

Abstract
Vascular oxidative injury accompanies many common conditions associated with hypertension. In the present study, we employed mouse models with excessive vascular production of ROS (tgsm/p22phox mice, which overexpress the NADPH oxidase subunit p22phox in smooth muscle, and mice with vascular-specific deletion of extracellular SOD) and have shown that these animals develop vascular collagen deposition, aortic stiffening, renal dysfunction, and hypertension with age. T cells from tgsm/p22phox mice produced high levels of IL-17A and IFN-γ. Crossing tgsm/p22phox mice with lymphocyte-deficient Rag1–/– mice eliminated vascular inflammation, aortic stiffening, renal dysfunction, and hypertension; however, adoptive transfer of T cells restored these processes. Isoketal-protein adducts, which are immunogenic, were increased in aortas, DCs, and macrophages of tgsm/p22phox mice. Autologous pulsing with tgsm/p22phox aortic homogenates promoted DCs of tgsm/p22phox mice to stimulate T cell proliferation and production of IFN-γ, IL-17A, and TNF-α. Treatment with the superoxide scavenger tempol or the isoketal scavenger 2-hydroxybenzylamine (2-HOBA) normalized blood pressure; prevented vascular inflammation, aortic stiffening, and hypertension; and prevented DC and T cell activation. Moreover, in human aortas, the aortic content of isoketal adducts correlated with fibrosis and inflammation severity. Together, these results define a pathway linking vascular oxidant stress to immune activation and aortic stiffening and provide insight into the systemic inflammation encountered in common vascular diseases.
Introduction
The normal aorta expands to accommodate a portion of the ejected blood during cardiac contraction and then recoils during cardiac relaxation. This capacitance, or Windkessel function, of the aorta reduces systolic pressure and maintains pressure and peripheral perfusion during diastole. In several conditions — including aging, autoimmune diseases, cigarette smoking, diabetes, obesity, and hypertension — the aorta stiffens, leading to loss of the Windkessel function and systolic hypertension. Pulse wave velocity (PWV), a surrogate measure of aortic stiffness, predicts untoward outcomes in aged and hypertensive humans (1, 2). A recent analysis of the Framingham population indicates that aortic stiffening precedes hypertension and that higher forward pressure wave amplitude, a hallmark of aortic stiffening, is associated with increased risk for incident cardiovascular diseases (3, 4).
A common feature of many conditions associated with aortic stiffening is vascular oxidant stress. Clinically, plasma levels of myeloperoxidase and F2-isoprostanes correlate with arterial stiffening in humans (5). Studies in experimental animals have implicated ROS in fibrotic changes in blood vessels, lungs, skin, and other tissues (6, 7). In addition to oxidative events, chronic inflammation is associated with arterial stiffness in humans (8–10). Patients with systemic lupus erythematosus, rheumatoid arthritis, and psoriasis exhibit increased aortic stiffness, and TNF-α antagonists improve parameters of aortic stiffness in patients with rheumatoid arthritis (11, 12). We have shown that hypertension is associated with the infiltration of immune cells into the adventitia and periadventitial fat, as well as the activation of T cells that release proinflammatory cytokines such as IL-17A, IFN-γ, and TNF-α (13, 14). IL-17A induces collagen synthesis in fibroblasts via activation of p38 MAP kinase (15). IFN-γ also promotes hypertension-related fibrosis in the heart, arteries, and the kidney (16).
A potential mechanism linking oxidative injury to immune activation and inflammation relates to modification of self-proteins. In particular, isoketals — alternatively referred to as isolevuglandins or γ-ketoaldehydes — are formed from fatty acid oxidation and react with protein lysines to form oxidized pyrrole adducts. These modified proteins become auto-antigens and can elicit antibody formation (17). We have recently shown that hypertension is associated with accumulation of isoketal-adducted proteins within DCs. These adducts markedly enhance DC surface expression of the costimulatory molecules CD80 and CD86, as well as cytokine production (18). Moreover, DCs modified in this fashion robustly drive T cell activation and can prime hypertension when adoptively transferred to naive mice. The mechanisms leading to DC acquisition of isoketal-adducted proteins remain undefined. In particular, it is unknown if DCs can acquire oxidatively modified proteins produced by other cells; however, such a process could link oxidative injury in other cells to immune activation and might explain how oxidative stress leads to vascular inflammation and, ultimately, aortic stiffening.
In the present studies, we employed mice with chronic vascular oxidative stress to define a pathway that links oxidative damage to immune activation via formation of immunoreactive isoketal-protein adducts, vascular inflammation, aortic stiffening, renal dysfunction, and hypertension. These experiments provide new insight into how diverse conditions diminish aortic compliance, alter renal function, and ultimately cause hypertension. These findings also show how vascular diseases associated with vascular oxidative injury can lead to systemic immune activation and inflammation.
Results
Chronic vascular oxidative stress induces aortic stiffening and hypertension.
As an initial approach to determine if vascular oxidative stress promotes aortic stiffening, we studied tgsm/p22phox mice. These animals have smooth muscle–targeted overexpression of p22phox, a docking subunit of all murine NADPH oxidase catalytic subunits (19). Vessels from these mice exhibit increased vascular Nox1 expression and a 2-fold increase in vascular superoxide and hydrogen peroxide production (19). Masson’s trichrome staining revealed minimal collagen staining in the aorta of WT mice at either 3, 6, or 9 months of age (Figure 1, A and B). In contrast, aortic adventitial collagen content was increased in the tgsm/p22phox mice at 6 and 9 months of age. Hydroxyproline analysis confirmed that aortic collagen content was increased by about 2-fold at 6 and 9 months of age in tgsm/p22phox mice (Figure 1C).
Figure 1. Tgsm/p22phox mice develop age-related aortic stiffening and hypertension.

(A) Effects of aging on vascular collagen deposition in WT and tgsm/p22phox mice. Perfusion-fixed sections of the thoracic aortas were sectioned (6 μm) and stained with Masson’s trichome to highlight collagen (blue). Areas within the red boxes were magnified (×20) and shown below the original images (×6). Scale bars: 100 μm. (B) Adventitial collagen area was quantified by planimetry. #P < 0.05 vs. WT; **P < 0.01 vs. 3 months. (C) Aortic collagen quantification by hydroxyproline assay. (D and E) Freshly isolated aortas were mounted on a myograph system in Ca2+-free buffer to determine pressure-diameter relationships. Stress-strain relationships were constructed from intraluminal pressure, wall thickness, and inner and outer diameters. These parameters were measured at 25 mmHg step changes in pressure from 0–200 mmHg. **P < 0.01 vs. Tg 3 months; ##P < 0.01 vs. WT. (F and G) Telemetry blood pressure of WT and tgsm/p22phox mice at 3, 6, and 9 months of age. †P < 0.05 vs. Tg 6 months; ††P <0.01 vs. Tg 6 months. Data were analyzed using 2-way ANOVA; n = 6–9.
Mechanical properties of the aortas of WT and tgsm/p22phox mice were studied ex vivo in a pressurized vessel apparatus. Aortas from WT mice at 3, 6, and 9 months of age demonstrated identical increases in diameter as intraluminal pressure was increased (Figure 1D). Aortas from 3-month-old tgsm/p22phox mice were similar to those of WT mice, but at 6 and 9 months of age, aortas from these mice demonstrated depressed pressure-diameter relationships. This was reflected by a leftward shift in the stress-strain curves (Figure 1E), indicating increased stiffness of the aortas of tgsm/p22phox mice at 6 and 9 months of age.
To confirm aortic stiffening in vivo, we measured PWV as previously described (20). At 3 months of age, PWV in WT and tgsm/p22phox mice was similar; however, with aging, this parameter increased in tgsm/p22phox mice but did not significantly change in WT mice (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI80761DS1).
Although aortic stiffening developed at 6 months of age in tgsm/p22phox mice, their blood pressure remained normal at this time. At 9 months of age, however, tgsm/p22phox mice developed moderate hypertension, while WT mice remained normotensive (Figure 1, F and G). Analysis of heart rate variability showed an increase in the low-frequency to high-frequency ratio (LF/HF) in both WT and tgsm/p22phox mice at 6 and 9 months of age, but this was not different between the genotypes (Supplemental Figure 2). Thus, in tgsm/p22phox mice, aortic stiffening precedes blood pressure elevation, mimicking the sequence of these events in humans. This increase in blood pressure does not seem to be related to increased sympathetic outflow.
The effects of oxidative stress on aortic stiffening and age-related hypertension were confirmed in mice with vascular smooth muscle–targeted deletion of the extracellular SOD (Sod3fl/fl Smmhc-Cre mice, another model of vascular oxidative stress (21). Deletion of Sod3 was induced at 10 weeks of age by injection of tamoxifen. As controls, Sod3fl/fl mice lacking Cre recombinase were also treated with tamoxifen. At 3 months of age, shortly after Sod3 deletion, there was no significant difference in aortic collagen between these 2 groups of animals, as estimated by Masson’s trichrome staining or quantified by hydroxyproline content (Figure 2, A and B). By 6 months of age, mice with Sod3 deletion had a marked increase in adventitial collagen compared with Sod3fl/fl mice lacking Cre recombinase. This was associated with development of aortic stiffening (Figure 2, C and D) but no increase in blood pressure (Figure 2, E and F). By 9 months of age, the SOD3-deficient mice exhibited not only aortic stiffening, but also a significant elevation of blood pressure (Figure 2, A–F). Collectively, these data recapitulate our finding in tgsm/p22phox mice and support a role of vascular ROS in promoting vascular fibrosis, aortic stiffening, and age-related hypertension.
Figure 2. Vascular oxidative stress induced by deletion of smooth muscle Sod3 promotes age-related aortic stiffening and hypertension.

