

Abstract
Reliance on Ca2+ signaling has been well-preserved through the course of evolution. While the complexity of Ca2+ signaling pathways has increased, activation of transcription factors including CREB by Ca2+/CaM-dependent kinases (CaMKs) has remained critical for long-term plasticity. In C. elegans, the CaMK family is made up of only three members, and CREB phosphorylation is mediated by CMK-1, the homologue of CaMKI. CMK-1 nuclear translocation directly regulates adaptation of thermotaxis behavior in response to changes in the environment. In mammals, the CaMK family has been expanded from three to ten members, enabling specialization of individual elements of a signal transduction pathway and increased reliance on the CaMKII subfamily. This increased complexity enables private line communication between Ca2+ sources at the cell surface and specific cellular targets. Using both new and previously published data, we review the mechanism of a γCaMKII-CaM nuclear translocation. This intricate pathway depends on a specific role for multiple Ca2+/CaM-dependent kinases and phosphatases: α/βCaMKII phosphorylates γCaMKII to trap CaM; CaN dephosphorylates γCaMKII to dispatch it to the nucleus; and PP2A induces CaM release from γCaMKII so that CaMKK and CaMKIV can trigger CREB phosphorylation. Thus, while certain basic elements have been conserved from C. elegans, evolutionary modifications offer opportunities for targeted communication, regulation of key nodes and checkpoints, and greater specificity and flexibility in signaling.
Keywords: Calcium, Calmodulin, Signaling to the nucleus, CaM kinase
1. Introduction
It is a privilege and pleasure to honor Ernesto Carafoli by contributing an article on one of his favorite subjects: Ca2+ signaling and the cell nucleus. Just as Ca2+ signaling has both local and global aspects, Ernesto’s interest in nuclear Ca2+ signaling is a local and dramatic expression of his many global passions. These range from biochemistry in the service of biophysical function to the molecular modulation of Ca2+ handling systems in the surface membrane and various cellular elements – plasma membrane, mitochondrion, endoplasmic reticulum and nucleus.
There are many organelles in the Carafoli portfolio, but hardly any that could be deemed more fascinating than the nucleus. It has both location and function going for it: the nucleus resides deep within the cell, marks the transition from prokaryotes to eukaryotes, contains the DNA, and harbors transcription and alternative splicing. By regulating gene expression, the nucleus is well equipped to play a dominant role in cellular plasticity and homeostasis. Nevertheless, the cellular position of the nucleus poses a problem: how are events at the surface membrane swiftly and reliably communicated to the nucleus? Such information transfer has long fascinated many scientists, Ernesto Carafoli among them, and is the topic of this review. Other papers in this volume will attest to the importance of intracellular calcium (Ca2+), calmodulin (CaM), and Ca2+/CaM-dependent protein kinases (CaMKs) in other aspects of cellular function. CaM kinase cascades can also drive phosphorylation of CREB and other transcription factors (TFs) in animal species ranging from the nematode worm C elegans to mammals like Homo sapiens. This article reviews our recent work on such signaling in mammalian neurons [1], presents new data from rat cortical excitatory neurons (Figs. 2, 4 and 5), and weaves our collected observations into the broader context of Ca2+ signaling, CaM-dependent kinases & phosphatases, and learning & memory, as illuminated by the pioneering work of others.
Fig. 2.
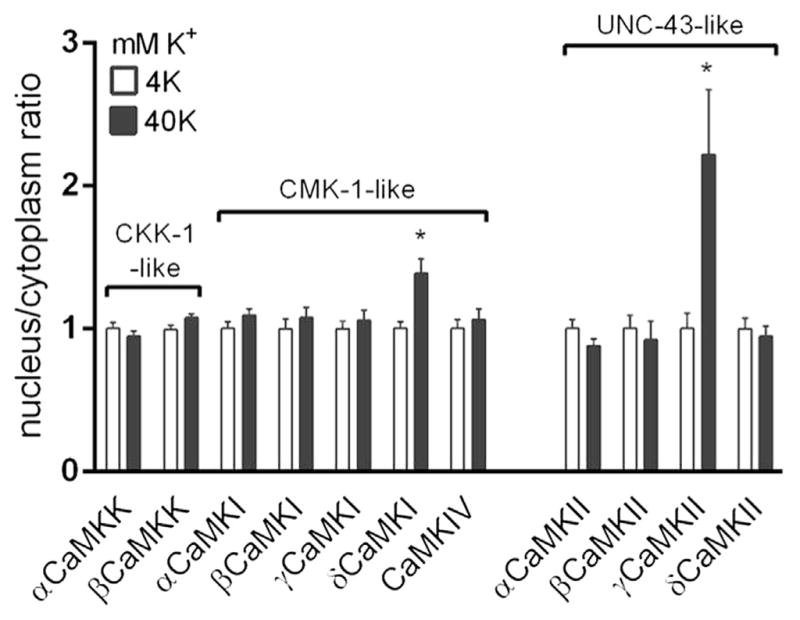
In cortical excitatory neurons, 40 mM K+ depolarization triggers nuclear translocation of only δCaMKI and γCaMKII, but not other CaM Kinase family members. The C. elegans homologue of each protein is noted. All values of nucleus/cytoplasm ratio are normalized to the baseline ratio.
Fig. 4.

A γCaMKII-dependent pathway triggers pCREB and gene expression in cortical excitatory neurons. (A) CREB phosphorylation was measured in cortical excitatory neurons following (Ai) field stimulation at various frequencies for 60 s, (Aii) 10 Hz field stimulation for variable duration, or (Aiii) 40 mM K+ stimulation for variable duration. pCREB intensity depends on both the frequency and duration of stimulation.(B) Nuclei isolated from cultured cortical neurons and subjected to western blot analysis to probe for γCaMKII. 40 mM K+ stimulation of intact cells induced an increase of nuclear γCaMKII that was prevented by Nim. Lamin B is a nuclear marker. (C) Cortical neurons transfected with either γCaMKII shRNA or nonsilencing control shRNA, stimulated with 40 mM K+ for 60 s and stained for CaM. γCaMKII knockdown prevented CaM translocation. (D) Cortical neurons expressing either γCaMKII shRNA or a nonsilencing control shRNA. Stimulated pCREB response prevented by γCaMKII knockdown. (E) pCREB response was prevented by CaV1 inhibitor Nim, CaM Kinase inhibitor KN93, calcineurin inhibitor CsA, and CaMKIV shRNA. KN92, the inactive congener of KN93, had no effect. (F) CREB phosphorylation in response to stimulation was reduced by inhibition of PP2A with 20 nM okadaic acid (OA), and increased with 2 μM OA. (G) 40 mM K+ depolarization for 90 s, followed by a 90 min incubation at rest, triggered an increase in c-fos immunostaining intensity. This increase was prevented by Nim and KN93 and reduced by KN92 (although c-fos with KN93 treatment was significantly lower than c-fos with KN92). Panels B–D modified from ref. [1].
Fig. 5.

Nuclear delivery of Ca2+/CaM is sufficient to drive CREB phosphorylation. (A) Testing the sufficiency of Ca2+/CaM to trigger pCREB, by using a nuclear-localized, caged CaM. (B) HA-tagged NLS-Nrgn or NLS-Nrgn S36D (not shown; lacks apoCaM binding) successfully targeted to the nucleus. Scale bar represents 10 μm. (C) SCG neurons expressing γCaMKII shRNA, stimulated with 40 mM K+ for 10 s. NLS-Nrgn (middle column) but not NLS-Nrgn S36D (right column) was able to restore the pCREB response (top row), consistent respectively with the ability or inability to harbor CaM for release upon nuclear Ca2+ elevation. Bottom row shows fluorescently labeled Nrgn constructs, if present. Under basal conditions, overexpressing NLS-Nrgn or NLS-Nrgn S36D did not affect pCREB levels. (D) In SCG neurons expressing γCaMKII shRNA, the restoration of pCREB by NLS-Nrgn was prevented by specific CaMKK inhibitor STO-609, consistent with the idea that CaM delivery operates upstream of the activation of nuclear CaMKK-CaMKIV-pCREB cascade.
