

Abstract
Oncogenic activations by mutations in key cancer genes such as EGFR and KRAS are frequently associated with human cancers. Molecular targeting of specific oncogenic mutations in human cancer is a major therapeutic inroad for anti-cancer drug therapy. In addition, progressive developments of oncogene mutations lead to drug resistance. Therefore, the ability to detect and continuously monitor key actionable oncogenic mutations is important to guide the use of targeted molecular therapies to improve long-term clinical outcomes in cancer patients. Current oncogenic mutation detection is based on direct sampling of cancer tissue by surgical resection or biopsy. Oncogenic mutations were recently shown to be detectable in circulating bodily fluids of cancer patients. This field of investigation, termed liquid biopsy, permits a less invasive means of assessing the oncogenic mutation profile of a patient. This paper will review the analytical strategies used to assess oncogenic mutations from biofluid samples. Clinical applications will also be discussed.
Introduction: The Clinical Application and Context of Liquid Biopsy
In recent years, pharmaceutical drugs such as gefitinib, erlotinib, afatnib, everolimus, sorafenib, pembrolizomab, and sunitinib have been used as interventions to slow the proliferation of cancerous cells, and these drugs have been found to have their efficacy linked to the oncogenic mutation status of patients1, 2, 3,4. The presence or absence of these oncogenic mutations indicates if drug therapy will effectively limit the spread of cancer and improve patient survival rate, and thus testing prior to treatment serves as a useful tool for precision medicine5, 6, 3. The monitoring of these oncogenic mutations is not only useful prior to treatment, but monitoring continues to remain important during and after the treatment process to assess for developed drug resistance7, 8, 9 and cancer recurrence10. The clinical practice to assess genetic mutations in cancer has historically been through direct sampling of cancerous tissue with biopsy or surgical resection. Over the past decade investigations have been made into evaluating cancer mutations using physiological biofluids. This emerging field that examines physiological biofluids and performs analysis on them for improving cancer management has been termed liquid biopsy11.
There are multiple factors that motivate the exploration of liquid biopsy as an alternative to the gold standard methods of direct tissue sampling via biopsy and resection: First, it is desirable to have an alternative to the direct sampling of tissue through resection and biopsy, as the genetic mutations present in a patient may alter following treatment12, 13, 14, and conducting additional biopsies and resection may present risk to the health status of the patient. Second, tumor heterogeneity is a concern10,15 in oncogenic mutation detection: certain sections within a tumor may have the mutation while other sections do not, and testing a single tumor sample may lead to a false negative reading for oncogenic mutations. Finally, an alternative to direct tissue sampling may also lessen the fiduciary and resource strain on caregivers and patients (the cost of a fine needle aspiration biopsy has been estimated to be approximately $1300 per procedure16, while a company such as Pathway Genomics have been marketing the ability to perform liquid biopsy panel tests for half the cost from 20 mL of blood17).
Liquid biopsy as a strategy for oncogenic mutation begins with the selection of an appropriate panel of mutations that must be tested for. A notable catalog of oncogenic gene mutations was begun by Wellcome Trust Sanger Institute in 200418. This catalog, titled COSMIC (catalog of somatic mutations in cancer) has collated and organized information about cancer state and mutations associated with each cancer state, curating the results of 20,000 scientific studies19 into a comprehensive and easily searchable database. Some clinically relevant mutations can be covered with a small panel of mutations, such as mutations in the EGFR domain for non-small cell lung cancer (NSCLC). In EGFR mutations for NSCLC, 90% of mutations are covered by the deletion mutations in exon 19 and the L858R point mutation8. However, other cancers require monitoring for a larger panel: The KRAS mutation occurs in 40% of patients with colorectal adenocarcinoma, but there are at least 7 different mutations in two adjacent codons20 that must be used in order to get a satisfactory coverage of the forms of mutation that exist. Distribution of mutations is also correlated to race and gender factors (e.g. EGFR mutation occurring in 51.4% of adenocarcinomas for individuals from Asian populations21), and these various population composition factors may also play into the development of an appropriate panel of biomarkers. Based on clinical needs and information from the COSMIC database, panel of tests that cover key oncogenic mutations22, 23, 24 are designed and targeted for detection in liquid biopsy.
The presence of mutated genetic content in biofluids
A variety of different biofluids have been examined for oncogenic mutation detection. Serum25, sputum26, 27, cerebrospinal fluid28, broncho-alveolar lavage fluid29, urine30, stool23, and saliva31,32 have all been investigated as possible avenues for oncogenic mutation analysis. A number of these studies seem to yield fruitful results, with some studies having high sensitivities and specificities when benchmarked with direct tissue sampling methods32, 29, 33. In most strategies of detecting mutated content from biofluids, processing steps must be taken to extract and purify out genomic content from the sampled biofluids. Two main approaches seem are taken when it comes to isolating and extracting mutated genetic content from biofluids.
Circulating Tumor Cells (CTC)
This form of biofluid analysis captures cells shed from a primary tumor site that are freely circulating in the body biofluid, and performs mutation analysis on the cells after they have been captured and concentrated12, 34. This selective concentration of tumor cells is potentially beneficial because it allows one to extract more complete DNA from tumor cells, instead of other approaches that may only extract degraded DNA35. In order to facilitate the capture of circulating tumor cells, which are in low abundance (as the amount of CTCs are relatively low in proportion to a biofluid sample, with 1–10 CTC cells occur per 10 mL of blood36), various strategies have been adopted in order to concentrate the tumor cells in the biofluid. The most commonly used strategies for capture of CTC for molecular analysis appear to be microfluidic channels12, 37 and magnetic beads38 coated with capturing antibodies, but other techniques such as the usage of electric fields (using the phenomena dielectrophoresis)39 and centrifugation40 have also been explored for capturing of CTC. This approach was effectively used in a study by Maheswaran12, in which EGFR mutation was successfully detected in 11 out of 12 patients (92%) using CTC cells that had been captured.