(A) Masson’s trichrome blue staining in 3-, 6-, and 9-month-old Sod3fl/fl Cre– and Sod3fl/fl Smmhc-Cre mice. Tamoxifen was administered at 3 mg/20 kg in corn oil via i.p. injection at 10 weeks of age. Images are ×20 magnification. Scale bars: 100 μm. (B) Aortic hydroxyproline content. (C and D) Pressure diameter relationship and stress-strain relationships. (E and F) Blood pressure measured by radiotelemetry (n = 6–8). #P < 0.05 vs. Cre–/–; *P < 0.05 vs. Sod3fl/fl Smmhc Cre 3 month; **P < 0.05 vs. Sod3fl/fl Smmhc-Cre 3 month. Aortic hydroxyproline content was analyzed using 2-way ANOVA. Pressure diameter relationships, stress-strain relationships, and blood pressures were analyzed using 1-way ANOVA with repeated measures.
Oxidative stress–induced vascular inflammation contributes to age-related hypertension.
We and others have previously demonstrated that vascular inflammation contributes to the development of hypertension and aortic stiffening (13–16, 22). We therefore sought to determine if chronic oxidative stress promotes vascular inflammation in tgsm/p22phox mice. Flow cytometry of single cell suspensions revealed that total leukocytes, monocyte/macrophages, B cells, total T cells, and CD4+ and CD8+ T cell subsets were all increased in aortas of 6- and 9-month-old tgsm/p22phox mice compared with similarly aged WT mice (Figure 3, A–N). IHC using anti-CD3 indicated that T lymphocytes were predominantly located in the adventitia and perivascular fat of thoracic aortas in Tgsm/p22phox mice at 6 and 9 months of age, compared with age-matched WT mice (Supplemental Figure 3).
Figure 3. Flow cytometry analysis of inflammatory cells in the aorta of WT and tgsm/p22phox mice.

Single cell suspensions were prepared from freshly isolated mouse aortas via enzymatic digestion and mechanical dissociation. Live cell singlets were analyzed for vascular inflammatory cells. (A–H) CD45+ total leukocytes, F4/80+ macrophages, CD19+ B lymphocytes, CD3+ T lymphocytes, and CD4+/CD8+T cell subsets were identified in the aorta of 9-month-old WT (A–D) and tgsm/p22phox mice (E–H). (I–N) Quantification of infiltrating leukocyte subsets using 2-way ANOVA (n = 6–8). **P < 0.01 vs. Tgsm/p22phox; #P < 0.05; ##P < 0.01.
In additional experiments, we examined the production of cytokines by aortic T cells using intracellular staining. At 3 months of age, the few T cells in the aorta of tgsm/p22phox mice produced minimal amounts of IFN-γ, IL-17A, or TNF-α; however, by 6 and 9 months, the number of both CD8+ and CD4+ T cells in the aorta producing these cytokines was significantly increased (Figure 4, A–G).
Figure 4. Production of cytokines by aortic T cells using intracellular staining.

One million splenocytes were stimulated with ionomycin and phorbol myristate acetate and brefeldin A at 37°C for 5 hours. Intracellular staining was then performed with the Cytofix/Cytoperm Plus fixation and permeabilization solution kit using anti–IL-17A and anti–IFN-γ antibodies. (A) Representative images of IFN-γ–producing CD8+ T cells of WT and tgsm/p22phox mice at 3, 6, and 9 months of age. (B–D) Quantification of CD8+ T cells producing IFN-γ, IL-17A, and TNF-α. (E–G) Quantification of CD4+ T cells producing IFN-γ, IL-17A, and TNF-α. *P < 0.01 vs. WT; **P < 0.01 vs. WT; #P < 0.05 vs. 6 months; ##P <0.01 vs. 3 months. These data were analyzed using 2-way ANOVA.
Chronic vascular oxidative stress promotes age-related renal inflammation, fibrosis, and dysfunction.
We considered the hypothesis that chronic vascular oxidative stress might predispose to renal inflammation and dysfunction. Flow cytometry of kidney single cell suspensions showed a significant increase in total leukocytes, monocyte/macrophages, DCs, total T cells, and both CD4+ and CD8+ T cells at 9 months of age but not at earlier time points in tgsm/p22phox mice (Figure 5, A–G). IHC showed that CD3+ cells accumulate in the periglomerular and perivascular regions of 9-month-old tgsm/p22phox mice. In contrast, there was minimal accumulation of immune cells, as assessed by either flow cytometry or IHC in aged WT mice (Figure 6A). In parallel with T cell infiltration, Masson’s trichrome staining revealed perivascular collagen deposition in the kidney of 9-month-old tgsm/p22phox mice but not WT mice (Figure 6B). These changes in renal inflammation and fibrosis were paralleled by a decline in the capacity of mice to excrete a volume and sodium challenge. Mice received an i.p. injection of normal saline equal to 10% of their body weight, and the urine volume and sodium excreted in the subsequent 4 hours was monitored as previously described (23, 24). This analysis revealed that tgsm/p22phox mice exhibited an accelerated decline in their ability to excrete this volume and sodium load with aging (Figure 6, C and D). Thus, chronic vascular oxidative stress initially causes vascular inflammation and subsequent renal inflammation, fibrosis, and dysfunction. These perturbations of renal function temporally correspond to the elevation of blood pressure observed in these mice.
Figure 5. Flow cytometry analysis of renal leukocytes.

Single cell suspensions were prepared from freshly isolated mouse kidneys via enzymatic digestion and mechanical dissociation. Live cell singlets were analyzed. (A) CD45+ total leukocytes, F4/80+ macrophages, CD11b+CD11c+ DCs, CD3+ T lymphocytes, and CD4+/CD8+ T cell subsets were identified in the aorta of 9-month-old WT and tgsm/p22phox mice. (B–G) Quantification of infiltrating leukocyte subsets using 2-way ANOVA (n = 6–8). *P < 0.05; **P <0.01; #P < 0.05; ##P < 0.01.
Figure 6. Development of age-related renal inflammation, fibrosis, and dysfunction in tgsm/p22phox mice.

(A and B) Consecutive 6-micron sections were obtained from paraffin-embedded mouse kidneys and were stained with anti-CD3 and Masson’s trichrome blue. Images are ×20 magnification. Scale bars: 100 μm. (C and D) Mice received a single i.p. injection of normal saline equal to 10% of body weight, and urine/sodium excretion in the subsequent 4 hours were monitored. These data were analyzed with 2-way ANOVA (n = 6–8).
T lymphocytes are required for collagen deposition, aortic stiffening, and hypertension in mice with chronic vascular oxidative stress.
Because T cells contribute to hypertension in other models, we crossed tgsm/p22phox mice with mice lacking the Rag-1–/– mice, to generate tgsm/p22phox × Rag-1–/– mice. Unlike tgsm/p22phox mice, tgsm/p22phox × Rag-1–/– mice did not develop collagen deposition (Figure 7, A–C), aortic stiffening (Figure 7, D and E), or hypertension (Figure 7, F and G). To further examine a role of T cells in aortic stiffening and hypertension in response to vascular oxidative stress, we adoptively transferred 2 × 107 T cells to tgsm/p22phox × Rag-1–/– mice at 3 months of age and allowed them to survive until 9 months of age. Adoptive transfer of T cells restored collagen deposition, aortic stiffening, and blood pressure elevation in the tgsm/p22phox × Rag-1–/– mice, supporting an essential role of T cells in the development of aortic fibrosis and hypertension induced by chronic vascular oxidative stress. Likewise, deficiency of Rag-1 protected mice from a decline in their ability to excrete a sodium and volume load at 9 months of age, while this protection was lost in mice that had undergone adoptive transfer of T cells (Supplemental Figure 4).
Figure 7. T cells mediate age-related aortic collagen deposition, aortic stiffening, and elevation of blood pressure in tgsm/p22phox mice.

