

ABSTRACT
A vaccine capable of protecting at-risk persons against infections due to Cryptococcus neoformans and Cryptococcus gattii could reduce the substantial global burden of human cryptococcosis. Vaccine development has been hampered though, by lack of knowledge as to which antigens are immunoprotective and the need for an effective vaccine delivery system. We made alkaline extracts from mutant cryptococcal strains that lacked capsule or chitosan. The extracts were then packaged into glucan particles (GPs), which are purified Saccharomyces cerevisiae cell walls composed primarily of β-1,3-glucans. Subcutaneous vaccination with the GP-based vaccines provided significant protection against subsequent pulmonary infection with highly virulent strains of C. neoformans and C. gattii. The alkaline extract derived from the acapsular strain was analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS), and the most abundant proteins were identified. Separation of the alkaline extract by size exclusion chromatography revealed fractions that conferred protection when loaded in GP-based vaccines. Robust Th1- and Th17-biased CD4+ T cell recall responses were observed in the lungs of vaccinated and infected mice. Thus, our preclinical studies have indicated promising cryptococcal vaccine candidates in alkaline extracts delivered in GPs. Ongoing studies are directed at identifying the individual components of the extracts that confer protection and thus would be promising candidates for a human vaccine.
IMPORTANCE
The encapsulated yeast Cryptococcus neoformans and its closely related sister species, Cryptococcus gattii, are major causes of morbidity and mortality, particularly in immunocompromised persons. This study reports on the preclinical development of vaccines to protect at-risk populations from cryptococcosis. Antigens were extracted from Cryptococcus by treatment with an alkaline solution. The extracted antigens were then packaged into glucan particles, which are hollow yeast cell walls composed mainly of β-glucans. The glucan particle-based vaccines elicited robust T cell immune responses and protected mice from otherwise-lethal challenge with virulent strains of C. neoformans and C. gattii. The technology used for antigen extraction and subsequent loading into the glucan particle delivery system is relatively simple and can be applied to vaccine development against other pathogens.
INTRODUCTION
Virtually all cases of cryptococcosis are caused by Cryptococcus neoformans and the closely related species Cryptococcus gattii (1, 2). C. neoformans and C. gattii are unique among medically important fungi in that their major virulence factor is a capsule composed primarily of glucuronoxylomannan (GXM) (3). C. neoformans has a worldwide distribution, with human exposure mainly occurring following inhalation of airborne organisms. The global burden of cryptococcal meningitis in people with AIDS has been estimated to be about 1 million cases annually, with a 60% mortality rate (4). Other immunosuppressed persons are also at high risk, e.g., about 1 to 5% of solid organ transplant recipients will develop cryptococcosis in their lifetimes (5). C. gattii has historically been found in tropical and subtropical regions, but hypervirulent strains have emerged on Vancouver Island, Canada, and spread to the mainland, including the U.S. Pacific Northwest (2, 6, 7). This habitat expansion has been theorized to be due to climate change, raising concerns about further spread (2, 8).
Despite the prevalence of cryptococcosis, there are no licensed cryptococcal vaccines. Populations that could be targeted for vaccination include the following: (i) HIV-infected persons; (ii) persons on medications which suppress T cells (particularly transplant recipients); (iii) persons living in regions where C. gattii is endemic; and (iv) persons with other high-risk diseases (e.g., sarcoidosis, lymphoma). Preclinical approaches to developing cryptococcal vaccines have included immunization with protein-GXM conjugates and peptide mimetics of GXM designed to elicit antibody-mediated protection, as well as live and killed C. neoformans strains (9–15). Other work has focused on the identification of C. neoformans antigens that stimulate protective T cell responses. Much of this effort has focused on mannoproteins (MPs; defined by their affinity for the mannose-binding lectin concanavalin A), which comprise a major antigenic fraction recognized by T cells of mice and humans (16–18). The extensive mannosylation of MPs promotes uptake by mannose receptors on antigen-presenting cells, which leads to adaptive immune responses (18–21). However, vaccination with MPs admixed via the Ribi adjuvant system (Abcam; which biases toward antibody responses) resulted in only modestly prolonged survival of C. neoformans-challenged mice (22). Moreover, vaccination with the flowthrough fraction (defined as the antigens which did not bind concanavalin A) also elicited modest protection. These data suggest Cryptococcus vaccines designed to elicit protective T cell responses will require novel adjuvant and/or antigen compositions in order to be successful.
