

Abstract
Biodistribution analysis is a key step in the evaluation of adeno-associated virus (AAV) capsid variants, whether natural isolates or produced by rational design or directed evolution. Indeed, when screening candidate vectors, accurate knowledge about which tissues are infected and how efficiently is essential. We describe the design, validation, and application of a new vector, pTR-UF50-BC, encoding a bioluminescent protein, a fluorescent protein and a DNA barcode, which can be used to visualize localization of transduction at the organism, organ, tissue, or cellular levels. In addition, by linking capsid variants to different barcoded versions of the vector and amplifying the barcode region from various tissue samples using barcoded primers, biodistribution of viral genomes can be analyzed with high accuracy and efficiency.
Introduction
Adeno-associated virus (AAV)-derived vectors are among the most promising gene therapy tools because of their safety and ability to mediate long-term transgene expression.1 AAV capsid variants, whether naturally occurring isolates, generated by rational design or selected by directed evolution from combinatorial libraries, typically exhibit differential tissue tropisms.2 These are usually analyzed using a combination of techniques such as in vivo imaging (bioluminescence generated by luciferase expression) at the organism level, quantification of viral nucleic acids (typically by quantitative polymerase chain reaction (qPCR)) at the organ or tissue level, and finally fluorescence detection (expression of green fluorescent protein or other fluorescent protein) at the tissue and cellular levels. Each of these techniques typically requires a distinct vector. Here we describe the design and validation of a universal vector, pTR-UF50-BC, that can be used interchangeably in all these applications. The vector encodes a bioluminescent protein (firefly luciferase) and a fluorescent protein (mApple), as well as 6-nt unique sequence identifier—a DNA barcode. Forty-five versions of pTR-UF50-BC were produced, each containing a distinct barcode. We investigated the utility of pTR-UF50-BC in analyzing the biodistribution of barcoded viral genomes in mice after systemic administration. Traditionally, biodistribution analysis has been a costly and time-consuming process, especially when large numbers of variants are to be screened. Linking each capsid variant to a particular DNA barcode and each organ or tissue to another barcode allows a considerable simplification of this process, saving time and efforts as well as animal and financial resources.
Results
pTR-UF50-BC vector design
A cDNA fragment formed by firefly luciferase and mApple proteins in the same open reading frame was constructed. Open-reading frame coding for mApple3 was chemically synthesized and sequence-verified. A foot-and-mouth disease virus-derived 2A sequence (APVKQTLNFDLLKLAGDVESNPGP)4 with a furin cleavage site (RAKR)5 immediately upstream of 2A (F2A) was used to link two cDNAs by splice-overlap extension polymerase chain reaction. A map of the transgene cassette is shown in Figure 1a. The list of the 45 available barcodes is shown in Table 1.
Figure 1.

Overview of the experiment. Diagram of barcoded AAV transgene cassette (a). CBA promoter, CMV-b-actin promoter; 2A peptide, foot-and-mouth disease virus 2A ribosomal skip peptide; Barcode: 6-nt unique sequence identifier; ITR, AAV2 terminal repeat; PolyA, bovine growth hormone gene polyadenylation site. Note that elements are not drawn to scale. Global flowchart (b). The barcoded AAV mixture (top left) is injected into animals; tissue samples are harvested (bottom left); DNA is isolated from all samples; DNA is quantified and used as a template in quantitative polymerase chain reaction (qPCR) to titer total AAV in each sample (bottom right); DNA is also used as PCR template, as well as the original AAV mixture, to amplify AAV-specific barcodes using sample-specific barcoded primers; all PCR products are then combined and sequenced in a single reaction to determine the variant composition in each sample; sequencing data are then used, in conjunction with viral titers to compute biodistribution for each variant (top right).
Table 1. pTR-UF50-BC barcode sequences.