Circulating Tumor DNA (ctDNA)
As opposed to specifically targeting the cancer cells present in the biofluid and analyzing their genomic content, this approach takes biofluid samples and extracts the nucleic acid using column based techniques41, 42, 43. Circulating tumor DNA as an approach has been noted to be advantageous in that it has been found to appear in the serum before circulating tumor cells33,34, and because biofluid sampling is more easily performed than CTC extraction43 (In a study performed by Sozzi et al44, 318 ng/mL of ctDNA could be extracted from 1 mL of lung cancer patients serum). In relation to clinical effectiveness, Thierry et al.33 reported a total clinical sensitivity of 92% and a clinical specificity of 98% for all KRAS mutations when ctDNA for 95 specimens when benchmarked against tissue based genotyping. In regards to a direct comparison of CTC and cTDNA approaches, studies have appeared in the two years comparing CTC to ctDNA35, 45 have found that ctDNA was able to perform higher sensitivity detection (Freidin et al.35 reported that CTC had a clinical sensitivity of 52% while ctDNA had a clinical sensitivity of 96%). The specific mechanism of which this extracellular DNA is transported in the biofluid is under investigation, and studies suggest that this circulating tumor DNA with mutations may be sequestered and protected by microvesicle46 structures such as exosomes47,48, which are 50–150 nm microvesicular structures. Investigations conducted by Kahlert et al.48 on serum exosomes demonstrated that double stranded DNA with genetic mutations in the KRAS and p53 domains can be detected in these vesicles.
It is evident from the body of work extant that both CTC DNA and ctDNA techniques have potential: but there are disadvantages49 that must be overcome. Alix-Panabieres and Pantel50 note technical obstacles of CTC analysis such as the large sample volumes requirements, the potential inadequacy of epithelial markers such as EpCam for identifying the most aggressive CTC types, and the difficulty in differentiate tumor cells from epithelial cells in patients with benign colon diseases such as diverticulosis. In regards to ctDNA, Pantel and Alix-Panabieres51 note that DNA from lysed cells may add nucleic acid content has the potential to interfere with detection, and Ilie et al52 note that inherent in the ctDNA approach is the inability of performing morphological analysis of cells. Time will tell whether these disadvantages will be significantly hinder the usage of these techniques, but at present it seems that emerging new platforms50 and refinements to existing protocols51 can aid in mitigating these disadvantages.
Molecular Detection Platforms for Liquid Biopsy
Genomic content that is collected from a biofluid must be tested with an appropriate analytical method in order to identify the mutated genetic sequences. The Sanger method of DNA sequencing for mutation analysis53 using gel or capillary electrophoresis and terminating dideoxynucleotides to acquire the sequence of DNA is considered the benchmark gold standard for identification of oncogenic gene mutations in tissue. However, Sanger sequencing sensitivity levels leaves room for improvement54, inasmuch as it does not possess the sensitivity adequate for detection in the context of liquid biopsy. Sanger sequencing is able to detect mutated sequences if the mutated allele to wildtype frequency is 20%55. This analytical sensitivity of 20% Diaz and Bardelli34 observe to be inadequate for analysis. As a result of this analytical sensitivity requirement various different technologies have emerged to try to see if greater discriminatory methods for detection of mutations could be accomplished. The key diagnostic techniques presently used for liquid biopsy are:
Beads, Emulsions, Amplification, and Magnetics (BEAMing)
This technique utilizes a magnetic microparticle and oil-water emulsions in order to enhance sensitivity. In this procedure, DNA that has been extracted and preamplified is dispersed into multiple 3–9 μm diameter water droplets. Each droplet has the primers, enzymes, and free nucleotide components necessary to amplify the targeted mutated sequence. However, one of the primers is attached to a magnetic microparticle, and if the mutated DNA sequence is present then the bead will eventually be coated with the amplified mutant sequence after the PCR is run on the emulsion. The emulsion droplets are then broken, the mutated sequence on the magnetic beads labeled with fluorescent probes, and all the beads that were used run through flow cytometry28, 56. If the beads have the mutated sequence present on their surface, their fluorescence will be readily observable during flow cytometry. This technique has been noted to have a sensitivity of up to 1 in 10,000 DNA molecules57 (0.01% analytical sensitivity), reaching the levels necessary for the detection of oncogenic mutations. In the context of liquid biopsy, this has been applied to the study of ctDNA for the detection of PIK3CA58 from plasma samples, with a perfect concordance between tissue sequencing and BEAMing assay of the collected blood samples.
BEAMing has recently been applied in a work by Tabonero et al59 for the examination of PIK3CA, KRAS, and BRAF mutations on genomic DNA isolated from 2mL of plasma, with a 76% KRAS concordance, a 88% PIK3CA concordance, and a 97% BRAF concordance when comparing archival tumor samples with patient plasma. These preliminary studies of BEAMing are suggestive, and further tests may help establish this as an extremely viable method for practical liquid biopsy of oncogenic mutations.
Polymerase Chain Reaction (PCR) Based Techniques
This technique involves the measurement of a nucleic acid sequence by making a live measurement of fluorescence expression levels during the PCR reaction process. This expression of fluorescence is done through two means: either through a double stranded DNA intercalating agent (the SYBR® Green method) or through a specially designed primer sequence that has both a quencher and a fluorescent particle on it (The Taqman® based method). In the SYBR Green case, as the target sequence is amplified, the amount of double stranded DNA increases, and the intercalating agent then goes between the doubled-stranded DNA particles and detection machinery can allow for live measurement of the fluorescing that occurs during the amplification process. In the Taqman® case, as the amplification occurs, the fluorescent particle on the probe that is normally quenched is cleaved off of the primer sequence and is then capable of being detected in real time. This ability to quantify in real time in conjunction with the fact that different sequences possess different melting points can aid in identification and differentiation of a wild type and mutation sequences54. RT-qPCR as a technique has been utilized in a variety of studies of mutations in the BCR-ABL kinase domain60, BRAF61, and KRAS35. This technique has been applied with success in the liquid biopsy context: The testing of ctDNA for the KRAS mutation, for example, was reported to be able to have an analytic sensitivity of 0.025% mutation to wild type ratio, and there was a clinical sensitivity of 96% for detecting mutations in liquid biopsy samples when benchmarked to tissue samples35.
