of EMSA showing inhibition of RAD52 by 60 μM of indicated small molecules (middle panels). Plot of EMSA data shown as percent inhibition of RAD52. Data shown as average ± s.d. from 3 independent experiments (right).
We screened a diverse set of 18,304 drug-like compounds and the Sigma Lopac collection of 1,280 pharmacologically active compounds and identified 10 small molecules that consistently inhibited RAD52 ssDNA binding by more than 60% and exhibited a relatively low IC50 (≤ 5μM) (Fig. 1b,c). The small molecules were confirmed by HPLC-mass spectrophotometry or newly purchased. Small molecule inhibition of RAD52 ssDNA binding was confirmed using an orthogonal electrophoresis mobility shift assay (EMSA)(Fig. 1d). Similar to previous studies, RAD52 shifted the ssDNA probe to the wells of the polyacrylamide gel due to its 323 kDa heptamer ring conformation and tendency to form higher-order heptamer ring superstructures(Benson et al., 1998; Lloyd et al., 2002; Van Dyck et al., 1998). As a control, we showed that these large RAD52-ssDNA complexes were resolved in an agarose gel as in previous studies (Supplementary Fig. 1)(Benson et al., 1998). Select compounds that inhibited RAD52 by more than 70% were further analyzed in cells.
Small Molecule Inhibition of RAD52 Activity in Cells
We examined the effects of select small molecules on SSA which is one of the main activities of RAD52(Kagawa et al., 2002; Krogh and Symington, 2004; Singleton et al., 2002). To measure SSA, a previously characterized GFP reporter system was used which detects activation of GFP expression as a result of SSA repair of an I-SceI induced DSB(Gunn et al., 2011; Gunn and Stark, 2012). As a control, we demonstrated that suppression of RAD52 via siRNA inhibited SSA as expected (Fig. 2a). Surprisingly, only one of the small molecules, 6-hydroxy-dopa (6-OH-dopa), a precursor of the catecholaminergic neurotoxin 6-hydroxydopamine, consistently inhibited SSA (Fig. 2b). The remaining compounds showed either no effect, slight inhibition, or stimulation of SSA which was probably due to off-target effects. As a control, we showed that the effect of 6-OH-dopa was dose dependent (Fig. 2c). Inhibition of SSA by 6-OH-dopa was additionally confirmed in vitro (Supplementary Fig. 1), and was dependent on RAD52 expression (Supplementary Fig. 2). We also confirmed that 6-OH-dopa does not interact with ssDNA (Supplementary Fig. 1). Importantly, we found that small molecules similar in structure to 6-OH-dopa, including L-DOPS which is in the same catechol chemical class, failed to inhibit SSA (Fig. 2d) and RAD52 ssDNA binding (Supplementary Fig. 1). Thus, these data indicate that a specific interaction between 6-OH-dopa and RAD52 is responsible for the observed inhibitory effects in vitro and in vivo.
Figure 2. 6-OH-dopa selectively inhibits RAD52 mediated recombination in vivo.

a, Plot showing SSA in U20S cells treated with scrambled and RAD52 siRNA. Data represent mean ± s.e.m from triplicates. **P = 0.00036; two-tailed Student’s t-test (left). Western blots of protein extracts from U20S cells treated with scrambled and RAD52 siRNA (right). b, Plot showing SSA in U20S cells following treatment with 5 μM of the indicated small molecules. Data represent mean ± s.e.m. from 3 separate experiments with triplicates in each experiment. **P = 0.00154; two-tailed Student’s t-test. c, Plot showing SSA in U20S cells following treatment with 0 μM, 10 μM and 20 μM of 6-OH-dopa. Data represent mean ± s.e.m from triplicates. ***P = 0.00039, ****P = 0.00009; two-tailed Student’s t-test. d, Plot showing SSA in U20S cells following treatment with 5 μM of the indicated small molecules. 2 = L-DOPS, 3 = DL-o-tyrosine, 4 = Beta-(2-hydroxy-4-methylphenyl) alanine, 5 = 6-OH-dopa. Data represent mean ± s.e.m from triplicates. *P = 0.00175; two-tailed Student’s t-test. e, Plot showing HR in U20S cells following treatment with 5 μM of the indicated small molecules. Data represent mean ± s.e.m. from 3 separate experiments with triplicates in each experiment. f, Plots showing percent inhibition of indicated protein as a function of 6-OH-dopa concentration. Data shown as average ± s.e.m. from 3 independent experiments. g, Plot showing NHEJ in U20S cells following treatment with or without 5 μM of 6-OH-dopa. Data represent average ± s.e.m. from two separate experiments with triplicates in each experiment.
We further examined the specificity of 6-OH-dopa for RAD52 in cells by investigating its effects on other DSB repair pathways. First, we tested whether 6-OH-dopa inhibits HR which shares the same upstream DNA damage response and resection pathways as SSA. Since HR functions mostly independently of RAD52 in BRCA proficient cells, we anticipated that inhibition of RAD52 would have little effect on HR(Feng et al., 2011; Lok et al., 2012). Indeed, using a previously characterized HR GFP reporter(Gunn et al., 2011; Gunn and Stark, 2012), we found that 6-OH-dopa treatment resulted in little or no reduction in HR (Fig. 2e), and had no effect on HR when RAD52 was suppressed (Supplementary Fig. 2). Thus, these data are consistent with the ability of 6-OH-dopa to inhibit RAD52 with a considerable amount of specificity. In support of this, we found that 6-OH-dopa had little or no effect on RAD51 (IC = n.d.) compared to RAD52 (IC = 1.1 μM) in vitro (Fig. 2f). 6-OH-dopa also had a significantly higher IC50 for S. cerevisiae RAD59 (IC > 10 μM) which shares 31.5% sequence identity with human RAD52 and performs a similar SSA activity (Fig. 2f)(Supplementary Fig. 3)(Krogh and Symington, 2004; Petukhova et al., 1999; Wu et al., 2006). We note that the small molecules that inhibited HR (RU-0180081, RU-0096909) showed stimulation of SSA which is expected based on the ability of HR to suppress SSA (Fig. 2e and Fig. 2b)(Stark et al., 2004; Tutt et al., 2001). To further analyze the specificity of 6-OH-dopa for RAD52 in cells, we tested its effect on NHEJ. Using another previously characterized GFP reporter(Gunn et al., 2011; Gunn and Stark, 2012), we found that 6-OH-dopa only slightly inhibited NHEJ (Fig. 2g). Considering that HR and NHEJ each require a host of proteins involved in nucleic-acid processing, signaling, and protein post-translational modification, the ability of 6-OH-dopa to selectively inhibit SSA in cells demonstrates a considerable amount of specificity of the small molecule for RAD52(Ciccia and Elledge, 2010; Deriano and Roth, 2013; Moynahan and Jasin, 2010). Thus, although 6-OH-dopa is a catechol and has the potential to interfere with some assays non-specifically, the exhaustive in vitro and cell-based data presented herein show that its mechanism on RAD52 is specific.
