

Abstract
Sepsis is a systemic inflammatory response induced by an infection, leading to organ dysfunction and mortality. Historically, sepsis-induced organ dysfunction and lethality were attributed to the interplay between inflammatory and antiinflammatory responses. With advances in intensive care management and goal-directed interventions, early sepsis mortality has diminished, only to surge later after “recovery” from acute events, prompting a search for sepsis-induced alterations in immune function. Sepsis is well known to alter innate and adaptive immune responses for sustained periods after clinical “recovery,” with immunosuppression being a prominent example of such alterations. Recent studies have centered on immune-modulatory therapy. These efforts are focused on defining and reversing the persistent immune cell dysfunction that is associated with mortality long after the acute events of sepsis have resolved.
Introduction
Sepsis is the constellation of symptoms occurring when an infection leads to a systemic inflammatory response (1), including fever, leukocytosis or leukopenia, decreased vascular resistance frequently leading to hypotension (septic shock), organ failure (severe sepsis), and death (2). Despite progress in antibiotic therapy, ventilator management, resuscitative strategies, and blood glucose maintenance, severe sepsis remains the leading cause of death in the intensive care unit (ICU) (3). Even more alarming is the escalating cost of sepsis-associated medical care, which is estimated at $17 billion annually in the United States (4). Given the expanding elderly population with immune senescence (5), overall sepsis mortality is expected to grow at an alarming rate during the next two decades (6).
Despite the more than 100 therapeutic clinical trials that have been conducted, no FDA-approved treatment options currently exist for sepsis. Even though substantial advances in our understanding of immune pathophysiology have resulted in improvements in survival, long-term sepsis mortality rates remain at 20% to 50% (7). Although there are sundry editorials, reviews, and commentaries opining a multitude of possible explanations, such as impaired cellular metabolism, tissue oxygenation, and myocardial dysfunction (8), one of the most accepted postulates describes a complex immune/inflammatory process that is still ill defined (8, 9). Historically, most research focused on ameliorating the hyperinflammatory response by attempting to block TNF and IL-1 (10); however, steady improvements in clinical treatment protocols (11) have resulted in more patients surviving the initial inflammatory and concomitant antiinflammatory response, only to manifest varying states of prolonged immune dysfunction, immune suppression (12), and persistent inflammation and catabolism (13), all of which are characterized by nosocomial infections, immune paralysis, and protracted events leading to death (14). Despite recent improvements in 30-day sepsis mortality rates, multiple reports confirm a dramatic and concerning increase in sepsis-associated mortality that escalates with time and particularly impacts the elderly population (7, 14). These data suggest that short-term gains in sepsis survival have been forfeited to escalating long-term sepsis mortality.
The sepsis death distribution has historically been biphasic, with an initial early peak at several days due to inadequate resuscitation, resulting in cardiac and pulmonary failure, and a late peak at several weeks due to persistent organ injury or failure (ref. 15 and Figure 1A). Considering the recent trend in mounting long-term mortality, a trimodal distribution is more indicative of the current death distribution (refs. 14, 15, and Figure 1B). The early peaks in mortality exist, albeit of much lower magnitude, and the third upswing occurs after 60 to 90 days and continues to soar over time (14, 16). These deaths are probably the consequence of more sophisticated ICU care that keeps elderly and comorbidly challenged patients alive longer in spite of ongoing immune, physiologic, and biochemical aberrations (17). Although the precise causes of long-term sepsis mortality are still uncertain, several reports suggest that advanced age, comorbidities, and persistent organ injury synergize to generate a deleterious state of chronic disease characterized by immune dysfunction, immune suppression, and persistent catabolism and inflammation (12, 13, 18). Furthermore, persistent inflammation, chronic immobility, catabolic drugs, and extended paralytics all contribute to a protracted state of immune dysregulation and chronic deterioration. Thus, investigators have been forced to refocus their efforts on the underlying innate and adaptive immune derangements that facilitate impaired sepsis recovery and survival, especially over the long term (19, 20). In this Review, we will highlight the multitude of sepsis-induced alterations in innate and adaptive immune cell function as well as the clinical implications and potential therapeutic interventions.
Figure 1. Historical and current sepsis mortality distribution.

(A) Historically, sepsis deaths have occurred in a biphasic distribution, with an initial early peak at several days due to inadequate resuscitation, resulting in cardiac and pulmonary failure, and a late peak at several weeks due to persistent organ injury and failure. Considering the recent trend in sepsis outcomes, the elderly population, and mounting long-term mortality, a trimodal distribution may be more indicative of the current sepsis-associated death distribution. (B) The two early peaks in mortality exist, albeit with much lower magnitude than in the past. The third upswing occurs approximately 60 to 90 days after sepsis and continues to soar as time progresses. This delay in sepsis mortality is thought to be the consequence of the more sophisticated ICU care that keeps elderly and comorbidly challenged patients alive longer in spite of ongoing immune, physiologic, and biochemical aberrations.
Immune cell dysfunction in sepsis
Sepsis affects the immune system by directly altering the lifespan, production, and function of the effector cells responsible for homeostasis (21, 22). The hematopoietic and effector cells responsible for the maintenance of immune surveillance against offending pathogens are also involved in tissue regeneration and wound healing. Over the past two decades, a debate has persisted as to whether innate and adaptive immunity or inflammatory and antiinflammatory processes are more detrimental to sepsis survival (23). The inflammatory response was formerly thought to drive early mortality in the initial days of sepsis, and the compensatory antiinflammatory response was thought to induce mortality several days to weeks later through immune suppression and organ failure (24). However, new insights garnered using genomic analysis of tissue samples from septic patients (12) and severely injured trauma patients have identified an enduring inflammatory state driven by dysfunctional innate and suppressed adaptive immunity that culminates in persistent organ injury (25) and death of the patient (refs. 26, 27, and Figure 2). Although drawbacks in each of these studies exist, when the collective results are juxtaposed with contemporary patient outcomes, it is clear that a new paradigm is necessary to explain the long-term mortality surge following sepsis. It may be that inflammatory and antiinflammatory responses and innate and adaptive immune systems are each equally important and represent potential targets for immune therapy to improve sepsis outcomes (20–22, 28). The following discussion provides an overview of the sepsis-induced immune alterations in innate and adaptive cell types, along with the most promising immune modifiers being considered for future therapy in human sepsis. Although this discussion will focus on human sepsis, many of the current insights are gleaned from murine sepsis models. The authors recognize the ongoing debate about the efficacy of murine models to accurately reflect human disease processes (29). The authors’ opinion is that both human and animal models are necessary for continued progress in the science of sepsis.
Figure 2. Immune dysregulation in sepsis.