Another form of PCR based techniques that has been applied to liquid biopsy is Droplet Digital PCR (ddPCR). This technique involves performing the PCR reaction in a large amount of individual picoliter-sized droplets28 and measuring fluorescence in real time from individual droplets during the PCR process. Droplet Digital PCR is similar to the BEAMING technique in that digital PCR breaks the sample into numerous small droplets to allow for a greater ability to specifically and sensitively detect mutated sequences that may be present. But there are key differences in the technique, as described by Pekin et al62: First, BEAMing requires preamplification of the DNA sample prior to dispersing the nucleic acid into the droplet form, while in digital PCR amplification can be performed within each droplet. Second, the beads must be processed to fluorescently tag each allele before processing in flow cytometry, while in digital PCR the amplification and detection processes are integrated into a disposable unit. The analytical sensitivity that has been achieved using this real time droplet digital PCR technique is 0.0045% mutant to wild type molecules63. Exploratory techniques using microfluidics to further advance the effectiveness of digital PCR have also been made, with reports of 1 mutation in 200,000 non-mutated wild type sequences62 being detectable in proof of concept systems.
This approach of utilizing droplet digital PCR has been applied to the study of a 19 patient cohort with colorectal cancer and KRAS mutations20. This study purified plasma DNA and performed a multiplexed digital PCR assay on the 19 patient cohort, finding a concordance of approximately 73% between patient plasma samples and mutations previously identified from tumor sample DNA.
Next generation sequencing (NGS)
This technique involves sequencing DNA in a highly rapidly and parallelized fashion (compared to the original Sanger sequencing method)64. The target patient samples first have their DNA isolated from a tissue sample or liquid biopsy sample and then the fragmented into shorter fragments. During this library preparation procedure the target sequences are also subject to a selective amplification of specific portions of the sequence (these specific portions are targeted because their relation to a known oncogenic gene mutation, which will enrich the mutated sequence and increase the likelihood of detection. Following the library preparation, the prepared library of DNA fragments is immobilized on a solid surface (typically microparticle beads or a slide) through ligands65, and these ligated DNA fragments on a solid are subjected to highly parallelized readout through a next generation sequencing platform.
There are a wide variety of techniques utilized for performing next generation sequencing in liquid biopsy. A historical and technical survey of the different platforms has been made by Mousadi-Nejad et al64 which provides an overview of the different mechanisms used to perform NGS, such as fluorescence or monitoring pH changes. In the field of NGS based liquid biopsy mutation detection, a cohort of lung cancer patients serum was analyzed using NGS IonTorrent sequencing (a pH based detection technique) by Couruad et al66 demonstrating a clinical sensitivity of 58% and a clinical specificity of 87% when comparing analysis of serum ctDNA to tissue samples. In this NGS study, the analytical sensitivity of this deep sequencing NGS approach was stated to be detect the mutated DNA even when it was only 0.2% of total sample DNA66.
Direct comparisons between different mutation detection platforms such as NGS and PCR Based Techniques have been conducted for tissue54, 55, but additional direct comparisons of these detection platforms will be necessary for a thorough evaluation of the most optimal method in the liquid biopsy context.
PCR enhancement techniques for Liquid Biopsy
Inasmuch as a large majority of the existing techniques are based on polymerase chain reaction (PCR), a variety of methods have been implemented in an attempt to selectively amplify the mutated sequence and increase the probability that it can be detected when in low abundance relative to the wildtype sequence. A body of review literature67,68, 69 addresses these different techniques, and among the multitude of different enrichment techniques, some general approaches are discernable:
Allele specific primer amplification: In this method, primers are designed so that the mutated sequence is at the end of the 3′ end of the primer sequence. Through this design, the target that possesses the mutation that perfectly complements the primer will be preferentially amplified by polymerase compared to the sequence that does not possess it. This seems to have high sensitivity but lower selectivity67. Thierry et al33 applied this allele-specific amplification approach in a quantitative PCR based study of the KRAS and BRAF mutations in ctDNA, reporting a 100% concordance between tissue and circulating DNA extracted from serum or plasma.
Enzyme based digestion of sequences: In this class of methods, endonuclease70 and primers are utilized together to enrich the mutant target and digest the sequences that are not mutated. The disadvantage of this method is that it requires an appropriate design and accommodation to the endonucleases available71, the enzyme may not be completely effective in digesting the wild type sequences68. This method of using digestion enzymes was used by Asano et al72 in a study of lung pleural fluid for EGFR mutation, and this method reported an analytical sensitivity of 0.05% mutant to wild type genes.
Preferential Homoduplex formation assay (PHFA): This method of selecting for the mutated sequences works by using competitive reactions during the PCR process in order to detect a specific mutated sequence73. This original technique involves adding a biotinylated and FITC double labeled amplicon in solution with the sample (that has no labels present). If the unlabeled sample matches the double-labeled amplified sequence, then the signal from the double-labeled amplicon will be diluted out during the PCR process and will not be able to measure. If the sample does not match the amplicon, then the double-labeled amplicon will continue to exist and be duplicated. A modified form of this PHFA using fluorescence resonance energy transfer (FRET) has also been explored: a fluorescent dye (such as FAM) and a quencher are combined together in the doubled labeled amplicon, and if there is a match between the sample and the doubled labeled amplicon DNA sequences, fluorescence will be observed because less quencher will complexed to suppress the fluorescent probe74. On the side of clinical liquid biopsy, this technique was used in the detection of the 1DH1 mutation (associated with glioma) serum and cerebrospinal fluid28 and the APC mutation (associated with colorectal cancer) in serum samples75.
Clamped Based PCR Technique: This technique utilizes blockading sequences during the PCR process. Two probes are introduced into a reaction during this process: The first is a primer that will be used to amplify the targeted sequence (such as the mutated sequence), the second is a sequence that is specifically meant to inhibit sequences that are similar but not identical to the targeted sequences (such as the wild type form). An example of this is the Competitive Allele Specific Hydrolosis (CAST) PCR76 technique, a form of clamped-based PCR technique that integrates with the Taqman qPCR that was successfully used to examine a panel of mutations in tissue for KRAS, EGFR, and BRAF mutations at a reported sensitivity of 0.5%77, 76. CastPCR was able to be used in the detection of BRAF in a case of melanoma78. Alternative inhibition of nucleic acids such as locked nucleic acids79 and peptide nucleic acids80 have been demonstrated to increase specificity of binding67. The usage of locked nucleic acids have been applied to liquid biopsy in a work by Breitenbuecher et al.81, where it was found that locked nucleic acids enhanced sensitivity levels and mutation in blood samples enriched for CTC81. This work found that the sensitivity levels using lock nucleic acid as inhibiting sequences was 0.01% of mutation to wild type allele. Shinozaki et al also applied an LNA clamped PCR system to detect circulating tumor DNA isolated from serum samples82. Finally, Spindler61 et al. used special hydrophobic nucleic acid based blocking sequences in order to make real time quantitative PCR more effective.