Glucan particles (GPs) are highly purified, hollow, porous cell wall shells manufactured by treating baker’s yeast (Saccharomyces cerevisiae) with a series of alkaline, acid, and solvent extractions (23–26). They are composed primarily of β-1,3-glucan and are devoid of proteins, lipids, and mannans. GPs are recognized by the C-type lectin receptor Dectin-1, but also potently activate the alternative pathway of complement (27, 28). In vivo, phagocytosis of GPs is mediated by both complement receptors and Dectin-1 (29). GPs stimulate dendritic cells (DCs) to produce cytokines associated with beneficial responses in vaccine models of protection (27). Importantly, polymer-complexed “payload” classes, including proteins, small interfering RNA, DNA, and other small molecules can be constructed within the hollow GPs, enabling the cellular delivery of a wide variety of “payload” classes (23, 29–31). Immunization of mice with GPs loaded with the model antigen ovalbumin (OVA) results in potent and long-lasting antigen-specific antibody and Th1/Th17-biased CD4+ T cell responses, even when using low doses of ovalbumin (23, 24). Moreover, beneficial effects have been noted in experimental Ascomycota infections, including aspergillosis and coccidioidomycosis, following vaccination of mice with fungal antigens in GPs (32, 33).
In the present study, we tested GPs as a vaccine delivery system in murine models of cryptococcosis. As a source of antigens, the GPs were loaded with soluble extracts obtained following mild alkali treatment of cryptococcal strains. Mice that received extracts encased in GPs mounted antigen-specific CD4+ T cell recall responses and were protected from subsequent lethal challenges with C. neoformans and C. gattii.
RESULTS
Establishment of a C57BL/6 mouse model of cryptococcosis using C. neoformans strains derived from H99 for vaccination and challenge studies.
A robust test of vaccine efficacy should demonstrate protection of genetically susceptible hosts against challenge with fully virulent pathogens. Therefore, when setting up our cryptococcosis vaccine model, we chose to use the C57BL/6 mouse strain. This inbred strain has been extensively studied and has been found to be particularly sensitive to infection by C. neoformans (34, 35). C. neoformans Kn99α (here referred to as Kn99), a hypervirulent α mating-type strain which has been backcrossed 10 times to the well-studied clinical H99 strain (36), was selected for the fungal challenges. To mimic natural infection, mice were inoculated intranasally under conditions promoting pulmonary aspiration. As expected, C57BL/6 mice rapidly succumbed to intranasal challenge doses of 103, 104, or 105 CFU Kn99 (Fig. 1A). Median survival was extended by only a few days as the challenge dose decreased. Even at the lowest challenge dose of 103 CFU, all mice succumbed. For the subsequent vaccination studies, we used 105 CFU of Kn99 as the challenge dose.
FIG 1 .

Protection of C57BL/6 mice by vaccination with live cap59 and cda123 strains. (A) Naive C57BL/6 mice were inoculated intranasally with 103, 104, or 105 CFU of strain Kn99, and their survival was monitored. P < 0.01 comparing animals that received 104 versus 103 or 105 CFU; P < 0.0001 comparing animals that received 103 versus 105 CFU. (B) An intranasal challenge with 105 CFU of Kn99 was used to monitor survival of mice that were vaccinated three times with 106 CFU of live C. neoformans strain cap59 or cda123. Survival curves are from two independent experiments, each with 5 mice per group. Unvaccinated (No Vac) mice served as controls. P < 0.01 and P < 0.0001 comparing No Vac animals versus animals vaccinated with cda123 or cap59, respectively. (C and D) CFU in the lungs and brains of unvaccinated and vaccinated mice 2 weeks postchallenge with 105 CFU of Kn99. Mice with no CFU were assigned a value of 100 CFU, which was the lower limit of detection of the assay. P < 0.05 comparing lung CFU of unvaccinated versus cap59-vaccinated mice.