New insights into immune dysregulation have been gained using samples from deceased septic patients as well as from severely injured trauma patients. These studies demonstrate an enduring inflammatory state driven by dysfunctional innate and suppressed adaptive immunity that culminates in persistent organ injury and death of the patient. Although the initial inflammatory process, if unabated, contributes to organ failure and early mortality, this process is largely ameliorated by improvements in patient management protocols. However, considering that the vast majority of sepsis survivors are elderly with highly comorbid conditions, the short-term gains in survival have merely been pushed back by several months to a year. Although theories about the processes underlying this observation are numerous, the widespread consensus is that persistent derangements in innate and adaptive immune system cellular function are the main culprits driving long-term mortality.
Neutrophils.
Neutrophils are a fundamental component of innate immunity and essential for microbial containment and eradication and for sepsis survival (30). They comprise the majority of the cells in the BM and are the first responders to foreign invaders (31). Sepsis induces a state of delayed neutrophil apoptosis (32), leading to persistent neutrophil dysfunction, compounded by the release of immature neutrophils from BM that culminates in neutrophil deficits in oxidative burst (33), cell migration (34, 35), complement activation, and bacterial clearance (36), all of which contribute to ongoing immune dysfunction and inflammation persistence. These findings, coupled with insufficiencies in TLR signaling (37), chemokine-induced chemotaxis (38), altered apoptotic pathways, and neutrophil senescence (39), result in functional deficiencies that persist even after sepsis symptoms have disappeared. Accumulating evidence also indicates that neutrophils function as antigen-presenting cells (APCs) in a broad array of infections and control innate and adaptive responses through activation of CD4+ and CD8+ T cells (40, 41). More important, multiple studies in humans have implicated the complex array of neutrophil deficits in the development of nosocomial and secondary infections (42). Sepsis patients with the most pronounced derangements in neutrophil function are the most predisposed to develop ventilator-associated pneumonia and other nosocomial infections (43). The majority of patients who succumb to sepsis have ongoing infections (12), suggesting that defects in neutrophil-mediated bacterial clearance could serve as therapeutic targets to stimulate improved neutrophil production, maturation, and function.
Monocytes and macrophages.
The impact of sepsis on monocyte subpopulations has been the subject of intense investigation over the past four decades. It has long been known that diminished mononuclear cell HLA-DR expression correlates with sepsis lethality in humans (44). Furthermore, the diminished ability of blood monocytes from septic patients to release proinflammatory cytokines after endotoxin (LPS) challenge has been described as “endotoxin tolerance,” which contributes to poor outcomes (45, 46). The major impact of endotoxin tolerance on monocytes and macrophages translates to decreased antigen presentation related to decreased HLA-DR expression (47). Moreover, blood monocytes from septic patients exhibit a diminished capacity to release the proinflammatory cytokines TNF, IL-1, IL-6, and IL-12 after LPS challenge, indicating that intracellular signaling has shifted toward production of the antiinflammatory mediators associated with nosocomial infections and increased mortality.
Low monocyte HLA-DR expression levels serve as a surrogate marker of monocyte “anergy,” development of nosocomial infections, and death (48–50). Several studies correlate low monocyte HLA-DR expression levels with diminished monocyte function and reduced antigen-specific lymphocyte proliferation (51, 52), suggesting that monocyte anergy and immune suppression independently contribute to the increased risk of adverse events in sepsis. Although the mechanisms underpinning LPS tolerance are not fully understood, sepsis-induced monocyte epigenetic reprogramming may play a pivotal role in the suppressive monocyte phenotype (53). Monocyte mRNA analysis demonstrates an increased expression of inhibitory cytokine genes and reduced expression of proinflammatory chemokine genes (54). Recent reports of studies done in humans make a convincing case for epigenetic reprogramming as the epicenter of monocyte anergy, although the functional impact of these epigenetic alterations is unknown (55).
NK cells.
NK cells were formerly viewed as rudimentary killers of cells that either lacked self-identification or were infected by viruses. However, we now understand that NK cells act as immune regulators. NK cells are divided into different subpopulations on the basis of CD16 and CD56 expression (56). Human sepsis data indicate that both CD56hi and CD56lo NK cell subpopulations are altered during sepsis. These alterations appear to be associated with increased mortality in septic humans (57–59). Additionally, NK cell cytotoxic function is decreased (60). Similar to monocyte LPS tolerance, NK cell ex vivo production of IFN-γ in response to TLR agonists is diminished. This observation suggests that NK cell tolerance may be responsible for the reactivation of latent viruses such as CMV, which is frequently described in critically ill patient populations and may serve as a potential target for therapeutic intervention (61).
DCs.
DCs are classified as either conventional DCs (cDCs) or plasmacytoid DCs (pDCs). The former are similar to monocytes and secrete IL-12, while the latter are similar to plasma cells and secrete IFN-α. cDCs and pDCs are of particular interest because of their enhanced apoptosis in sepsis patients (62) and in those who develop nosocomial infections (63). Similar to the reduction in HLA-DR expression observed on monocytes, DCs also show reduced HLA-DR expression and produce increased amounts of immune-suppressive IL-10 (64). Furthermore, coculture of DCs with T cells facilitates T cell anergy or Treg proliferation, both of which are associated with immune dysfunction. Multiple reports have shown that prevention of sepsis-induced DC apoptosis or augmentation of DC function enhances sepsis survival (65, 66). One report demonstrated that immune suppression can be ameliorated by DC treatment with growth factor FMS-like tyrosine kinase 3 ligand (FLT3L) (67). FLT3L treatment in models of burn sepsis enhances DC cytokine secretion (IL-12, IL-15, and IFN-γ) and augments CD4+ T cell, NK cell, and neutrophil function (67). Further investigations suggest that the improvements in sepsis survival gained from DC therapy occur through TLR signaling and increased expression of MHC class II antigens and the costimulatory molecules CD80 and CD86 (68). These observations have led researchers to postulate that DC numbers and functional improvements may be primary targets for therapeutic interventions in sepsis (65, 66).
Myeloid-derived suppressor cells, stem cells, and myelopoiesis.