These variant forms of PCR demonstrate the ability to achieve high analytical sensitivity of the detection process of mutated sequences. Some of these enhancements to the process of the PCR have also been able to achieve high clinical sensitivity33.
Improving Workflows and Emerging Point of Care Methods for Oncogenic Mutation Detecting
As the field of oncogenic mutation pushes the boundaries of sensitivity in detecting mutations using traditional sequencing or PCR based techniques, a point of inquiry is the appropriate workflow for mutation detection in a clinical setting. Apart from the ability to sensitively identify mutated DNA when they are in low abundance relative to wild type DNA, factors such as clinical benefit, sample volume, test turnaround time, and cost play a role that must be considered in the application of mutation detection. Modifications have been made along different junctures in the traditional sample-collection and molecular testing workflow for oncogenic mutation detection in an attempt to simplify mutation detection for clinical practical.
Progress been made in the direction of creating point-of-care diagnostic tools for rapid mutation detection without a large amount of analysis equipment using novel techniques such as:
Nanoparticle Based Mutation Capture and Visualization: In recent years, it appears that attempts have been made to use nanoparticle strategies to detect genetic mutations: The advantage of using nanoparticles are the efficiency of capturing and concentrating target mutations, and the fact that nanoparticles can be manipulated to create optical effects that can be evaluated with the naked eye (making them more practical tools for point-of-care mutation detection). Latore et al84 using a gold nanoparticle system that is capable of detecting SNP using the aggregation effects of gold nanoparticles coated with oligonucleotides, which precipitate out if the specific mutated target sequence attaches to gold nanoparticles and changes the hydrophobicity of the nanoparticle. An additional example of this approach is a study made by Valentini85 et al, which used of a combination magnetic nanoparticle and gold nanoparticle strategy: This system was successful demonstrating the detection of KRAS mutations with a test system that can be read with the naked eye.
Microfluidic platforms for sample processing and reading: In efforts to create more compact and integrated systems for analysis of oncogenic mutations, efforts have been made to integrate genetic analysis with microfluidic systems86. A notable example of this is in a work by Wang et al.87, where an integrated microfluidic system that could isolate genomic DNA from whole blood, perform selective amplification of the desired sequences, and perform visual readout with the eye in one hour was fabricated. Wang et al’s87 work performed detection on JAK2-V617F mutation (associated with hematologic malignancies) with 90% concordance between the microfluidic system and melting curve analysis based detection of mutated sequences, and an analytical sensitivity of 1% mutant to wild type molecules sequence. Further examples of microfluidic based techniques in the mutation detection process are presented in a helpful review article from Handal and Ugaz88.
While it would be necessary to expend significant effort to further refine these novel analytical strategies and evaluate their applicability to oncogenic mutation analysis in biofluids, they offer an intriguing view of the future. Current techniques for collection and processing of tissue and biofluid specimens are complex and require a fair amount of investment for analytical devices, but emerging methods suggest that there may be a future for rapid and convenient evaluation of mutation status.
Emerging Method: EFIRM (Electric Field Induced Release and Modification) and its advantages
An emerging platform for the detection of oncogenic gene mutation that has demonstrated high clinical sensitivity and specificity for mutation detection is the electric field induced release and measurement method (EFIRM)89. This technique is based on the principle that nucleic acid hybridization can be facilitated through applying electric fields90, 91. This electric field can be applied to hybridize sequences selectively, whether SNP or deletion mutations92. By applying these electric fields, the mutated sequences present in a biofluid can be actively hybridized to an oligonucleotide capture probe that has been immobilized with a conducting polymer93 to an electrode surface. Following this active hybridization capture of the mutated sequence, an additional detector probe sequence with a fluorescein label is hybridized to the remaining portions of mutation sequence that are unbound to the capture probe at the electrode surface. Finally, a reporter enzyme and tetramethylbenzidine based substrate solution is used to generate oxidation and reduction reactions. These oxidation and reduction reactions that occur at the surface of the electrode are subsequently measured and used for the quantitation of the target sequence present94. If target sequences are not freely present in the biofluid but are instead contained in microvesicles, the EFIRM technique is also capable of being used to lyse the exosomes using an electric field and rapidly capture the molecular content present before significant degradation occurs from constituents of the extracellular biofluid environment89.
In a blinded pilot study, 40 patients with NSCLC had their saliva samples collected for testing using the EFIRM method. These 40 patient saliva samples analyzed for mutation using EFIRM and compared to tissue based oncogenic analysis. Characterizing the performance of EFIRM using area-under-the-curve metric (AUC, a composite score of clinical sensitivity and specificity), EFIRM performed with a high clinical sensitivity and specificity. An AUC score of 0.94 and 0.96 was achieved for detecting exon-19 deletion and the L858R mutations, respectively, in saliva samples. A comparison analysis of salivary samples with plasma samples also showed R values of 0.98 and 0.99 for the relationship between serum and saliva for the Exon-19 deletion and L858R mutation, respectively.
The advantage of the EFIRM technique is that it allows for the rapid analysis of oncogenic gene mutations in a small volume of biofluid (50 μL) in an integrated and efficient fashion. Without steps such as DNA isolation and amplification of sequences, the EFIRM platform can robustly perform detection for a mutant sequence with great effectiveness in less than one hour. This EFIRM method allows for a simplified workflow that may carry benefit to care providers that wish to include gene mutation analysis into their clinical workflow. The results of this study indicating that plasma and saliva measurements are correlated with each other also presents the possibility that non-invasive detection of oncogenic gene mutations in saliva is an emerging frontier that can offer benefits to rapid assessment of patient health status for precise treatment of cancer.