Pan T cells were isolated from the spleen of 3-month-old WT mice and adoptively transferred to age-matched tgsm/p22phox × Rag-1–/– mice to reconstitute T cell population. Mice were subsequently studied at 9 months of age. (A) Examples of aortic Masson’s trichrome blue staining. (B) Quantification of collagen staining by planimetry. (C) Quantification of aortic hydroxyproline levels. #P < 0.05. (D and E) Pressure diameter curves and stress strain relationships obtained using isolated aortic segments studied as in Figure 1. *P < 0.05 vs. Tgsm/p22phox × Rag-1–/–; **P < 0.01 vs. Tgsm/p22phox × Rag-1–/–; ##P < 0.01 vs. WT. (F and G) Telemetry recordings of blood pressures over 3 days. #P < 0.05 vs. WT. Collagen deposition was quantified using 1-way ANOVA. Aortic stiffness and blood pressure were analyzed using 1-way ANOVA with repeated measures (n = 6–8).
Isoketals accumulate in the aorta and antigen presenting cells of tgsm/p22phox mice.
Because pyrrole-modified proteins can act as autoantigens and stimulate inflammatory responses (17, 18), we sought to determine if chronic vascular oxidant stress promotes their formation. We used stable isotope dilution liquid chromatography/mass spectrometry (LC/MS) analysis of isoketal-lysyl-lactam to quantify isoketal-protein adducts in thoracic aortic homogenates of 9-month-old mice (Figure 8A, red arrows). Aortic isoketal-protein adducts were markedly elevated in tgsm/p22phox mice compared with WT mice (Figure 8B). Flow cytometry of single cell suspensions confirmed a higher content of isoketal adducts in CD45– cells of the tgsm/p22phox aortas compared with aortas of WT mice at 3, 6, and 9 months of age. These tended to increase in the transgenic mice, but this did not reach statistical significance (Supplemental Figure 5).
Figure 8. Chronic vascular oxidative stress promotes isoketal-adduct formation.

(A and B) Stable isotope dilution multiple reactions monitoring MS analysis of isoketal-lysine-lactam (IsoK-Lys) adducts in aortas of 9-month-old animals. Representative LC/MS chromatographs from pooled samples for each group (A). The left panel shows chromatographs for IsoK-Lys in samples, while the right panel shows chromatographs for [13C615N2] internal standards in the same samples. Mann-Whitney tests were employed for analysis. The bar graph in B shows mean data. (C) Fix-perfused mouse aortas were embedded in paraffin and subjected to IHC for isoketal staining. Images are ×20 magnification. Scale bars: 100 μm. (D–I) Intracellular staining for isoketals in spleen macrophages (D and E), DCs (F and G), and monocytes (H and I). These data were analyzed using 1-way ANOVA, n = 6. *P < 0.05; **P < 0.01. (J) DCs isolated from 9-month-old WT and tgsm/p22phox mice were exposed to weak acid to elute peptides presented on MHC-I complexes. The presence of isoketal-adducted peptides in the eluent was visualized by dot blot. Isoketal adducts were present in 3 of 4 tgsm/p22phox mice but not in WT mice (see Supplemental Figure 5C for full uncut gel).
To further confirm the presence of isoketal-protein adducts in vessels of tgsm/p22phox mice, we performed immunohistochemical staining using the single chain antibody D11 (18). Abundant isoketal-protein adducts were identified in aortas of 9-month-old tgsm/p22phox mice but not WT mice (Figure 8C). Treatment of mice from 4–9 months of age with either the SOD mimetic tempol or the isoketal scavenging agent 2-hydroxybenzylamine (2-HOBA) eliminated isoketal staining in the tgsm/p22phox mouse vessels.
In additional experiments, we found that isoketals also accumulate in antigen presenting cells (APCs) of tgsm/p22phox mice. Using a recently described gating strategy (25), macrophages were identified as CD64+MerTK+, and DCs were selected from the remaining cells as CD11c+ and MHC-II+ cells (Supplemental Figure 6). Monocytes were identified as CD11b+CD64+F4/80loSSClo cells. Virtually all monocytes were Ly-6C+, and most expressed MHC-II. Intracellular staining for isoketals using D11 conjugated to Alexa Fluor 488 revealed that DCs, macrophages, and to a smaller extent monocytes of 9-month-old tgsm/p22phox mice accumulate isoketal-protein adducts, while cells of age-matched WT animals do not (Figure 8, D–I). This was prevented by chronic treatment with either tempol or 2-HOBA.
We also analyzed the presence of isoketal adducts in peptides eluted from MHC-I using a dot blot analysis and the D11 antibody. Peptides from 9-month-old WT mice were devoid of such adducts; however, those eluted from DCs of aged tgsm/p22phox mice were clearly modified by isoketals (Figure 8J). Thus, DCs from mice with chronic vascular oxidative stress present oxidatively modified peptides.
Scavenging of ROS prevents vascular inflammation, aortic stiffening, renal dysfunction, and hypertension in tgsm/p22phox mice.
To determine if scavenging superoxide or isoketals prevents vascular oxidative stress–induced aortic stiffening and hypertension, we treated mice with either tempol or 2-HOBA from 4–9 months of age and monitored blood pressure with telemetry. Both of these therapies prevented aortic fibrosis in tgsm/p22phox mice at 6 and 9 months of age (Figure 9, A–C). Similarly, treatment with either tempol or 2-HOBA prevented aortic stiffening and age-related hypertension (Figure 9, D–G). Treatment with either tempol or 2-HOBA also preserved the ability to excrete a sodium and volume challenge at 9 months of age (Supplemental Figure 7). These data support a role of vascular ROS in promoting vascular fibrosis, aortic stiffening, and age-related hypertension. Despite the effectiveness of tempol and 2-HOBA in preventing aortic stiffening and hypertension, we found that these agents did not reverse these abnormalities if introduced at 7 months of age (Supplemental Figure 8).
Figure 9. Scavenging superoxide or isoketals prevents aortic collagen deposition, aortic stiffening and hypertension in tgsm/p22phox mice.

Tgsm/p22phox mice received either no treatment, tempol, or 2-HOBA in the drinking water from 3–9 months of age. (A) Masson’s trichrome staining at ×20 magnification. Scale bars: 100 μm. (B and C) Mean data for quantification of collagen staining (B) and hydroxyproline content (C). *P < 0.05; **P < 0.01. (D and E) Pressure diameter and stress strain relationships for isolated aortic segments studied as in Figure 1. ##P < 0.01 vs. WT; **P < 0.01 vs. 2-HOBA. (F and G) Effect of antioxidant treatment on blood pressure in aged tgsm/p22phox mice. Collagen deposition was quantified using 1-way ANOVA. Aortic stiffness and blood pressure were analyzed with 1-way ANOVA with repeated measurements (n = 6–8).
To determine if superoxide or isoketal scavenging reduced vascular inflammation, we performed flow cytometry on the aortas of tgsm/p22phox mice treated with vehicle, tempol, or 2-HOBA from 4–9 months of age. Both tempol and 2-HOBA markedly reduced the aortic infiltration of leukocytes (Figure 10, A–C). The infiltrating CD45+ total leukocytes, F4/80+ monocyte/macrophages, CD19+ B cells, CD3+ total T cells, and CD4+ and CD8+ T cell subsets were significantly reduced by either of these treatments (Figure 10, D–I).
Figure 10. Role of ROS and isoketals in vascular inflammation.

Tgsm/p22phox mice were treated with tempol or 2-HOBA from 4–9 months of age. (A–C) Flow cytometric examples for leukocytes measured in single cells suspensions of aortas. (D–I) Mean data analyzed using 1-way ANOVA (n = 6–8). *P < 0.05; **P < 0.01.
Vascular oxidative stress promotes cytokine production by splenic T cells.
We and others have previously shown that IL-17A and IFN-γ contribute to hypertension (14, 16, 26) and that IL-17A plays a critical role in aortic stiffening (15). We therefore sought to determine if T cells of mice with vascular oxidative stress produce IL-17A and IFN-γ. Splenic T cells from 9-month-old WT and tgsm/p22phox mice were analyzed by flow cytometry using intracellular staining for these cytokines (Figure 11A). While WT T cells produced minimal amounts of IL-17A, 5% of CD4+ T cells and 10% of double-negative (CD3+/CD4–/CD8–) T cells of tgsm/p22phox mice produced this cytokine (Figure 11, B and D–F). Likewise, IFN-γ–producing cells were doubled in all 3 subsets of T lymphocytes in tgsm/p22phox mice compared with WT (Figure 11, C and G–I). Treatment with tempol or 2-HOBA normalized T cell production of IL-17A and IFN-γ in tgsm/p22phox mice. These data suggest that vascular oxidative stress systemically activates T cells and promotes their proinflammatory cytokine production.
Figure 11. Cytokine production of T cells in the spleen of mice with chronic vascular oxidative stress.