1.1. Evolutionary perspective on CaM kinase cascades and transcriptional regulation
It is intriguing to take an evolutionary view of the mechanisms that power CaMK-dependent communication between neuronal activity and nuclear gene expression. In both worm and human, signaling to the nucleus involves the same pattern of basic cellular processes: surface/cytoplasmic initiation, cytonuclear translocation and nuclear TF phosphorylation (Fig. 1A). What is not conserved is the assignment of CaM kinases for these various functions. It appears that the expansion of the CaMK family (Fig. 1B) has enabled the assignment of different family members for these processes. Whereas two kinds of CaMK sufficed for signaling to the nucleus in worm neurons, mammalian neurons rely on at least five different isoforms of CaMK. Some obvious questions come to mind. Do each of these family members play distinct roles? How do their actions fit together in an overall cascade? What is the functional benefit of the increased complexity?
Fig. 1.

CaM Kinase-dependent pathways of signaling to the nucleus across evolution. (A) Communication between the cell surface/cytoplasm and the nucleus via translocation of CaMKs is a common feature of neurons in C. elegans and mammals. In worm neurons, CKK-1 conveys the signal by phosphorylating CMK-1, which translocates to the nucleus to phosphorylate nuclear transcription factors. In mammalian excitatory neurons, more CaMKs are involved. Multiple CaMKs (CaMKK, CaMKIV, CaMKII), including the dedicated shuttle protein γCaMKII, work synergistically to promote nuclear signaling. The possible functional advantages for using multiple CaMKs in mamals are briefly listed in italics. “Tagging” refers to the proposed marking of synapses for subsequent capture of newly recruited resources. (B) CaM Kinases have been conserved across evolution. Mammalian α/ βCaMKK are homologous to C. elegans, CKK-1; Mammalian α/β/γ/δCaMKI and CaMKIV are homologous to C. elegans CMK-1; Mammalian α/β/γ/δCaMKII are homologous to C. elegans UNC-43. Red font indicates proteins that are involved in a cytoplasm to nuclear signaling pathway, either in worm or mammal. In the simplified representation of CaMK structures, “Catalytic” refers to the catalytic domain and “CaM” refers to the Ca2+/CaM binding/regulatory domain, both present in all three subfamilies. The association domain supports multimerization and is exclusive to the CaMKII subfamily.
The well-known mammalian CaM kinase family (Fig. 1B) is comprised of the CaMKII subfamily (α, β, γ, and δ genes), the CaMKK subfamily (α and β genes), and the CaMKI/IV subfamily (CaMKIα, -β, - γ, and - δ genes, together with CaMKIV, a structurally similar homolog) [2]. Through evolution, this family has been expanded from an ancestral state where each of these subfamilies consisted of only one member. For example, in C. elegans, the single homologues of CaMKK, CaMKI/IV and CaMKII are, respectively, CKK-1, CMK-1 and UNC-43 (where UNC stands for “uncoordinated”).
Both C. elegans and mammals use a CaMK cascade to support learning and memory, but they appear to do so in different ways. In nematode neurons (see Box 1), CKK-1 (the CaMKK equivalent) transduces temperature-dependent cytoplasmic signals and phosphorylates CMK-1 (the CaMKI/IV equivalent). CMK-1 then plays two roles, as the cytonuclear translocator and the kinase that activates nuclear gene expression. By phosphorylating a nuclear-resident transcription factor (CRH-1, a CREB homolog, and HSF-1 are two of the best studied), a memory of an environmental state is encoded.
BOX 1. Signaling to the Nucleus in C. elegans Neurons.
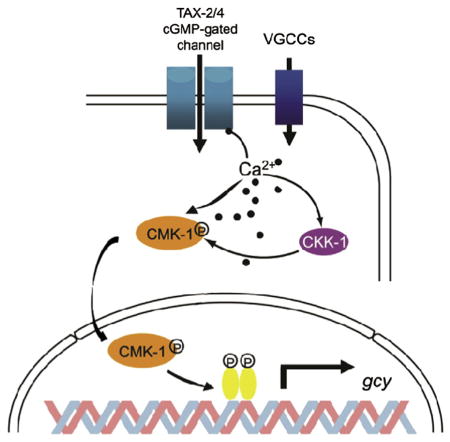
The CaM Kinase-dependent nuclear signaling pathway in nematodes is inherently interesting and serves as motivation for this review of signaling in mammalian neurons. On December 3, 2014, two research articles [59,60] were published in Neuron on the subject of C. elegans’ ability to alter thermotaxis based on past experience. These studies built on years of work showing that CaM Kinase cascade signals to CREB and other transcription factors in the nematode [61,62]. Both studies characterized experience-dependent response properties and homeostasis in thermosensory neurons, within a full behavioral context.
In the worm’s head, all the elements of the CaM Kinase cascade operate in individual cells that mediate specific behaviors. AFD neurons are known to mediate thermotaxis toward a preferred temperature, while the FLP neurons modulate thermotaxis away from noxious thermosensory stimuli. CMK-1 was identified through a forward genetic screen, where a gain of function mutant demonstrated abnormally low thermal avoidance. Remarkably, these studies demonstrate that activity-dependent translocation of a CaM kinase is directly responsible not only for transcriptional regulation, but also for a clear behavioral response to the external environment.
Schild et al. and Yu et al. demonstrated, in two separate neuron types and to two different physiological ends, that this CaMK signaling is functionally important in experience-dependent thermotaxis and heat avoidance. In AFD neurons, a threshold for temperature-evoked activity is set based on the animal’s cultivation temperature. Nematodes adapted their thermotaxis behavior, tolerating warmer temperatures if they had been incubated at higher temperatures. CMK-1 regulates this physiological set point via modulation of guanylyl cyclase gene (gcy) expression levels [60]. In FLP nociceptors, cytoplasmic CMK-1 promotes thermal avoidance in the face of noxious temperatures. In contrast, nuclear CMK-1 produces analgesia and a decrease in thermal avoidance [59]. Interestingly, the mechanism of Ca2+ signaling is conserved between these two scenarios. CMK-1 translocates to the nucleus in a CKK-1 dependent manner, regulating gene transcription that controls behavioral output (portrayed in the schematic figure above, adapted from Ref. [60]).
Information transfer between a surface membrane and the nucleus is clearly a key to neuronal adaptation, and these studies are an elegant example of one such mechanism of nuclear communication. This CaMK cascade is particularly notable for its direct regulation of a behavioral output, a discovery for which we thank the relative simplicity of the nematode nervous system.
We have discovered a biochemical pathway for nuclear signaling in mammalian excitatory neurons [1], but it is more multilayered than in the worm – the various steps in signaling call for the participation of at least 5 different CaMKs (players listed in Fig. 1). Some features seem roughly similar to that in the nematode. The enzyme cascade of CKK-1 phosphorylating CMK-1 exists in the form of CaMKK phosphorylating CaMKI or CaMKIV in mammalian neurons [3,4]. CaMKIV mainly stays within the nucleus and is the CREB Kinase of mammalian neurons, as has been demonstrated in hippocampal, cortical and sympathetic neurons [1,5]. In contrast, CaMKI is capable of phosphorylating CREB in vitro, but it does not appear active as a CREB kinase in mammalian excitatory neurons. In cortical glutamatergic neurons subjected to shRNA knockdown of each of the individual CaMKI isoforms, CREB phosphorylation was no different than with scrambled shRNA controls (S.M. Cohen, unpublished data). Thus, in contrast to CMK-1 in C. elegans, in mammalian excitatory neurons CaMKIV, not CaMKI, is required for activating CREB.
Additional features are quite different in mammalian neurons, as depicted in Fig. 1A. First, whereas there is no known role for worm CaMKII (UNC-43) in signaling to the nuclear CaMKs, various mammalian CaMKIIs can do so [1,6]. Second, worm and mammalian neurons use different strategies for conveying information to the nucleus. Whereas CMK-1 plays two roles in the worm, both as nuclear translocator and CREB kinase (denoted by gray bar in Fig. 1A), these tasks have been relegated to two different CaMKs: CaMKII and CaMKIV. Thus, the functions of translocator and TF kinase appear to have been split up in mammals and given to separate players. Later in this paper, we will argue that the assignment of specific roles to individual CaMKs, enabled by expansion of the CaMK family, confers important functional advantages.