| Clone | Barcode | Clone | Barcode | Clone | Barcode | Clone | Barcode | Clone | Barcode |
|---|---|---|---|---|---|---|---|---|---|
| BC01 | TTGTTG | BC11 | AACATC | BC21 | ACCACG | BC31 | TGACGC | BC41, 18 | ACGTTC |
| BC02 | TCCCCG | BC12 | TGGTTC | BC22 | TACTAC | BC32, 33 | ATAATG | BC42 | TGGCCG |
| BC03 | TTCCAC | BC13 | ACTCCG | BC23 | TTTTGG | BC33, 32 | ATAATG | BC43 | TTGAAC |
| BC04, 05 | AGGGAC | BC14 | ATCCCC | BC24, 19 | AGCTCC | BC34, 28 | ATGCTG | BC44 | TGGTCG |
| BC05, 04 | AGGGAC | BC15 | AACCCC | BC25 | TCCGAC | BC35 | TCGCCC | BC45 | TAATCC |
| BC06 | TCCTAG | BC16 | TAGAAG | BC26 | ACACCC | BC36 | TCTCAC | BC46 | TGCCCG |
| BC07 | AGTACC | BC17 | ATGTAC | BC27 | ACCCAC | BC37 | TCTCCC | BC47 | AGTGCC |
| BC08 | ATATGG | BC18, 41 | ACGTTC | BC28, 34 | ATGCTG | BC38 | ATCACC | BC48 | ATACCG |
| BC09 | AGCAGG | BC19, 24 | AGCTCC | BC29 | TTGGCC | BC39 | TCAGCG | BC49 | TGCGAG |
| BC10 | ACAAGC | BC20 | AGCTTC | BC30 | ACTATG | BC40 | ACAACC | BC50 | ACTGAC |
Vector validation
In order to evaluate both the bioluminescence and fluorescent reporter components of our vector, recombinant AAV consisting of the luciferase-mApple construct pseudotyped with AAV-5 capsid was injected in the right striatum of the mouse brain (Figure 2b,d) or in the muscle of the right posterior leg (Figure 2a,c). Luciferase activity was assayed 2 weeks later. After intraperitoneal injection of luciferin, strong bioluminescence signal was detected at the AAV injection site (Figure 2d: skull, Figure 2c: leg).
Figure 2.
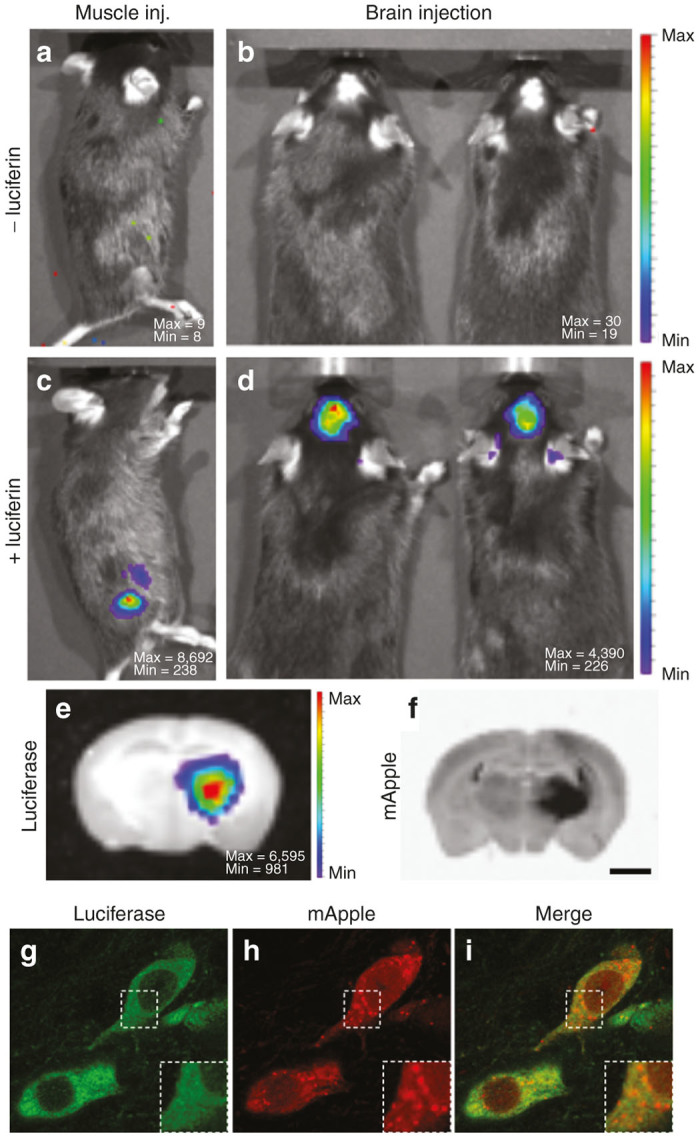
Evaluation of luciferase and mApple signals in the mouse model. Intense BLI signal was detected in the right leg of the mouse 2 weeks after intramuscular injection of AAV5-luciferase-mApple (c). BLI signal located in the skull was detected in a mouse that received a stereotactic injection of AAV5-luciferase-mApple in the right striatum (d). Note the absence of BLI signal before the intraperitoneal injection of luciferin (a,b). The pseudo-color scale represents the intensity of light emitted in number of counts. Max and Min are the maximum and the minimum number of counts respectively. Further analysis of a coronal section of the brain confirms that the BLI signal comes from the right striatum (e). Fluorescent signal examination reveals that mApple signal is located in the same anatomic location as luciferase (f). Fluorescent immunohistochemistry of luciferase and subsequent confocal analysis validates colocalization of luciferase (g) and mApple (h) at the cellular level. Image i is a merge of g and h. Scale bars: 1.8 mm (e,f), 11.7 µm (g–i), 6 µm (g–i magnifications).