(A) Gating strategy for identifying T cell populations among total spleenocytes. Single cell suspensions were prepared from freshly isolated mouse spleens via enzymatic digestion and mechanical dissociation. (B–I) Intracellular staining indicating IL-17A (B, D–F) and IFN-γ (C, G–I) production in T cell subsets in WT and Tgsm/p22phox mice. Bar graphs were analyzed using 1-way ANOVA (n = 5–7). **P < 0.01.
Protein homogenates of tgsm/p22phox mouse vessels are immunogenic.
We have shown that isoketal-protein adducts promote maturation and activation of DCs in angiotensin II–induced hypertension and that these activated DCs can drive proliferation of T cells from angiotensin II–treated mice (18). To determine if proteins from the aortas of tgsm/p22phox mice are immunogenic, we prepared aortic homogenates from WT and tgsm/p22phox mice at 9 months of age. These were used to pulse naive DCs from WT mice for 2 hours and were then washed from the cells (Figure 12A). The pulsed DCs were then cocultured with 1 × 107 T cells isolated from the spleen of either WT or tgsm/p22phox mice and prelabeled with CFSE at a ratio of 1:10. Of note, T cells isolated from tgsm/p22phox mice, but not those from WT mice, survived and proliferated in response to the stimulation of immunogenic protein homogenate of tgsm/p22phox mice, suggesting that preexposure to isoketal-adducted antigens in vivo primed these cells for clonal expansion upon a second challenge (Figure 12, B–I). Aortic homogenates of tgsm/p22phox mice, but not WT mice, supported survival of CD3+ (Figure 12, B and F), CD4+, and CD8+ T cells (Figure 12, C and G) when presented to T cells by DCs. DCs pulsed with tgsm/p22phox aortic homogenates also induced proliferation of CD4+ (Figure 12, D and H) and to a lesser extent CD8+ T cells (Figure 12, E and I), and increased the release of IL-17A/F, IFN-γ, and TNF-α into the medium (Table 1). Chronic treatment of tgsm/p22phox mice with tempol or 2-HOBA reduced the immunogenicity of their aortic protein, as indicated by the diminished survival, proliferation, and cytokine production of T cells in this assay.
Figure 12. Tgsm/p22phox aortic homogenates prime DCs to promote T cell proliferation and activation.

(A) Experimental design. Aortic homogenates from WT or tgsm/p22phox mice were used to prime naive DCs, which were then cocultured with T cells from these mice in various combinations. (B, C, F, and G) Flow cytometry and bar graphs identifying live CD3+ T cells and CD4+/CD8+ T cell subsets from cocultured DCs and T cells. (D, E, H, and I) Representative flow cytometry images and bar graphs showing percentages and numbers of proliferating CD4+ (D and H) and CD8+ (E and I) T cells, as determined using CSFE dilution (n = 6, 1-way ANOVA). Homo, homogenates. Black lines indicate original data, orange peaks indicate initial population, and purple peaks indicate generations of cells undergoing division as simulated by FlowJo 7.6.4 software. *P < 0.05 compared with Tgsm/p22phox homogenate + Tgsm/p22phox T cells; **P < 0.01 Tgsm/p22phox homogenate + Tgsm/p22phox T cells.
Table 1. Vascular oxidative stress–induced DC activation and cytokine production.
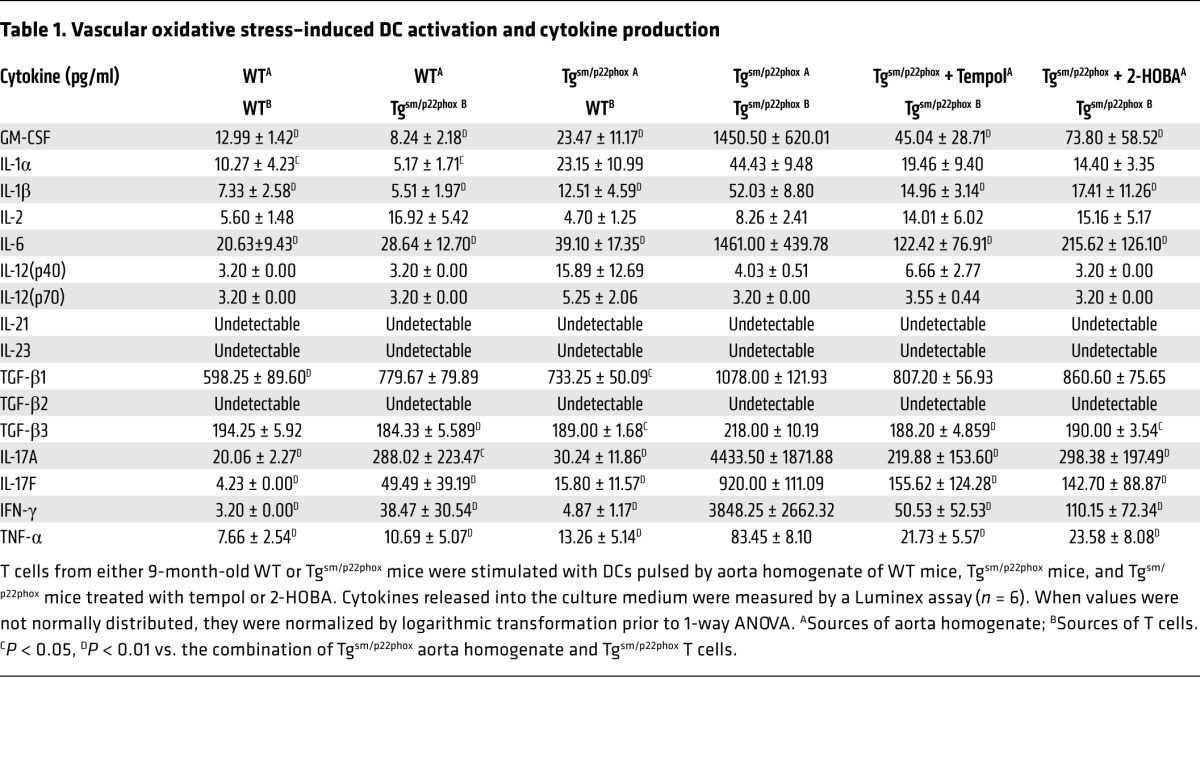
To determine if DCs were functionally activated in these conditions, we also evaluated the polarizing cytokines released into the medium. When DCs exposed to tgsm/p22phox aortic homogenates were cocultured with T cells isolated from tgsm/p22phox mice, there was a marked increase of GM-CSF, IL-1α, IL-1β, IL-6, TGF-β1, and TGF-β3 (Table 1). These increases were blunted when DCs were primed with aortic homogenates from tgsm/p22phox mice chronically treated with tempol or 2-HOBA, suggesting that superoxide and isoketals contribute to immunogenicity of vascular proteins. Interestingly, tgsm/p22phox mouse aortic homogenates failed to induce DCs to produce these cytokines if they were cocultured with T cells from WT mice, suggesting that interactions with isoketal-primed T cells also promote DC activation. Other cytokines — such as IL-2, IL-12 (p40), IL-12 (p70), IL-21, IL-23, and TGF-β2 — were either undetectable or not differentially produced across treatment groups.
Evidence that the isoprostane/isoketal pathway contributes to aortic stiffening and inflammation in humans.
To determine if our findings in mice have relevance to humans, we performed 2 additional studies. First, we examined the degree of fibrosis and the presence of isoketal adducts and T cells in sections of human aortas using staining with Masson’s trichrome, D11, and anti-CD3, respectively. Unlike mouse vessels, human aortas were often noted to contain collagen and T cells not only in the adventitia, but also in the media (Figure 13A). Despite this difference in species, we found that the severity of fibrosis was significantly correlated with the presence of isoketal adducts (Figure 13, A and B) and T cells (Figure 13, A and C) in human aortas. Additionally, there was also correlation between the presence of T cells and isoketal adducts (Figure 13, A and D).
Figure 13. Role of isoketal and oxidation in aortic collagen deposition and stiffening in humans.