1.2. Cycling of the CREB kinase itself?
Before writing off a worm-like scenario for mammalian neurons, we must consider a hypothesis proposed by Tony Means. Having pointed out that CaMKIV is the CREB kinase [7,8], he writes further [9]: “However, it has been unclear where in the cell CaMKIV becomes activated (CaMKIV is primarily nuclear, and CaMKK is primarily cytoplasmic). One possibility is that CaMKIV (unaccompanied or within a complex), cycling in and out of the nucleus, becomes activated in the cytoplasm upon a Ca2+ signal. Activated CaMKIV could then translocate back into the nucleus to potentiate transcription. Because activated CaMKIV would no longer require elevated Ca2+ levels for its activity, the implication is that cytoplasmic Ca2+ signals, even if they never crossed into the nucleus, could lead to activation of CaMKIV and nuclear CaMKIV functions.”
That CaMKIV seems to be nucleus-resident, with much lower immunoreactivity in the cytoplasm, does not exclude this cycling hypothesis. If the flux of activated CaMKIV into the nucleus were precisely balanced by a countermovement of deactivated CaMKIV cycling back out, immunocytochemistry would detect no net change in CaMKIV level upon depolarization, and only an increase in pCaMKIV signal, just as observed [1,6]. However, because the hypothesis calls for “activated CaMKIV [that would] no longer require elevated Ca2+ levels for its activity”, the cytonuclear signaling should be unaffected by a nucleus-restricted Ca2+/CaM trap. On the contrary, our lab and Mark Mayford’s group found that a nucleus-restricted Ca2+/CaM sponge protein, devised by John Dedman [10], was effective in preventing CREB phosphorylation [11,12]. This provides compelling evidence against the sufficiency of CaMKIV cycling as mechanism of cytonuclear communication.
With this possibility ruled out, remaining candidates for the translocating agent that mediates CREB activation are: free Ca2+ itself, a CREB kinase kinase, or Ca2+/CaM. We will now address these remaining possibilities, summarizing the evidence for or against them, and considering their functional implications.
1.3. Inward spread of the rise in free Ca2+?
Many years ago, our lab teamed up with Roger Tsien’s to visualize the changes in free cytoplasmic Ca2+ in vertebrate neurons with fura-2 [13,14]. Viewed even with early imaging approaches, the inward spread of Ca2+ from the cell periphery was both visually striking and apparently simple. So much so that it continues to dominate thinking about the spatiotemporal spread of cellular information within cells. In the case of excitation-transcription coupling, what could be more direct than relying on the inward spread of free Ca2+ to propagate the signal to transcribe genes? On the contrary, multiple observations weigh against this possibility. First, experiments using the Ca2+ chelators EGTA and BAPTA provided evidence against the global Ca2+ hypothesis [6,15]. CREB phosphorylation was not prevented when cells were loaded with EGTA (a slow on-rate Ca2+ chelator) but was fully eliminated by BAPTA (a fast on-rate Ca2+ chelator). Because EGTA very effectively buffers all global Ca2+, including nuclear Ca2+ increases, rises in nuclear Ca2+ could be excluded as decisive in governing CREB phosphorylation. As a consequence of their different kinetics of binding free Ca2+, EGTA only allows Ca2+ rises close to Ca2+ sources (~1 μm), while BAPTA suppresses Ca2+ rises even closer (~0.1 μm) [11]. The differential effect of the Ca2+ chelators implied that the critical Ca2+ rise takes place close to the mouth of the Ca2+channel and not at the nucleus.
1.4. Translocation of CREB kinase kinase?
Several prominent reviews have highlighted translocation of CaMKK as a way to connect cytoplasmic Ca2+ increases and CaMKIV activation [4,16,17]. However, our data indicate that CaMKK activation of CaMKIV is critical, but neither αCaMKK nor βCaMKK undergo a net redistribution [1,11]. Indeed, there is no net translocation of any of the 11 multifunctional CaMK family members except for γCaMKII and possibly δCaMKI (Fig. 2). δCaMKI, whose nuclear translocation was first proposed by Sakagami et al. [18], cannot phosphorylate CaMKIV. δCaMKI-mediated CREB phosphorylation is also unlikely, in light of data using δCaMKI-directed shRNA (S.M. Cohen, unpublished data). Furthermore, γCaMKII-kinase dead experiments have shown that it is not working as a CREB kinase or CREB kinase kinase [1]. Taken together, these considerations weigh against the idea that nuclear translocation of a CREB kinase kinase is directly responsible for CREB activation in mammalian excitatory neurons.
1.5. Translocation of Ca2+/CaM?
Another commonly held view is that CaM is ubiquitous and stands ready to respond to a rise in Ca2+ signal. To the contrary, measurements using Anthony Persechini’s FRET-based probe demonstrate that the amount of free Ca2+/CaM can be limiting, presumably because of Ca2+/CaM binding to high affinity binding partners [19,20]. A hypothesis proposing the translocation of free Ca2+/CaM followed naturally from availability of an indicator for it. Following the finding of CaM nuclear translocation following seizures [21], Deisseroth et al. [11] used controlled stimulation to demonstrate CaM translocation in three ways: immunocytochemistry, nuclear isolation and Western blotting, and live imaging of meroCaM. Experimental evidence linking CaM translocation and pCREB came from 1) the correlation between increased nuclear CaM and nuclear pCREB on an individual cell basis following stimulation, and 2) the abolition of CREB phosphorylation in neurons transfected with a nuclear-localized CaM binding protein [10,11]. Stimulus-driven CaM translocation was soon confirmed in other tissues. The major difficulty that remained with the hypothesis was to understand what drove the CaM translocation.
Translocation of free Ca2+/CaM?
One biochemical rationale for CaM translocation was that it operated by a diffusion trap mechanism, with diffusion driven by the low free CaM in the nucleus [22]. Indeed, Teruel et al. found that the net movement of fluorescently labeled CaM was directed toward the nucleus but could be reversed by deliberately increasing the CaM buffering power in the cytoplasm [22]. This lent credibility to the notion that free Ca2+/CaM can move on its own, but still left the puzzle of how the activator function of CaM could be preserved over the tens of seconds or minutes required for diffusional redistribution to occur.
Shuttling of Ca2+/CaM by a dedicated co-transporter?
The idea of Ca2+/CaM translocation via co-transport with a targeted carrier protein was also proposed [11,23]. This idea was attractive because co-transport offers the possibility of specificity in information transfer by directing Ca2+/CaM to a selected target. The existence of a binding partner was supported by experimental evidence showing that fusion of the Ca2+/CaM binding peptide M13 to CaM prevents redistribution [23]. While promising, this hypothesis was difficult to test directly because, if Ca2+/CaM travels in an encapsulated form, it cannot readily be detected with a proteinaceous indicator, in contrast to what is possible with free Ca2+ or free Ca2+/ CaM. One must simply know the nature of the encapsulator.
Despite much effort, the search for the binding partner was in vain; CaMKK α and β, CaMKI α, β, and γ, CaMKII α, β, δ, and CaMKIV failed to move. Until recently, δCaMKI was a lone exception [18]. Among CaM Kinases, αCaMKII (and to a lesser extent, βCaMKII) captured the interest of neuroscientists and cell biologists because of their role in plasticity at glutamatergic synapses. Meanwhile, γCaMKII received little attention, possibly because its wide tissue distribution was taken as weighing against an important role in the brain.
The recent discovery of γCaMKII as the carrier protein has shed new light on intracellular information transfer. Ma et al. demonstrated in sympathetic neurons that γCaMKII translocates to the nucleus following 10 Hz stimulation or depolarization with 40 mM K+ (Fig 3A) [1]. The nucleus to cytoplasm ratio of γCaMKII was correlated with that of CaM, suggesting that γCaMKII may indeed shuttle Ca2+/CaM to the nucleus (Fig 3B). Indeed, shRNA knockdown of γCaMKII prevented the nuclear redistribution of CaM (Fig 3C). As CaM translocation is relevant in terms of communicating information to the nuclear transcriptional apparatus, the next question was whether γCaMKII is critical for CREB phosphorylation. shRNA knockdown of γCaMKII prevented pCREB in response to 40 K depolarization, and the response was rescued by an shRNA-resistant γCaMKII construct (Fig 3D).