Acapsular strains of C. neoformans are not only hypovirulent, but also, when used as a live vaccine delivered subcutaneously, they confer some protection against subsequent challenge with fully encapsulated cryptococcal isolates (11). Consistent with these findings, mice that received a subcutaneous vaccination with live acapsular strain cap59 were partially protected from lethal challenge with Kn99 (Fig. 1B). Protection, albeit not as robust, was also observed following vaccination with the live cda1Δ cda2Δ cda3Δ mutant strain (cda123 strain), an attenuated strain which lacks cell wall chitosan (37–39). Strains cap59 and cda123 were derived from wild-type strains H99 and Kn99, respectively (39, 40). Two weeks after fungal challenge, CFU counts in lungs were significantly lower in the mice vaccinated with cap59 (Fig. 1C). In contrast, at this time point few mice had CFU in their brain tissue (Fig. 1D).
Extraction of antigens from strains cap59 and cda123.
The vaccination studies with the live attenuated strains suggested that these strains contained antigens that conferred protection. A mild alkaline extraction technique, described in Materials and Methods, was developed to test whether protective antigens could be recovered from the two mutant fungal strains for use as subunit vaccines. Typically, a purified extract from 500 ml of culture contained about 7.5 mg of protein. The endotoxin concentration ranged from 0.5 to 18 endotoxin units (EU)/mg of protein in four lots of the cap59 extract and 0.02 to 0.05 EU/mg of protein in two extracts of cda123. The alkaline extracts contained numerous protein and carbohydrate bands when resolved by SDS-PAGE (Fig. 2). Despite being derived from the same genetic background, many of the bands were unique to only one of the strains. As expected, the carbohydrate stain periodic acid-Schiff (PAS) demonstrated that the extracts from the cda123 strain, but not the cap59 strain, had a very-high-molecular mass band consistent with the GXM component of the capsule.
FIG 2 .
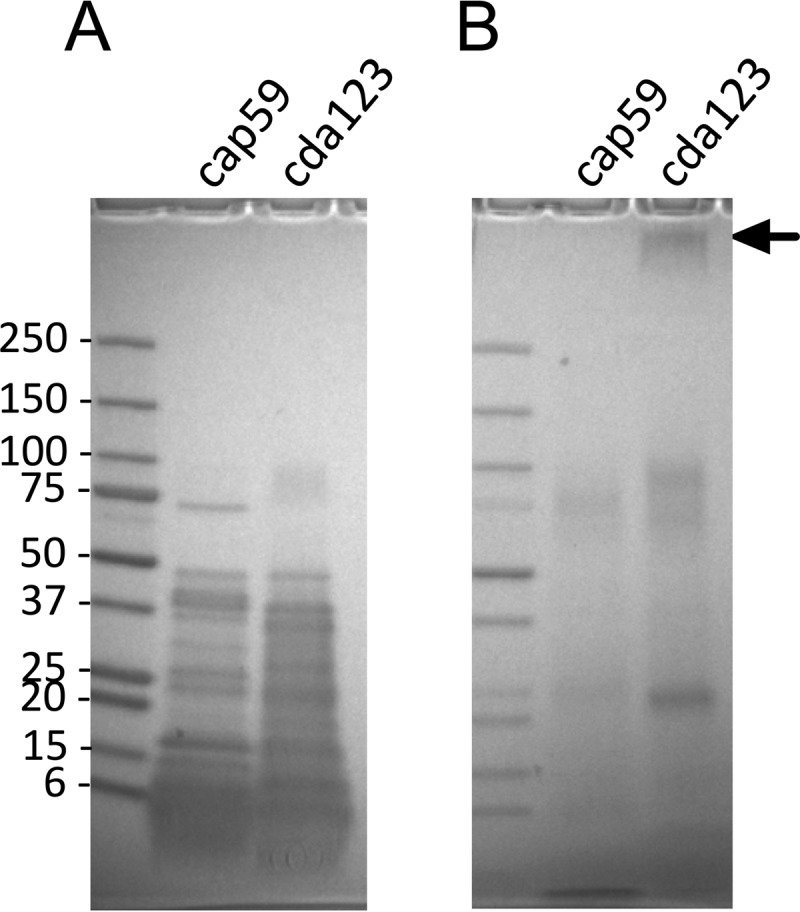
SDS-PAGE results for cap59 and cda123 extracts. Alkaline extracts were prepared from C. neoformans strains cap59 and cda123. Each lane was loaded with 50 µg of protein. The left lanes show molecular mass size markers (in kilodaltons). (A) Gels were stained for protein with Coomassie blue. (B) Replicate lanes from the same gel were stained for carbohydrate with PAS. The arrow identifies the region expected for GXM.