(A–D) Data from human aortas. (E) Comparison of PWV and isoprostane content in patients. Consecutive 6 μm sections were stained with anti-CD3, D-11 ScFv, and Masson’s trichrome blue (A). Images are ×20 magnification. Scale bars: 100 μm. Image J was used to quantify the area of D-11 staining for isoketal adducts and the area of blue in the Masson’s trichrome stains. Human T cells were quantified by counting positively stained cells per ×20 field (B and C). Spearman’s correlations comparing PWV and 8-isoprostanes in normotensive and treated hypertensive humans (E). M, media; A, adventitia.
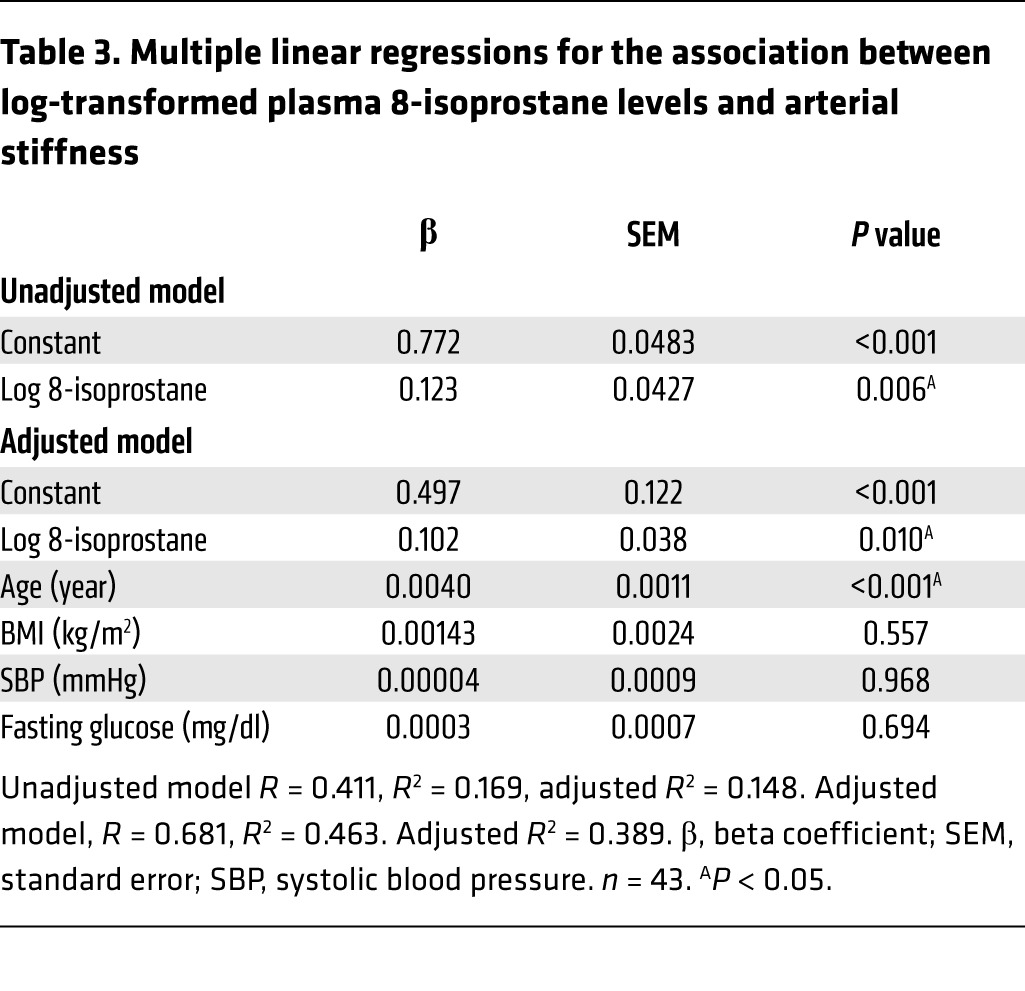
Because isoketals are formed upon spontaneous rearrangement of isoprostanes, we performed an additional study in which we measured plasma levels of F2-isoprostanes in 15 normotensive subjects and 28 patients with well-controlled hypertension. The general characteristics of these subjects are shown in Table 2. Age, BMI, fasting glucose, HbA1C, and systolic blood pressure were greater in the patient group than with the normotensive volunteers. Subjects in the hypertensive group were receiving between 2 and 4 antihypertensive agents, including diuretics, calcium channel blockers, angiotensin I–converting enzyme inhibitors, angiotensin type 1 receptor antagonists, and β-blockers. Overall, the hypertensive patients had increased levels of plasma F2-isoprostanes compared with normotensive subjects (10.7 ± 1.6 ng/ml vs. 17.7 ± 2.2 ng/ml, P < 0.05, Table 2). Importantly, there was highly significant correlation between levels of plasma F2-isoprostanes and PWV for the entire cohort (Figure 13E), and this remained significant after adjusting for age, BMI, fasting glucose, and systolic blood pressure (Table 3).
Table 2. Characteristics of study groups.
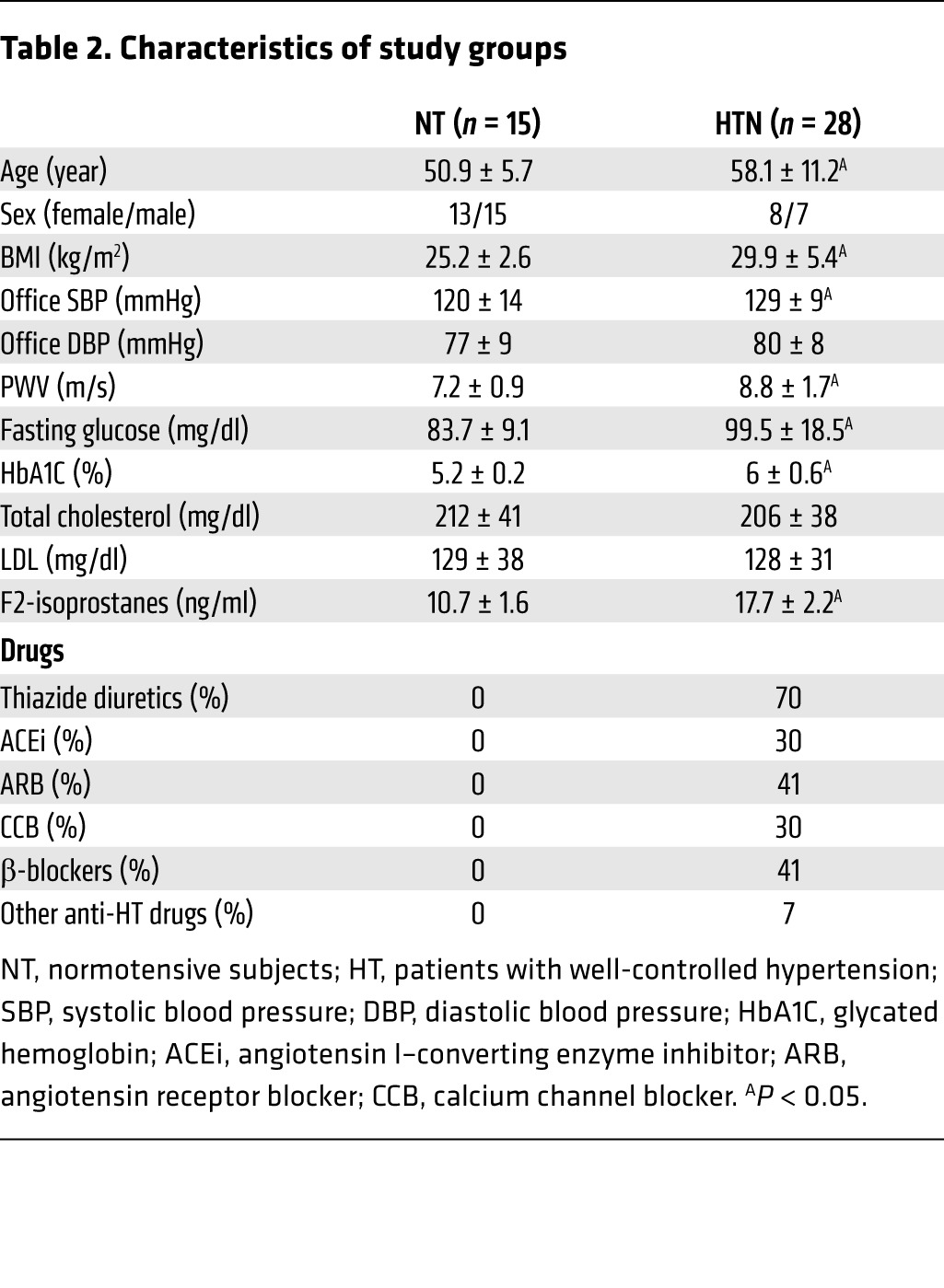
Table 3. Multiple linear regressions for the association between log-transformed plasma 8-isoprostane levels and arterial stiffness.
Discussion
Our present study defines a new pathway in which vascular oxidative stress promotes systemic inflammation via formation of immunogenic isoketal-protein adducts. These act as autoantigens that are presented to T lymphocytes by APCs, leading to T cell activation. These activated T cells then infiltrate the vascular adventitia and release cytokines that induce collagen deposition, leading to aortic stiffening, renal inflammation and dysfunction, and ultimately hypertension (Figure 14). Aortic stiffening accompanies several common conditions, including aging, hypertension, atherosclerosis, overt diabetes, obesity, and cigarette smoking (5, 27–29). In many of these conditions, vascular oxidative stress is known to exist and might initiate the pathway delineated in this study.
Figure 14. Proposed pathway.