Myeloid-derived suppressor cells (MDSCs) are a population of immature myeloid cells that expand dramatically in sepsis, suppress adaptive immune responses, and signal through TLR-mediated pathways (40, 69). Sepsis-associated MDSCs are phenotypically similar to the MDSCs described in advanced cancer (40, 70). Although MDSCs can inhibit CD8+ T cell function, the actual impact of MDSCs in human sepsis is unknown. The collective reports suggest a beneficial role of MDSCs centered on replenishing innate cell function and immune surveillance through emergency granulopoiesis (35). We found that, before MDSC expansion occurs, there is a window of susceptibility to the secondary infections and subsequent mortality that are associated with reduced BM cell numbers and reduced blood and tissue neutrophil numbers and function (33). Optimal MDSC expansion through enhanced granulopoiesis confers long-term immunity to secondary infections in sepsis (71). Because of the inherent difficulty of immune phenotyping immature myeloid cells in humans versus the relative ease of doing so in mice, few clinical studies have investigated the roles of MDSCs in septic patients (72). Nonetheless, there is considerable interest in myelopoiesis, MDSC expansion, and hematopoietic cell function (33, 40, 71, 73, 74). Given the importance of an efficient regeneration of functioning neutrophils, monocytes, and DCs, it is no surprise that MDSCs expand to meet the continual need for functional innate immune cells. And given that it takes the BM five to seven days to produce a mature neutrophil, an expansive pool (~18 × 1011) of immature myeloid precursors in BM and secondary lymphoid organs (75) is a necessity. In humans, approximately 16 × 1010 neutrophils are produced daily, and this cell population can be rapidly increased by 5- to 10-fold in response to infection. Our work has shown that myeloid expansion involving hematopoietic stem cells (HSCs) occurs through c-KIT–, type I IFN– (IFN-I), and CXCL10-dependent mechanisms that involve IFN-I–secreting B cells (73, 74). Moreover, impaired HSC proliferation and development in human BM transplant models is associated with mortality due to secondary infections (76). Humans with suppressed granulopoiesis clearly experience infections, demonstrating the essential requirement for effective neutrophil production. Conversely, overzealous MDSC proliferation may facilitate a physiologic syndrome of persistent inflammation, such as that seen in adult respiratory distress syndrome (ARDS) or persistent inflammation, immunosuppression, and catabolism syndrome (PICS), causing poor outcomes in patients with sepsis (13). The specific contributions of myelopoiesis and MDSCs to sepsis recovery versus those of persistent inflammation and catabolism remain poorly understood.
γδ T cells.
γδ T cells comprise a small subset of T cells that possess a distinct T cell receptor (TCR) on their cell surface. Most T cells have a TCR composed of two α and β glycoprotein chains, while γδ T cells have a TCR that is made up of one γ chain and one δ chain. This group of T cells exists chiefly in the gut mucosa within a population of intraepithelial lymphocytes (77). Although the antigens to which γδ T cells respond are unknown, it is suspected that these cells recognize lipid antigens from pathogens present on mucosal surfaces within the intestine (78). Upon activation, γδ T cells release IFN-γ, IL-17, and other chemokines. In septic patients, the number of circulating γδ T cells is significantly reduced, and the reductions correlate with the highest rates of mortality (79). The reduction in γδ T cells in the intestinal mucosa may serve to potentiate otherwise noninvasive intestinal bacteria to become invasive and translocate into the systemic circulation, causing infections following sepsis (80).
CD4+ T cells and Th cell subpopulations.
Th cells assist other cells with immunologic processes, including B cell differentiation, cytotoxic T cell activation, and monocyte stimulation. When presented with peptide antigens by MHC class II molecules expressed on APCs, CD4+ T cells become activated, rapidly divide, and secrete cytokines that regulate immune responses. Once activated, CD4+ T cells can differentiate into one of several subsets including Th1, Th2, Th3, Th17, Th22, Th9, or T follicular helper (Tfh) cells, which facilitate differing immune responses through cytokine production (81). Although numerous reports detail the effects of sepsis on circulating and peripheral CD4+ T cell subsets (82), we will highlight only selected reports to convey to the reader the common themes and potential areas of therapeutic interest.
One of the most notable T cell defects induced by sepsis is the development of apoptosis, which decimates CD4+ cell populations (12, 83). In humans who succumbed to sepsis, there was a much greater magnitude of lymphocyte (specifically CD4+) apoptosis than was found in cells from sepsis survivors (12). Of the CD4+ cells that persist, multiple reports demonstrate that both Th1- and Th2-associated cytokine production is diminished during sepsis (84). Marked reductions in the transcription factors T-bet and GATA3, which modulate the Th1 and Th2 responses, respectively, support the notion that CD4+ subsets are suppressed during sepsis (85). There are many factors that regulate CD4+ Th cell subpopulation differentiation, including histone methylation and chromatin remodeling, which, together, are postulated to suppress Th1 and Th2 CD4+ T cell functions (86). However, the sepsis-induced immune impact is not only relegated to Th1 and Th2 CD4+ T cells, but also to Th17 cell subsets and probably other Th cell subsets as well. The Th17 cytokine response is diminished in sepsis and may negatively impact mortality (87). Given the fundamental role of Th17 in the eradication of fungal infections, reduced Th17 cytokine production in sepsis may be responsible for the increased susceptibility to fungal infections frequently encountered in critically ill patient populations (88). Moreover, IL-7 treatment has been demonstrated to increase Th17 cell responsiveness and reduce mortality from secondary fungal infections, making IL-17 a potential therapeutic agent (89).
Tregs.
Tregs are a component of adaptive immunity that suppresses responses of other effector T cell subsets, helping to maintain tolerance to self-antigens and suppress autoimmune disease (90). In states of sepsis and critical illness, Tregs may potentiate undesirable effector T cell (Teff) suppression that prolongs recovery. Increased Treg ratios have been described early after sepsis and remained elevated in those patients who died. Further studies revealed that the increase in Tregs was due to the loss of effector Th cells rather than an absolute increase in Treg numbers (91). This observation suggests that Tregs are more resistant to sepsis-induced apoptosis, thereby preventing the recovering immune system from mounting excessive autoimmune responses. Moreover, heat shock proteins and histones that induce mononuclear cell epigenetic changes have also been implicated as inducers of Tregs in sepsis (92). Recent mouse studies demonstrated that Tregs are detrimental to Teff proliferation and function (93). This effect was mitigated by siRNAs that inhibited Treg differentiation (91). In other reports, glucocorticoid-induced TNF receptor–related protein (GITR) inhibitory Abs were used to block Treg function, resulting in improved immune function and microbial killing (94). Treg-associated immune dysfunction in sepsis has also been linked to increased solid tumor growth, probably from diminished cytotoxic T lymphocyte (CTL) and mononuclear cell effects (95). Therefore, the notion exists that Tregs persist in sepsis, augment immune dysregulation, contribute to poor outcomes, and may serve as targets for immune modulation.
B cells.