Conclusion
This brief synopsis of major genetic mutation detection techniques demonstrates that a wide variety of techniques exist for capturing oncogenic mutations, and a large number of these studies have already demonstrated the viability of a liquid biopsy based oncogenic mutation strategy. It is evident that with the continual march towards more efficient genetic sequencing and advanced PCR based technologies the field of liquid biopsy will continue to grow. New announcements seem to emerge weekly about industrial and academic efforts to improve the sensitivity of liquid biopsy and develop streamlined panels for monitoring oncogenic mutation. Though a large amount of studies appear to be oriented towards the usage of circulating tumor DNA over circulating tumor cells, further investigations in the next years will elucidate which method may be the most appropriate for a clinical detection of oncogenic mutations from liquid biopsy samples. Technologies are also readily being explored to see whether highly sensitive systems such as EFIRM can also play a role in serving as robust platforms for oncogenic mutation. EFIRM-based liquid biopsy delivers the near perfect concordance performance (sensitivity) with biopsy-based genotyping. Together with the 100% specific specificity of the tumor mutation signature, eLB is the holy grail of liquid biopsy.
Supplementary Material
Figure 1.
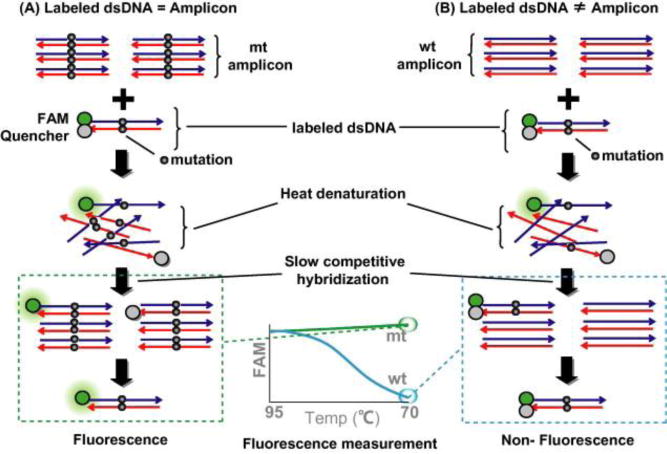
The homoduplex mechanism of detecting mutated signal using a fluorescence resonance energy transfer based strategy. (A) If the mutant signal is present in the DNA, during the PCR process the quencher unit will be gradually diluted out and a signal will be observed. (B) If the wildtype sequence is present in the DNA, there will be no dilution of the quencher unit during the PCR process and no significant amounts of fluorescing will occur. (Reprinted from Kitano et al74, with permission from Elsevier 2015).
Figure 2.
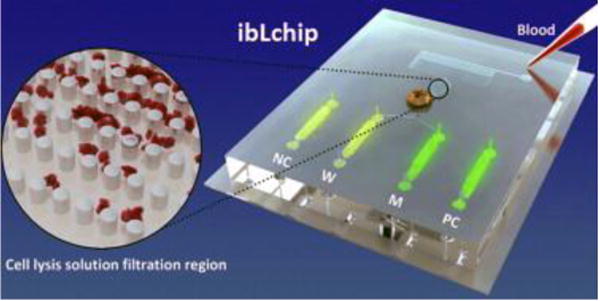
Example of integrated microfluidic mutation analysis in work. This work allowed the collection of whole blood samples, removal of red blood cells, DNA amplification, and fluorescent reporting that could be performed in one hour. (Reprinted from Wang et al87, with permission from Elsevier 2015).
Figure 3.
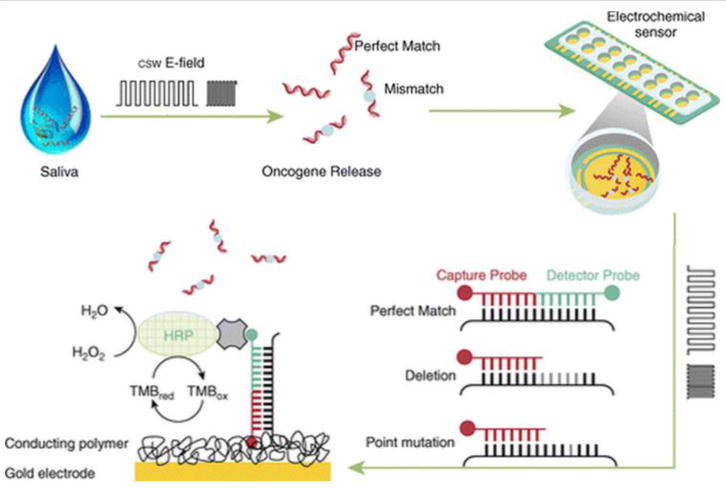
A schematic overview of the EFIRM technique. This method involves the novel application of electric potentials to facilitate hybridization of mutated sequences to a set of probes on the surface of an electrode and the usage of these probes to create readout of the signal. (Reprinted with permission of the American Thoracic Society Copyright © 2015 American Thoracic Society. From Wei et al32. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society).
Acknowledgments
Research reported in this publication was supported by the National Institute Of Dental & Craniofacial Research of the National Institutes of Health under Award Number T90DE022734. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosures
David T. Wong is co-founder of RNAmeTRIX Inc., a molecular diagnostic company. He holds equity in RNAmeTRIX, and serves as a company director and scientific advisor. The University of California also holds equity in RNAmeTRIX. Intellectual property that David T. Wong invented and which was patented by the University of California has been licensed to RNAmeTRIX. Additionally, he is a consultant to PeriRx.
Contributor Information
Michael Tu, Email: emtu@ucla.edu, School of Dentistry, University of California, Los Angeles, Los Angeles, CA.
David Chia, Email: dchia@mednet.ucla.edu, Department of Pathology, UCLA David Geffen School of Medicine at UCLA, Los Angeles, California.
Fang Wei, Email: frada@ucla.edu, School of Dentistry, University of California, Los Angeles, Los Angeles, CA.
David Wong, Email: dtww@ucla.edu, School of Dentistry, University of California, Los Angeles, Los Angeles, CA.