Chronic vascular oxidative stress leads to formation of immunogenic isoketal-protein adducts, which accumulate in DCs and promote T cell activation. Activated T cells accumulate in perivascular tissues and release cytokines that enhance collagen deposition and aortic stiffening. This enhances pulse wave velocity. Renal inflammation and fibrosis follows these events, leading to overt hypertension.
A recent analysis of the Framingham population indicates that aortic stiffening precedes development of overt hypertension (3). In this regard, the tgsm/p22phox mouse recapitulates this sequence of events. At 6 months, aortic compliance is reduced in these animals prior to development of hypertension. At 9 months, these mice develop modest elevations of blood pressure, while WT mice remain normotensive at this age. We also observed identical chronological changes in aortic stiffening and hypertension in mice with targeted vascular smooth muscle deletion of Sod3. Hemodynamic changes caused by aortic stiffening, including increased PWV and higher forward pressure wave amplitude, contribute separately to the risk of adverse cardiovascular outcomes (3, 4). Of note, PWV is elevated in tgsm/p22phox mice at 6 months of age, prior to the development of renal inflammation, fibrosis, and dysfunction, supporting the notion that aortic stiffening directly causes renal damage and, ultimately, hypertension. Taken together, data from these 2 models suggest that mice with chronic vascular oxidative stress recapitulate age-related aortic stiffening and hypertension in humans and suggest that chronic vascular oxidative stress might contribute to the human disease.
Studies by our laboratory and others have implicated T cell–derived cytokines in the development of tissue fibrosis and target organ damage in hypertension (13–16, 30). In the present study, we found that vascular oxidative stress induces production of IL-17A by splenic T cells. Moreover, DCs primed with aortic homogenates from tgsm/p22phox mice markedly activate T cells from this same genotype but not T cells from WT mice. This activation profile included not only proliferation, but also robust production of IL-17A, IL-17F, TNF-α, and IFN-γ. These observations support a pathway in which vascular oxidative stress leads to formation of isoketal-protein adducts that are taken up by DCs that, in turn, can activate T cells. Of note, we have previously shown that IL-17A induces fibroblast production of collagen via activation of p38 MAP kinase (15). Similarly, Valente et al. have shown that IL-17A stimulates mouse cardiac fibroblast migration and proliferation via a p38 MAP kinase pathway (31). IFN-γ also mediates cardiac inflammation and fibrosis in angiotensin II–infused WT mice, and IFN-γ receptor–deficient mice are protected from these changes (16). In addition, TNF-α has been implicated in angiotensin II–induced hypertension, and TNF-α antagonist treatment improves parameters of aortic compliance in humans with autoimmune disease (11, 13). Thus, the milieu of Th1/Th17 cytokines stimulated by isoketal-adducted proteins likely promotes age-related aortic stiffening and hypertension.
Related to the above considerations, a striking finding in the current study is the marked production of cytokines — including IL-1β, IL-6, TGF-β1, and GM-CSF — by DCs primed with tgsm/p22phox aortic homogenates when exposed to T cells from tgsm/p22phox mice. This suggests that not only do DCs stimulate T cell proliferation and cytokine production, but also that T cells with T cell receptors that respond to isoketal adducts reciprocally activate DCs. One such signal might involve costimulatory engagement of DC B7 ligands by T cell CD28, which we have previously shown to be critical for hypertension and T cell activation in this disease (32). In this regard, Orabona et al. have shown that CD28 can stimulate DCs to produce IL-6, IFN-γ, and to a lesser extent IL-23 in a B7 and p38 MAP kinase–dependent fashion (33).
Macrophages and monocytes can also function as APCs and have the capability to trigger adaptive immune responses in addition to professional DCs (25). In the present study, we found an increase of total F4/80+ cells in the aorta of tgsm/p22phox mice with aging. In addition, we found that splenic macrophages and monocytes of tgsm/p22phox mice accumulate isoketal adducts. We have previously shown that monocyte/macrophage-like cells do not increase expression of the B7 ligands, CD80 and CD86, in concert with their acquisition of isoketal adducts as robustly as DCs, suggesting that they might not be as effective in T cell stimulation (18).
There is increasing awareness that posttranslational protein modification, such as citrullination and carbamylation, can lead to immune responses (34, 35). Our current and prior studies indicate that the covalent modification of self-proteins by isoketals might represent a similar phenomenon (36) and provide a mechanism by which chronic oxidative stress can lead to systemic immune activation. Despite the plethora of studies implicating ROS in diseases such as atherosclerosis, hypertension, and diabetes, several large clinical trials using either vitamin E or combinations of antioxidants have failed to show any decrease in cardiovascular morbidity or mortality (37, 38), and high doses of vitamin E have paradoxically been associated with increased cardiovascular event rates (39), perhaps because ROS can have important signaling functions that modulate vascular tone, cell growth, and survival (40–42) and are critical for innate immunity. We have previously shown that these higher concentrations of vitamin E are required to suppress plasma F2 isoprostanes, which form from the same precursors as isoketals (43). It is therefore possible that agents such as 2-HOBA, which directly scavenge isoketals with a high rate constant and do not interfere with other ROS, will prove beneficial in treatment of disorders such as hypertension and aortic stiffening.
Methods
Animals.
Male WT and tgsm/p22phox mice were studied at 3, 6, and 9 months of age. For measurement of blood pressure, 6 mice at each age were implanted with telemetry units. Ten days later, blood pressure was recorded for 10 minutes every hour for 3 days. In additional tgsm/p22phox mice, tempol (n = 6) or 2-HOBA (n = 6) was administered in the drinking water from 4–9 months of age. In some experiments, 3-month-old tgsm/p22phox × Rag-1–/– mice underwent adoptive transfer of T cells as described previously (13, 15, 32). Nine-month-old tgsm/p22phox × Rag-1–/– mice, with or without T cell reconstitution, were used for blood pressure and vascular stiffness measurements. Sod3fl/fl Smmhc-Cre mice and Sod3fl/fl × Cre– mice were treated with tamoxifen (3 mg/20 kg, dissolved in corn oil) via 5 consecutive i.p. injections at 10 weeks of age and were studied at 9 months of age (21).
Power spectral analysis of heart rate variability.
Blood pressure was recorded by telemetry for 5 minutes every hour at a sampling rate of 500 Hz, and data were extracted from waveform pressure. Heart rate variability was analyzed from multiple artifact-free blood pressure records of each mouse by LabChart Pro software (ADInstruments). Power spectra were calculated as previously described (21).
Ex vivo measurement of aortic stiffness.
For studies of vascular stiffness, the descending thoracic aorta was mounted on cannulas at the in situ length in calcium-free buffer (44). Intraluminal pressure was increased in a step-wise fashion while video microscopy was used to follow wall thickness and vessel diameter. Diameters were recorded at increments of 25 mmHg from 0–200 mmHg. Stress-strain relationships were calculated as described by Baumbach et al. (44). Hydroxyproline was measured in isolated thoracic aortas as previously described (45–47).
In vivo measurement of PWV.
PWV was measured using the transit time method with Doppler ultrasound (Vevo 2100, VisualSonics) as previously described (20). Briefly, mice were anesthetized with 2% isoflurane/air and maintained under light anesthesia. Mice were kept at supine positions on a heated platform (38°C) with ECG electrodes taped to their paws for continuous recordings. Thoracic and abdominal hair was removed. Pressure waveforms were imaged 1 mm below the exit to the left subclavian artery and 1 mm above the exit to left renal artery on the descending aorta. Arrival times were calculated based on the distances between the foot of the pressure waveform and the peak of R wave for both the proximal and distal locations (Supplemental Figure 1B). The transit time was calculated as the difference in these arrival times and was averaged over 5 cardiac cycles. The in situ distances between these 2 positions were measured postmortem with a ruler. PWV (m/s) was calculated by dividing the distance by the transit time.
Flow cytometry.