Fig. 3.

γCaMKII is a CaM shuttle, critical for signaling to pCREB in mammalian sympathetic neurons. (A) SCG neurons were stimulated at 10 Hz for 60 s or exposed to 40 mM K+ for 300 s and stained with a γCaMKII antibody. Scale bar represents 10 μm.(B) Single-cell correlation of nuclear:cytoplasmic intensity ratio between CaM and γCaMKII (R = 0.7) in response to 40 mM K+ stimulation for 300 s(C) SCG neurons expressing γCaMKII shRNA or nonsilencing control shRNA stimulated as in (B). Increased nuclear:cytoplasmic ratio for CaM was prevented by γCaMKII knockdown.(D) SCG neurons expressing γCaMKII shRNA or nonsilencing control shRNA were stimulated with 40 mM K+ for 10 s and stained for pCREB. The pCREB response was prevented by γCaMKII knockdown and rescued by overexpressing the shRNA-resistant γCaMKII construct γCaMKIIR. (E) Lentiviral-transduced SCG neurons expressing βCaMKII shRNA with βCaMKII shRNA resistant βCaMKIIR were stimulated with 40 mM K+ for 300 s and stained for pCREB. Nuclear increase of pCREB response was effectively rescued by shRNA-resistant βCaMKIIR. (F) Immunocytochemistry of SCG neurons examining localization of Ca2+ channels and signaling CaM kinase. Arrowheads, sites at the thin neuronal edge where overexpressed γA′ CaMKII S334E form puncta and colocalize with CaV1.3 channels upon 40 mM K+ stimulation for 60 s. Scale bar represents 1 μm. (G) SCG neurons expressing γA′ CaMKII or γA′ CaMKII S334E stimulated with 40 mM K+ for 300 s; γA′, but not γA′ S334E translocated to the nucleus upon stimulation. Inset, position of S334E point mutation, just carboxyl to the nuclear localization sequence (NLS). (H) Cartoon describing how the CaV1 channel acts as a signaling hotspot. Upon stimulation, γCaMKII is recruited to CaV1, where it is phosphorylated by βCaMKII at T287 and dephosphorylated by CaN at S334 before proceeding to the nucleus. Modified from Ref. [1].
How does γCaMKII shuttle Ca2+/CaM to the nucleus? The pathway begins at the CaV1 channel, where newly formed puncta of Ca2+/CaM-activated α/βCaMKII gather, along with γCaMKII and prebound calcineurin. α/βCaMKII phosphorylates γCaMKII at T287 to lock in Ca2+/CaM by a mechanism called “CaM trapping” [24,25]. Either βCaMKII shRNA knockdown, or replacement of native γCaMKII with a T287A variant, prevents CREB phosphorylation, indicating the importance of γCaMKII phosphorylation at T287 (Fig 3E). γCaMKII is sent to the nucleus upon dephosphorylation at S334 by calcineurin, which unveils a downstream nuclear localization signal (NLS). If this dephosphorylation is prevented by creating a phosphomimetic S334E mutation on γCaMKII, a bottleneck forms and the mutant γCaMKII accumulates in puncta at the CaV1 (Fig 3F). This mutation prevents pCREB, as the γCaMKII-Ca2+/CaM complex never reaches the nucleus (Fig 3G). Thus, CaV1 channel seems to work as a check point on the membrane surface, where γCaMKII gets phosphorylated by activated βCaMKII and dephosphorylated by CaN (Fig 3H).
This subsurface signaling mechanism appears tuned for sensitivity, speed and efficiency. Even modest stimulus trains (10 s at 5 Hz -> 50 spikes) can initiate a clear elevation of CREB phosphorylation (50% over basal levels) [6]. Such a system is necessary in a system as complex as the mammalian central nervous system, where the same story indeed holds true. In cortical excitatory neurons, γCaMKII translocates to the nucleus and is critical for CREB phosphorylation and gene expression. Cortical neurons can be activated by evoking action potentials with field stimulation or with 40 K depolarization. These manipulations triggered pCREB (Fig 4Ai–iii), and 40 K depolarization caused an increase of γCaMKII in the nuclear extract of neurons (Fig 4B). This nuclear increase was prevented by Nim, suggesting that the CaV1 channels serve as the key Ca2+ source for initiating γCaMKII translocation. As in sympathetic neurons, γCaMKII shRNA prevents both CaM nuclear redistribution (Fig 4C) and the resulting CREB phosphorylation (Fig 4D). Pharmacological and shRNA studies have confirmed many elements of this pathway (Fig 4E). Nim and KN93 prevent CREB phosphorylation, demonstrating the importance of the CaV1 channel and CaM Kinases, respectively. KN92, the inactive congener of KN93, shows no effect. γCaMKII must be dephosphorylated by CaN to unveil its NLS, as CsA prevented pCREB. The CREB kinase is also the same: CaMKIV- and shRNA knockdown of this protein prevented pCREB.
Further insights into the pathway are suggested by the effect of the drug Okadaic Acid (OA) [26–28]. At 20 nM, OA inhibits PP2A. PP2A resides in the nucleus and associates with CaMKIV. It can in principle dephosphorylate γCaMKII at T287, reversing the action of α/βCaMKII and lowering the affinity of γCaMKII for Ca2+/ CaM. Thus, we have proposed the following scenario [1]: upon nuclear entry, γCaMKII is dephosphorylated by PP2A, which induces the release of Ca2+/CaM molecules that immediately activate CaMKIV. Inhibition of PP2A with OA 20 nM would oppose the release of Ca2+/CaM, thereby dampening the rise in pCREB, just as observed (Fig 4F). Consistent with our previous findings that implicate PP1 as a critical CREB phosphatase [5], OA at 2 μM results in increased pCREB in cortical neurons because it inhibits both PP1 and PP2A.
The γCaMKII pathway seems not only critical for CREB phosphorylation, but also necessary for activity-dependent gene expression in cortical neurons. To test this, we tracked the CREB-dependent gene c-fos [29]. Depolarizing neurons with 40 K led to a dramatic increase in c-fos level (Fig 4G). Importantly, this increase in gene expression was prevented when the γCaMKII pathway was blocked (by inhibiting the activity of CaV1 channels or CaM Kinases) (Fig 4G).
1.6. Delivery of nuclear Ca2+/CaM is not only necessary, it is sufficient
To examine whether nuclear delivery of CaM is sufficient for CREB phosphorylation, a “nuclear CaM uncaging” experiment was designed by Ma and colleagues (Fig. 5) [1]. We utilized a key feature of an endogenous CaM binding protein, neurogranin (Nrgn): Nrgn sequesters apoCaM but liberates CaM when it is activated [30]. Fusing Nrgn with a nuclear localization sequence (NLS) (NLS-Nrgn) was effective in targeting Nrgn (and apoCaM) to the nucleus (Fig. 6B). The expression of NLS-Nrgn did not trigger pCREB response under basal conditions; however, it rescued CREB phosphorylation when γCaMKII was knocked down to prevent CaM translocation. In sharp contrast, the nuclear targeting of an S36D variant of Nrgn that could not bind apoCaM (NSL-Nrgn S36D), failed to restore CREB phosphorylation in the absence of γCaMKII and CaM translocation. Importantly, we established that the nuclear uncaged Ca2+/CaM acts through nuclear CaMK cascade because the CaMKK inhibitor STO-609 prevented the rescue action of NLS-Nrgn [1] (Fig. 6D). Thus, our results strongly suggest that nuclear delivery of CaM is not only necessary, but also sufficient for triggering CREB phosphorylation through a nuclear CaMK cascade Fig. 7.
Fig. 6.