The cap59 extracts were then subjected to LC-MS/MS. Proteins were identified using the C. neoformans var. grubii database and analyzed for their relative abundance. Phosphatidylglycerol transfer protein was the most abundant protein, followed closely by superoxide dismutase. Proteins whose relative abundance was at least 5% that of phosphatidylglycerol transfer protein are listed in Table 1. Importantly, a whole transcriptome shotgun sequencing (RNA-Seq)-based analysis of C. neoformans isolates obtained from the cerebrospinal fluid of two patients with cryptococcal meningitis (41) showed mRNA encoding all 23 proteins. Several had high RNA-Seq read numbers, suggesting that these extracted proteins are likely produced during infection.
TABLE 1 .
LC-MS/MS analysis of protein in C. neoformans cap59 extracts
| CNAG designation | Description | Abundancea | Signal peptideb | No. of RNA-Seq readsc | Homologyd |
|---|---|---|---|---|---|
| CNAG_07442 | Phosphatidylglycerol transfer protein | 100 | Yes | 25,623 | 0.39 |
| CNAG_01019 | Superoxide dismutase [Cu-Zn] | 97 | No | 70,771 | 5E−62 |
| CNAG_06432 | Acetate kinase | 50 | No | 18,367 | None |
| CNAG_00581 | Saccharopepsin | 27 | Yes | 97,106 | 2E−107 |
| CNAG_01230 | Chitin deacetylase 2/ MP98 | 24 | Yes | 5,835 | None |
| CNAG_04625 | Cerevisin | 23 | Yes | 115,412 | 3E−27 |
| CNAG_00919 | Carboxypeptidase D | 19 | Yes | 934 | 6E−19 |
| CNAG_00450 | 3-Isopropylmalate dehydrogenase | 16 | No | 1,129 | 9E−30 |
| CNAG_00734 | Dihydroorotase, homodimeric type | 16 | No | 1,814 | 1.5 |
| CNAG_03232 | Lactamase | 14 | No | 23,885 | 3E−04 |
| CNAG_05097 | YjeF family protein | 13 | No | 8,699 | 1E−72 |
| CNAG_01896 | Alcohol dehydrogenase (NADP) | 11 | No | 87,413 | 2E−71 |
| CNAG_00264 | Nuclease I | 11 | Yes | 51,905 | None |
| CNAG_01348 | Cyanate hydratase | 9 | No | 8,774 | 2.2 |
| CNAG_04981 | Catalase | 7 | No | 101,133 | 2E−102 |
| CNAG_03223 | Hypothetical protein | 6 | Yes | 48,826 | None |
| CNAG_00575 | Catalase | 6 | No | 21,202 | 3E−81 |
| CNAG_01498 | Arylsulfatase | 6 | Yes | 2,938 | 3E−43 |
| CNAG_01137 | Aconitate hydratase | 6 | No | 19,654 | 0 |
| CNAG_04443 | Hypothetical protein | 5 | No | 19,418 | 8E−10 |
| CNAG_04269 | Leucyl aminopeptidase | 5 | Yes | 15,297 | 0.62 |
| CNAG_07745 | Alcohol dehydrogenase, propanol-preferring | 5 | No | 46,734 | 3E−12 |
The relative abundance of protein, averaged for two cap59 extracts. The most abundant protein, phosphatidylglycerol transfer protein, was arbitrarily set at 100.
Determined using SignalP 4.0.
