

Abstract
Type 1 diabetes (T1D) patients show abnormalities in early B cell tolerance checkpoints, resulting in the accumulation of large numbers of autoreactive B cells in their blood. Treatment with rituximab, an anti-CD20 mAb that depletes B cells, has been shown to preserve β cell function in T1D patients and improve other autoimmune diseases, including rheumatoid arthritis and multiple sclerosis. However, it remains largely unknown how anti–B cell therapy thwarts autoimmunity in these pathologies. Here, we analyzed the reactivity of Abs expressed by single, mature naive B cells from 4 patients with T1D before and 52 weeks after treatment to determine whether rituximab resets early B cell tolerance checkpoints. We found that anti–B cell therapy did not alter the frequencies of autoreactive and polyreactive B cells, which remained elevated in the blood of all patients after rituximab treatment. Moreover, the limited proliferative history of autoreactive B cells after treatment revealed that these clones were newly generated B cells and not self-reactive B cells that had escaped depletion and repopulated the periphery through homeostatic expansion. We conclude that anti–B cell therapy may provide a temporary dampening of autoimmune processes through B cell depletion. However, repletion with autoreactive B cells may explain the relapse that occurs in many autoimmune patients after anti–B cell therapy.
Introduction
Autoantibody production is a characteristic of most autoimmune diseases including type 1 diabetes (T1D), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE). The contribution of autoantibodies to T1D remains elusive, but an involvement of B cells in disease development was initially evidenced in nonobese diabetic (NOD) mice backcrossed with Igμ-null animals that lacked B cells and did not develop significant insulitis. Treatment of hyperglycemic NOD mice with an anti–B cell Ab reversed diabetes, demonstrating a role for B cells in the development of the disease in mice (1, 2). Further analyses revealed the autoantigen-presenting cell function of B cells in T1D; B cell receptor (BCR) recognition of T1D self-antigens allows B cells to present them through MHC class II molecules to T cells that they also activate via CD80/CD86-CD28 interactions (3).
In humans, we previously reported that T1D patients display defective central and peripheral B cell tolerance checkpoints that result in the accumulation in their blood of self-reactive, mature naive B cells (4). Increased numbers of circulating autoreactive mature naive B cells that may contain clones recognizing disease-specific autoantigens may therefore increase the probability of initiating T1D and contribute to disease pathogenesis. In line with this hypothesis, out of all the X-linked agammaglobulinemia patients who display severely decreased numbers of B cells due to BTK mutations, only one has been reported with T1D, suggesting a role for B cells in T1D pathogenesis (5, 6).
Anti–B cell therapy with anti-CD20 mAb (rituximab) that depletes B cells was shown to reduce the decline in C-peptide secretion in the year following diagnosis of T1D and to reduce requirements for exogenous insulin and lower glycosylated hemoglobin levels (7). However, rituximab efficiency in T1D was not sustained and lost significance 2 years after anti–B cell therapy (8). Nonetheless, rituximab has shown efficacy in several other autoimmune diseases (9, 10). The mechanism of rituximab treatment in autoimmunity remains poorly understood. Here, we investigated whether rituximab-mediated anti–B cell therapy modifies the frequencies of autoreactive B cells in T1D.