Single cell suspensions were prepared from aortas as previously described (15, 32). Briefly, the entire aorta with surrounding perivascular fat was minced with fine scissors and digested with 1 mg/ml collagenase A, 1 mg/ml collagenase B, and 100 μg/ml DNAase I in phenol-free RPMI 1640 medium with 5% FBS for 30 minutes at 37°C, with intermittent agitation. Fc receptors were blocked with CD16/CD32 for 20 minutes at 4°C (clone 2.4G2, BD Biosciences) prior to the staining of surface markers. The antibodies used were: Amcyan anti-CD45 (clone 2D1, BD Biosciences), PE anti-F4/80 (clone BM8, eBioscience), Pacific Blue anti-CD19 (clone 6D5, BioLegend), APC-Cy7 anti-CD3e (clone 145-2C11, BD Biosciences), APC anti-CD4 (clone GK1.5, eBioscience), and PE-Cy7 anti-CD8a (clone 53-6.7, BD Biosciences). Cells (1 × 106) were incubated with 1.5 μl of each antibody in 100 μl of FACS buffer for 35 minutes. The cells were then washed twice with FACS buffer and immediately analyzed on a FACSCanto flow cytometer with DIVA software (BD Biosciences). Dead cells were eliminated from analysis using 7-AAD (catalog 559925, BD Biosciences). For each experiment, we performed flow minus one (FMO) controls for each fluorophore to establish gates. Data analysis was performed using FlowJo software (Tree Star Inc.).
Intracellular staining of splenocytes.
Splenocytes (1 × 106) were resuspended in RPMI medium supplemented with 5% FBS and stimulated with ionomycin and phorbol myristate acetate and brefeldin A at 37°C for 5 hours. Dead cells were excluded from analysis with a fixable Violet dead cell stain. The following surface antibodies were employed for staining T cells: BV510 anti-CD45 (clone 2D1, BD Biosciences), PerCP-Cy5.5 anti-CD3e (clone 145-2C11, BD Biosciences), PE-Cy7 anti-CD8a (clone 53-6.7, BD Biosciences), and APC-H7 anti-CD4 (clone GK1.5, BD Biosciences). Intracellular staining was then performed with the Cytofix/Cytoperm Plus fixation/permeabilization solution kit (BD Biosciences) using PE anti–IL-17A (clone eBio17B7, eBioscience), and FITC anti–IFN-γ antibodies (clone XMG1.2, eBioscience). In some experiments, macrophages, DCs, and monocytes were gated using APC anti-CD11b (clone M1/70, BD Biosciences), PE anti-MerTK (clone HMER5DS, eBioscience), APC-Cy7 anti-CD11c (clone HL3, BD Biosciences), PerCP-Cy5.5 MHC-II/I-A b (clone M5/114.15.2, BD Biosciences), PE-Cy7 anti-CD64 (clone X54-5/7.1, BioLegend), AmCyan anti-F4/80 (clone BM8, BioLegend), and Alexa Fluor 700 anti–Ly-6C (clone AL-21, BD Biosciences) as previously described (25). A single chain antibody, D11 ScFv, was conjugated to Alexa Fluor 488 (catalog A10468, Invitrogen) for the detection of isoketals in these cells.
Measurement of isoketal adducts.
Isoketals in mouse aortas were also measured as the isoketal-lysyl-lactam adducts using MS analysis as previously described (18, 48). To detect DC isoketal adducted peptides by dot blot analysis, DCs from the spleen of 9-month-old WT or tgsm/p22phox mice were sorted using a Miltenyi Biotec DC isolation kit to positively select IAb+/CD11b+/CD11c+ cells. The cells were placed in citrate-phosphate buffer at pH 3.3 containing a protease inhibitor cocktail and iodoacetamide (1:100) to elute MHC-bound peptides. The peptides were then passed through ultrafiltration devices (Amicon Ultra, Millipore) to isolate peptides <5,000 Da and to remove β2-microglobulin proteins. Dot blot analysis was carried out on the resulting flow-through to detect isoketal-adducted peptides using D11 ScFv.
T cell proliferation assay.
Aorta homogenates were obtained from WT mice, tgsm/p22phox mice, or tgsm/p22phox mice chronically treated with tempol or 2-HOBA. Protein concentrations were determined in these samples using Bradford assay. CD11c+ DCs were isolated from total splenocytes of WT mice using a magnetic cell sorter and a commercially available kit (Miltenyi Biotec). This is an acceptable method to isolate essentially all DCs from mouse splenocytes. One million DCs were exposed to 100 mg aortic protein for 2 hours in RPMI 1640 with 10% FBS, 5% penicillin/streptomycin/fungizone, and 5% sodium pyruvate. These DCs were washed twice before being cocultured for 7 days with pan T cells prelabeled with CFSE (catalog C34554, Invitrogen). T cells were obtained from 9-month-old WT mice or tgsm/p22phox mice that had either been untreated or chronically treated with tempol or 2-HOBA in the drinking water. Various combinations of aorta homogenate and T cells were employed as described in Figure 9, A and B. These cells were subjected to flow cytometry analysis, and cytokines in the culture medium were measured by luminex.
IHC of mouse tissues.
Consecutive 6-micron sections were obtained from paraffin-embedded mouse thoracic aorta and kidney. These sections were stained with anti-CD3 (clone 145-2C11, BD Biosciences), D-11 ScFv, and Masson’s trichrome blue as previously described (18, 49). Image J was used to quantify the area of D-11 staining for isoketal adducts and the area of blue in the Masson’s trichrome stains per 20× field.
Human studies.
PWV was measured using the SphygmoCor System (AtCor Medical). Venous blood samples were collected in tubes containing heparin. Plasma 8–isoprostane was detected by an enzyme immunoassay EIA kit (Cayman Chemical).
Human aortic tissue samples were obtained by one of the authors (J.A. Curci) during the course of open revascularization procedures for atherosclerotic occlusive disease of the aorta or at the time of renal transplantation at Washington University, St Louis, Missouri, USA. All specimens were full thickness infrarenal aortic tissue that was discarded at the time of the procedure by the clinical team and were anonymously transferred to the laboratory for study. Portions of these tissue samples were fixed in formalin, embedded in paraffin, and sliced into 6-micron sections. Consecutive sections were stained with anti-CD3 (clone HIT3a, BD Biosciences), D-11 ScFv, and Masson’s trichrome blue as previously described (18, 49). Human T cells were quantified by counting the number of positively stained cells per 20× field. Five representative fields were quantified for each sample.
Statistics.
Data in the manuscript are represented as mean ± SEM. Comparisons of blood pressure over time and aortic mechanics were made using 1-way ANOVA for repeated measures, followed by a Bonferroni post hoc test when significance was indicated. To compare the effect of age on various mouse strains, 2-way ANOVA was employed. To compare the effects of antioxidant treatments and the effect of Rag-1 deficiency, 1-way ANOVA was performed. The values of cytokines were normalized with logarithmic transformation prior to 1-way ANOVA. Correlations between immune-staining parameters were made using Spearman’s correlation analysis. Multivariate regression was used for analysis of human data. P values are reported in the figures.
Study approval.
All animal procedures were approved by Vanderbilt University’s Institutional Animal Care and Use Committee, and the mice were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services. The institutional review boards of Washington University and the University of Campinas approved the respective human studies. Subjects enrolled at Campinas signed a written informed consent form before enrolling in the study.
Author contributions
JW, MAS, AK, HAI, KRCM, LX, WC, SSD, and NRB performed experiments, analyzed data, and participated in manuscript preparation. SSD and LJR performed MS. JJG, CMW, KEB, LJR, HM, HC, MSM, and DGH participated in study design and manuscript preparation and obtained funding for the study. JAC provided human samples and assisted in manuscript preparation. HM supervised and obtained funding for human studies of PWV. RLM produced and provided the D11 antibody and assisted in experimental design.
Supplementary Material
Acknowledgments
We are grateful to the Transitional Pathology Shared Resource (TPSR) core and the Epithelial Biology Center at Vanderbilt University for assistance with imaging and quantification of immunohistochemical staining. We also thank the Vanderbilt University Medical Center Hormone Assay and Analytical Services Core (supported by NIH grants DK059637 and DK020593), who performed the cytokine measurements. We thank William Zackert for performing the MS and IHC for isoketal in the aorta samples. We also thank Zhizhang Wang in the Division of Cardiovascular Medicine at Vanderbilt University for his assistance in conducting in vivo measurement of PWV. This work was supported by NIH R01 grants HL105294, HL039006, and HL108701; VITA contract HHSN268201400010C; Program Project grants P01 HL58000 and GM015431; an American Heart Association predoctoral fellowship to J. Wu (13PRE14480008); and postdoctoral fellowship awards to A. Kirabo and M.A. Saleh. M.S. Madhur is the recipient of NIH K08 award (1K08HL121671) and a Vanderbilt Physician Scientist Development Award. H.A. Itani is supported by NIH F32 training grant (1F32HL124972-01).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(1):50–67. doi:10.1172/JCI80761.