Average numbers of normalized RNA-Seq reads recorded for two strains of Cryptococcus isolated directly from infected human patients (GEO accession number GSE51573) (41).
Homology to the human protein with the highest similarity (BLASTp analysis); the smaller the value, the greater the similarity.
Protection of mice vaccinated with cap59 and cda123 strain extracts in GPs.
Soluble alkaline extracts from cap59 and cda123 were packaged into GPs (here designated GP-cap59 and GP-cda123). Mice were then vaccinated subcutaneously (three times, at 2-week intervals, with 10 µg of protein/dose) and challenged with C. neoformans Kn99. Control mice that were left unvaccinated or were vaccinated with mouse serum albumin in GPs (GP-MSA) all died within 27 days (Fig. 3A). However, the mice that received GP-cap59 or GP-cap123 vaccination had significantly prolonged survival; 60% of the vaccinated mice were still alive when the experiment was terminated at 50 days. CFU counts from lungs of the euthanized survivors were determined (Fig. 3B). While CFU in most mice were below the limit of detection, lungs from two mice had CFU greater than 106.
FIG 3 .

Survival curves following vaccination with GP-cap59 and GP-cda123. (A) Mice were left unvaccinated (No Vac) or vaccinated three times with GP-MSA, GP-cap59, or GP-cda123. Mice were then challenged intranasally with 105 CFU of Kn99. Each experiment had five mice per group. The results with GP-cap59 and GP-cda123 are combined from two independent experiments, one with “No Vac” and one with GP-MSA as the control. P < 0.01 comparing No Vac or GP-MSA with GP-cap59 or GP-cda123. (B) The mice that survived 50 days were euthanized and the CFU of Kn99 in the lungs were measured. (C) Five distinct lots of GP-cap59 vaccine were manufactured and tested as described for panel A. Each experiment had five mice per group. The cumulative results of the five studies are shown. P < 0.0001 comparing GP-MSA with GP-cap59. (D) CFU of Kn99 in lungs of mice surviving 50 days (shown in panel C). The lower limit of detection of the fungal load in the lung was 200 CFU (B) or 20 CFU (D). Samples below detection were assigned 200 CFU (B) or 20 CFU (D). The dotted lines denote the inoculum of the challenge dose.
Chitin deacetylases (CDAs), particularly CDA2, have been shown to be targets of the T cell response (42, 43). Moreover, CDA2 was among the top antigens identified in the cap59 extracts (Table 1). Therefore, to avoid losing potentially protective CDA antigens and to circumvent potential inhibitory effects of GXM (44), we elected to focus on the alkaline extracts from the cap59 strain. To determine the reproducibility of the vaccination system, five distinct lots of GP-cap59 were made and separately tested. Overall, each lot behaved similarly and, taken in aggregate, significant protection compared to GP-MSA controls was seen (Fig. 3C and D). As in Fig. 3B, within the 50-day period following infection with 105 CFU of Kn99, the median fungal load in the lungs of survivors was reduced more than 100-fold (Fig. 3D); in several mice, CFU were below the detection limit.
Ex vivo CD4+ T cell responses following vaccination.
In previous studies, we demonstrated that vaccination of mice with GPs containing the model antigen ovalbumin (GP-OVA) induced potent CD4+ T cell responses, even when relatively low doses of antigen were used as ex vivo stimuli (23, 24). This raised the possibility that ex vivo assays could be used to determine the antigen components of cap59 responsible for T cell stimulation. CD4+ T cells, purified from mice that had been vaccinated with GP-cap59, were stimulated with a range of concentrations of GP-cap59, and lymphoproliferation (LP) was determined. Significant ex vivo responses were seen at concentrations of cap59 as low as 1 ng/well (Fig. 4).
FIG 4 .

Amount of cap59 extract in GPs needed to induce an ex vivo CD4 T cell response. CD4+ T cells were purified from the lymph nodes and spleen of mice that had been vaccinated three times with GP-cap59. The cap59 extract, quantified by protein concentration, was loaded in 10-fold decrements into GPs. GPs in the “0.0” group contained MSA without cap59 extract. The T cells (105/well) were added to wells containing 104 mitomycin C-treated BMDCs and 105 GPs containing the indicated amount of cap59 extract. Lymphoproliferation was assayed by [3H]thymidine incorporation. Data are means ± standard errors of the means of results from three independent experiments, each performed in triplicate. P < 0.01 and P < 0.001 comparing 0.0 versus 1.0 and 10 ng cap59 extract, respectively.