We further examined the ability of 6-OH-dopa to inhibit RAD52 activity in cells by testing its effects on RAD52 foci formation at DNA damage caused by cisplatin and ionizing radiation (Fig. 3). eGFP-RAD52 was stably expressed in BCR-ABL transformed murine hematopoietic 32Dcl3 cells, which are known to be deficient in BRCA1(Cramer-Morales et al., 2013; Podszywalow-Bartnicka et al., 2014). Previous studies have demonstrated that eGFP-RAD52 is functional in cells(Essers et al., 2002; Feng et al., 2011), and we found that the purified protein acts like wild-type RAD52 in vitro (Supplementary Fig. 1). The eGFP-RAD52 expressing cells were incubated with DMSO or 6-OH-dopa then further treated with cisplatin which causes intra- and inter-strand crosslinks that arrest replication forks. Consistent with the ability of the small molecule to inhibit RAD52 ssDNA binding, 6-OH-dopa suppressed eGFP-RAD52 foci formation in a dose dependent manner (Fig. 3a; Supplementary Fig. 4). We further investigated the effect of 6-OH-dopa on eGFP-RAD52 foci in HEK293T cells exposed to ionizing radiation. The small molecule again decreased the number of eGFP-RAD52 foci in a dose dependent manner (Fig. 3b; Supplementary Fig. 4). 6-OH-dopa also inhibited eGFP-RAD52 foci in MDA-MB-436 cells complemented with BRCA1 following exposure to ionizing radiation (Fig. 3c; Supplementary Fig. 4). Together, these results demonstrate that 6-OH-dopa effectively inhibits RAD52 ssDNA binding in vivo.
Figure 3. 6-OH-dopa inhibits RAD52 foci formation.

a, Cells stably expressing eGFP-RAD52 were exposed to cisplatin and 6-OH-dopa. Representative images of nuclei counterstained with DAPI (blue) and eGFP-RAD52 (green)(left). Plot showing quantification of cells with eGFP-RAD52 foci following treatment with cisplatin and 6-OH-dopa (right). Data represent mean ± s.e.m from triplicates. *P = 0.01, **P = 0.007; two-tailed Student’s t-test. b, Representative fluorescent images of HEK293T cells visualizing DAPI stain (blue) and eGFP fluorescence (green) following treatment with ionizing radiation (IR)(left). Plot showing percent HEK293T cells with ≥ 5 eGFP-RAD52 foci following treatment with IR and 6-OH-dopa (right). Data shown as average ± s.e.m. from 3 independent experiments. *P = 0.05, **P = 0.03; two-tailed Student’s t-test. c, Representative fluorescent images of MDA-MB-436 BRCA1 complemented cells visualizing DAPI stain (blue) and eGFP RAD52 (green) following treatment with or without ionizing radiation (IR)(left). Plot showing percent MDA-MB-436 BRCA1 complemented cells with ≥ 5 eGFP-RAD52 foci following treatment with IR and 6-OH-dopa (right). Data shown as average ± s.e.m. from 3 independent experiments. *P = 0.03, **P = 0.02; two-tailed Student’s t-test.
Small Molecule Dissociation of RAD52-ssDNA Complexes
We next considered whether 6-OH-dopa was capable of dissociating pre-formed RAD52-ssDNA complexes. Given that RAD52 bound tightly to ssDNA (Kd = 25 nM; Fig. 1a), we anticipated that RAD52-ssDNA complexes would be resistant to 6-OH-dopa treatment. To test this, RAD52-ssDNA complexes were formed and allowed to reach equilibrium after 30 min of incubation. 6-OH-dopa was then added and FP was measured 2 min later. Remarkably, the small molecule fully dissociated RAD52-ssDNA complexes as indicated by a reduction in mP levels nearly identical to those observed for the DNA control (Fig. 4a). 6-OH-dopa dissociation of RAD52-ssDNA complexes was also observed by EMSA (Fig. 4b).
Figure 4. 6-OH-dopa dissociates RAD52-ssDNA complexes and acts as a non-competitive inhibitor.

a, Schematic of FP assay (top). Plot showing dissociation of RAD52-ssDNA complexes by 30 μM 6-OH-dopa (bottom). Data shown as average ± s.d. from 3 independent experiments. mP = milipolarization. b, Schematic of EMSA (top). Non-denaturing gel showing dissociation of RAD52-ssDNA complexes by 30 μM 6-OH-dopa (bottom). c, Schematic of EMSA (top). Non-denaturing gel showing 6-OH-dopa (25 μM) inhibition of RAD52 in the presence of excess of Cy3-ssDNA substrate (bottom).
The ability of the small molecule to rapidly dissociate RAD52-ssDNA complexes suggested that it might act as a non-competitive inhibitor. To test this, we investigated whether the inhibitory activity of the small molecule was diminished by simultaneously adding a large of excess of ssDNA substrate. Strikingly, the small molecule continued to inhibit RAD52 in the presence of a ~100-fold excess of the ssDNA substrate (Fig. 4c). Hence, these data suggest that the small molecule binds to a different site than the ssDNA binding channel and thus acts a non-competitive inhibitor.