B cells represent a heterogeneous cell population with varying functional and phenotypical properties. Until recently, the function of B cells in sepsis was relegated to the production of Abs and the development of memory plasma B cells (96). However, many recent reports demonstrated that B cells play a more important role in sepsis than previously thought. Although patients with septic shock demonstrate diminished overall B cell numbers, the largest deficits in B cell number is in CD5+ B1a-type cells, which can be predictive of survivors and nonsurvivors (97). In murine sepsis models, B cells are needed to improve cytokine production, reduce bacteremia, and improve outcomes through IFN-I (98). Others have recently described an innate response activator (IRA) B cell population that is phenotypically and functionally distinct from B1a cells, depends on pattern recognition receptors (PRRs), and produces granulocyte-macrophage CSF (GM-CSF). Deletion of IRA B cells impairs bacterial clearance, causes a cytokine storm, and precipitates septic shock. These observations position IRA B cells as gatekeepers of bacterial infection and identify new treatment targets for patients with sepsis (99). Additionally, IRA B cell production of IL-3 has been shown to potentiate inflammation in sepsis, induce myelopoiesis of Ly-6Chi mononuclear cells, and augment the cytokine storm, while high plasma IL-3 levels are independently associated with increased mortality in patients with sepsis (100). Taken together, these reports not only further our understanding of the role of B cells in immune activation and emergency myelopoiesis, but also identify IL-3 as a potential therapeutic target in sepsis (101).
Potential immune-modulatory therapies
Granulocyte CSF and GM-CSF in sepsis.
Granulocyte CSF (G-CSF) is a glycoprotein that stimulates the production of stem cells and granulocytes (102). G-CSF is also highly efficacious in reducing the incidence of sepsis in patients with low absolute neutrophil counts, such as those undergoing BM transplantation, chemotherapy, or radiation (103). Two randomized clinical trials involving recombinant G-CSF have been conducted in an attempt to increase neutrophil production and function. Investigators hypothesized that administration of G-CSF would improve neutrophil function and microbial eradication in such circumstances. While there was an increase in total leukocyte numbers in blood, there was no improvement in 28-day mortality rates (104, 105). Although the initial conclusion of these two clinical studies was discouraging, the fact remains that most of the sepsis-induced mortality occurs in a protracted process beyond 90 days (14). The impact of G-CSF on mortality beyond 28 days is unknown. Given that persistent derangements in myelopoiesis, granulopoiesis, and neutrophil function are hallmarks of long-term sepsis mortality, especially in diabetic and elderly populations, prolonged G-CSF administration might prove efficacious for infection eradication and survival in future clinical trials.
GM-CSF is a cytokine that stimulates stem cells to produce neutrophils, monocytes, and macrophages (106). In the immunosuppressive phase of sepsis, patients who were ventilator dependent were treated with recombinant GM-CSF and had fewer ventilator and ICU days (107, 108). Recombinant GM-CSF therapy in immunosuppressed pediatric patients with sepsis restored TNF production in lymphocytes and reduced nosocomial infections (109). Moreover, a meta-analysis of more than 12 clinical studies involving either G-CSF or GM-CSF demonstrated that either therapy significantly reduced the rate of infection (110). Considering that 70%–80% of patients who die from sepsis harbored persistent infections (12), GM-CSF and/or G-CSF, in combination with other immune-modulatory agents, may prove valuable for infection eradication during sepsis and improved long-term survival after sepsis.
IFN-γ.
IFN-γ is the only member of the type II IFN family and is crucial for immune function against viral, bacterial, and protozoal infections. Moreover, IFN-γ is a key activator of macrophages, inducing class I MHC expression (47). When treated with recombinant IFN-γ, patients with sepsis and decreased monocyte HLA-DR expression showed reversal of monocyte dysfunction and improved sepsis survival (111). Although the majority of the interventional studies with IFN-γ were done in burn and severely injured trauma cohorts, the largest of these trials reported a decrease in the number of infection-related deaths among patients treated with IFN-γ (112). Furthermore, a recent study of severely injured trauma patients revealed that 42 of 63 genes identified as being differentially expressed in uncomplicated and complicated trauma patients were specifically associated with IFN signaling. The authors found that IFN-associated genes were suppressed in trauma patients with complicated outcomes (26, 27), implying that this set of genes may be useful for identifying patients at risk for complications after trauma and that these patients might respond positively to therapies utilizing IL-7, IL-15, IFN-γ, and GITR agonists. IFN-γ may be more effective in sepsis populations if targeted to patients who demonstrate or are at risk for immune suppression, decreased monocyte HLA-DR expression, adaptive immune dysfunction, or chronic inflammation during prolonged hospital stays. Though IFN-γ offers promise as a potential immune therapy due to its ability to rejuvenate monocyte function and adaptive immunity, IFN-γ may be more efficacious if administered in a time-phased approach in conjunction with GM-CSF and G-CSF, or even IL-7 and/or IL-15, in order to bolster specific immune function, reduce secondary and nosocomial infections, and improve long-term survival as sepsis recovery evolves.
Programmed cell death protein 1 and ligand.
As a result of steady progress in tumor biology, a new class of drugs has been developed that inhibit programmed cell death protein 1 (PD-1), a protein that under normal circumstances sends an inhibitory signal that reduces CD8+ T cell proliferation and accumulation in lymph nodes. PD-1 is expressed on T and B lymphocytes and myeloid cells. The PD-1 ligand PD-L1 is expressed on epithelial and endothelial cells, monocytes, macrophages, and DCs (113). Given that PD-1 is upregulated on CD4+ and CD8+ T cells in states of viral infection and cancer, it is often associated with the phenomenon of “T cell exhaustion,” which is thought to result from prolonged periods of antigen exposure (114). Both anti–PD-1 and anti–PD-L1 treatments have shown great promise in trials against cancer and viral infection. Hence, it has been postulated that anti–PD-1 and anti–PD-L1 therapies could have similar beneficial effects in patients suffering from sepsis-induced immune dysfunction (115). Patients with severe sepsis show increased levels of PD-1 and PD-L1 on their monocyte and T lymphocyte populations (116). Recent studies demonstrated neutrophil PD-L1 upregulation on neutrophils from septic mice and humans, resulting in the potentiation of lymphocyte apoptosis through contact inhibition, which correlated with outcome (117). Moreover, in clinically relevant animal models of bacterial sepsis, inhibition of PD-1 and PD-L1 signaling improved survival and reduced the incidence of fungal infections (118). Considering the beneficial impact on adaptive immunity and tumor eradication strategies, it makes sense that PD-1 and PD-1L could concomitantly serve as biomarkers of sepsis-initiated immune suppression as well as prospective therapeutic targets to reverse adaptive immune dysfunction and improve long-term survival.
Recombinant human IL-3, IL-7, and IL-15.