To localize luciferase expression at the tissue level, some animals were sacrificed immediately after luciferin injection and their brains were sliced. BLI analysis of coronal sections showed an intense signal emanating from the striatum, on the side of AAV injection (Figure 2e). Subsequent fluorescent analysis of the brain sections revealed mApple signal coming from the exact same region of the brain (Figure 2f).
To determine whether both luciferase and mApple reporter genes are expressed efficiently within one cell, immunostaining of luciferase with an antibody (Figure 2g) and subsequent confocal analysis were performed. Luciferase was found homogeneously distributed in the cytosol (Figure 2g). On the other hand, mApple was detected both in the nucleus and the cytoplasm, displaying prevalent vesicular accumulation (Figure 2h). The distribution patterns of luciferase and mApple suggested that both reporter genes are efficiently expressed from the same promoter (Figure 2i).
Together, these results demonstrate that our AAV dual-reporter combination of bioluminescence and fluorescence provides full spectrum of coverage needed for any reporter gene application, from the organism level to the cellular level.
Design of biodistribution analysis experiment
In order to analyze the biodistribution of a particular AAV variant, samples of various tissues or organs are typically harvested a few days after systemic injection in a set of animals, and the presence of viral genomes in each sample is quantified by qPCR. In parallel, the amount of total DNA in each sample is measured, so that the distribution of viral genomes can be normalized and expressed as viral genomes per certain quantity of DNA or even per cell (assuming a correspondence between amount of total DNA and number of cells). The same procedure would be performed individually for each additional variant.
In the method presented here (Figure 1b), each AAV variant is produced using a different barcoded version of the pTR-UF50-BC vector (Table 1). All variants are then mixed at about the same final titer and the mixture is injected into a single set of animals. Days or weeks later, animals are sacrificed, tissue samples are harvested, and DNA is isolated from each of them. In addition to quantification of viral genomes and genomic DNA, a region of the vector sequence that includes the barcode is amplified by PCR using a different pair of barcoded primers (Table 2) for each sample. An aliquot of the viral mixture that was injected is amplified as well using yet a different pair of primers. All PCR products are then pooled and analyzed in a single sequencing reaction. Each sequencing read contains two types of barcode: one that specifies the AAV variant (Table 1) and one that specifies the PCR template, thus identifying a tissue sample and animal or the original viral mixture (Table 2). Sequence analysis then reveals, on the one hand, the exact composition of the viral mixture that was injected (relative amounts of all capsid variants), and on the other hand the viral composition (relative numbers of variant-specific barcodes) in each sample (detected by the sample-specific barcodes). The composition of each sample is then corrected, knowing the actual composition of the original mixture. In addition, as in the traditional method, the total viral load as well as the total DNA content have been determined for each sample. Combining all this information allows to determine the precise biodistribution of each viral variant across all tissue samples.
Table 2. Oligonuceotide sequences.