Small Molecule Disruption of RAD52 Rings
To gain more insight into the mechanism of 6-OH-dopa action, we examined its interaction with RAD52. Since the small molecule inhibits RAD52 ssDNA binding, we envisaged that it acted on the ssDNA binding domain (residues 1-209; RAD52 1-209). X-ray crystallographic studies demonstrate that the ssDNA binding domain forms an undecamer ring with a large central channel(Kagawa et al., 2002; Singleton et al., 2002). The eleven subunits form a symmetrical pinwheel like conformation with a positively charged channel circling the outer surface that likely interacts with ssDNA(Kagawa et al., 2002; Singleton et al., 2002). We found that the small molecule inhibited RAD52 1-209 with a comparable IC50 (1.6 μM) as wild-type RAD52 (1.1 μM), indicating that it interacts with the ssDNA binding domain (Fig. 5a,b). Isothermal calorimetry (ITC) confirmed that 6-OH-dopa interacts with the ssDNA binding domain with a 17.7 μM Kd (Fig. 5c). Unexpectedly, these data suggested a stoichiometry of 5.04 small molecules per RAD52 1-209 ring (Fig. 5c). This suggests the possibility that one small molecule binds to every other subunit around the ring (Fig. 5c), which is plausible based on the slightly different orientation of subunits(Kagawa et al., 2002; Singleton et al., 2002). Alternatively, the small molecule may bind at the interface of every other subunit pair which would also account for ~5 binding sites.
Figure 5. 6-OH-dopa targets the RAD52 ssDNA binding domain and disrupts RAD52 rings.

a, Schematic of EMSA (top). Non-denaturing gel of EMSA showing 6-OH-dopa inhibition of RAD52 1-209 (bottom). b, Schematic of FP assay (top). Plot showing percent inhibition of RAD52 1-209 ssDNA binding as a function of 6-OH-dopa concentration. IC50 = 1.6 μM. Data shown as average ± s.d. from 3 independent experiments (bottom). c, Plot of isothermal calorimetry (ITC) data showing 6-OH-dopa interaction with RAD52 1-209. Kd = 17.8 μM, n = 5. Model of 6-OH-dopa binding to RAD52 1-209 (inset). d, Silver stained non-denaturing gel of RAD52 1-209 following incubation with 6-OH-dopa (left). Western blot of non-denaturing gel of RAD52 1-209 following incubation with 6-OH-dopa (right). e, Plots showing light scattering data of RAD52 1-209 following incubation with (right panel) and without (left panel) 6-OH-dopa with 1 M NaCl. Model of 6-OH-dopa action on RAD52 1-209 with high salt (right). f, Plots showing light scattering data of RAD52 1-209 following incubation with (right panel) and without (left panel) 6-OH-dopa with 0.15 M NaCl. Model of 6-OH-dopa action on RAD52 1-209 with low salt (right). g, Silver stained non-denaturing gel of RAD52 WT following incubation with 6-OH-dopa (left). h, Gel filtration profiles of RAD52 WT following incubation with (red line) and without (black line) 6-OH-dopa (left). Model of 6-OH-dopa action on RAD52 WT (right).
Considering that 6-OH-dopa may inhibit RAD52 by acting at multiple subunit interfaces, we wondered whether the small molecule altered the structure or composition of the RAD52 1-209 undecamer ring. To test this, the mobility of RAD52 1-209 was analyzed in a native gel following incubation with increasing amounts of the small molecule. Remarkably, 6-OH-dopa enabled the protein to enter the gel which appeared as multiple discrete bands, the smallest of which was in the range of ~50–100 kDa (Fig. 5d, left). Western blot analysis of RAD52 1-209 following native gel electrophoresis confirmed the identity of the bands as RAD52 1-209 (Fig. 5d, right). Together, these results indicate that 6-OH-dopa dissociates RAD52 complexes. As a control, we showed that a structurally similar small molecule, DL-o-tyrosine, which failed to inhibit RAD52 (Supplementary Fig. 1) had no effect on RAD52 1-209 composition (Supplementary Fig. 1). We also showed that another potent inhibitor of RAD52 1-209 (RU-0098062; Fig. 1b-1d) had no effect on the composition of the protein (Supplementary Fig. 1). Thus, these results clearly show that a specific interaction between 6-OH-dopa and the ssDNA binding domain facilitates dissociation of RAD52 complexes.
Light scattering was used to confirm small molecule dissociation of RAD52 complexes and to determine the size of the resulting protein species. First, light scattering was performed under high salt conditions, similar to the reactions resolved in native gels. In the absence of the small molecule, the molecular weight of RAD52 1-209 was ~439 kDa which likely includes a mixture of single, double and triple undecamer rings (Fig. 5e, left panel). The addition of 6-OH-dopa, however, produced a discrete 48.3 kDa species (Fig. 5e, right panel) that was not observed in the buffer or small molecule controls (Supplementary Fig. 5). We note that the 439 kDa peak slightly increased to 595.6 kDa which may be due to some precipitation of RAD52 1-209 by 6-OH-dopa. Considering that 48.3 kDa is nearly identical in mass to the sum of two monomers of RAD52 1-209 (48 kDa), these data demonstrate that the small molecule dissociates the undecamer ring into dimers (Fig. 5e, right). To confirm this result, we repeated the light scattering experiment under low salt conditions which reduce hydrophobic interactions, such as those that promote RAD52 superstructures. Indeed, under low salt conditions RAD52 1-209 was mostly detected at 270 kDa which closely corresponds to its undecamer form (263 kDa)(Fig. 5f, left panel). Addition of the small molecule again produced a 48.3 kDa species (Fig. 5f, right panel) that was not observed in the buffer or 6-OH-dopa controls (Supplementary Fig. 5). Remarkably, under these conditions 100% of the undecamers (270 kDa) were converted into dimers (48.3 kDa), demonstrating that the small molecule is more active when hydrophobic interactions are suppressed by low salt (Fig. 5f, right).