Bearing in mind the significant loss of lymphocytes in severe sepsis, several investigators have postulated that IL-7 administration is a potential therapeutic strategy. IL-7 is a hematopoietic cytokine produced by stromal cells and is required for B and T cell production, development, homeostasis, and maintenance (119). IL-7 is an attractive molecule due to its ability to upregulate expression of the antiapoptotic protein BCL2, which causes increased numbers of blood CD4+ and CD8+ lymphocytes. Moreover, IL-7 enhances TCR diversity, which is lost in septic patients and is associated with increased nosocomial infections and mortality (89, 120). Low doses of recombinant human IL-7 preferentially activate Teffs from patients with sepsis (121). Although evidence exists that IL-7 can increase PD-1 expression in lymphocytes, which is a hallmark of lymphocyte exhaustion, cytokine-driven peripheral T cell expansion and survival remain intact (122). However, when IL-7 was administered to patients with HIV, lymphocyte expression of PD-1 was decreased (123). Although IL-7 has the potential to work synergistically with therapies targeting PD-1 or PD-L1 to improve the functional aspects or T cell functions that are presumed to be lost in sepsis, the impact in human sepsis is still unclear. Although clinical trials support the use of IL-7 in HIV-infected patients receiving antiretroviral therapy, no sepsis trials exist to date. Given the promise that IL-7 has shown in other disease states of lymphopenia and immune suppression, sepsis trials with IL-7 alone or in combination with other immune modulators such as anti–PD-1 should be seriously considered.
Although still in preclinical studies, it is worth mentioning IL-3 and IL-15 as potential sepsis therapies. Considering the integral role of IL-15 in the development and activation of effector and memory T, NK, and NKT cells and neutrophils, it has become a promising candidate for immune therapy (124). In mouse models of sepsis, treatment with IL-15 diminished immune dysfunction and improved survival (125). The synergistic role that IL-3 plays in HSC and progenitor development along with IL-7 makes IL-3 an appealing therapy to augment the potential impact of IL-7 (100, 101). However, there is a paucity of data relating to the effects of either IL-3 or IL-15 in states of sepsis, and further discussion is speculative.
Immune-modulatory intervention.
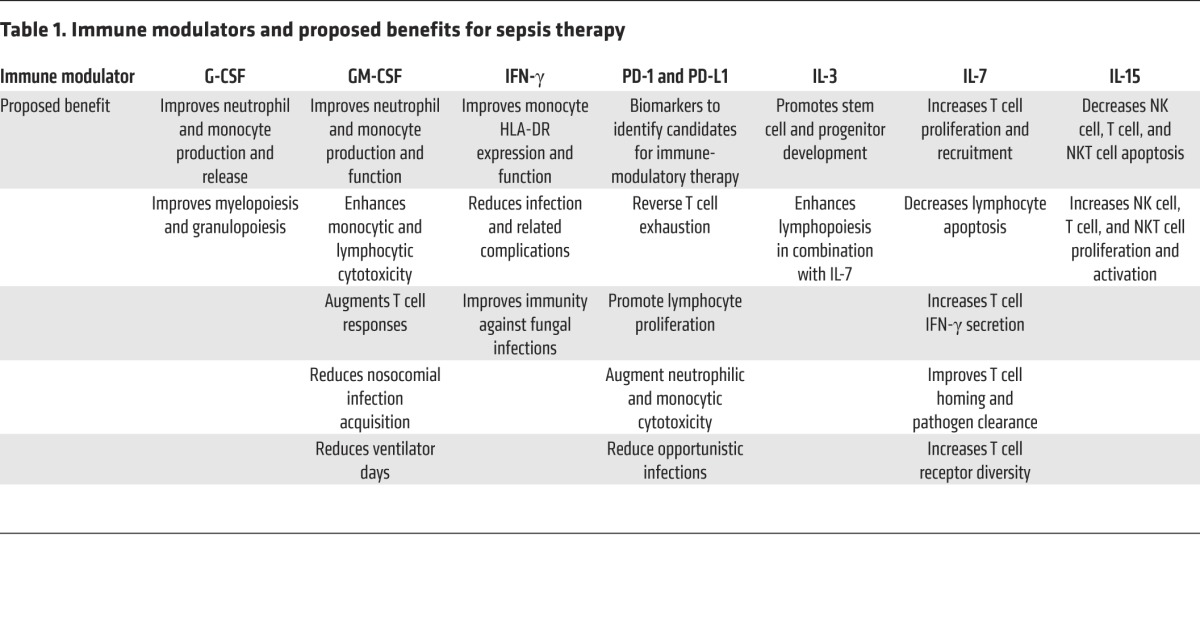
The sepsis landscape is riddled with failed single-agent therapeutic interventions to block particular pathways or processes in humans. Although there are as many explanations as failures, continued attempts to augment immune processes with single agents at early time points, with the expectation that sepsis mortality will be halved at 6 or 12 months, are doomed to fail. However, if the sepsis-induced immune derangements are juxtaposed with other immune-mediated disease processes, such as cancer, autoimmune disease, or HIV, in which immune-modulatory therapy has improved patient outcomes, it is clear that single-agent therapy is not ideal. Rather, it is preferable to use a combination of multiple agents that are introduced and altered over time, according to disease-specific progression, patient immune responses, and defined host/pathogen genomic interactions (126). Cancer chemotherapy strategies use a personalized combination of agents to induce, maintain, and prolong cancer remission on the basis of patient disease progression and tumor-specific genetic patterns. We believe that the same strategy could be applied to sepsis therapy with a high probability of success if the interventions are tailored to specific host/pathogen genomic patterns and immune perturbations that occur in the elderly and in patients with comorbidities as post-sepsis recovery evolves (18). Given the ease of modern genomic determination, screening patients for specific genetic variations that influence microbial eradication could be used to develop a personalized treatment plan. For example, GM-CSF, in combination with IL-3, may bolster monocyte and neutrophil production and function early after sepsis when the mature pool of these cells is depleted. Next, a combination of anti–PD-1 or anti–PD-L1, coupled with IFN-γ, may prove beneficial for lymphocyte activation and augmentation of innate immune surveillance to prevent secondary and nosocomial infections (Table 1). Last, a low-dose combination of oxandrolone, testosterone, and propranolol, or even dronabinol, may ameliorate protein catabolism and persistent inflammation and promote anabolism, which has already been implemented to promote recovery in severely burned patients. Moreover, with improvements in biomarkers, cellular function determination, and host/pathogen genomic prediction models, strategically engineered combinations of immune modulators could be used in a goal-directed manner on the basis of the patient’s comorbidity profile, immune function, and recovery course. For example, poorly maintained type 2 diabetic patients recovering from sepsis are predisposed to develop secondary and nosocomial infections associated with poor neutrophil and lymphocyte function. This group of patients may benefit from the concomitant administration of G-CSF, GM-CSF, and anti–PD-1 or anti–PD-1L early in the sepsis recovery phase to prevent ongoing infection and subsequent mortality, followed by IFN-γ therapy to facilitate an infection-free period and promote more durable recovery. Using this strategy in sepsis would allow for tailored and monitored interventions that vary over time with specific patient populations, minimizing the human physiologic heterogeneity that has plagued previous sepsis clinical trials.