| Name | Sequence (5′ to 3′) | Strand |
|---|---|---|
| CBA-P | [6-FAM]TGCC CAGGAGCTGTA GGAAAAAGA A[BHQ1a-6FAM] | Reverse |
| CBA-F | TGCTAACC ATGTTC ATGCC | Forward |
| CBA-R | TGATGAGACA GCACAATAACC | Reverse |
| BC1F | CCATCTCATCC CTGCGTGTCTCC GACTCAGAAGGAC GAGCTGTACA AGTAAATCG | Forward |
| BC2F | CCATCTCATC CCTGCGTGTCTCCGAC TCAGTTGGACGAGCTGTAC AAGTAAATCG | Forward |
| BC3F | CCATCTCATCCCT GCGTGTCTCCGACTCAGCA GGACGAGCTGTACA AGTAAATCG | Forward |
| BC4F | CCATCTCATCCC TGCGTGTCTCCGACTCA GTAGGACGAGCTGTACA AGTAAATCG | Forward |
| BC5F | CCATCTCATC CCTGCGTGTCTCCGA CTCAGTCGGACGAGCTGT ACAAGTAAATCG | Forward |
| BC1R | CCTCTCTATGGGCA GTCGGTGATAACC ATTATAAGCTG CAATAAACAAG | Reverse |
| BC2R | CCTCTCTATGGGCA GTCGGTGATGGCCATT ATAAGCTGCAAT AAACAAG | Reverse |
| BC3R | CCTCTCTATGGGCA GTCGGTGATTTCCATTA TAAGCTGCAATA AACAAG | Reverse |
| BC4R | CCTCTCTATGGG CAGTCGGTGATCTCCA TTATAAGCTGCA ATAAACAAG | Reverse |
| BC5R | CCTCTCTATGGGCA GTCGGTGATCA CCATTATAAGCT GCAATAAACAAG | Reverse |
| BC6R | CCTCTCTATGGGCA GTCGGTGATATCCA TTATAAGCTGCAA TAAACAAG | Reverse |
| BC7R | CCTCTCTATGGGC AGTCGGTGATGTCCA TTATAAGCTGC AATAAACAAG | Reverse |
| IT-F | TCGTCGGCAGCGTCA GATGTGTATAAGAGACAGCCATCTCATCCCTGCGTGTCTC | Forward |
| IT-R | GTCTCGTGGGCTCGGA GATGTGTATAAGAGACA GCCTCTCTATGGGCAGTCGGTGA | Reverse |
DNA-barcode script
A Python script was written to analyze sequencing data generated by either Ion Torrent or Illumina platforms. Given variant-specific and sample-specific barcode definitions as well as information on the expected barcode locations in the sequencing reads, the script outputs both raw numbers and percentages of each variant (identified by the vector-derived barcode) in each sample (identified by the primer-derived barcodes).
Biodistribution analysis of nine AAV capsid variants
An approximately equimolar (based on qPCR measurements) mixture of nine viral preparations of barcoded capsid variants was injected into a set of three mice. Three weeks later, mice were euthanized, total DNA was isolated from eight tissue samples per mouse, and the presence of AAV genomes in each sample was quantified by qPCR (Figure 3). The capsid-specific barcode region was amplified from the tissue samples as well as from an aliquot of the injected viral mixture using a total of 25 distinct barcoded primer pairs. A mixture of all PCR products was then sequenced using the IonTorrent platform. Barcode sequences were unambiguously detected in 2.08 million reads out of a total of 3.43 million reads. Distributions of variants in all samples were calculated, and the precise composition of the initial mix (Table 3) was used to normalize the results. The mixture turned out not to be as equimolar as was expected, as the fraction of only five out of the nine variants was within 10% of the expected value.
Figure 3.
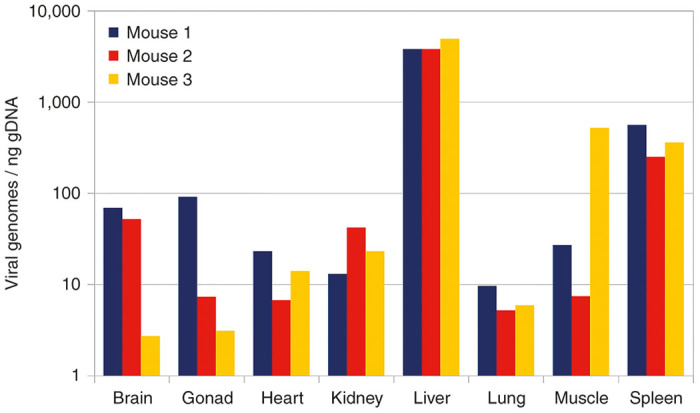
Global biodistribution. Distribution of unspecified viral genomes in tissue samples from three animals, derived from quantitative polymerase chain reaction titers.
Table 3. Sequencing-derived composition of viral mix.
| Variant | Number of reads | % |
|---|---|---|
| V1 | 18,807 | 8.59 |
| V2 | 22,362 | 10.21 |
| V3 | 51,280 | 23.42 |
| V4 | 21,870 | 9.99 |
| V5 | 24,078 | 10.99 |
| V6 | 19,930 | 9.10 |
| V7 | 24,609 | 11.24 |
| V8 | 12,329 | 5.63 |
| V9 | 23,727 | 10.83 |
A three-dimensional chart showing the average distribution of each variant across tissue samples, obtained by combining sequencing results with qPCR data, is displayed in Figure 4, allowing easy comparison of the different variants. For example, variants V2 and V3 appear to have the highest liver tropism, while variant V4 has both the lowest liver tropism and the highest spleen tropism. More detailed results are shown in Supplementary Figure S1.
Figure 4.