Results and Discussion
The impact of anti–CD20 rituximab therapy on T and B cell populations was assessed in the blood of newly diagnosed T1D patients selected from a subset of subjects enrolled in the ancillary TrialNet TN-02 study. Participants with new-onset T1D were randomized to receive placebo or rituximab (total n = 87). Drug treatment showed efficacy with regard to the primary endpoint, which was a significant reduction in the decline in the C-peptide AUC response to a mixed meal 1 year after receiving 4 weekly infusions of rituximab (7). We performed flow cytometry to analyze T and B cell subsets before treatment and 13, 26, and 52 weeks after treatment in 19 subjects who were selected by the coordinating center and who showed changes in B cell populations and metabolic responses that were similar to those reported in the original trial (Figure 1A and Table 1). All B cell populations including total CD19+, naive CD19+CD27–, and memory CD19+CD27+ cells, as well as other B cell subpopulations, displayed robust depletion when analyzed at 13 weeks after treatment in all patients. B cells were reemerging 26 weeks after treatment before reaching numbers at 52 weeks that were similar to those seen with pretreatment (Figure 1A). As expected, cells not bearing CD20 (total CD3+ T cells and Tregs) and total wbc numbers showed constant population sizes from pretreatment through the post-treatment period, confirming rituximab’s CD20-dependent targeted depletion (Figure 1A). To determine the extent and qualitative changes of B cell depletion induced by anti–B cell therapy and to more specifically characterize the proportion of several B cell subsets, we purified B cells from frozen peripheral blood mononuclear cells (PBMCs) obtained from 13 T1D patients before and 52 weeks after injection of rituximab (Figure 1B). We found that CD19+CD10+IgMhiCD21loCD27– transitional B cells that recently emigrated from the BM had significantly increased 1 year after treatment, whereas CD19+CD10–IgM+CD21+CD27– mature, naive CD19+CD10–IgM+CD21+CD27+ IgM memory, and CD19+CD10–IgM–CD21+CD27+ isotype-switched memory B cells all had significant decreases in population sizes after treatment, suggesting B cell repletion by newly generated B cells (Figure 1B). We conclude that rituximab treatment caused B cell depletion, followed by a predominance of naive B cells during the repletion period, 1 year after therapy.
Figure 1. Rituximab treatment results in transient reduced numbers of peripheral B cells.

(A) Populations of lymphocytes from 19 T1D patients 0, 13, 26, and 52 weeks after rituximab treatment were assessed by flow cytometry. CD3, CD3+ T cell counts; Tregs, CD3+CD4+CD25hiCD62L+ cell counts; CD19CD38, CD24+/–IgD+/–CD38+CD19+CD10+/– cell counts; CD19CD38IgD, CD24+/–IgD+CD38+CD19+CD10+/– cell counts; CD19CD27, CD1c+/–IgD+/–CD27+CD19+IgM+/– cell counts; CD19, CD1c+/–CD95+/– CD27+/–CD19+CD20+/– cell counts; Naive B, CD24+IgD+CD38–CD19+CD10– cell counts. Data represent the mean ± SEM. (B) Comparative frequencies of CD19+CD10+IgMhiCD27– new emigrant/transitional B cells (top left), CD19+CD10–IgM+CD27– mature naive B cells (top right), CD19+CD10–CD27+ memory B cells (bottom left), and switched memory B cells (bottom right) were determined for T1D patients before and after treatment with rituximab. Bold horizontal lines represent averages, and the number of T1D patients analyzed is indicated. *P < 0.05 and **P < 0.01, by 2-tailed paired Student’s t test. Tx, treatment.