References
- 1.Meaume S, Benetos A, Henry OF, Rudnichi A, Safar ME. Aortic pulse wave velocity predicts cardiovascular mortality in subjects > 70 years of age. Arterioscler Thromb Vasc Biol. 2001;21(12):2046–2050. doi: 10.1161/hq1201.100226. [DOI] [PubMed] [Google Scholar]
- 2.Laurent S, et al. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. 2003;34(5):1203–1206. doi: 10.1161/01.STR.0000065428.03209.64. [DOI] [PubMed] [Google Scholar]
- 3.Kaess BM, et al. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308(9):875–881. doi: 10.1001/2012.jama.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper LL, et al. Components of hemodynamic load and cardiovascular events: the Framingham Heart Study. Circulation. 2015;131(4):354–361. doi: 10.1161/CIRCULATIONAHA.114.011357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kals J, et al. Inflammation and oxidative stress are associated differently with endothelial function and arterial stiffness in healthy subjects and in patients with atherosclerosis. Scand J Clin Lab Invest. 2008;68(7):594–601. doi: 10.1080/00365510801930626. [DOI] [PubMed] [Google Scholar]
- 6.Stephen EA, Venkatasubramaniam A, Good TA, Topoleski LD. The effect of oxidation on the mechanical response and microstructure of porcine aortas. J Biomed Mater Res A. 2014;102(9):3255–3262. doi: 10.1002/jbm.a.34998. [DOI] [PubMed] [Google Scholar]
- 7.Soskel NT, Watanabe S, Sandberg LB. Mechanisms of lung injury in the copper-deficient hamster model of emphysema. Chest. 1984;85(6 suppl):70S–73S. doi: 10.1378/chest.85.6_supplement.70s. [DOI] [PubMed] [Google Scholar]
- 8.Galarraga B, Khan F, Kumar P, Pullar T, Belch JJ. Etanercept improves inflammation-associated arterial stiffness in rheumatoid arthritis. Rheumatology. 2009;48(11):1418–1423. doi: 10.1093/rheumatology/kep251. [DOI] [PubMed] [Google Scholar]
- 9.Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, Manzi S. Vascular stiffness in women with systemic lupus erythematosus. Hypertension. 2001;37(4):1075–1082. doi: 10.1161/01.HYP.37.4.1075. [DOI] [PubMed] [Google Scholar]
- 10.Gisondi P, et al. Chronic plaque psoriasis is associated with increased arterial stiffness. Dermatology. 2009;218(2):110–113. doi: 10.1159/000182256. [DOI] [PubMed] [Google Scholar]
- 11.Kume K, Amano K, Yamada S, Hatta K, Ohta H, Kuwaba N. Tocilizumab monotherapy reduces arterial stiffness as effectively as etanercept or adalimumab monotherapy in rheumatoid arthritis: an open-label randomized controlled trial. J Rheumatol. 2011;38(10):2169–2171. doi: 10.3899/jrheum.110340. [DOI] [PubMed] [Google Scholar]
- 12.Angel K, Provan SA, Gulseth HL, Mowinckel P, Kvien TK, Atar D. Tumor necrosis factor-α antagonists improve aortic stiffness in patients with inflammatory arthropathies: a controlled study. Hypertension. 2010;55(2):333–338. doi: 10.1161/HYPERTENSIONAHA.109.143982. [DOI] [PubMed] [Google Scholar]
- 13.Guzik TJ, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madhur MS, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, et al. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114(4):616–625. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marko L, et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60(6):1430–1436. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 17.Miyashita H, et al. Lysine pyrrolation is a naturally-occurring covalent modification involved in the production of DNA mimic proteins. Sci Rep. 2014;4: doi: 10.1038/srep05343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirabo A, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124(10):4642–4656. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laude K, et al. Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice. Am J Physiol Heart Circ Physiol. 2005;288(1):H7–H12. doi: 10.1152/ajpheart.00637.2004. [DOI] [PubMed] [Google Scholar]
- 20.Weisbrod RM, et al. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62(6):1105–1110. doi: 10.1161/HYPERTENSIONAHA.113.01744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension. 2011;58(2):232–239. doi: 10.1161/HYPERTENSIONAHA.111.172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304(6):R407–R414. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trott DW, et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. 2014;64(5):1108–1115. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saleh MA, et al. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. 2015;125(3):1189–1202. doi: 10.1172/JCI76327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakubzick C, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97(4):696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell GF, et al. Cross-sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation. 2007;115(20):2628–2636. doi: 10.1161/CIRCULATIONAHA.106.667733. [DOI] [PubMed] [Google Scholar]
- 28.Dietrich T, Schaefer-Graf U, Fleck E, Graf K. Aortic stiffness, impaired fasting glucose, and aging. Hypertension. 2010;55(1):18–20. doi: 10.1161/HYPERTENSIONAHA.109.135897. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell GF, et al. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham Heart Study. Hypertension. 2004;43(6):1239–1245. doi: 10.1161/01.HYP.0000128420.01881.aa. [DOI] [PubMed] [Google Scholar]
- 30.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27(12):2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 31.Valente AJ, Yoshida T, Gardner JD, Somanna N, Delafontaine P, Chandrasekar B. Interleukin-17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual-specificity phosphatase MKP-1/DUSP-1. Cell Signal. 2012;24(2):560–568. doi: 10.1016/j.cellsig.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinh A, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122(24):2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orabona C, et al. CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat Immunol. 2004;5(11):1134–1142. doi: 10.1038/ni1124. [DOI] [PubMed] [Google Scholar]
- 34.Menard HA, Lapointe E, Rochdi MD, Zhou ZJ. Insights into rheumatoid arthritis derived from the Sa immune system. Arthritis Res. 2000;2(6):429–432. doi: 10.1186/ar122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mydel P, et al. Carbamylation-dependent activation of T cells: a novel mechanism in the pathogenesis of autoimmune arthritis. J Immunol. 2010;184(12):6882–6890. doi: 10.4049/jimmunol.1000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirabo A, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124(10):4642–4656. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Czernichow S, et al. Effect of supplementation with antioxidants upon long-term risk of hypertension in the SU. J Hypertens. 2005;23(11):2013–2018. doi: 10.1097/01.hjh.0000187259.94448.8a. [DOI] [PubMed] [Google Scholar]
- 38.Myung SK, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;346: doi: 10.1136/bmj.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297(8):842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 40.Matoba T, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun. 2002;290(3):909–913. doi: 10.1006/bbrc.2001.6278. [DOI] [PubMed] [Google Scholar]
- 41.Arnold RS, et al. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci U S A. 2001;98(10):5550–5555. doi: 10.1073/pnas.101505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem. 2008;283(45):31153–31162. doi: 10.1074/jbc.M805056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts LJ. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radic Biol Med. 2007;43(10):1388–1393. doi: 10.1016/j.freeradbiomed.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumbach GL, Siems JE, Heistad DD. Effects of local reduction in pressure on distensibility and composition of cerebral arterioles. Circ Res. 1991;68(2):338–351. doi: 10.1161/01.RES.68.2.338. [DOI] [PubMed] [Google Scholar]
- 45.Mecham RP. Methods in elastic tissue biology: elastin isolation and purification. Methods. 2008;45(1):32–41. doi: 10.1016/j.ymeth.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Starcher B. A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal Biochem. 2001;292(1):125–129. doi: 10.1006/abio.2001.5050. [DOI] [PubMed] [Google Scholar]
- 47.Hofman K, Hall B, Cleaver H, Marshall S. High-throughput quantification of hydroxyproline for determination of collagen. Anal Biochem. 2011;417(2):289–291. doi: 10.1016/j.ab.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 48.Davies SS, Amarnath V, Brame CJ, Boutaud O, Roberts LJ. Measurement of chronic oxidative and inflammatory stress by quantification of isoketal/levuglandin γ-ketoaldehyde protein adducts using liquid chromatography tandem mass spectrometry. Nat Protoc. 2007;2(9):2079–2091. doi: 10.1038/nprot.2007.298. [DOI] [PubMed] [Google Scholar]
- 49.Davies SS, et al. Localization of isoketal adducts in vivo using a single chain antibody. Free Radic Biol Med. 2004;36(9):1163–1174. doi: 10.1016/j.freeradbiomed.2004.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.