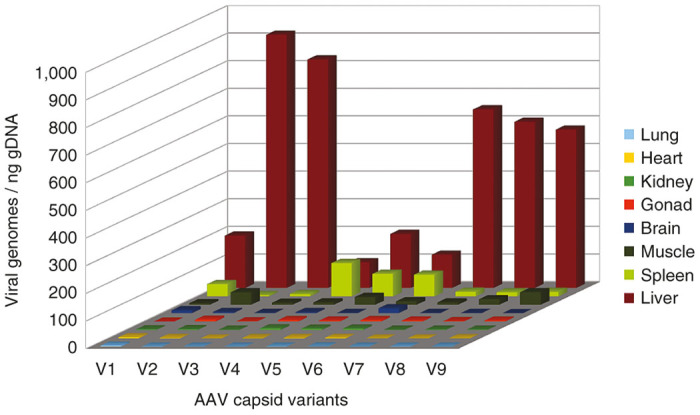
Comprehensive biodistribution. Distribution of viral genomes in tissue samples derived from global biodistribution combined with barcode sequencing (mean values from three animals). AAV, adeno-associated virus.
Evaluation of PCR-induced bias
Confidence in our method is based on the assumption that relative amounts of variant-specific barcode sequences, as calculated from the sequencing data, accurately reflect the actual relative amounts in the animal tissues. However, as the sequencing data are obtained after PCR amplification of tissue samples, it might seem reasonable to question this assumption. In order to test the possible effect of PCR on variant composition, a DNA mixture derived from 10 AAV variants was amplified for 20–35 PCR cycles. In order to save the cost of a dedicated sequencing reaction, the resulting samples were added to an existing Illumina sequencing project and analyzed using the same DNA-barcode script. Sequencing results are shown in Figure 5 and were obtained by analyzing a total of 438,392 recovered sequences. Remarkably, very little difference can be seen between the original template and the various PCR products. In addition, no correlation between number of cycles and deviation from original values is detected.
Figure 5.
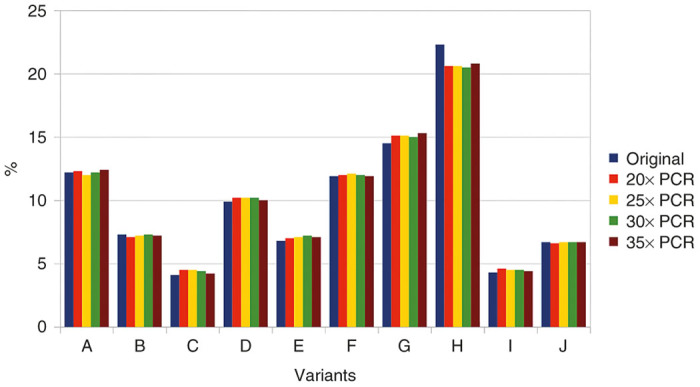
Effects of PCR cycles on variant composition. PCR, polymerase chain reaction.
Discussion
Productivity gains
To illustrate the advantages of our method, one should compare it with the traditional method. Consider a typical example of a biodistribution analysis involving 10 capsid variants to be screened across 10 mouse organs or tissues, with an animal group size of 3 and qPCR being done in triplicate. The traditional method would require 30 mice, 300 DNA isolations and 900 qPCR. In contrast, our method would only use 3 mice and necessitate 30 DNA isolations and 90 qPCR. In addition, 30 PCR and one deep-sequencing reaction would need to be performed. Thus, the improvement in terms of animal use, time and efforts is significant. The cost benefit might seem less clear and may vary depending on local rates for the different services involved. The cost could be further decreased by combining the analysis with an existing sequencing project, since less than 100,000 reads are really needed while the most basic deep sequencing systems typically generate millions or tens of millions of reads.
Accuracy
A major advantage is the gain in accuracy due to the precise knowledge of variant composition in the injected mixture, as calculated from sequencing data, allowing correcting variant composition in the tissue samples. Although qPCR is the method of choice to titer AAV, it has often been reported to suffer from poor reproducibility, which spurred the search for alternative titering methods.6,7 Indeed, our example (Table 3) shows such discrepancy between qPCR titers and sequencing-derived relative titers. In addition, significant variations in actual amount of injected AAV may occur due to pipetting errors or loss during tail vein injection (especially when injecting one variant per animal). Therefore, relative titering by deep sequencing of the actual injected mixture is a fundamental feature of our method that allows superior accuracy in biodistribution analysis. Obviously, confidence in that accuracy is crucially dependent on the quality of the plasmid used to generate the vectors, which should be validated by sequencing to verify barcode integrity. However, the DNA-barcode script incorporates an “after the fact” quality check by giving the option to list, for each sample, in addition to the variant-defined barcode sequences, all additional sequences that are detected at the variant-specific barcode location, together with their frequency. For example, the presence of such a sequence at a higher frequency than other nonvariant sequences would indicate a lower confidence in the results, particularly if it differs from one of the variant barcode sequences by just one nucleotide substitution, suggesting that the actual frequency of that particular variant is higher than the calculated one.