Table 1. Immune modulators and proposed benefits for sepsis therapy.
Conclusions
Sepsis induces a multitude of defects in immunity that cause protracted inflammation, immune suppression, susceptibility to infections, and death. Although there are new cell-based methodologies available to identify patients with post-sepsis immune dysregulation, it is still unclear which interventions targeting cell-specific deficits will be most beneficial. Considering the interrelated and interdigitating complexity of immune derangements as well as the protracted and convoluted road to mortality, we believe that single-agent immune-modulatory intervention, as attempted in past sepsis trials, will probably fail. Conversely, the notion of more thorough and rigorous patient selection, coupled with frequent monitoring of immune function and goal-directed immune-modulatory therapy involving multiple agents, may, over time, provide optimal clinical benefit.
Acknowledgments
M.J. Delano acknowledges the Research and Education Foundation Scholarship that supported this work and was generously provided by the American Association for the Surgery of Trauma. Additional support was provided by NIH grants GM-29507 and GM-61656 and by the Godfrey D. Stobbe Endowment (to P.A. Ward).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(1):23–31. doi:10.1172/JCI82224.
References
- 1.Kaukonen KM, Bailey M, Pilcher D, Cooper DJ, Bellomo R. Systemic inflammatory response syndrome criteria in defining severe sepsis. N Engl J Med. 2015;372(17):1629–1638. doi: 10.1056/NEJMoa1415236. [DOI] [PubMed] [Google Scholar]
- 2.[No authors listed] American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20(6):864–874. doi: 10.1097/00003246-199206000-00025. [DOI] [PubMed] [Google Scholar]
- 3.Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014;5(1):4–11. doi: 10.4161/viru.27372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coopersmith CM, et al. A comparison of critical care research funding and the financial burden of critical illness in the United States. Crit Care Med. 2012;40(4):1072–1079. doi: 10.1097/CCM.0b013e31823c8d03. [DOI] [PubMed] [Google Scholar]
- 5.Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med. 2006;34(1):15–21. doi: 10.1097/01.CCM.0000194535.82812.BA. [DOI] [PubMed] [Google Scholar]
- 6.Kahn JM, et al. The epidemiology of chronic critical illness in the United States*. Crit Care Med. 2015;43(2):282–287. doi: 10.1097/CCM.0000000000000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41(5):1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 8.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40(4):463–475. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Ward PA, Bosmann M. A historical perspective on sepsis. Am J Pathol. 2012;181(1):2–7. doi: 10.1016/j.ajpath.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tracey KJ, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330(6149):662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 11.Schorr CA, Dellinger RP. The Surviving Sepsis Campaign: past, present and future. Trends Mol Med. 2014;20(4):192–194. doi: 10.1016/j.molmed.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Boomer JS, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306(23):2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gentile LF, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012;72(6):1491–1501. doi: 10.1097/TA.0b013e318256e000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winters BD, Eberlein M, Leung J, Needham DM, Pronovost PJ, Sevransky JE. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med. 2010;38(5):1276–1283. doi: 10.1097/CCM.0b013e3181d8cc1d. [DOI] [PubMed] [Google Scholar]
- 15.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995;75(2):257–277. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 16.Needham DM, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502–509. doi: 10.1097/CCM.0b013e318232da75. [DOI] [PubMed] [Google Scholar]
- 17.Yende S, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242–1247. doi: 10.1164/rccm.200712-1777OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elliott D, et al. Exploring the scope of post-intensive care syndrome therapy and care: engagement of non-critical care providers and survivors in a second stakeholders meeting. Crit Care Med. 2014;42(12):2518–2526. doi: 10.1097/CCM.0000000000000525. [DOI] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Moldawer LL. Parallels between cancer and infectious disease. N Engl J Med. 2014;371(4):380–383. doi: 10.1056/NEJMcibr1404664. [DOI] [PubMed] [Google Scholar]
- 20.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delano MJ, Moldawer LL. Magic bullets and surrogate biomarkers circa 2009. Crit Care Med. 2009;37(5):1796–1798. doi: 10.1097/CCM.0b013e3181a09440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34(3):129–136. doi: 10.1016/j.it.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15(5):496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bone RC, Grodzin CJ, Balk RA. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest. 1997;112(1):235–243. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- 25.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao W, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208(13):2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuenca AG, et al. Development of a genomic metric that can be rapidly used to predict clinical outcome in severely injured trauma patients. Crit Care Med. 2013;41(5):1175–1185. doi: 10.1097/CCM.0b013e318277131c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. 2014;20(4):224–233. doi: 10.1016/j.molmed.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gentile LF, et al. A better understanding of why murine models of trauma do not recapitulate the human syndrome. Crit Care Med. 2014;42(6):1406–1413. doi: 10.1097/CCM.0000000000000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6(3):173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 31.Link DC. Neutrophil homeostasis: a new role for stromal cell-derived factor-1. Immunol Res. 2005;32(1-3):169–178. doi: 10.1385/IR:32:1-3:169. [DOI] [PubMed] [Google Scholar]
- 32.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6(11):813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 33.Delano MJ, et al. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J Immunol. 2011;186(1):195–202. doi: 10.4049/jimmunol.1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120(7):2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delano MJ, et al. Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J Immunol. 2011;187(2):911–918. doi: 10.4049/jimmunol.1100588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grailer JJ, Kalbitz M, Zetoune FS, Ward PA. Persistent neutrophil dysfunction and suppression of acute lung injury in mice following cecal ligation and puncture sepsis. J Innate Immun. 2014;6(5):695–705. doi: 10.1159/000362554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lerman YV, et al. Sepsis lethality via exacerbated tissue infiltration and TLR-induced cytokine production by neutrophils is integrin alpha3beta1-dependent. Blood. 2014;124(24):3515–3523. doi: 10.1182/blood-2014-01-552943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alves-Filho JC, et al. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106(10):4018–4023. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nacionales DC, et al. Aged mice are unable to mount an effective myeloid response to sepsis. J Immunol. 2014;192(2):612–622. doi: 10.4049/jimmunol.1302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delano MJ, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204(6):1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davey MS, et al. Microbe-specific unconventional T cells induce human neutrophil differentiation into antigen cross-presenting cells. J Immunol. 2014;193(7):3704–3716. doi: 10.4049/jimmunol.1401018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris AC, et al. C5a-mediated neutrophil dysfunction is RhoA-dependent and predicts infection in critically ill patients. Blood. 2011;117(19):5178–5188. doi: 10.1182/blood-2010-08-304667. [DOI] [PubMed] [Google Scholar]