Table 1. Clinical characteristics of the research subjects.
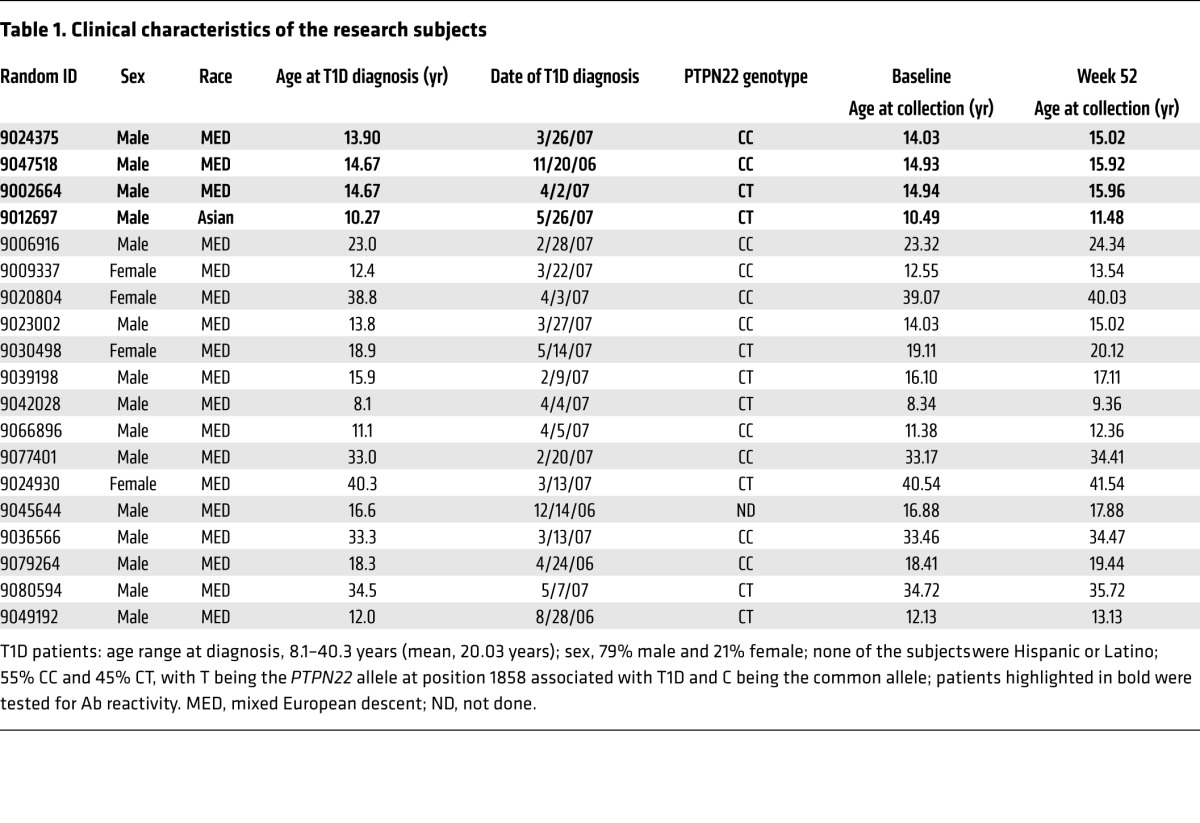
To determine whether anti–B cell therapy restores defective early B cell tolerance checkpoints in T1D, we analyzed the reactivity of Abs expressed by single, mature naive B cells from 4 T1D patients who were designated by the TrialNet study as “responders” on the basis of C-peptide responses before and 52 weeks after rituximab treatment (ref. 11, Table 1, Supplemental Figure 1, and Supplemental Tables 1–9; supplemental material available online with this article; doi:10.1172/JCI83840DS1). All pretreatment T1D patients had high frequencies of HEp-2–reactive Abs that ranged from 35.0% to 50.0% of the clones compared with an average of 20.1% in healthy donors (Figure 2, A and B). These elevated frequencies of HEp-2–reactive, mature naive B cells were similar to those previously determined in 8 T1D patients, further attesting to the notion that defective early B cell tolerance checkpoints are characteristic of this autoimmune disease (Figure 2B and ref. 4). Of note, 2 of the 4 patients carry the T1858 PTPN22 risk allele that results in naive B cell selection defects (4). Mature naive B cells 52 weeks after treatment expressed a high proportion of HEp-2–reactive Abs (29.6%–50.0%) that was similar to the levels observed pretreatment and was also statistically significantly elevated compared with the proportion detected in healthy donors (P < 0.001) (Figure 2, A and B). Defective early B cell tolerance checkpoints in T1D after rituximab treatment were also illustrated by the elevated frequencies of polyreactive clones (18.5%–26.1%) in mature naive B cells that were similar to pretreatment values (14.3%–38.5%) and contained some antinuclear clones (Figure 2C, P < 0.001, Figure 2D, and Supplemental Figure 2). Elevated frequencies of autoreactive B cells after rituximab did not result from increased serum concentrations in B cell–activating factor (BAFF), a critical B cell survival factor that controls peripheral B cell numbers (Supplemental Figure 3 and ref. 12). Hence, rituximab does not reduce the elevated numbers of circulating autoreactive B cells present in the blood of T1D patients.
Figure 2. High frequencies of autoreactive B cells in T1D patients after anti–B cell therapy.

(A) Abs cloned from mature naive B cells from a healthy donor (HD27) and 4 T1D patients before and after rituximab treatment were tested by ELISA for HEp-2 cell reactivity. Dotted lines represent the ED38+ control, and solid lines show binding for each cloned recombinant Ab. Horizontal lines define the cutoff OD405nm for positive reactivity. Frequency of HEp-2 cell–reactive clones (solid black area) is summarized in the pie charts, with the total number of clones tested indicated in the centers. Frequencies of HEp-2–reactive (B), polyreactive (C), and antinuclear (D) mature naive B cells were higher in T1D patients compared with frequencies in the healthy donor and remained unchanged after treatment with rituximab. *P < 0.05 and ***P < 0.001, by 2-tailed unpaired Student’s t test for the healthy donor versus T1D patients. HD, healthy donor.