PCR-induced bias
The experiment illustrated in Figure 5 suggests that possible PCR-induced variations in the variant composition should not be a concern and are not dependent on the number of PCR cycles. However, it could be argued that PCR bias is inevitable at very low template concentrations. Indeed, if the PCR mixture only contains a few dozen template molecules, the variant composition on the PCR product will not be representative of the actual variation in the tissue sample. In practice, however, this is irrelevant, as illustrated in Figure 4: the purpose of biodistribution analysis is to determine which variant performs better in a given organ or tissue. For example, variants V2 or V3 would be the best candidates to target the liver and V4 would be best for the spleen, but it does not matter which variant is more present than others in the lungs, heart or kidneys, because the viral load in these organs is so low that it is irrelevant.
Scale-up potential
The method described here can potentially have a wide range of applications. We have created 45 barcoded variants of the pTR-UF50-BC vector (Table 1) so far, but this number could potentially be extended to 4,096 without changing the size (6 bp) of the barcode region. Hundreds of variants could therefore be analyzed simultaneously. Likewise, we have designed 5 forward and 7 reverse barcoded primers (Table 2), which allow 35 primer pair combinations, but the numbers could be extended to 16 each, or 256 primer pair combinations, without even changing the barcode size (2 nt). Therefore, large numbers of organs or tissue types could be tested and allow a higher-resolution biodistribution analysis, or larger numbers of animals, even of different species, could be tested simultaneously.
Applicability to transgene expression analysis
As discussed, in the pTR-UF-BC50 vector, a 6-nt barcode sequence is placed downstream of the luciferase-mApple ORF to provide an identifier for biodistribution analysis. The terminology, however, could be misleading because viral vector presence in a given tissue is not necessarily equivalent to the expression of a transgene, i.e., a transduction event. The method described here could easily be modified to quantify gene expression, since the barcode sequence is transcribed as part of the mRNA encoding luciferase-mApple (Figure 1). Global transgene expression levels would be determined by real-time quantitative reverse transcription PCR (qRT-PCR) targeting that particular mRNA, and relative expression of the different variants would be determined by sequencing RT-PCR products.
Comparison with existing AAV Barcode-Seq method
Another group has recently developed a method, designated AAV Barcode-Seq,8 to collect high-throughput data on correlation between AAV capsid sequence and phenotype and used it to draw a high-resolution functional map of the AAV capsid protein. Although the applications might seem quite different, the method actually presents significant similarity to our own. The specificity of our approach, however, is that it provides a comprehensive set of easy to implement tools and procedures for one particular application, which is the biodistribution analysis of AAV capsid variants. Our method can directly be used in a vector development pipeline, between directed evolution and final vector evaluation, to quickly select the best variants among several candidates. A key feature is that the variant capsid genes are provided in trans while the barcodes are part of the viral genomes in which viral genes have been replaced by reporter genes. After barcode sequencing-mediated biodistribution analysis, selected variants can be used immediately for bioluminescence or fluorescence studies without needing any subcloning or repackaging.
Competition
One possible concern that was not addressed in this study is that potential competition among capsid variants for cell receptors might affect the results. This issue could even be exacerbated if some viral preparations contain large amounts of empty capsids. In order to prevent such possible competition effects, we would recommend limiting the injected dose, or injecting different amounts in different sets of animals.
Materials and Methods
Vector
The complete sequence of vector pTR-UF50-BC has been deposited in GenBank under accession number KF926476.
AAV production
HEK-293 cells were cotransfected with three plasmids: one derived from pACG-2 (ref. 9) containing the AAV2 rep gene and one of the variant cap genes, one of the pTR-UF50-BC barcoded vectors, and pHelper (Agilent, Santa Clara, CA). AAV was purified from both cells and culture medium 3 days later on iodixanol gradient10 and quantified by qPCR.
Animals
Four- to five-week-old male C57BL/6J mice (University of Florida Animal Care Services, Gainesville, FL) were used for vector validation experiments. Ten-week-old male BALB/c mice (Charles River Laboratories, Wilmington, MA) were used for biodistribution analysis. All animal procedures described in this article were approved by the University of Florida Institutional Animal Care and Use Committee.