- 43.Wilkinson TS, et al. Ventilator-associated pneumonia is characterized by excessive release of neutrophil proteases in the lung. Chest. 2012;142(6):1425–1432. doi: 10.1378/chest.11-3273. [DOI] [PubMed] [Google Scholar]
- 44.Cheadle WG, Wilson M, Hershman MJ, Bergamini D, Richardson JD, Polk HC., Jr Comparison of trauma assessment scores and their use in prediction of infection and death. Ann Surg. 1989;209(5):541–545. doi: 10.1097/00000658-198905000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livingston DH, Appel SH, Wellhausen SR, Sonnenfeld G, Polk HC., Jr Depressed interferon gamma production and monocyte HLA-DR expression after severe injury. Arch Surg. 1988;123(11):1309–1312. doi: 10.1001/archsurg.1988.01400350023002. [DOI] [PubMed] [Google Scholar]
- 46.Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88(5):1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Docke WD, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3(6):678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 48.Cazalis MA, et al. Decreased HLA-DR antigen-associated invariant chain (CD74) mRNA expression predicts mortality after septic shock. Crit Care. 2013;17(6): doi: 10.1186/cc13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheron A, et al. Lack of recovery in monocyte human leukocyte antigen-DR expression is independently associated with the development of sepsis after major trauma. Crit Care. 2010;14(6): doi: 10.1186/cc9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landelle C, et al. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. 2010;36(11):1859–1866. doi: 10.1007/s00134-010-1962-x. [DOI] [PubMed] [Google Scholar]
- 51.Venet F, et al. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol. 2006;177(9):6540–6547. doi: 10.4049/jimmunol.177.9.6540. [DOI] [PubMed] [Google Scholar]
- 52.Monneret G, Venet F, Pachot A, Lepape A. Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol Med. 2008;14(1–2):64–78. doi: 10.2119/2007-00102.Monneret. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. 2011;6(3):273–283. doi: 10.4161/epi.6.3.14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishii M, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114(15):3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saeed S, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345(6204): doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009;126(4):458–465. doi: 10.1111/j.1365-2567.2008.03027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halstead ES, Carcillo JA, Schilling B, Greiner RJ, Whiteside TL. Reduced frequency of CD56 dim CD16 pos natural killer cells in pediatric systemic inflammatory response syndrome/sepsis patients. Pediatr Res. 2013;74(4):427–432. doi: 10.1038/pr.2013.121. [DOI] [PubMed] [Google Scholar]
- 58.Giamarellos-Bourboulis EJ, et al. Early changes of CD4-positive lymphocytes and NK cells in patients with severe Gram-negative sepsis. Crit Care. 2006;10(6): doi: 10.1186/cc5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Souza-Fonseca-Guimaraes F, Parlato M, Philippart F, Misset B, Cavaillon JM, Adib-Conquy M. Toll-like receptors expression and interferon-gamma production by NK cells in human sepsis. Crit Care. 2012;16(5): doi: 10.1186/cc11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiche L, et al. Interferon-gamma production by natural killer cells and cytomegalovirus in critically ill patients. Crit Care Med. 2012;40(12):3162–3169. doi: 10.1097/CCM.0b013e318260c90e. [DOI] [PubMed] [Google Scholar]
- 61.Limaye AP, et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA. 2008;300(4):413–422. doi: 10.1001/jama.300.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hotchkiss RS, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol. 2002;168(5):2493–2500. doi: 10.4049/jimmunol.168.5.2493. [DOI] [PubMed] [Google Scholar]
- 63.Grimaldi D, et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med. 2011;37(9):1438–1446. doi: 10.1007/s00134-011-2306-1. [DOI] [PubMed] [Google Scholar]
- 64.Pastille E, et al. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J Immunol. 2011;186(2):977–986. doi: 10.4049/jimmunol.1001147. [DOI] [PubMed] [Google Scholar]
- 65.Efron PA, et al. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol. 2004;173(5):3035–3043. doi: 10.4049/jimmunol.173.5.3035. [DOI] [PubMed] [Google Scholar]
- 66.Scumpia PO, et al. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol. 2005;175(5):3282–3286. doi: 10.4049/jimmunol.175.5.3282. [DOI] [PubMed] [Google Scholar]
- 67.Toliver-Kinsky TE, Lin CY, Herndon DN, Sherwood ER. Stimulation of hematopoiesis by the Fms-like tyrosine kinase 3 ligand restores bacterial induction of Th1 cytokines in thermally injured mice. Infect Immun. 2003;71(6):3058–3067. doi: 10.1128/IAI.71.6.3058-3067.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benjamim CF, Lundy SK, Lukacs NW, Hogaboam CM, Kunkel SL. Reversal of long-term sepsis-induced immunosuppression by dendritic cells. Blood. 2005;105(9):3588–3595. doi: 10.1182/blood-2004-08-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ochoa JB, et al. Arginase I expression and activity in human mononuclear cells after injury. Ann Surg. 2001;233(3):393–399. doi: 10.1097/00000658-200103000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cuenca AG, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17(3–4):281–292. doi: 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176(4):2085–2094. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 73.Kelly-Scumpia KM, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med. 2010;207(2):319–326. doi: 10.1084/jem.20091959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scumpia PO, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. 2010;184(5):2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cartwright GE, Athens JW, Wintrobe MM. The kinetics of granulopoiesis in normal man. Blood. 1964;24:780–803. [PubMed] [Google Scholar]
- 76.McCabe A, Zhang Y, Thai V, Jones M, Jordan MB, MacNamara KC. Macrophage-lineage cells negatively regulate the hematopoietic stem cell pool in response to interferon γ at steady state and during infection. Stem Cells. 2015;33(7):2294–2305. doi: 10.1002/stem.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holtmeier W, Kabelitz D. γΔ T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151–183. doi: 10.1159/000086659. [DOI] [PubMed] [Google Scholar]
- 78.Andreu-Ballester JC, et al. Association of gammadelta T cells with disease severity and mortality in septic patients. Clin Vaccine Immunol. 2013;20(5):738–746. doi: 10.1128/CVI.00752-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grimaldi D, et al. Specific MAIT cell behaviour among innate-like T lymphocytes in critically ill patients with severe infections. Intensive Care Med. 2014;40(2):192–201. doi: 10.1007/s00134-013-3163-x. [DOI] [PubMed] [Google Scholar]