Bibliography
- 1.Kris MG, Natale RB, Herbst RS, Lynch TJ, Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC. JAMA. 2003;290:2149. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 2.Soulieres D. J Clin Oncol. 2003;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 3.Neychev V, Steinberg SM, Cottle-Delisle C, Merkel R, Nilubol N, Yao J, Meltzer P, Pacak K, Marx S, Kebebew E. BMJ Open. 2015;5:e008248–e008248. doi: 10.1136/bmjopen-2015-008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, Miller KD. J Clin Oncol. 2008;26:1810–1816. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 5.Muller PA, Vousden KH. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paez JG. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. Proc Natl Acad Sci. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gazdar AF. Oncogene. 2009;28:S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 10.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Nat Rev Clin Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 11.Steen HB. Carcinogenesis. 2000;21:1773–1776. doi: 10.1093/carcin/21.10.1773. [DOI] [PubMed] [Google Scholar]
- 12.Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, Settleman J, Tompkins RG, Lynch TJ, Toner M, Haber DA. N Engl J Med. 2008;359:366–377. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DWY, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, Brenton JD, Rosenfeld N. Sci Transl Med. 2012;4:136ra68–136ra68. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 14.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. J Clin Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedard PL, Hansen AR, Ratain MJ, Siu LL. Nature. 2013;501:355–364. doi: 10.1038/nature12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenssen C, Dietrich CF. Best Pract Res Clin Gastroenterol. 2009;23:743–759. doi: 10.1016/j.bpg.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Pathway Genomics Company website. https://www.pathway.com/cancer-intercept-detect/#pricing, (accessed November 2015)
- 18.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taly V, Pekin D, Benhaim L, Kotsopoulos SK, Le Corre D, Li X, Atochin I, Link DR, Griffiths AD, Pallier K, Blons H, Bouche O, Landi B, Hutchison JB, Laurent-Puig P. Clin Chem. 2013;59:1722–1731. doi: 10.1373/clinchem.2013.206359. [DOI] [PubMed] [Google Scholar]
- 21.Shi Y, Au JSK, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, Heeroma K, Itoh Y, Cornelio G, Yang PC. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. 2014;9:154–162. doi: 10.1097/JTO.0000000000000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas RK, Baker AC, DeBiasi RM, Winckler W, LaFramboise T, Lin WM, Wang M, Feng W, Zander T, MacConnaill LE, Lee JC, Nicoletti R, Hatton C, Goyette M, Girard L, Majmudar K, Ziaugra L, Wong KK, Gabriel S, Beroukhim R, Peyton M, Barretina J, Dutt A, Emery C, Greulich H, Shah K, Sasaki H, Gazdar A, Minna J, Armstrong SA, Mellinghoff IK, Hodi FS, Dranoff G, Mischel PS, Cloughesy TF, Nelson SF, Liau LM, Mertz K, Rubin MA, Moch H, Loda M, Catalona W, Fletcher J, Signoretti S, Kaye F, Anderson KC, Demetri GD, Dummer R, Wagner S, Herlyn M, Sellers WR, Meyerson M, Garraway LA. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 23.Ahlquist DA, Skoletsky JE, Boynton KA, Harrington JJ, Mahoney DW, Pierceall WE, Thibodeau SN, Shuber AP. Gastroenterology. 2000;119:1219–1227. doi: 10.1053/gast.2000.19580. [DOI] [PubMed] [Google Scholar]
- 24.Su Z, Dias-Santagata D, Duke M, Hutchinson K, Lin YL, Borger DR, Chung CH, Massion PP, Vnencak-Jones CL, Iafrate AJ, Pao W. J Mol Diagn. 2011;13:74–84. doi: 10.1016/j.jmoldx.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuang TCY, Chuang AYC, Poeta L, Koch WM, Califano JA, Tufano RP. Head Neck. 2009 doi: 10.1002/hed.21178. NA–NA. [DOI] [PubMed] [Google Scholar]
- 26.Keohavong P. Carcinogenesis. 2004;26:303–308. doi: 10.1093/carcin/bgh328. [DOI] [PubMed] [Google Scholar]
- 27.Gao W, Keohavong P. In: Molecular Toxicology Protocols. Keohavong P, Grant SG, editors. Vol. 1105. Humana Press; Totowa, NJ: 2014. pp. 325–344. [Google Scholar]
- 28.Chen WW, Balaj L, Liau LM, Samuels ML, Kotsopoulos SK, Maguire CA, LoGuidice L, Soto H, Garrett M, Zhu LD, Sivaraman S, Chen C, Wong ET, Carter BS, Hochberg FH, Breakefield XO, Skog J. Mol Ther Acids. 2013;2:e109. doi: 10.1038/mtna.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills NE, Fishman CL, Scholes J, Anderson SE, Rom WN, Jacobson DR. JNCI J Natl Cancer Inst. 1995;87:1056–1060. doi: 10.1093/jnci/87.14.1056. [DOI] [PubMed] [Google Scholar]
- 30.Buyru N, Tigli H, Ozcan F, Dalay N. J Biochem Mol Biol. 2003;36:399–402. doi: 10.5483/bmbrep.2003.36.4.399. [DOI] [PubMed] [Google Scholar]
- 31.Qiu W, Tong GX, Turk AT, Close LG, Caruana SM, Su GH. BioMed Res Int. 2014;2014:1–7. doi: 10.1155/2014/810487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei F, Lin CC, Joon A, Feng Z, Troche G, Lira ME, Chia D, Mao M, Ho CL, Su WC, Wong DTW. Am J Respir Crit Care Med. 2014;190:1117–1126. doi: 10.1164/rccm.201406-1003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, Del Rio M, Lamy P-J, Bibeau F, Nouaille M, Loriot V, Jarrousse A-S, Molina F, Mathonnet M, Pezet D, Ychou M. Nat Med. 2014;20:430–435. doi: 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- 34.Diaz LA, Bardelli A. J Clin Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freidin MB, Freydina DV, Leung M, Montero Fernandez A, Nicholson AG, Lim E. Clin Chem. 2015 doi: 10.1373/clinchem.2015.242453. [DOI] [PubMed] [Google Scholar]