AAV injections
All surgical procedures were performed using aseptic techniques and isoflurane gas anesthesia. Brain surgeries were performed as previously described.11 Briefly, once anesthetized, mice were placed in the stereotactic frame (Kopf Instruments, Tujunga, CA), and 2 µl of AAV vector (1 × 1010 vg) were injected into the right striatum (anterior-posterior −0.3 mm, lateral −2.0 mm, dorsoventral −3.0 mm), through a glass micropipette with an inner diameter of 30–40 µm at a rate of 0.5 µl/minute. The needle was left in place for 5 minutes prior to withdrawal from the brain. For muscle injections, a single dose of 100 µl of AAV vector was administrated in the leg muscle of anesthetized mice using a 30G syringe over a period of 15 seconds. For biodistribution analysis, mice were administered a dose of 2 × 1011 vg of AAV vector mix in a 250 µl volume through tail vein injection.
Bioluminescence imaging
Mice were imaged as previously described12 using a Xenogen IVIS imaging system (Perkin Elmer, Santa Clara, CA), 5 minutes after intraperitoneal injection of D-luciferin (126 mg/kg) dissolved in phosphate-buffered saline (PBS). For ex vivo brain bioluminescence imaging, mice were sacrificed by cervical dislocation immediately after being imaged, decapitated and had their brain dissected. Subsequently, brains were sliced into 1.0-mm thick coronal sections and imaged. The BLI data are reported as raw data, as the total number of counts reaching the charge-coupled device detector.
Brain tissue preparation
Animals were deeply anesthetized with pentobarbital (Beuthanasia-D) and perfused through the ascending aorta. Brains were perfused with 10 ml of saline solution, followed by 10 ml of ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Brains were removed and postfixed overnight at 4 °C in paraformaldehyde solution. Fifty micrometer-thick coronal sections were cut on a vibratome stage. Sections were stored in 0.02% azide in PBS until assayed.
Immunohistochemistry for confocal microscopy
Immunostaining was carried out on free-floating sections as follows: incubation with 0.1% Triton-X100 and 3% donkey serum in PBS for 1 hour at room temperature; incubation with anti-firefly luciferase antibody (Abcam, Cambridge, MA) diluted 1:1,000 at 4 °C overnight; three washes with PBS for 10 minutes each wash; incubation with Cy2 donkey anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hours at room temperature. Sections were then incubated with 4’,6-diamidino-2-phenylindole at 1 µg/ml for 10 minutes, washed with PBS three times for 10 minutes each wash, mounted on slides with Mowiol 4–88 (Sigma-Aldrich, St. Louis, MO) and examined with a confocal microscope TCS SP5 (Leica Microsystems, Wetzlar, Germany).
Isolation of genomic DNA
Three weeks after injection, mice were euthanized by cervical dislocation after being anesthetized with isoflurane. Organs and tissue samples were immediately harvested and placed on ice in 1.5 ml tubes. Samples were then homogenized using disposable pestles, and aliquots of the resulting paste were then transferred into new tubes for DNA isolation. GeneJET Genomic DNA Purification kit (Thermo Fisher Scientific, Waltham, MA) was used according to the manufacturer’s instructions. Total DNA was quantified on a NanoDrop 1000 (Thermo Fisher Scientific) by averaging three readings for each sample.
qPCR
Vector genomes were quantified by qPCR on a LightCycler 480 instrument (Roche, Basel, Switzerland), using dual color hydrolysis probe CBA-P and oligonucleotide primers CBA-F and CBA-R (Table 2) targeting a region of the promoter sequence. Reactions were performed in triplicate in 20 µl volumes containing 0.5 µmol/l each primer, 0.23 µmol/l probe and 50% LightCycler 480 Probes Master reagent (Roche). Linearized plasmid DNA encoding the vector genome was used as a standard. Cycling conditions were as follows: 7 minutes initial activation at 97 °C followed by 45 cycles of 10 seconds at 95 °C denaturation, 25 seconds at 58 °C annealing, and 5 seconds at 72 °C extension.
IonTorrent sequencing
The barcode region of the pTR-UF50-BC vector was amplified individually from each DNA sample as well as from an aliquot of the viral mixture that was used for injection, using barcoded primers BCn-F and BCn-R (where n designates a number identifying a particular barcode, Table 2). Each reaction was performed using a different barcode pair. The primer regions upstream of the barcode are the IonTorrent A and trP1 sequences respectively for the forward and reverse primers, allowing PCR products to be used as sequencing templates. PCR was performed using Q5 DNA polymerase (New England Biolabs, Ipswich, MA) following manufacturer’s instructions in 20 µl reactions. Cycling conditions were: 30 seconds initial denaturation at 98 °C, followed by 30 cycles of 10 seconds denaturation at 98 °C, 25 seconds annealing at 61 °C and 10 seconds extension at 72 °C, followed by 1 minute final extension at 72 °C. For difficult templates, the number of cycles was increased up to 35, and reaction volume was increased up to 50 µl if necessary. Aliquots (3.5 µl) of PCR products were analyzed by agarose gel electrophoresis to visually estimate relative amplification yields. PCR products were then pooled in approximately equimolar amounts and the resulting mixture was purified using a DNA clean and concentrator kit (Zymo Research, Irvine, CA). Purified DNA was then sequenced on an Ion PGM (Thermo Fisher Scientific) instrument by the NextGen DNA sequencing ICBR core at the University of Florida.