- 80.Tomasello E, Bedoui S. Intestinal innate immune cells in gut homeostasis and immunosurveillance. Immunol Cell Biol. 2013;91(3):201–203. doi: 10.1038/icb.2012.85. [DOI] [PubMed] [Google Scholar]
- 81.Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest. 2007;117(5):1119–1127. doi: 10.1172/JCI31720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Venet F, et al. Regulatory T cell populations in sepsis and trauma. J Leukoc Biol. 2008;83(3):523–535. doi: 10.1189/jlb.0607371. [DOI] [PubMed] [Google Scholar]
- 83.Hotchkiss RS, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001;166(11):6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 84.O’Sullivan ST, Lederer JA, Horgan AF, Chin DH, Mannick JA, Rodrick ML. Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg. 1995;222(4):482–490. doi: 10.1097/00000658-199522240-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pachot A, et al. Longitudinal study of cytokine and immune transcription factor mRNA expression in septic shock. Clin Immunol. 2005;114(1):61–69. doi: 10.1016/j.clim.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 86.Romani L. Immunity to fungal infections. Nat Rev Immunol. 2011;11(4):275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- 87.Wu HP, Chung K, Lin CY, Jiang BY, Chuang DY, Liu YC. Associations of T helper 1, 2, 17 and regulatory T lymphocytes with mortality in severe sepsis. Inflamm Res. 2013;62(8):751–763. doi: 10.1007/s00011-013-0630-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smeekens SP, et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat Commun. 2013;4: doi: 10.1038/ncomms2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Unsinger J, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol. 2010;184(7):3768–3779. doi: 10.4049/jimmunol.0903151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–165. doi: 10.1038/nri3605. [DOI] [PubMed] [Google Scholar]
- 91.Venet F, et al. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (–)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 2009;35(4):678–686. doi: 10.1007/s00134-008-1337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116(7):2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 93.Scumpia PO, et al. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol. 2006;177(11):7943–7949. doi: 10.4049/jimmunol.177.11.7943. [DOI] [PubMed] [Google Scholar]
- 94.Scumpia PO, et al. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood. 2007;110(10):3673–3681. doi: 10.1182/blood-2007-04-087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cavassani KA, et al. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood. 2010;115(22):4403–4411. doi: 10.1182/blood-2009-09-241083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 97.Monserrat J, et al. Early alterations of B cells in patients with septic shock. Crit Care. 2013;17(3): doi: 10.1186/cc12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kelly-Scumpia KM, et al. B cells enhance early innate immune responses during bacterial sepsis. J Exp Med. 2011;208(8):1673–1682. doi: 10.1084/jem.20101715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rauch PJ, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335(6068):597–601. doi: 10.1126/science.1215173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weber GF, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. 2015;347(6227):1260–1265. doi: 10.1126/science.aaa4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hotchkiss RS, Sherwood ER. Immunology. Science. 2015;347(6227):1201–1202. doi: 10.1126/science.aaa8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lieschke GJ, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84(6):1737–1746. [PubMed] [Google Scholar]
- 103.Petit I, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3(7):687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 104.Nelson S, et al. A randomized controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. J Infect Dis. 1998;178(4):1075–1080. doi: 10.1086/515694. [DOI] [PubMed] [Google Scholar]
- 105.Root RK, et al. Multicenter, double-blind, placebo-controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit Care Med. 2003;31(2):367–373. doi: 10.1097/01.CCM.0000048629.32625.5D. [DOI] [PubMed] [Google Scholar]
- 106.Francisco-Cruz A, et al. Granulocyte-macrophage colony-stimulating factor: not just another haematopoietic growth factor. Med Oncol. 2014;31(1): doi: 10.1007/s12032-013-0774-6. [DOI] [PubMed] [Google Scholar]
- 107.Paine R. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med. 2012;40(1):90–97. doi: 10.1097/CCM.0b013e31822d7bf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meisel C, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009;180(7):640–648. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]
- 109.Hall MW, et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med. 2011;37(3):525–532. doi: 10.1007/s00134-010-2088-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care. 2011;15(1): doi: 10.1186/cc10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med. 2012;185(1):110–112. doi: 10.1164/ajrccm.185.1.110. [DOI] [PubMed] [Google Scholar]
- 112.Dries DJ, et al. Effect of interferon gamma on infection-related death in patients with severe injuries. A randomized, double-blind, placebo-controlled trial. Arch Surg. 1994;129(10):1031–1041. doi: 10.1001/archsurg.1994.01420340045008. [DOI] [PubMed] [Google Scholar]
- 113.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 115.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y, et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care. 2011;15(1): doi: 10.1186/cc10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang JF, et al. Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression: an animal study and a prospective case-control study. Anesthesiology. 2015;122(4):852–863. doi: 10.1097/ALN.0000000000000525. [DOI] [PubMed] [Google Scholar]
- 118.Chang KC, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care. 2013;17(3): doi: 10.1186/cc12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011;11(5):330–342. doi: 10.1038/nri2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Perales MA, et al. Recombinant human interleukin-7 (CYT107) promotes T-cell recovery after allogeneic stem cell transplantation. Blood. 2012;120(24):4882–4891. doi: 10.1182/blood-2012-06-437236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Demaret J, et al. STAT5 phosphorylation in T cell subsets from septic patients in response to recombinant human interleukin-7: a pilot study. J Leukoc Biol. 2015;97(4):791–796. doi: 10.1189/jlb.5AB1114-545R. [DOI] [PubMed] [Google Scholar]
- 122.Kinter AL, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008;181(10):6738–6746. doi: 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 123.Levy Y, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis. 2012;55(2):291–300. doi: 10.1093/cid/cis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pelletier M, Ratthe C, Girard D. Mechanisms involved in interleukin-15-induced suppression of human neutrophil apoptosis: role of the anti-apoptotic Mcl-1 protein and several kinases including Janus kinase-2, p38 mitogen-activated protein kinase and extracellular signal-regulated kinases-1/2. FEBS Lett. 2002;532(1–2):164–170. doi: 10.1016/S0014-5793(02)03668-2. [DOI] [PubMed] [Google Scholar]
- 125.Inoue S, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. 2010;184(3):1401–1409. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sironi M, Cagliani R, Forni D, Clerici M. Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat Rev Genet. 2015;16(4):224–236. doi: 10.1038/nrg3905. [DOI] [PMC free article] [PubMed] [Google Scholar]