Although electron microscopy studies demonstrate that RAD52 WT forms heptamer rings and large heptamer ring superstructures(Stasiak et al., 2000; Van Dyck et al., 1998; Van Dyck et al., 2001), the atomic structure of full-length RAD52 has not been solved. Thus, it remains unknown whether the RAD52 WT heptamer ring possesses a ssDNA binding channel like that observed in the RAD52 1-209 undecamer ring. However, considering that 6-OH-dopa exhibits a comparable IC50 for RAD52 WT (1.1 μM) and RAD52 1-209 (1.6 μM), the ssDNA binding domain is likely to have a similar conformation in the heptamer and undecamer forms. We therefore examined whether 6-OH-dopa had similar effects on the composition of RAD52 WT heptamer rings. Indeed, the small molecule enabled RAD52 WT to enter a native gel as discrete higher molecular weight complexes in a similar manner as observed for RAD52 1-209 (Compare Fig. 5g and Fig. 5d). The lower molecular weight complex appeared slightly larger than 670 kDa which corresponds to a double heptamer and is consistent with previous gel filtration studies (Fig. 5g)(Lloyd et al., 2005). The higher molecular weight complexes likely represent heptamer ring superstructures (Fig. 5g). In contrast to the results obtained with RAD52 1-209, 6-OH-dopa failed to produce smaller molecular weight complexes from RAD52 WT (Compare Fig. 5g and Fig. 5d). To more accurately determine the size of RAD52 WT complexes following incubation with 6-OH-dopa, we analyzed the protein by gel filtration. Similar to previous studies, the molecular weight of RAD52 WT in the absence of 6-OH-dopa was around 670 kDa, which corresponds to a double heptamer ring (Fig. 5h, black line)(Lloyd et al., 2005). Notably, only a small fraction of the loaded protein entered the column which we attribute to the tendency of RAD52 WT to form large superstructures that are excluded from the column. Consistent with this interpretation, most of RAD52 WT failed to enter the native gel in the absence of the small molecule (Fig. 5g). Similar to the results obtained from native gel analysis, the small molecule enabled a significantly higher concentration of RAD52 WT to enter the column as multiple discrete higher molecular weight complexes (Fig. 5h, red line). The smaller peak corresponds to a double heptamer, whereas the larger peak migrated as larger superstructures, probably composed of 3-4 heptamer rings (Fig. 5h, red line). Taken together, these experiments demonstrate that 6-OH-dopa disrupts RAD52 heptamer and undecamer ring superstructures, and further dissociates undecamer rings into dimers.
Selective Killing of BRCA Deficient Cells by Small Molecule Inhibition of RAD52
Since RAD52 ssDNA binding activity has been shown to be essential for the survival of BRCA deficient cells(Cramer-Morales et al., 2013), we examined whether 6-OH-dopa selectively reduces the viability of cells harboring mutations in BRCA1/2. As a control, we tested whether suppression of RAD52 reduced the viability of the BRCA1 deficient triple negative breast cancer cell line MDA-MB-436. Similar to previous studies using other BRCA1 deficient cells(Lok et al., 2012), we found that suppression of RAD52 via siRNA (Supplementary Fig. 2) resulted in reduced viability after a short 3 day time course (Fig. 6a). In contrast, suppression of RAD52 caused a slight increase in the viability of the same cell line complemented with wild-type BRCA1 (Fig. 6a); scrambled siRNA had negligible effects on viability (Fig. 6a). As a further control, we showed that olaparib selectively reduced the viability of the BRCA1 mutated cells as expected (Fig. 6a). Remarkably, 6-OH-dopa treatment resulted in a nearly identical reduction in viability of the BRCA1 deficient cells (Fig. 6a). 6-OH-dopa also selectively halted the proliferation of the Chinese hamster cell line VC8 (Fig. 6b) and the pancreatic cancer cell line CAPAN-1 (Fig. 6c), which are both defective in BRCA2 due to truncation mutations. Similar growth defects were observed following olaparib treatment (Fig. 6b and Fig. 6c), demonstrating that the ability of 6-OH-dopa to selectively inhibit the proliferation of BRCA deficient cells is comparable to olaparib. 6-OH-dopa also selectively reduced the viability of the BRCA1 deficient triple negative breast cancer cell line HCC1937 (Fig. 6d).
Figure 6. 6-OH-dopa selectively inhibits the growth of BRCA deficient cells.

a-d, Plots showing relative viability of BRCA proficient (black) and deficient (grey) cells following treatment with 20 μM (a), 75 μM (b), 10 μM (c), 5 μM (d) 6-OH-dopa, 20 μM olaparib (c), or indicated siRNA. Data shown as average ± s.e.m. from triplicates a: ****P = 0.0008, *P = 0.02, **P = 0.005, b: ***P = 0.0002, *P = 0.03, c: *P = 0.01, *P = 0.01, d: *P = 0.05; two-tailed Student’s t-test. e-h, Plots showing clonogenic survival of indicated BRCA proficient and deficient cells following treatments with 6-OH-dopa or olaparib. Data shown as average ± s.e.m. from 3 independent experiments. Data points in (g,h) represent 3 different patient cells pooled together. f: *P = 0.03; two-tailed Student’s t-test. i, Plots showing viability of BRCA1 deficient MDA-MB-436 cells following transfection with scrambled (black) or RAD52 (white) siRNA and treatment with or without 6-OH-dopa. Data shown as average ± s.e.m. from triplicates. *P = 0.0004; two-tailed Student’s t-test. j, Plots showing percent increase in γH2AX foci in BRCA1 proficient (black) and deficient (grey) MDA-MB-436 cells following treatment with 6-OH-dopa (left). Percent increase was determined from the number of nuclei with greater than five γH2AX foci. Data shown as average ± s.e.m. from 3 independent experiments. Cell images show DAPI staining (blue) and γH2AX foci (green)(right). **P = 0.002, ***P = 0.0078; two-tailed Student’s t-test. k, Plots showing percent of BRCA2 deficient (grey; VC8) and proficient (black; V79) cells positive for annexin V following treatment with 6-OH-dopa. *P = 0.01, **P = 0.003; two-tailed Student’s t-test (left). Plots showing percent of BRCA1 deficient (grey) and proficient (black) HCC1937 cells positive for annexin V following treatment with 6-OH-dopa (right).