Illumina sequencing
Primers IT-F and IT-R (Table 2) were used to amplify a barcoded mixture that was a leftover from a previous Ion Torrent sequencing project. A first PCR was performed to generate the template (BC0) for subsequent PCRs. Using same primers, BC0 was then used as a template to generate PCR products BC20, BC25, BC30, and BC35 corresponding to 20, 25, 30, and 35 amplification cycles respectively. In all cases, Q5 DNA polymerase (New England Biolabs) was used according to the manufacturer’s instructions. Template dilution factor was 50 in all reactions. Cycling conditions were as follows: 30 seconds initial denaturation at 98 °C, followed by 20–35 cycles of 10 seconds denaturation at 98 °C, 20 seconds annealing at 70 °C and 10 seconds extension at 72 °C, followed by 1 minute final extension at 72 °C. All five PCR products BC0 to BC35 were then indexed using the Nextera XT Index kit (Illumina, San Diego, CA) following manufacturer’s instructions, before being purified using DNA clean and concentrator kit (Zymo Research). They were then pooled into an existing Illumina sequencing project as less than 1% of the project’s DNA amount. Sequencing was performed on a MiSeq (Illumina) instrument by the NextGen DNA sequencing ICBR core at the University of Florida.
Sequencing data analysis
Sequencing data was analyzed using the script DNA-barcode that was written in Python 2.7 and can be downloaded from http://sourceforge.net/projects/dnabarcode/. Detailed instructions are included in the package.
Acknowledgments
This work was funded by NIH NHLBI ROI HL097088 and NIH R01 GM082946. Special thanks to Barbara Locke from ACS for performing tail vein injections and to David Moraga Amador and his team at ICBR for expert sequencing services.
The authors declared no conflict of interest.
References
- Hastie, E and Samulski, RJ (2015). Adeno-associated virus at 50: a golden anniversary of discovery, research, and gene therapy success–a personal perspective. Hum Gene Ther 26: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y, Siriwon, N, Rohrs, JA and Wang, P (2015). Generation of Targeted Adeno-Associated Virus (AAV) Vectors for Human Gene Therapy. Curr Pharm Des 21: 3248–3256. [DOI] [PubMed] [Google Scholar]
- Shaner, NC, Lin, MZ, McKeown, MR, Steinbach, PA, Hazelwood, KL, Davidson, MW et al. (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods 5: 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, J, Qian, JJ, Yi, S, Harding, TC, Tu, GH, VanRoey, M et al. (2005). Stable antibody expression at therapeutic levels using the 2A peptide. Nat Biotechnol 23: 584–590. [DOI] [PubMed] [Google Scholar]
- Thomas, G (2002). Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3: 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock, M, Alvira, MR, Chen, SJ and Wilson, JM (2014). Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum Gene Ther Methods 25: 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedra, J, Ontiveros, M, Miravet, S, Penalva, C, Monfar, M and Chillon, M (2015). Development of a rapid, robust, and universal picogreen-based method to titer adeno-associated vectors. Hum Gene Ther Methods 26: 35–42. [DOI] [PubMed] [Google Scholar]
- Adachi, K, Enoki, T, Kawano, Y, Veraz, M and Nakai, H (2014). Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat Commun 5: 3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J, Samulski, RJ and Xiao, X (1997). Role for highly regulated rep gene expression in adeno-associated virus vector production. J Virol 71: 5236–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin, S, Byrne, BJ, Mason, E, Zolotukhin, I, Potter, M, Chesnut, K et al. (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6: 973–985. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk, OS, Li, S, Sullivan, LF, Chen, W, Kondrikova, G, Manfredsson, FP et al. (2008). The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci USA 105: 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroose, CM, Reumers, V, Gijsbers, R, Bormans, G, Debyser, Z, Mortelmans, L et al. (2006). Noninvasive monitoring of long-term lentiviral vector-mediated gene expression in rodent brain with bioluminescence imaging. Mol Ther 14: 423–431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.