Fractions of cap59 extracts responsible for T cell stimulation and protection.
cap59 extracts were separated by size exclusion chromatography into 1-ml fractions. Each fraction was then assayed for protein and carbohydrate concentrations as well as the capacity to stimulate ex vivo CD4+ T cell lymphoproliferation (Fig. 5A). Lymphoproliferative responses were strongest with the higher-molecular-size fractions. Moreover, lymphoproliferative responses correlated with carbohydrate concentrations to a much greater extent than to protein concentrations.
FIG 5 .

Immunostimulatory and protective fractions of the cap59 extract following size exclusion chromatography. (A) The cap59 extract was separated on a Sephadex G-100 column. One-milliliter fractions were collected and assayed for protein and carbohydrate. A 260-µl portion of each fraction was then loaded into GPs and used as a stimulus of CD4+ T cell LP (as in Fig. 4). The shaded area indicates the pool of fractions 8 to 11 that best protected mice as a GP vaccine. (B) Pools of four sequential fractions were loaded into GPs and used to vaccinate mice. The fraction numbers (F#) of the pools are indicted in shaded column. GPs loaded only with MSA served as the control GP vaccine. Mice (5/group) were vaccinated three times and challenged with 105 CFU of Kn99. P < 0.01 comparing F#8 to 11 with MSA.
We next sought to determine which fractions were responsible for protection in mice. Due to the small amount of antigen in each fraction, we combined fractions into pools of 4 fractions and concentrated them 30-fold. These were then loaded into GPs and used to vaccinate mice. Only five mice per group could be studied, on account of the limited amount of material obtained. Nevertheless, the results were striking, with 4/5 mice in the group containing pooled fractions 8 to 11 surviving (Fig. 5B). In contrast, except for one survivor in the fractions 4 to 7 group, all the other mice succumbed to infection. Interestingly, survival did not directly correlate with ex vivo lymphoproliferation.
Immune recall responses in the pulmonary compartment.
Natural exposure to C. neoformans most commonly occurs following inhalation, and the lungs are a common site of clinical infection with this fungus. We postulated that vaccination with GP-cap59 would stimulate robust lung recall responses following pulmonary challenge with C. neoformans. To test this, four groups of mice were studied: unvaccinated uninfected, vaccinated uninfected, unvaccinated infected, and vaccinated infected. Lungs were harvested 5 days after infection. Readouts were the numbers of lung leukocytes, CD4+ T cells, and CD8+ T cells, as well as cytokine skewing after ex vivo stimulation with GP-cap59. As expected, infection was associated with an influx of leukocytes into the lung, which was more robust in the mice that had been vaccinated (Fig. 6A). CD4+ and CD8+ T cells also increased following infection, although the absolute numbers were similar regardless of whether the mice had been vaccinated (Fig. 6B).
FIG 6 .

Analysis of leukocytes from lungs of vaccinated and/or infected mice. In each of three studies, two mice were vaccinated with GP-cap59 three times at 2-week intervals, and two mice were unvaccinated. Two or more weeks following the third vaccination, a naive and a vaccinated mouse were infected with 105 CFU of KN99 and a naive and a vaccinated mouse were not uninfected. Five days postinfection, the lungs were collected and single-cell suspensions were prepared. (A) The leukocytes at the interface of a 67% and 40% Percoll gradient were collected and counted. Purified leukocytes were stimulated with GP-cap59 ex vivo (at a ratio of 3 GPs per 10 leukocytes) for 24 h, and brefeldin A was added for the last 5 h of the incubation, as described in Materials and Methods. Then, the cells were collected, stained for CD3, CD4, CD8, IFN-γ, IL-4, and IL-17a, and analyzed by polychromatic FACS. (B) The numbers of CD4+ (CD3+ CD4+ CD8−) and CD8+ (CD3+ CD4− CD8+) T cells were calculated by multiplying the percentage of each population times the total leukocyte counts. (C) The numbers of CD4+ T cells producing IFN-γ, IL-4, IL-17a, or IFN-γ/IL-17a were similarly calculated. (D) As described for panel C, the numbers of CD8+ T cells producing cytokines were calculated. Results shown are means ± standard errors of the means of three independently performed experiments with four mice for each experiment. One-way ANOVA with Tukey multiple comparison correction was performed to compare cell numbers among four groups (unvaccinated uninfected, vaccinated uninfected, unvaccinated infected, and vaccinated infected). *, P < 0.05; **, P < 0.01.