The ability of 6-OH-dopa to target BRCA deficient cells was further examined in clonogenic survival assays. We observed that 6-OH-dopa inhibited the survival of BRCA deficient cell lines, while having little or no effect on BRCA proficient cells at the given doses (Fig. 6e and Fig. 6f). Considering that previous studies have revealed BRCA deficiencies in acute myeloid leukemia (AML) and chronic myelogenous leukemia (CML)(Cramer-Morales et al., 2013; Podszywalow-Bartnicka et al., 2014), we examined the ability of 6-OH-dopa to halt the growth of AML and CML patient cells that were previously identified as expressing low levels of BRCA1/2 (Supplementary Fig. 6)(Cramer-Morales et al., 2013; Podszywalow-Bartnicka et al., 2014). Remarkably, treatment with 6-OH-dopa or olaparib resulted in a similar reduction in colony formation by BRCA deficient AML patient cells with little effects on AML patient cells expressing normal levels of BRCA1/2 (Fig. 6g). We also observed a significant reduction in the survival of BRCA1 deficient CML patient cells treated with 6-OH-dopa (Fig. 6h). To rule out the possibility that the selective killing of BRCA deficient cells was due to an off-target effect, we examined the effect of 6-OH-dopa on the viability of cells deficient in both the BRCA pathway and RAD52. For example, in the event that 6-OH-dopa selectively halted the proliferation of BRCA deficient cells by inhibiting a target other than RAD52, the small molecule would still substantially reduce the viability of BRCA deficient cells that lacked RAD52. The small molecule, however, lost its ability to significantly reduce the viability of BRCA1 deficient cells when RAD52 expression was suppressed (Fig. 6i)(Supplementary Fig. 2). Thus, these data indicate that the selective growth inhibition of BRCA deficient cells by 6-OH-dopa results from its ability to inactivate RAD52.
Small Molecule Inhibition of RAD52 Causes DNA Damage and Apoptosis in BRCA Deficient Cells
Since RAD52 is recruited to DNA breaks caused by replicative stress (Fig. 3a)(Lisby et al., 2001; Wray et al., 2008), the reduced viability of BRCA deficient cells due to RAD52 inhibition is likely caused by insufficient repair of DNA breaks caused by replication stress. Consistent with this, we observed that 6-OH-dopa treatment resulted in a substantial increase in DNA damage indicated by γH2AX foci, which was significantly more pronounced in BRCA deficient cells (Fig. 6j). We also found that 6-OH-dopa specifically increased apoptosis in BRCA deficient cells, indicated by annexin V staining (Fig. 6k). Taken together, our data indicate that in BRCA deficient cells 6-OH-dopa inactivation of RAD52 causes cell death due to insufficient repair of DNA breaks caused by regular replicative stress (Fig. 7a, right). In contrast, 6-OH-dopa inactivation of RAD52 in BRCA proficient cells has little or no effect on cell proliferation due to efficient HR repair of replication forks by the primary BRCA pathway (Fig. 7a, left).
Figure 7. Models of small molecule inhibition of RAD52.

a, Models of 6-OH-dopa action. b, Model of small molecule disruption of RAD52 undecamers. c, Models of small molecule disruption of RAD52 WT heptamer superstructures (left) and stacked heptamer superstructures (right).
DISCUSSION
Although previous genetic studies have identified RAD52 as a potential drug target for personalized medicine in BRCA deficient cancers(Feng et al., 2011; Lok et al., 2012), it remained unknown whether RAD52 could be inactivated by a small molecule which is a necessary step towards therapeutic development. Here, we identify and characterize 6-OH-dopa as a major allosteric inhibitor of RAD52 that selectively halts the growth of BRCA deficient cancer cells including those obtained from AML and CML patients. Our findings validate RAD52 as a therapeutic target for precision medicine in BRCA deficient cancers and showcase a novel allosteric mechanism of inhibition.
Although we initially identified multiple inhibitors of RAD52 in vitro, only 6-OH-dopa consistently inhibited SSA in cells while having minimal effects on HR and NHEJ. Considering that HR and NHEJ each require a host of proteins involved in nucleic-acid processing, signaling, and post-translational modifications, these data show that 6-OH-dopa exhibits a considerable amount of specificity for RAD52 in cells. Consistent with this, 6-OH-dopa had no effect on RAD51 which forms a similar heptamer ring structure with nearly identical dimensions as RAD52(Shin et al., 2003). Intriguingly, this characteristic ring-shape is common among replication and repair proteins including the proto-typical RecA recombinase(Kagawa et al., 2002; Patel and Picha, 2000; Shin et al., 2003; Singleton et al., 2002; Stasiak et al., 2000; Yu and Egelman, 1997). 6-OH-dopa, however, had very little effect on RecA (Supplementary Fig. 1) and exhibited a 10-fold higher IC50 for yeast Rad59 which shares 31.5% sequence identity with RAD52. Thus, the small molecule also showed a considerable amount of specificity for RAD52 in vitro. Importantly, 6-OH-dopa inhibited RAD52 foci in multiple cell lines, which unequivocally demonstrates its ability to inactivate RAD52 in vivo. Thus, although 6-OH-dopa is a catechol and has the potential to interfere with some assays non-specifically, the in vitro and cell data presented herein show that its mechanism on RAD52 is specific.
We discovered an unprecedented allosteric mechanism whereby multiple small molecules bind to RAD52 undecamer rings and promote their dissociation into dimers. To our knowledge, such a dramatic molecular transformation induced by a small molecule has never been reported. Considering that the putative ssDNA binding channel observed in crystal structures is constructed from all eleven subunits and encircles the entire undecamer ring(Kagawa et al., 2002; Singleton et al., 2002), small molecule dissociation of the ring is certain to abolish the ssDNA binding channel. Since a different potent inhibitor of RAD52 (RU-0098062) failed to dissociate the undecamer, the allosteric mechanism exhibited by 6-OH-dopa is clearly very specific. Small molecule inhibition of Mre11 ssDNA binding by an allosteric mechanism has also been reported, demonstrating the success of this inhibitory method(Shibata et al., 2014).