EGTA and BAPTA differentially affect Ca2+ signaling. EGTA is a slow Ca2+ chelator which allows local Ca2+ increases, but is still powerful enough to affect local signaling near the CaV1 channel. As fewer activated CaMKII molecules are recruited to CaV1, the dynamics of signaling slows down, resulting in retarded kinetics of CREB phosphorylation. BAPTA, a fast Ca2+ chelator, prevents all CREB phosphorylation. This is shown in cortical excitatory neurons (A) and in superior cervical ganglion neurons (B, from Ref. [6]). (C) EGTA confines Ca2+ entry to within ~1 μm of the channel pore, restricting the catchment area of activating CaMKII.
Fig. 7.
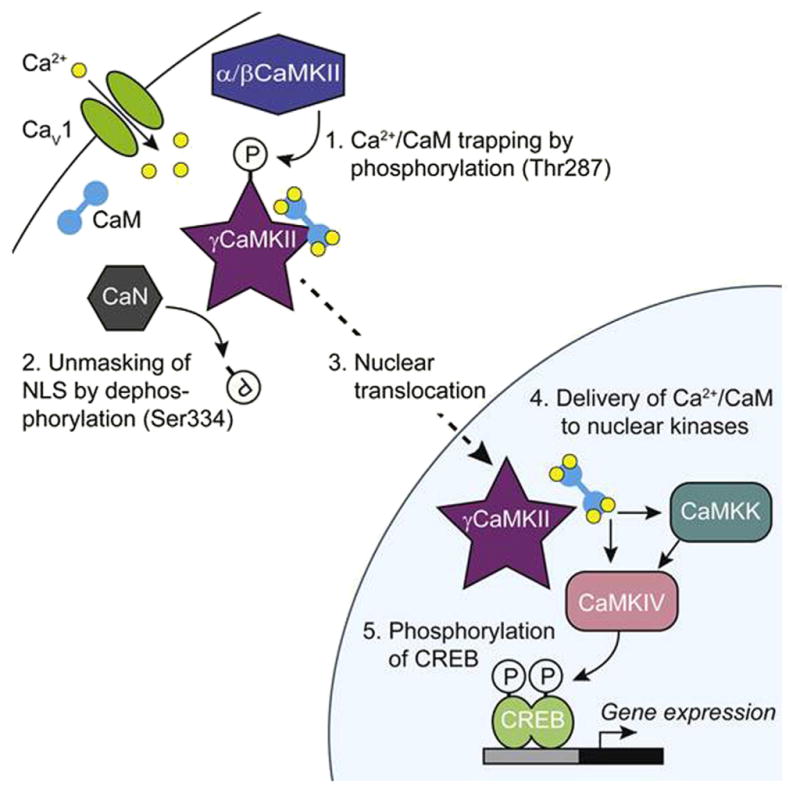
A schematic depiction of the pathway for CaMK-dependent signaling. Graphical abstract from Ref. [1]. Cartoon representations of the various players are used for the sake of simplicity. The dodecameric structure of the CaMKII enables intramultimer autophosphorylation and persistent activation in the case of α/βCaMKII, as shown in the work of Schulman and colleagues [24], and provides a large capacity to trap and transport Ca2+/CaM in the case of γCaMKII. Thus, the evolutionary development of CaMKII involvement for signaling upstream of the classic CaMK cascade (Soderling and colleagues and Means and associates [3,4]) offers multiple functional advantages.
2. Discussion
2.1. γCaMKII shuttling of Ca2+/CaM to the nucleus: a hybrid of known mechanisms
Our recent discovery of the γCaMKII-CaM shuttle (Fig. 7; [1]) provides a compelling solution to the problem of how to rapidly link neuronal activity to gene expression. The overall biochemical pathway is novel, but it has some key features reminiscent of known mechanisms discovered by other investigators. Here, we compare the steps in the shuttle pathway to previous work and point out what aspects are new and different. Bearing in mind the simpler signaling in nematode neurons, we return to questions about the functional advantages the γCaMKII-CaM shuttle may offer to a neuron in the mammalian brain.
2.2. Mobilization of α/βCaMKII promotes CaM sequestration by γCaMKII
Remarkably, the shuttle pathway in mammalian neurons mobilizes all three subclasses of CaMK (Fig. 1). Before our studies, there was no precedent for a biochemical linkage between signaling by α/ βCaMKII and by CaMKK and CaMKIV. However, the early steps in mobilizing α/βCaMKII are reminiscent of classic observations of Shen and Meyer. They showed that heteromultimers of α/βCaMKII associate with F-actin under basal conditions, specifically mediated by βCaMKII. Upon synaptic stimulation, Ca2+/CaM binding liberates these heteromultimers and enables them to translocate to the synapse where Ca2+ entry pathways are concentrated (NMDA receptors in their case, CaV1 channels in ours), and where CaMKII plays an important role in synaptic plasticity [31]. α/βCaMKII also play a pivotal role in signaling to nucleus as is evident from our shRNA experiments [6,32–34]. What seems counterintuitive about this arrangement is that α and β CaMKII move toward the plasma membrane and form puncta near CaV1 channels upon stimulation, rather than moving toward the nucleus. This mobilization might seem to be in the wrong direction. However, the spatial concentration of CaMKII may serve an additional purpose, as a marker for intensely active postsynaptic spines. Available evidence favors the notion that large molecular clusters of CaMKII can act as a “synaptic tag” [35–37]. Using the same mechanism for local synaptic tagging and signaling to the nucleus might make functional sense if tagging needs to be quantitatively coupled to transcriptional mobilization of “resources”, to be captured later at tagged sites. While the exact molecular mechanisms underlying α/β CaMKII puncta formation are unknown, our own work suggests the involvement of a voltage-induced conformational change of CaV1, over and above Ca 2+ flux per se (*Tadross M, *Li B and Tsien RW, unpublished, *denotes equal contribution).
The importance of recruiting α/βCaMKII to a local signaling hotspot at the CaV1 channel (described in Fig. 3H) is underlined by the ability of EGTA to manipulate the dynamics of Ca2+ signaling. EGTA-AM was loaded into cortical (Fig. 6A) and sympathetic (Fig. 6B) neurons prior to depolarization with 40 K. An assay of CREB phosphorylation over time shows that in both cell types CREB phosphorylation occurs, but that the kinetics are slowed in the presence of the exogenous Ca2+ buffer. This can be explained by the necessity of α/βCaMKII and/or γCaMKII recruitment to CaV1, both of which must occur to enable proper nuclear signaling. EGTA buffers Ca2+ up to ~1 μM from the channel pore, and attenuates Ca2+ even <1 μM from it, thereby restricting the “catchment” area from where various CaMKII molecules are recruited (Fig. 6C). With fewer CaMKII molecules being recruited to the channel, less γCaMKII-sequestered CaM is sent to the nucleus per unit time, resulting in reduced dynamics of signaling to CREB.
We have demonstrated that α/βCaMKII serve the purpose of phosphorylating γCaMKII at T287 and thus sequestering Ca2+/CaM in preparation for the journey to the nucleus. This calls for transmultimer phosphorylation, which goes beyond the well-described phenomenon of intramultimer phosphorylation that takes place even under highly dilute conditions. Ma et al., 2014 found that purified βCaMKII phosphorylates purified and immobilized γCaMKII K43R (kinase dead), indicating that transmultimer phosphorylation can take place in vitro when the players are localized together at high concentration. We regard formation of β/ γCaMKII heteromultimers and intramultimer phosphorylation as unlikely because nuclear translocation of βCaMKII was not detected. Nevertheless, more biochemistry and molecular biology experiments need to be done to demonstrate transmultimer phosphorylation in situ and to explain why β/γCaMKII heteromultimers are disfavored.