Vaccination did profoundly affect the quality of the pulmonary T cell response to infection, as assessed by intracellular cytokine staining for the Th1, Th2, and Th17 markers gamma interferon (IFN-γ), interleukin-4 (IL-4), and IL-17a, respectively. Mice that were vaccinated and infected had a large influx of CD4+ T cells that stained positive for IFN-γ or IL-17a (Fig. 6C). CD4+ T cells that expressed both IFN-γ and IL-17a were also observed; however, there was only a slight increase in cells staining for IL-4. Cytokine staining of CD8+ T cells from the vaccinated and infected mouse lungs revealed a somewhat different pattern (Fig. 6D). First, the total number of CD8+ T cells that stained for any of the three cytokines assayed was considerably lower. Second, the number of CD8+ T cells that stained for IL-4 was not significantly different from the number staining for IFN-γ or IL-17a.
Protection against C. gattii infections.
In the final set of experiments, we examined whether a GP-cap59 vaccine would protect against challenge with C. gattii. An alkaline extract was made from the C. gattii cap59 (acapsular) strain (45) and packaged into GPs. Mice received a prime and 2 boosts of GP-cap59 and then were challenged with 105 wild-type C. gattii strain R265 cells (46). Controls included unvaccinated mice and mice that received GP-MSA. Mice vaccinated with GP-cap59 had significantly prolonged survival compared with the control groups (Fig. 7A). Four vaccinated mice survived the 70-day observation period; however, following euthanasia, we were able to detect C. gattii CFU in each of those mice (Fig. 7B).
FIG 7 .

Protection against challenge with C. gattii provided by the GP-cap59 (C. gattii) vaccine. An alkaline extract from C. gattii strain cap59 was loaded into GPs (GP-cap59 [Cg], containing 10 µg extract protein per vaccine dose). Mice were left unvaccinated (No Vac) or vaccinated three times with GP-MSA or GP-cap59. Mice were then inoculated intranasally with 105 CFU of C. gattii strain R265. (A) Survival curves combined from two independent experiments. The first experiment had two groups of five mice, one of which received GP-cap59 and the other no vaccination. The second experiment had two groups. One group contained nine mice which received GP-cap59. The other group had five mice which received GP-MSA. P < 0.0001 comparing GP-cap59 with either control group. (B) C. gattii CFU in the lung of each mouse (n = 4) that survived 70 days. The dotted line denotes the inoculum of the challenge dose.
DISCUSSION
Vaccination is one of the greatest public health successes, greatly reducing and in some cases eliminating many deadly diseases (47). Development of a vaccine to protect vulnerable people against cryptococcosis is needed, given the prevalence of this disease and its associated morbidity and mortality (2, 4). Here, we provide proof of principle for a vaccination approach that packages fungal antigens in a GP delivery system. Similar protection against a lethal challenge with a C. neoformans strain was observed in mice that received subcutaneous vaccinations of GPs containing alkaline extracts from either of two different mutant C. neoformans strains. Similarly, an alkaline extract derived from a C. gattii strain protected against subsequent lethal challenge with C. gattii. Protection did not appear to be due to the stimulatory effects that β-glucans have on innate and trained immunity (48, 49), as control mice that received immunizations with GPs containing only MSA had 100% mortality.