Since our data suggests that five small-molecules interact with the undecamer, we propose a model whereby one small-molecule binds to every other subunit interface (Fig. 7b). This scheme would account for approximately 5 small-molecule binding sites and provide a plausible mechanism by which 6-OH-dopa induces such a dramatic conformational change. For example, we propose that 6-OH-dopa binds to a region near the subunit interface that is critical for ring formation. This is supported by our data showing that the small molecule completely transforms undecamers into dimers when hydrophobic interactions are reduced by low salt. Such an interaction may induce a conformational change that inactivates ssDNA binding (Fig. 7b). A total of five small-molecule binding events at alternating interfaces may then trigger dissociation of RAD52 rings into dimers (Fig. 7b). In support of this model, previous studies suggest that particular residues involved in ssDNA binding also contribute to oligomerization or proper folding of the protein(Lloyd et al., 2005). Moreover, residues within the ssDNA binding domain promote self-association of RAD52 proteins in cells(Shen et al., 1996). Thus, 6-OH-dopa disruption of this region may also explain its ability to dissociate superstructures of the protein. A co-crystal structure of RAD52 1-209 bound to 6-OH-dopa is needed to identify the precise binding site.
Intriguingly, 6-OH-dopa exhibited a 31% lower IC50 for RAD52 WT (1.1 μM) compared to RAD52 1-209 (1.6 μM), indicating that fewer small molecules are required to inhibit the wild-type protein. Indeed, these data are consistent with the fact that RAD52 WT is composed of 36% fewer subunits than RAD52 1-209 and suggest that 36% fewer small molecules, for example ~3, bind the heptamer, presumably at alternating subunit interfaces (Fig. 7c, left). Since 6-OH-dopa precipitated RAD52 WT at high concentrations, we were unable to determine the number of binding sites by ITC.
Importantly, we found that 6-OH-dopa also altered the composition of RAD52 WT, which suggests it induces a similar conformational change as RAD52 1-209. For example, 6-OH-dopa enabled RAD52 WT to enter a native gel as multiple discrete protein complexes in a similar manner to RAD52 1-209. Since a previous report claimed that large RAD52 heptamer ring superstructures prohibit RAD52 WT from entering gel filtration columns(Van Dyck et al., 1998), we examined whether the small molecule affected the gel filtration profile of RAD52 WT. Indeed, 6-OH-dopa enabled a significantly higher concentration of RAD52 WT to enter the column as superstructures and double-heptamers. The small molecule, however, showed no evidence of dissociating heptamer rings into smaller complexes, which suggests the C-terminal region of RAD52 WT contributes to ring formation or stability. Together, these data demonstrate that 6-OH-dopa disrupts large superstructures of RAD52 heptamer rings that are normally excluded from native gels and gel filtration columns (Fig. 7c, left). Since a double heptamer was the smallest form of RAD52 WT detected by gel filtration, the possibility exists that RAD52 WT forms stacked double heptamers like RAD51 (Fig. 7c, right)(Shin et al., 2003). This modified model would explain why 6-OH-dopa is unable to fully disrupt these structures. Given that RAD52 superstructures and high density RAD52 repair centers have been shown to promote DSB repair in vitro and in vivo(Lisby et al., 2003; Lisby et al., 2001; Lloyd et al., 2002), respectively, and human RAD52 has been shown to self-associate in cells(Shen et al., 1996), the ability of the small molecule to disrupt heptamer ring superstructures likely contributes to its inhibitory effect in vivo.
Lastly, we demonstrated that 6-OH-dopa selectively halts the growth of a variety of BRCA deficient cancer cells. For example, 6-OH-dopa treatment caused significant growth defects in BRCA1 mutated triple negative breast cancer cell lines, but had little or no effect in the same cell lines complemented with wild-type BRCA1. Small molecule growth inhibition of BRCA2 deficient cells, including the CAPAN-1 pancreatic cancer cell line, was also observed. Importantly, 6-OH-dopa also selectively halted the proliferation of BRCA deficient AML and CML patient cells. Moreover, the small molecule caused a similar growth defect in these and other BRCA deficient cells as olaparib which was recently approved by the FDA. Hence, these data suggest that pharmacological inhibition of RAD52 may prove as a successful alternative for treating BRCA deficient cancers. Finally, we showed that 6-OH-dopa caused an increase in DNA damage and apoptosis in BRCA deficient cells. Considering that RAD52 is heavily recruited to stalled replication forks (Fig. 3a)(Lisby et al., 2001; Wray et al., 2008), our data support a model whereby 6-OH-dopa selectively kills BRCA deficient cells by preventing RAD52 repair of DNA breaks caused by replication stress (Fig. 7a).
In summary, our results demonstrate small molecule disruption of RAD52 rings as a promising mechanism for precision medicine in BRCA deficient cancers. Since RAD52 acts as a backup DNA repair factor in normal cells, and RAD52 null mice show no major phenotypes, we anticipate that pharmacological inhibition of RAD52 in patients will enable selective killing of BRCA deficient cancer cells while minimizing the risk of side effects in normal cells.
SIGNIFICANCE
Tumor suppressor proteins BRCA1 and BRCA2, which play major roles in homologous recombination (HR) and genome maintenance, are mutated in subsets of ovarian and breast cancers and such inactivating BRCA mutations strongly predispose women to breast and ovarian cancer. Since BRCA deficient cells are defective in HR, they are highly susceptible to drugs that cause DNA damage or suppress DNA repair. Therefore, BRCA deficient cancers can be specifically targeted for killing via so-called precision medicines. Because suppression of the backup recombination factor RAD52 has been shown to cause synthetic lethality in BRCA deficient cells, RAD52 is a promising drug target for precision medicine in HR defective cancers. Here, we identify and characterize 6-hydroxy-DL-dopa (6-OH-dopa) as an effective small-molecule inhibitor of RAD52 in vitro and in cells. Unexpectedly, 6-OH-dopa acts via an allosteric mechanism, whereby it dimerizes undecamer rings of the RAD52 single-strand DNA (ssDNA) binding domain whose activity is essential for the survival of BRCA deficient cells. 6-OH-dopa also disrupts super-structures of full-length RAD52 hepatamer rings, suppresses RAD52 foci formation at DNA damage, and selectively inhibits RAD52 mediated recombination in cells. Consistent with its ability to inhibit RAD52, 6-OH-dopa selectively kills BRCA deficient cancer cells including those obtained from leukemia patients. These findings indicate that RAD52 is a ‘druggable’ target and suggest that small-molecule disruption of RAD52 rings may be an effective form of precision medicine for BRCA deficient cancers.