- 36.Alix-Panabières C, Pantel K. Nat Rev Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- 37.Karabacak NM, Spuhler PS, Fachin F, Lim EJ, Pai V, Ozkumur E, Martel JM, Kojic N, Smith K, Chen P, Yang J, Hwang H, Morgan B, Trautwein J, Barber TA, Stott SL, Maheswaran S, Kapur R, Haber DA, Toner M. Nat Protoc. 2014;9:694–710. doi: 10.1038/nprot.2014.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Nat Rev Clin Oncol. 2014;11:129–144. doi: 10.1038/nrclinonc.2013.253. [DOI] [PubMed] [Google Scholar]
- 39.Gascoyne P, Shim S. Cancers. 2014;6:545–579. doi: 10.3390/cancers6010545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou HW, Warkiani ME, Khoo BL, Li ZR, Soo RA, Tan DSW, Lim WT, Han J, Bhagat AAS, Lim CT. Sci Rep. 2013;3 doi: 10.1038/srep01259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kopreski MS. J Natl Cancer Inst. 2000;92:918–923. doi: 10.1093/jnci/92.11.918. [DOI] [PubMed] [Google Scholar]
- 42.Kopreski M, Benko F, Kwee C, Leitzel K, Eskander E, Lipton A, Gocke C. Br J Cancer. 1997;76:1293–1299. doi: 10.1038/bjc.1997.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murtaza M, Dawson SJ, Tsui DWY, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong ASC, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, Caldas C, Rosenfeld N. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 44.Sozzi G, Conte D, Mariani L, Lo Vullo S, Roz L, Lombardo C, Pierotti MA, Tavecchio L. Cancer Res. 2001;61:4675–4678. [PubMed] [Google Scholar]
- 45.Punnoose EA, Atwal S, Liu W, Raja R, Fine BM, Hughes BGM, Hicks RJ, Hampton GM, Amler LC, Pirzkall A, Lackner MR. Clin Cancer Res. 2012;18:2391–2401. doi: 10.1158/1078-0432.CCR-11-3148. [DOI] [PubMed] [Google Scholar]
- 46.Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, Williams C, Rodriguez-Barrueco R, Silva JM, Zhang W, Hearn S, Elemento O, Paknejad N, Manova-Todorova K, Welte K, Bromberg J, Peinado H, Lyden D. Cell Res. 2014;24:766–769. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rak J, Guha A. BioEssays. 2012;34:489–497. doi: 10.1002/bies.201100169. [DOI] [PubMed] [Google Scholar]
- 48.Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, Zhang J, Weitz J, Chin L, Futreal A, Kalluri R. J Biol Chem. 2014;289:3869–3875. doi: 10.1074/jbc.C113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haber DA, Velculescu VE. Cancer Discov. 2014;4:650–661. doi: 10.1158/2159-8290.CD-13-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alix-Panabieres C, Pantel K. Clin Chem. 2013;59:110–118. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- 51.Pantel K, Alix-Panabieres C. Cancer Res. 2013;73:6384–6388. doi: 10.1158/0008-5472.CAN-13-2030. [DOI] [PubMed] [Google Scholar]
- 52.Ilie M, Hofman V, Long E, Bordone O, Selva E, Washetine K, Marquette CH, Hofman P. Ann Transl Med. 2014;2:107. doi: 10.3978/j.issn.2305-5839.2014.08.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCourt CM, McArt DG, Mills K, Catherwood MA, Maxwell P, Waugh DJ, Hamilton P, O’Sullivan JM, Salto-Tellez M. PLoS ONE. 2013;8:e69604. doi: 10.1371/journal.pone.0069604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. J Mol Diagn. 2010;12:425–432. doi: 10.2353/jmoldx.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ihle M, Fassunke J, König K, Grünewald I, Schlaak M, Kreuzberg N, Tietze L, Schildhaus HU, Büttner R, Merkelbach-Bruse S. BMC Cancer. 2014;14:13. doi: 10.1186/1471-2407-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson AL, Iglehart JD. Clin Cancer Res. 2012;18:3209–3211. doi: 10.1158/1078-0432.CCR-12-0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. Nat Methods. 2006;3:551–559. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 58.Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, Zorzi J, Jeter SC, Oliver GR, Fetting J, Emens L, Riley C, Stearns V, Diehl F, Angenendt P, Huang P, Cope L, Argani P, Murphy KM, Bachman KE, Greshock J, Wolff AC, Park BH. Clin Cancer Res. 2012;18:3462–3469. doi: 10.1158/1078-0432.CCR-11-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tabernero J, Lenz HJ, Siena S, Sobrero A, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Yoshino T, Goldberg RM, Sargent DJ, Wagner A, Laurent D, Teufel M, Jeffers M, Grothey A, Van Cutsem E. Lancet Oncol. 2015 doi: 10.1016/S1470-2045(15)00138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Branford S. Blood. 2004;104:2926–2932. doi: 10.1182/blood-2004-03-1134. [DOI] [PubMed] [Google Scholar]
- 61.Spindler KLG, Pallisgaard N, Vogelius I, Jakobsen A. Clin Cancer Res. 2012;18:1177–1185. doi: 10.1158/1078-0432.CCR-11-0564. [DOI] [PubMed] [Google Scholar]
- 62.Pekin D, Skhiri Y, Baret J-C, Le Corre D, Mazutis L, Ben Salem C, Millot F, El Harrak A, Hutchison JB, Larson JW, Link DR, Laurent-Puig P, Griffiths AD, Taly V. Lab Chip. 2011;11:2156. doi: 10.1039/c1lc20128j. [DOI] [PubMed] [Google Scholar]
- 63.Milbury CA, Zhong Q, Lin J, Williams M, Olson J, Link DR, Hutchison B. Biomol Detect Quantif. 2014;1:8–22. doi: 10.1016/j.bdq.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Masoudi-Nejad A, Narimani Z, Hosseinkhan N. Next Generation Sequencing and Sequence Assembly. Vol. 4. Springer New York; New York, NY: 2013. pp. 11–39. [Google Scholar]