To determine whether mature naive B cells after rituximab treatment were newly generated B cells or clones that had resisted anti–B cell therapy, we determined the B cell proliferative history by measuring κ-deleting recombination excision circles (KRECs) (13). The frequency of KRECs among new emigrant/transitional and mature naive B cells from T1D patients before treatment was similar to that seen in the healthy donor counterparts, with no division in new emigrant/transitional naive B cells and an average of two divisions for mature naive B cells as previously reported (Figure 3A and ref. 13, 14). In contrast, mature naive B cells from T1D patients after B cell depletion had undergone less proliferation, with an average of 0.928 divisions compared with 1.872 before treatment (Figure 3A). This low proliferative history revealed that most mature naive B cells after rituximab treatment were newly generated rather than clones that may have resisted depletion and undergone extensive homeostatic expansion. We propose that a decreased average number of divisions in mature naive B cells results from the substantial influx during B cell reconstitution after anti–B cell therapy of new emigrant/transitional B cells devoid of a proliferative history. The enhanced proportion after rituximab treatment of new emigrant/transitional B cells expressing Ki67, a marker for clones that have recently proliferated, likely at the pre–B cell stage, suggests a rapid transit of immature B cells in the BM and fast emigration to the periphery (Figure 3B). The increase in Ki67+ clones in mature naive B cells from T1D patients after rituximab treatment may reflect an increased homeostatic expansion occurring in this newly reconstituted peripheral compartment (Figure 3B). We conclude that B cells from T1D patients following rituximab treatment mostly contain newly generated clones with an unchanged frequency of autoreactive Abs compared with that seen before treatment.
Figure 3. Mature naive B cells after rituximab treatment are newly generated clones.

(A) Evaluation of the number of cell divisions undergone in vivo by KREC analysis in new emigrant/transitional (left) and mature naive (right) B cells from a healthy donor and T1D patients before and after treatment with rituximab. (B) Evaluation of the proliferative state in vivo by Ki67 staining of new emigrant/transitional (left) and mature naive (right) B cells from a healthy donor and T1D patients before and after treatment with rituximab. Bold horizontal lines represent averages, and the number of individuals analyzed is indicated. **P < 0.01 and ***P < 0.001, by 2-tailed Student’s t test.
We report herein that the accumulation of autoreactive clones in the mature naive B cell compartment of T1D patients is not corrected by anti–B cell therapy. Defects in early B cell tolerance checkpoints are a common feature in many autoimmune diseases such as T1D, RA, SLE, and multiple sclerosis (MS) and result in large numbers of circulating autoreactive B cells in the blood of these patients (4, 14–16). Early B cell tolerance checkpoints include a central B cell selection step in the BM that is largely dependent on sensing direct binding of self-antigens through BCR and potentially TLRs, whereas the peripheral B cell tolerance checkpoint appears to require functional Tregs to prevent the accumulation of autoreactive mature naive B cells (17, 18). Central B cell tolerance is consistently impaired in T1D and RA patients and generates many self-reactive new emigrant/transitional B cells, suggesting that gene variants such as the T1858 PTPN22 risk allele associated with these autoimmune diseases affect BCR and TLR function in the regulation of this checkpoint (4, 17, 19, 20). In contrast, most MS patients only have a specific defective peripheral B cell tolerance checkpoint that is likely associated with dysregulated Treg function in these patients (14). Hence, anti–B cell therapy in MS may result in more sustained remission, because newly generated B cells exiting the BM will be properly counterselected in most patients (10). In contrast, rituximab may not prevent the reappearance of autoreactive B cells in patients with broken central B cell tolerance, as seen in T1D, RA, and SLE, leading to relapse in patients. Hence, anti–B cell therapy in T1D, RA, and SLE patients may only temporary slow down autoimmune processes in the absence of B cells before newly generated autoreactive B cells can reinitiate or reactivate disease pathogenesis.
Methods
Patients.