EXPERIMENTAL PROCEDURES
Protein purification
RAD52 WT. A Histidine-tagged RAD52 WT expression vector (pFB581)(Benson et al., 1998) was transformed into Rosetta2(DE3)/pLysS cells (Stratagene). Freshly grown colonies were resuspended in 16 2.8 L Fernbach flasks each containing 1 L of LB broth supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. The flasks were shaken at room temp until the optical density at 600 nm reached 0.5, then 0.8 mM IPTG was added for an additional 4 h. The biomass was then collected by centrifugation and stored at −80°C. Fro zen pellets were thawed on ice and resuspended in lysis buffer (25 mM TrisHCl pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 10 mM imidazole pH 8, 0.05 % (v/v) NP-40 substitute (Thermo Scientific), 2 mM DTT, 10 mM PMSF and Complete cocktail protease inhibitors (Roche)). Resuspended cells were sonicated on ice with constant stirring then spun down twice at 35,000 rpm for 30 min at 4° C. The clarified cell lysate was loaded onto a 5 ml His-Trap column (GE Lifesciences) then washed in the following steps: 20 column volumes (CV) lysis buffer, 10 CV lysis buffer with 30 mM imidazole, 10 CV lysis buffer with 60 mM imidazole, and 5 CV lysis buffer with 80 mM imidazole. The bound protein was eluted in two steps with 200 mM and 500 mM imidazole, respectively, in buffer containing 25 mM TrisHCl pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.005% (v/v) NP-40 substitute, 1 mM DTT. Fractions containing RAD52 were pooled, dialysed against buffer A (25 mM TrisHCl 7.5, 75 mM NaCl, 10 % glycerol, 1 mM DTT, 0.001% NP-40) then loaded onto a 5 ml Q-Sepharose column and washed with the same buffer. The bound protein was then eluted in six steps 1 CV each with buffer A containing increasing NaCl concentration as follows: 100 mM, 125 mM, 150 mM, 200 mM, 250 mM, 300 mM. Fractions containing pure RAD52 were dialysed against storage buffer (25 mM TrisHCl pH 7.5, 100 mM NaCl, 10% glycerol and 1 mM DTT). RAD52 WT was concentrated to 2.5 mg/ml and stored in aliquots at -80° C. All steps were performed at 4° C. RAD52 1-209 was purified as described(Singleton et al., 2002).
High-throughput screening
2 μl of 350 nM RAD52 WT was added to 384-well plates (Greiner Bio-One) containing 16 μl of buffer B (31.3 mM TrisHCl pH 7.5, 1.25 mM DTT, 31.3 mM NaCl, 0.013% NP-40, 0.63 mM MgCl2, and 0.13 mg/mL BSA) and 18.75 μm of individual compounds from a highly diverse 18,304 small-molecule library and the Sigma Lopac collection. 2 μl of 100 nM FAM-conjugated ssDNA (RP316FAM) was then added and plates were centrifuged at 1,000 rpm for 30 s. After at least 30 min of incubation, fluorescence polarization was determined using a BioTek plate reader. High and low signal controls included and lacked RAD52, respectively. Reactions were performed at room temp and components were added using a Thermo MultiDrop Combi liquid handler.
IC50 determination
40 nM RAD52 WT, 85 nM RAD52 1-209, 280 nM RAD51, or 300 nM RAD59 was mixed with reaction buffer C (25 mM TrisHCl pH 7.5, 1 mM DTT, 25 mM NaCl, 0.01% NP-40, 0.5 mM MgCl2, and 0.1 mg/mL BSA), 10 nM FAM-conjugated ssDNA (RP316FAM) and indicated amounts of 6-OH-dopa in a total volume of 20 μl, then incubated at room temp for 30 min. Reactions with RAD51 additionally contained 1 mM ATP. Fluorescence polarization was then determined using a Clariostar (BMG Labtech) or BioTek plate reader. IC50 was determined by calculating the concentration of 6-OH-dopa required to inhibit ssDNA binding by 50% based on fluorescence polarization.
Supplementary Material
Highlights.
Small molecule inhibition of RAD52 activity in vitro and in vivo
Small molecule dimerization of RAD52 undecamer rings
Small molecule disruption of RAD52 ring superstructures
Small molecule inhibition of RAD52 selectively kills BRCA deficient cells
Acknowledgments
Research was funded by: Department of Defense (Breast Cancer Breakthrough Award W81XWH-14-1-0344), and in part by National Institutes of Health (grant 1R01CA190237-01) and Temple University School of Medicine start-up funds to R.T.P., and; National Institutes of Health (grant 1R01CA186238) to T.S. We are grateful to M. Jacobson (Temple University School of Medicine) for use of the Clariostar (BMG Labtech) plate reader, S.C. West (Cancer Research UK) for RAD52 WT and 1-209 expression vectors, J. Stark (Beckman Research Institute City of Hope) for U2OS reporter cell lines, P. Sung (Yale University) for RAD51 expression vector, S. Powell (Memorial Sloan-Kettering Cancer Center) for eGFP-RAD52 expression vector, N. Johnson (Fox Chase Cancer Center) for MDA-MB-436 cells, and S.C. Kowalczykowski (University of California at Davis) for Rad59 protein.
Footnotes
AUTHOR CONTRIBUTIONS
G.C., S.M., K.S., R.T.P. performed, designed and interpreted experiments. T.K. purified RAD52 WT, assisted with light scattering experiments, and provided conceptual and editorial input. A.L. was responsible for ITC experiments and analysis. M.A. performed light scattering experiments and provided editorial input. S.M. purified RAD52 1-209 and eGFP-RAD52, and provided conceptual and editorial input. J.F.G. provided editorial input. T.S. provided conceptual input and was responsible for experiments with patient cells. R.T.P. conceived the study and wrote the manuscript.