2.3. A dedicated shuttle protein enables specialization of Ca2+ entry pathways and private line communication to specific cellular targets
2.3.1. Specialization of Ca2+ entry pathways
This study illuminates the long-observed phenomenon of CaV1 privilege in E-T coupling [6,15,38,39]. Clusters of CaV1 channels help mark a nucleation point for molecular participants in surface-to-nucleus communication, including CaN and α/βCaMKII [6,32]. The localization of key players at CaV1 channels, whether sustained or normally fleeting, gives a specific explanation of the nanoscale relationship between these channels and their targets. In turn, that nanodomain signaling accounts for the differential effects of fast and slow on-rate Ca2+ buffers (BAPTA, EGTA) in preventing CaMKII relocation [6] and CREB phosphorylation [15]. The signaling system is geared to drive long distance communication to the nucleus, yet it is launched at a CaV1-centered Ca2+ nanodomain. This arrangement is highly advantageous for pathway specialization: E-T coupling thus comes under the control of a particular Ca2+ entry pathway, freeing up other Ca2+ entry pathways (CaV2) for other biological functions such as signaling to ER and mitochondria [6]. Given the multitude of Ca2+ entry and release mechanisms within cells [40,41], it will be interesting to test whether signals emanating from other Ca2+ sources, such as the NMDAR, may also mobilize this newly identified pathway.
2.3.2. Enables private line signaling for specificity
What would be the rationale for locally driven signaling? Clearly, signaling at a distance but with specificity calls for something other than a global Ca2+ signal. Ca2+ is an attractive candidate to mediate signaling because it provides specificity at short distance. The functional importance of a shuttle mechanism is to combine signaling at a distance with specificity. Ca2+/CaM harnesses the short-distance specificity of Ca2+, and mediates Ca2+ association with an NLS-containing binding partner, thus providing long-distance specificity. Such specificity may indeed concern the balance between life and death, as gene expression and cell death are alternative outcomes of a rise in nuclear Ca2+. As outlined by Carafoli et al. in their review of the Mediterranean nuclear Ca2+ meeting [42]: “These historical and general aspects of the role of calcium (and the more recent results on nuclear calcium involvement) in cell death were discussed by P. Nicotera. He discussed excitotoxicity, a term that defines the suprastimulation of glutamate receptor subtypes leading to neuronal calcium overload and activation of apoptosis or necrosis.” The CaM shuttle may operate like a private letter, rather than a loud shout from rooftop to rooftop.
Additional advantages of the shuttle system arise from its dynamic features. Once Ca2+/CaM is sequestered, signaling to the nucleus can proceed even after the increase in cytosolic Ca2+ has largely subsided. Although the offset of a brief depolarizing stimulus is followed by an immediate fall in Ca2+, pCREB continues to increase for 45 s before reaching a peak and then gradually decaying [32]. This ballistic action, set in motion by the previous activity, is enabled by Ca2+/CaM trapping by γCaMKII and maximizes the impact of neuronal activity while presumably minimizing the duration of the Ca2+ rise and potential Ca2+-dependent toxicity.
2.4. Phosphatases work together with kinases at both ends of the γCaMKII shuttle
While kinases always appear front and center in signaling to nucleus, phosphatases also play important roles—something that Claude Klee and Ernesto Carafoli would agree upon. In the γCaM-KII-CaM shuttle, one phosphatase, CaN, works near the channel as the shuttle dispatcher. This adds one more arrow to the extensive quiver of CaN actions [43]. The other, PP2A, appears to act in the nucleus to unload the cargo [1].
CaN resides close to CaV1 channels in the basal state and is rapidly activated upon Ca2+/CaM rise [44]. The proximity of CaN, together with the recruitment of α/βCaMKII, endows the CaV1 channel with the features of a signaling hotspot. CaM kinases and CaN work in close succession within the Ca2+ nanodomain of the CaV1 channel [45]. As mentioned earlier, upon activation CaN regulates the NLS of γCaMKII by dephosphorylation at S334. Precedent for regulation of CaMKII NLS by phosphorylation or dephosphorylation was first established by Heist and Schulman [46]. Dephosphorylation at S334 accounts for previous findings that CaN can work synergistically with CaMKII, rather than in opposition, to drive CREB phosphorylation [47] and CRE-mediated gene expression [48,49]. This CaN action takes place in parallel to CaN-dependent regulation of nuclear localization of the CREB coactivator CTRC1 [50]. CaN action on γCaMKII occurs within seconds of depolarization onset, distinguishing it from the much slower effect of nuclear CaN in promoting PP1-mediated dephosphorylation of pCREB. This takes place over several minutes, uses a distinct mechanism, and provides negative feedback on gene expression [5].
The other critical phosphatase is PP2A, which resides in the nucleus in a biochemical complex with CaMKIV, as discovered by Means and colleagues [51]. Our experiments with low-concentration OA (20 nM) to block PP2A support the following scenario. Upon arrival of γCaMKII-CaM, PP2A dephosphorylates the T287 site of γCaMKII, leading to local Ca2+/CaM desequestration. Free Ca2+/CaM can then activate CaMKK and CaMKIV in the classic nuclear CaMK cascade [3,4,52], thus leading to phosphorylation of CREB (see Ma et al., 2014 for supporting evidence). The co-localization of PP2A with CaMKIV, described by Anderson, Means and colleagues, elegantly ensures that Ca2+/CaM is delivered directly and locally to a key enzymatic target.
2.5. Steep coupling between neuronal activity, CaMKIV activation, and various nuclear events
Putting both CaMKK and CaMKIV within the nucleus, the destination of the Ca2+/CaM shuttle also makes sense because both CaMKs require Ca2+/CaM binding to drive full CaMKIV activation. The enzymes work in series, so CREB phosphorylation should increase in a steep relationship with the incremental increase in nuclear free Ca2+/CaM [16,52]. Indeed, we found that increases in CREB phosphorylation and downstream c-fos expression were proportionately larger than rises in Ca2+/CaM. A power-law dependence on free Ca2+/CaM is a likely contributing factor.
Ca2+/CaM delivery to the nucleus also provides an opportunity for integration. Progressive accumulation of nuclear Ca2+/CaM provides a means for piling up the effects of multiple weak stimuli [23]. Once CaM has translocated, rises in nuclear Ca2+ may also contribute in driving transcriptional activation [23].
Besides CREB, CaMKIV also phosphorylates and activates CREB binding protein (CPB) [53]. This cofactor works in coordination with CREB and other transcription factors such as CREST [54] to trigger gene expression. CBP also controls gene expression by regulating the nuclear localization of certain histone deacetylases [55]. CaMKIV also plays a pivotal role in the regulation of alternative splicing of pre-mRNA [56–58]. Thus, by regulating nuclear CaMKIV activation, the shuttle mechanism helps illuminate the hitherto-unexplained dominance of CaV1 channels in wide-ranging phenomena such as depolarization-induced activation of CBP, histone deacetylation and exon selection.
2.6. Concluding remarks
Clearly, signaling to the nucleus via CaMKs has ancient roots. Our main conclusion is that CaMK-based stimulus-transcription coupling has gained power as well as sophistication over the course of evolution. Key features of this system that distinguish mammalian excitatory neurons from worm neurons include (1) the participation of CaMKII isoforms, (2) the emergence of a dedicated shuttle for Ca2+/CaM, and (3) the localization of the entire cascade of nuclear CaMKK and CaMKIV within the nucleus. Each of these evolutionary elaborations offers compelling functional advantages.
3. Methods
3.1. Primary cultures of cortical neurons
Cortical neurons were cultured from postnatal day 0–1 male and female Sprague–Dawley rat pups. The frontal cortex was isolated and washed twice in ice-cold modified HBSS (4.2 mM NaHCO3 and 1 mM HEPES, pH 7.35, 300 mOsm) containing 20% fetal bovine serum (FBS; Hyclone, Logan, UT). Cortices were washed and digested for 30 min in a papain solution (2.5 ml HBSS + 145 U papain +40 μl DNase) at 37 °C with gentle shaking every 10 min. Digestion was stopped by adding 5 ml of modified HBSS containing 20% fetal bovine serum. After additional washing, the tissue was dissociated using Pasteur pipettes of decreasing diameter. The cell suspension was pelleted twice and filtered with a 70 μm nylon strainer, and plated on 10 mm coverslips coated with poly-D-lysine. The cultures were maintained in NbActiv4 (BrainBits, Springfield, IL). A 50% medium change was performed at 7 days, and once per week thereafter. Neurons were used 14–21 days after plating.