- 65.Grada A, Weinbrecht K. J Invest Dermatol. 2013;133:e11. doi: 10.1038/jid.2013.248. [DOI] [PubMed] [Google Scholar]
- 66.Couraud S, Vaca Paniagua F, Villar S, Oliver J, Schuster T, Blanche H, Girard N, Tredaniel J, Guilleminault L, Gervais R, Prim N, Vincent M, Margery J, Larive S, Foucher P, Duvert B, Vallee M, Le Calvez Kelm F, McKay J, Missy P, Morin F, Zalcman G, Olivier M, Souquet P-J, for the BioCAST/IFCT-1002 investigators Clin Cancer Res. 2014;20:4613–4624. doi: 10.1158/1078-0432.CCR-13-3063. [DOI] [PubMed] [Google Scholar]
- 67.Milbury CA, Li J, Makrigiorgos GM. Clin Chem. 2009;55:632–640. doi: 10.1373/clinchem.2008.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parsons BL, Heflich RH. Mutat Res Mutat Res. 1997;387:97–121. doi: 10.1016/s1383-5742(97)00026-4. [DOI] [PubMed] [Google Scholar]
- 69.Gocke CD, Benko FA, Kopreski MS, Evans DB. Ann N Y Acad Sci. 2006;906:31–38. doi: 10.1111/j.1749-6632.2000.tb06587.x. [DOI] [PubMed] [Google Scholar]
- 70.Ward R, Hawkins N, O’Grady R, Sheehan C, O’Connor T, Impey H, Roberts N, Fuery C, Todd A. Am J Pathol. 1998;153:373–379. doi: 10.1016/S0002-9440(10)65581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolff J, Gemmell N. BioTechniques. 2008;44:193–199. doi: 10.2144/000112719. [DOI] [PubMed] [Google Scholar]
- 72.Asano H. Clin Cancer Res. 2006;12:43–48. doi: 10.1158/1078-0432.CCR-05-0934. [DOI] [PubMed] [Google Scholar]
- 73.Oka T, Matsunaga H, Tokunaga K, Mitsunaga S, Juji T, Yamane A. Nucleic Acids Res. 1994;22:1541–1547. doi: 10.1093/nar/22.9.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitano S, Nakayama M, Yamane A, Tsukahara Y, Amano M. Anal Biochem. 2011;408:197–205. doi: 10.1016/j.ab.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 75.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Proc Natl Acad Sci. 2005;102:16368–16373. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Didelot A, Le Corre D, Luscan A, Cazes A, Pallier K, Emile JF, Laurent-Puig P, Blons H. Exp Mol Pathol. 2012;92:275–280. doi: 10.1016/j.yexmp.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 77.Bolton L, Reiman A, Lucas K, Timms J, Cree IA. PLOS ONE. 2015;10:e0115672. doi: 10.1371/journal.pone.0115672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ashida A, Uhara H, Mikoshiba A, Sakaizawa K, Kumagai N, Koga H, Okuyama R. Acta Derm Venereol. 2015 doi: 10.2340/00015555-2194. [DOI] [PubMed] [Google Scholar]
- 79.Dono M, Massucco C, Chiara S, Sonaglio C, Mora M, Truini A, Cerruti G, Zoppoli G, Ballestrero A, Truini M, Ferrarini M, Zupo S. Mol Med. 2012;18:1519–1526. doi: 10.2119/molmed.2012.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Däbritz J, Hänfler J, Preston R, Stieler J, Oettle H. Br J Cancer. 2005 doi: 10.1038/sj.bjc.6602319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Breitenbuecher F, Hoffarth S, Worm K, Cortes-Incio D, Gauler TC, Köhler J, Herold T, Schmid KW, Freitag L, Kasper S, Schuler M. PLoS ONE. 2014;9:e85350. doi: 10.1371/journal.pone.0085350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shinozaki M, O’Day SJ, Kitago M, Amersi F, Kuo C, Kim J, Wang HJ, Hoon DSB. Clin Cancer Res. 2007;13:2068–2074. doi: 10.1158/1078-0432.CCR-06-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Latorre A, Posch C, Garcimartín Y, Ortiz-Urda S, Somoza Á. Chem Commun. 2014;50:3018. doi: 10.1039/c3cc47862a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polak P, Zalevsky Z, Shefi O. Int J Biol Macromol. 2013;59:134–137. doi: 10.1016/j.ijbiomac.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 85.Valentini P, Fiammengo R, Sabella S, Gariboldi M, Maiorano G, Cingolani R, Pompa PP. ACS Nano. 2013;7:5530–5538. doi: 10.1021/nn401757w. [DOI] [PubMed] [Google Scholar]
- 86.Zhang HD, Zhou J, Xu ZR, Song J, Dai J, Fang J, Fang ZL. Lab Chip. 2007;7:1162. doi: 10.1039/b701649b. [DOI] [PubMed] [Google Scholar]
- 87.Wang H, Liu W, Zhang X, Xu X, Kang Z, Li S, Wu Z, Yang Z, Yao B, Guan M. J Chromatogr A. 2015;1410:28–34. doi: 10.1016/j.chroma.2015.07.079. [DOI] [PubMed] [Google Scholar]
- 88.Handal MI, Ugaz VM. Expert Rev Mol Diagn. 2006;6:29–38. doi: 10.1586/14737159.6.1.29. [DOI] [PubMed] [Google Scholar]
- 89.Wei F, Yang J, Wong DTW. Biosens Bioelectron. 2013;44:115–121. doi: 10.1016/j.bios.2012.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Edman CF, Raymond DE, Wu DJ, Tu E, Sosnowski RG, Butler WF, Nerenberg M, Heller MJ. Nucleic Acids Res. 1997;25:4907–4914. doi: 10.1093/nar/25.24.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fixe F, Branz HM, Louro N, Chu V, Prazeres DMF, Conde JP. Nanotechnology. 2005;16:2061–2071. doi: 10.1088/0957-4484/16/10/014. [DOI] [PubMed] [Google Scholar]
- 92.Wei F, Sun B, Liao W, Ouyang J, Sheng Zhao X. Biosens Bioelectron. 2003;18:1149–1155. doi: 10.1016/s0956-5663(02)00249-x. [DOI] [PubMed] [Google Scholar]
- 93.Wei F, Liao W, Xu Z, Yang Y, Wong DT, Ho CM. Small. 2009;5:1784–1790. doi: 10.1002/smll.200900369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wei F, Patel P, Liao W, Chaudhry K, Zhang L, Arellano-Garcia M, Hu S, Elashoff D, Zhou H, Shukla S, Shah F, Ho CM, Wong DT. Clin Cancer Res. 2009;15:4446–4452. doi: 10.1158/1078-0432.CCR-09-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.