Samples from T1D patients before and after rituximab treatment were obtained from the TrialNet study, and the studies described herein were ancillary to the primary clinical trial. All 19 T1D patients analyzed were characterized as “responders” in the rituximab study and were randomly selected by the TrialNet study among the 32 subjects who showed a significant reduction in the decline in the C-peptide AUC response to a mixed meal 1 year after receiving 4 weekly infusions of rituximab. Healthy donors analyzed for Ab reactivity were previously reported (17). Additional untreated patients with T1D as well as age-matched healthy donors were recruited for the assessment of B cell phenotype and proliferation history.
Cell staining and sorting, cDNA, RT-PCR, Ab production, ELISAs, and indirect fluorescence assays.
Single CD19+CD21+CD10–IgM+CD27– mature naive B cells from patients were sorted on a FACSAria flow cytometer (BD) into 96-well PCR plates, and Ab reactivity was tested as previously described (4, 14–16). Serum BAFF concentrations were determined by ELISA according to the manufacturer’s instructions (R&D Systems).
Flow cytometry.
The following Abs were used for flow cytometric stainings: anti–IgM-FITC (clone G20-127) and anti–CD21-APC (clone B-ly4) from BD Biosciences; anti–CD19-APC-Cy7 (clone HIB19), anti–CD27-PerCP-Cy5.5 (clone O323), anti–CD10-PE-Cy7 (clone HI10a), and anti–Ki67-PE (clone Ki67) from BioLegend.
KREC assay.
The ratio of KRECs to Jκ-Cκ recombination genomic joints (coding joints) was determined as previously described (13). The number of cell divisions was calculated by subtracting the cycle threshold of the PCR detecting the coding joint from that of the PCR detecting the signal joint.
Statistics.
Differences between patients and healthy donors were analyzed for statistical significance with a 2-tailed unpaired Student’s t test, whereas comparisons of T1D patients before and 52 weeks after rituximab treatment were assessed using 2-tailed paired Student’s t tests. P values of less than 0.05 were considered statistically significant.
Study approval.
All aspects of the study were approved by the Yale University School of Medicine Human Investigation Committee and the participating TrialNet study investigation centers. A complete list of the participating centers and the TrialNet study organization is provided in Appendix 1.
Author contributions
NC, CM, and TC performed the experiments. TO, KCH, and EM wrote the manuscript.
Supplementary Material
Acknowledgments
We thank L. Devine and C. Wang for cell sorting. This work was supported by grants AI061093, AI071087, AI082713, and AI095848 from National Institute of Allergy and Infectious Diseases (NIAID), NIH (to E. Meffre). The sponsor of the trial was the Type 1 Diabetes TrialNet Pathway to Prevention Study Group. See Supplemental Appendices 1 and 2 for Type 1 Diabetes TrialNet Pathway to Prevention Study Group details.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(1):282–287. doi:10.1172/JCI83840.
References
- 1.Serreze DV, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Igmunull mice. J Exp Med. 1996;184(5):2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161(8):3912–3918. [PubMed] [Google Scholar]
- 3.Hu CY, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117(12):3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menard L, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121(9):3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin S, et al. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345(14):1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 6.Winkelstein JA, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) 2006;85(4):193–202. doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 7.Pescovitz MD, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361(22):2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pescovitz MD, et al. B-lymphocyte depletion with rituximab and beta-cell function: two-year results. Diabetes Care. 2014;37(2):453–459. doi: 10.2337/dc13-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards JCW, et al. Efficacy of B cell-targeted therapy with Rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 10.Hauser SL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 11.Herold KC, et al. Increased T cell proliferative responses to islet antigens identify clinical responders to anti-CD20 monoclonal antibody (rituximab) therapy in type 1 diabetes. J Immunol. 2011;187(4):1998–2005. doi: 10.4049/jimmunol.1100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9(7):491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 13.van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med. 2007;204(3):645–655. doi: 10.1084/jem.20060964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinnunen T, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest. 2013;123(6):2737–2741. doi: 10.1172/JCI68775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med. 2005;201(10):1659–1667. doi: 10.1084/jem.20042321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yurasov S, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–711. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meffre E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci. 2011;1246:1–10. doi: 10.1111/j.1749-6632.2011.06347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinnunen T, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood. 2013;121(9):1595–1603. doi: 10.1182/blood-2012-09-457465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arechiga AF, et al. Cutting edge: The PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol. 2009;182(6):3343–3347. doi: 10.4049/jimmunol.0713370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. New Engl J Med. 2011;365(17):1612–1623. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.