3.2. Drug treatments and stimulation
To induce CREB phosphorylation, we stimulated cortical neurons with the indicated high [K+] solution at 37 °C for 10–300 s, and fixed the cells immediately after the stimulation (in 4% paraformaldehyde in PBS, with 20 mM EGTA and 4% (w/v) sucrose), or incubated the cells in culture media for 90 min at 37 °C before fixation. Where indicated, drugs were added 30 min before and included throughout the stimulation. All K+-rich stimulation solutions contained 0.5 μM TTX (Ascent Scientific) to block action potentials. In addition, when stimulating cortical neurons, 10 μM NBQX (Ascent Scientific) and 10 μM APV (Ascent Scientific) were included to block AMPA and NMDA receptors, respectively. 4 mM K Tyrode’s consisted of (in mM): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose, pH 7.4. When stimulating with elevated [K+], Na+ was adjusted to maintain osmolarity. To block: CaM Kinases, KN93 (Tocris) was used at 4uM; to block CaV1 channels, Nimodipine (abcam) was used at 10uM; to block CaMKK, STO609 (Tocris) was used at 3.3uM; to block PP2A, okadaic acid (Tocris) was used at 20 nM; to block PP1 and PP2A, okadaic acid (Tocris) was used at 2uM.
Cells were loaded with Ca2+ chelators EGTA-AM and BAPTA-AM (Life Technologies), used at 200uM, along with 1:1000 Pluronic F-127 (Life Technologies) 45 min before stimulation.
3.3. Field stimulation
Two platinum electrodes were placed with a distance of ~10 mm in the control (4 mM K+) Tyrode’s solution (4 K). The cultured cortical neurons on the coverslips were field stimulated at the specified frequency and duration for with square wave pulses (3 ms per pulse). Amplitude and duration of the pulse was controlled by a Grass S11 stimulator. The stimulus amplitudes were set to 20% above threshold.
3.4. DNA constructs
CaMKIV shRNA was kindly provided by Haruhiko Bito. A description of γCaMKII shRNA can be found in Ref. [1].
3.5. Transfection
Cortical neurons were transfected 7 days after plating using a high efficiency Ca2+-phosphate transfection method.
3.6. Immunocytochemistry
Cells were fixed in ice-cold 4% paraformaldehyde in phosphate buffer supplemented with 20 mM EGTA and 4% (w/v) sucrose. Fixed cells were then permeabilized with 0.1% Triton X-100, blocked with 6% normal donkey serum and incubated overnight at 4°C in primary antibodies: rabbit anti-δCaMKI (1:500, sc-134638, Santa Cruz); mouse anti-γCaMKI (1:1000, PA5-19661, Abcam); rabbit anti-γCaMKI (1:200; Thermo Scientific); mouse anti-αCaMKII (1:1000, 13-7300, Invitrogen); goat anti-αCaMKII (1:100, sc-5391, Santa Cruz); rabbit anti-αCaMKII (1:1000, Abcam); mouse anti-βCaMKII (1:1000, 13-9800, Invitrogen); goat anti-γCaMKII (1:500, sc-1541, Santa Cruz); goat anti-δCaMKII (1:500, sc-5392, Santa Cruz); mouse anti-CaMKIV (1:500, sc-136249, Santa Cruz); rabbit anti-αCaMKK (1:333, sc-11370, Santa Cruz); rabbit anti-βCaMKK (1:250, Santa Cruz); rabbit anti-c-fos (1:1000, 2250, Cell Signaling). The next day, cells were washed with PBS, incubated at room temperature for 50 min with Alexa secondary antibodies (1:1000, Molecular Probes), washed with PBS (3x5 min) and mounted using ProLong Gold (with or without Dapi) (Invitrogen). The cells were imaged with a 60X (1.3 NA) oil objective on an Axioplan epifluorescent microscope equipped with an AxioCam digital camera using AxioVision.
3.7. Image analysis
Images were analyzed using a custom-written macro in ImageJ (NIH) to analyze the pixel intensity of immunostaining [1]. The nuclear marker DAPI and an antibody against a molecular marker for a specific cell type were used to delineate the nucleus and cytoplasm, respectively. A region of interest adjacent to each neuron analyzed was used as an ‘off-cell’ background. The pCREB level in each neuron was calculated by subtracting the average pCREB channel intensity in the ‘off-cell’ background region of interest from the average intensity in the nuclear region of interest for each neuron. The nuclear: cytoplasmic ratio was calculated by comparing nuclear immunoreactivity with apical dendrite immunoreactivity (membrane immunoreactivity was not included).
3.8. Statistical analysis
Statistical analyses were performed using Prism 6.0 (Graph-Pad software). Student’s t-test was used for comparisons between two groups. One-or two-way analysis of variance (ANOVA) was used for analysis between three or more groups (notably, between replicates of each experiment). Pearson Correlation was used for correlation analysis. All data are shown as mean ± standard error of the mean (SEM), unless otherwise mentioned.
Supplementary Material
Acknowledgments
We thank people in the Tsien laboratory for helpful suggestions and discussions, and Katherine Eyring for useful comments on the text. This work was supported by research grants to R.W.T. from the National Institute of General Medical Sciences (GM058234), the National Institute of Neurological Disorders and Stroke (NS24067), the National Institute of Mental Health (MH071739), the Simons Foundation, the G. Harold & Leila Y. Mathers Foundation, and the Burnett Family Foundation. Samuel M. Cohen is supported by the MSTP Program of New York University.
Footnotes
Transparency document
Transparency document related to this article can be found online at http://dx.doi.org/10.1016/j.bbrc.2015.02.146
Conflict of interest
None.
Contributor Information
Richard W. Tsien, Email: richard.tsien@nyumc.org.
Huan Ma, Email: mahuan@gmail.com.