Supplemental Information includes Supplemental Experimental Procedures and six figures, and can be found with this article online at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benson FE, Baumann P, West SC. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature. 1998;391:401–404. doi: 10.1038/34937. [DOI] [PubMed] [Google Scholar]
- Ceccaldi RL, JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, O’Connor KWK, PA, Elledge SJ, Boulton SJ, Yusufzai YT, D’Andrea AD. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015:517. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Buechelmaier ES, Powell SN. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Molecular and cellular biology. 2013;33:387–395. doi: 10.1128/MCB.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng H, Roy D, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013 doi: 10.1182/blood-2013-05-501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, Vermeulen W, Hoeijmakers JH, Kanaar R. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 2002;21:2030–2037. doi: 10.1093/emboj/21.8.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:686–691. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis JC, McCarthy A, Thomsen MK, Ashworth A, Swain A. Brca2 and Trp53 deficiency cooperate in the progression of mouse prostate tumourigenesis. PLoS Genet. 2010;6:e1000995. doi: 10.1371/journal.pgen.1000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature reviews Molecular cell biology. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- Gunn A, Bennardo N, Cheng A, Stark JM. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. The Journal of biological chemistry. 2011;286:42470–42482. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- Ji Y, Tulin AV. The roles of PARP1 in gene control and cell differentiation. Curr Opin Genet Dev. 2010;20:512–518. doi: 10.1016/j.gde.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, Shibata T, Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Molecular cell. 2002;10:359–371. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell research. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Heyer WD. Who's who in human recombination: BRCA2 and RAD52. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:441–442. doi: 10.1073/pnas.1016614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd JA, Forget AL, Knight KL. Correlation of biochemical properties with the oligomeric state of human rad52 protein. The Journal of biological chemistry. 2002;277:46172–46178. doi: 10.1074/jbc.M207262200. [DOI] [PubMed] [Google Scholar]
- Lloyd JA, McGrew DA, Knight KL. Identification of residues important for DNA binding in the full-length human Rad52 protein. J Mol Biol. 2005;345:239–249. doi: 10.1016/j.jmb.2004.10.065. [DOI] [PubMed] [Google Scholar]
- Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2012 doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Powell SN. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin Cancer Res. 2012;18:6400–6406. doi: 10.1158/1078-0432.CCR-11-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- Mai PL, Chatterjee N, Hartge P, Tucker M, Brody L, Struewing JP, Wacholder S. Potential excess mortality in BRCA1/2 mutation carriers beyond breast, ovarian, prostate, and pancreatic cancers, and melanoma. PLoS One. 2009;4:e4812. doi: 10.1371/journal.pone.0004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature reviews Molecular cell biology. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Picha KM. Structure and function of hexameric helicases. Annual review of biochemistry. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- Petukhova G, Stratton SA, Sung P. Single strand DNA binding and annealing activities in the yeast recombination factor Rad59. The Journal of biological chemistry. 1999;274:33839–33842. doi: 10.1074/jbc.274.48.33839. [DOI] [PubMed] [Google Scholar]
- Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, Wolanin K, Skowronek K, Nieborowska-Skorska M, Dasgupta Y, Skorski T, Piwocka K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle. 2014;13:3727–3741. doi: 10.4161/15384101.2014.965013. [DOI] [PubMed] [Google Scholar]
- Rijkers T, Van Den Ouweland J, Morolli B, Rolink AG, Baarends WM, Van Sloun PP, Lohman PH, Pastink A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Molecular and cellular biology. 1998;18:6423–6429. doi: 10.1128/mcb.18.11.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, Garcia JA, Naidoo J, Longgood J, Frantz DE, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. 2013;9:271–276. doi: 10.1038/nchembio.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Peterson SR, Comeaux JC, Zastrow D, Moyzis RK, Bradbury EM, Chen DJ. Self-association of human RAD52 protein. Mutat Res. 1996;364:81–89. doi: 10.1016/0921-8777(96)00025-0. [DOI] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, et al. Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J. 2003;22:4566–4576. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. Structure of the single-strand annealing domain of human RAD52 protein. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Molecular and cellular biology. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasiak AZ, Larquet E, Stasiak A, Muller S, Engel A, Van Dyck E, West SC, Egelman EH. The human Rad52 protein exists as a heptameric ring. Curr Biol. 2000;10:337–340. doi: 10.1016/s0960-9822(00)00385-7. [DOI] [PubMed] [Google Scholar]
- Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature reviews Molecular cell biology. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Mol Aspects Med. 2013;34:1124–1137. doi: 10.1016/j.mam.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Hallmarks of 'BRCAness' in sporadic cancers. Nature reviews Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E, Hajibagheri NM, Stasiak A, West SC. Visualisation of human rad52 protein and its complexes with hRad51 and DNA. J Mol Biol. 1998;284:1027–1038. doi: 10.1006/jmbi.1998.2203. [DOI] [PubMed] [Google Scholar]
- Van Dyck E, Stasiak AZ, Stasiak A, West SC. Visualization of recombination intermediates produced by RAD52-mediated single-strand annealing. EMBO Rep. 2001;2:905–909. doi: 10.1093/embo-reports/kve201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P, Zong X, Laplana M, Wei Y, Han Y, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet. 2014;46:736–741. doi: 10.1038/ng.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J, Liu J, Nickoloff JA, Shen Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res. 2008;68:2699–2707. doi: 10.1158/0008-5472.CAN-07-6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sugiyama T, Kowalczykowski SC. DNA annealing mediated by Rad52 and Rad59 proteins. The Journal of biological chemistry. 2006;281:15441–15449. doi: 10.1074/jbc.M601827200. [DOI] [PubMed] [Google Scholar]
- Yu X, Egelman EH. The RecA hexamer is a structural homologue of ring helicases. Nat Struct Biol. 1997;4:101–104. doi: 10.1038/nsb0297-101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.