References
- 1.Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW. GammaCaMKII shuttles Ca(2+)/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell. 2014;159:281–294. doi: 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colomer J, Means AR. Physiological roles of the Ca2+/CaM-dependent protein kinase cascade in health and disease. Subcell Biochem. 2007;45:169–214. doi: 10.1007/978-1-4020-6191-2_7. [DOI] [PubMed] [Google Scholar]
- 3.Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- 4.Means AR. Regulatory cascades involving calmodulin-dependent protein kinases. Mol Endocrinol Baltim Md) 2000;14:4–13. doi: 10.1210/mend.14.1.0414. [DOI] [PubMed] [Google Scholar]
- 5.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 6.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, Means AR. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J Biol Chem. 1998;273:31880–31889. doi: 10.1074/jbc.273.48.31880. [DOI] [PubMed] [Google Scholar]
- 9.Chow FA, Anderson KA, Noeldner PK, Means AR. The autonomous activity of calcium/calmodulin-dependent protein kinase IV is required for its role in transcription. J Biological Chem. 2005;280:20530–20538. doi: 10.1074/jbc.M500067200. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Campos B, Jamieson GA, Jr, Kaetzel MA, Dedman JR. Functional elimination of calmodulin within the nucleus by targeted expression of an inhibitor peptide. J Biol Chem. 1995;270:30245–30248. doi: 10.1074/jbc.270.51.30245. [DOI] [PubMed] [Google Scholar]
- 11.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 12.Limback-Stokin K, Korzus E, Nagaoka-Yasuda R, Mayford M. Nuclear calcium/calmodulin regulates memory consolidation. J Neurosci. 2004;24:10858–10867. doi: 10.1523/JNEUROSCI.1022-04.2004. Official J. Soc. Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RW, Tsien RY. Imaging of cytosolic Ca2+ transients arising from Ca2+ stores and Ca2+ channels in sympathetic neurons. Neuron. 1988;1:355–365. doi: 10.1016/0896-6273(88)90185-7. [DOI] [PubMed] [Google Scholar]
- 14.Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RY, Tsien RW. Spatial distribution of calcium channels and cytosolic calcium transients in growth cones and cell bodies of sympathetic neurons. Proc Natl Acad Sci U S A. 1988;85:2398–2402. doi: 10.1073/pnas.85.7.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: post-synaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 16.Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S, Navarro AJ, Lyst MJ, Ekiert R, Bird AP, Greenberg ME. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakagami H, Kamata A, Nishimura H, Kasahara J, Owada Y, Takeuchi Y, Watanabe M, Fukunaga K, Kondo H. Prominent expression and activity-dependent nuclear translocation of Ca2+/calmodulin-dependent protein kinase Idelta in hippocampal neurons. Eur J Neurosci. 2005;22:2697–2707. doi: 10.1111/j.1460-9568.2005.04463.x. [DOI] [PubMed] [Google Scholar]
- 19.Persechini A, Cronk B. The relationship between the free concentrations of Ca2+ and Ca2+-calmodulin in intact cells. J Biol Chem. 1999;274:6827–6830. doi: 10.1074/jbc.274.11.6827. [DOI] [PubMed] [Google Scholar]
- 20.Persechini A, Stemmer PM. Calmodulin is a limiting factor in the cell. Trends Cardiovasc Med. 2002;12:32–37. doi: 10.1016/s1050-1738(01)00144-x. [DOI] [PubMed] [Google Scholar]
- 21.Vendrell M, Pujol MJ, Tusell JM, Serratosa J. Effect of different convulsants on calmodulin levels and proto-oncogene c-fos expression in the central nervous system. Brain Res Mol Brain Res. 1992;14:285–292. doi: 10.1016/0169-328x(92)90095-s. [DOI] [PubMed] [Google Scholar]
- 22.Teruel MN, Chen W, Persechini A, Meyer T. Differential codes for free Ca(2+)-calmodulin signals in nucleus and cytosol. Curr Biol. 2000;10:86–94. doi: 10.1016/s0960-9822(00)00295-5. [DOI] [PubMed] [Google Scholar]
- 23.Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW. Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci U S A. 2001;98:15342–15347. doi: 10.1073/pnas.211563998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 25.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Sci (New York, NY) 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 26.Cohen P, Holmes CF, Tsukitani Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci. 1990;15:98–102. doi: 10.1016/0968-0004(90)90192-e. [DOI] [PubMed] [Google Scholar]
- 27.Nagao M, Shima H, Nakayasu M, Sugimura T. Protein serine/threonine phosphatases as binding proteins for okadaic acid. Mutat Res. 1995;333:173–179. doi: 10.1016/0027-5107(95)00143-3. [DOI] [PubMed] [Google Scholar]
- 28.Shenolikar S. Protein serine/threonine phosphatases–new avenues for cell regulation. Annu Rev Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- 29.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 30.Baudier J, Deloulme JC, Van Dorsselaer A, Black D, Matthes HW. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. J Biol Chem. 1991;266:229–237. [PubMed] [Google Scholar]
- 31.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Sci (New York, NY) 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 32.Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma H, Cohen S, Li B, Tsien RW. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep. 2012;33 doi: 10.1042/BSR20120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma H, Groth RD, Wheeler DG, Barrett CF, Tsien RW. Excitation-transcription coupling in sympathetic neurons and the molecular mechanism of its initiation. Neurosci Res. 2011;70:2–8. doi: 10.1016/j.neures.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- 36.De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Sci (New York, NY) 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 37.Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998;21:181–188. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- 38.Greenberg ME, Ziff EB, Greene LA. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Sci (New York, NY) 1986;234:80–83. doi: 10.1126/science.3749894. [DOI] [PubMed] [Google Scholar]
- 39.Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- 40.Berridge MJ. Calcium signalling remodelling and disease. Biochem Soc Trans. 2012;40:297–309. doi: 10.1042/BST20110766. [DOI] [PubMed] [Google Scholar]
- 41.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 42.Carafoli E, Nicotera P, Santella L. Calcium signalling in the cell nucleus. Cell Calcium. 1997;22:313–319. doi: 10.1016/s0143-4160(97)90016-6. [DOI] [PubMed] [Google Scholar]
- 43.Aramburu J, Rao A, Klee CB. Calcineurin: from structure to function. Curr Top Cell Regul. 2000;36:237–295. doi: 10.1016/s0070-2137(01)80011-x. [DOI] [PubMed] [Google Scholar]
- 44.Oliveria SF, Dell’Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tadross MR, Tsien RW, Yue DT. Ca2+ channel nanodomains boost local Ca2+ amplitude. Proc Natl Acad Sci U S A. 2013;110:15794–15799. doi: 10.1073/pnas.1313898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heist EK, Srinivasan M, Schulman H. Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J Biol Chem. 1998;273:19763–19771. doi: 10.1074/jbc.273.31.19763. [DOI] [PubMed] [Google Scholar]
- 47.Hahm SH, Chen Y, Vinson C, Eiden LE. A calcium-initiated signaling pathway propagated through calcineurin and cAMP response element-binding protein activates proenkephalin gene transcription after depolarization. Mol Pharmacol. 2003;64:1503–1511. doi: 10.1124/mol.64.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kingsbury TJ, Bambrick LL, Roby CD, Krueger BK. Calcineurin activity is required for depolarization-induced, CREB-dependent gene transcription in cortical neurons. J Neurochem. 2007;103:761–770. doi: 10.1111/j.1471-4159.2007.04801.x. [DOI] [PubMed] [Google Scholar]
- 49.Schwaninger M, Blume R, Kruger M, Lux G, Oetjen E, Knepel W. Involvement of the Ca(2+)-dependent phosphatase calcineurin in gene transcription that is stimulated by cAMP through cAMP response elements. J Biol Chem. 1995;270:8860–8866. doi: 10.1074/jbc.270.15.8860. [DOI] [PubMed] [Google Scholar]
- 50.Ch’ng TH, Uzgil B, Lin P, Avliyakulov NK, O’Dell TJ, Martin KC. Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell. 2012;150:207–221. doi: 10.1016/j.cell.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson KA, Noeldner PK, Reece K, Wadzinski BE, Means AR. Regulation and function of the calcium/calmodulin-dependent protein kinase IV/protein serine/threonine phosphatase 2A signaling complex. J Biol Chem. 2004;279:31708–31716. doi: 10.1074/jbc.M404523200. [DOI] [PubMed] [Google Scholar]
- 52.Racioppi L, Means AR. Calcium/calmodulin-dependent kinase IV in immune and inflammatory responses: novel routes for an ancient traveller. Trends Immunol. 2008;29:600–607. doi: 10.1016/j.it.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 54.Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Sci (New York, NY) 2004;303:197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- 55.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001;410:936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- 57.Xie J, Jan C, Stoilov P, Park J, Black DL. A consensus CaMK IV-responsive RNA sequence mediates regulation of alternative exons in neurons. Rna. 2005;11:1825–1834. doi: 10.1261/rna.2171205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu G, Razanau A, Hai Y, Yu J, Sohail M, Lobo VG, Chu J, Kung SK, Xie J. A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J Biol Chem. 2012;287:22709–22716. doi: 10.1074/jbc.M112.357343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schild LC, Zbinden L, Bell HW, Yu YV, Sengupta P, Goodman MB, Glauser DA. The balance between cytoplasmic and nuclear CaM Kinase-1 signaling controls the operating range of noxious heat avoidance. Neuron. 2014;84:983–996. doi: 10.1016/j.neuron.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu YV, Bell HW, Glauser DA, Van Hooser SD, Goodman MB, Sengupta P. CaMKI-dependent regulation of sensory gene expression mediates experience-dependent plasticity in the operating range of a thermosensory neuron. Neuron. 2014;84:919–926. doi: 10.1016/j.neuron.2014.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eto K, Takahashi N, Kimura Y, Masuho Y, Arai K, Muramatsu MA, Tokumitsu H. Ca(2+)/Calmodulin-dependent protein kinase cascade in Caenorhabditis elegans. Implication in transcriptional activation. J Biol Chem. 1999;274:22556–22562. doi: 10.1074/jbc.274.32.22556. [DOI] [PubMed] [Google Scholar]
- 62.Kimura Y, Corcoran EE, Eto K, Gengyo-Ando K, Muramatsu MA, Kobayashi R, Freedman JH, Mitani S, Hagiwara M, Means AR, Tokumitsu HA. CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep. 2002;3:962–966. doi: 10.1093/embo-reports/kvf191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.