The cryptococcal strains used to make the alkaline extracts either lacked capsule entirely (cap59) or, in the case of the cda123 strain, the cells were loosely encapsulated (39). The lack of capsule facilitated loading of the cap59 extracts into GPs, as the extracts were less viscous than those from cda123. It is recognized that the cda123 strain is deficient in three proteins that could contribute to protection. One of the three deleted chitin deacetylases, CDA2 (also known as MP98), has been shown to be a major target of the T cell response in mice (42, 43). For these reasons, our focus was on the cap59 extracts.
The protection we observed was with highly virulent cryptococcal strains and use of a mouse strain, C57BL/6, that is highly susceptible to cryptococcosis (35, 50). Nevertheless, while protection was significant, it fell short of 100%, and sterilizing immunity was seen in only some of the survivors. Ongoing efforts are focused on improving vaccine efficacy. This includes identifying which of the many antigens in the alkaline extracts elicit the most robust protection and thus should be considered for inclusion in future subunit vaccines. The mild alkaline extraction technique we used is predicted to remove non-covalently linked cell wall proteins without lysing the cells. However, many of the proteins identified in the extracts do not have signal sequences, suggesting that the extraction procedure selectively recovers proteins located inside the cell membrane or in extracellular vesicles (51). While the most abundant proteins in our extracts are expressed by cryptococci in infected humans (41), the possibility of additional protective antigens not found in the extracts must be entertained. Thus, while alkaline extraction is a relatively simple technique for generating antigens from Cryptococcus, in future candidate vaccines we will also test proteins identified as immunogenic (18, 52–56) and not found in abundance in the alkaline extracts.
The arm(s) of the immune system responsible for GP vaccine-mediated protection remains to be determined. Interestingly, when the alkaline extracts were fractioned by size exclusion chromatography, the fractions that stimulated the greatest CD4+ lymphoproliferative responses were not those that afforded the greatest protection. However, a caveat for interpretation of these results is that the fractions were loaded into GPs based on volume rather than protein content. Interestingly, the protective fractions had a ratio of carbohydrate to protein characteristic of mannoproteins (18).
Mice that were vaccinated with GP-cap59 and then challenged with live C. neoformans responded with a robust recall T cell response in the lung. As was observed when immunizing with GP-OVA (23, 24), the CD4+ T cell response was Th1 and Th17 skewed, as determined by cellular IFN-γ and IL-17a production. In addition, a small but significant fraction of the CD4+ T cells expressed both IFN-γ and IL-17a. In contrast, significant numbers of CD4+ Th2 cells, defined by IL-4 expression, were not observed. In mouse models of cryptococcosis, protective responses are strongly associated with Th1-type cytokines, with a lesser role for Th17-type cytokines (57, 58). Th2-type cytokines are associated with exacerbation of disease.
Compared with the lung recall CD4+ T cell response, the CD8+ T cell response was considerably less robust, and Tc1, Tc2, and Tc17 cells were observed in approximately equal numbers. This does not preclude a role for GP-based cryptococcal vaccines in HIV-infected persons; using a live attenuated Blastomyces sp. strain, the Klein Lab has shown that while CD8+ T cells are only weakly primed in CD4+ T cell-sufficient hosts, robust CD8+ T cell responses occur in CD4-deficient mice (59, 60). Ongoing experiments are directed at determining whether GP-based vaccines can similarly protect mice depleted of CD4+ T cells. In addition, while we hypothesize that T cells are responsible for the protection conferred by our GP-based cryptococcal vaccines, the contribution of antibodies needs to be explored.
The ideal cryptococcal vaccine should protect against both C. neoformans and C. gattii, even in the setting of immunocompromise. However, in regions where C. gattii is endemic in the environment, there may be a role for a vaccine that specifically targets infections with this species. This may turn out to be particularly so if habitat expansion of C. gattii continues (2, 8). While the vast majority of persons who acquire symptomatic infection with C. neoformans are severely immunocompromised prior to exposure, those who acquire C. gattii infections often have no known or only mild immunodeficiency (61). Notably, while alkaline extracts packaged in GPs protected against both cryptococcal species, there was a trend toward enhanced protection against C. neoformans compared with C. gattii, despite preparing species-specific extracts. In a related approach, Chaturvedi et al. made cell wall and cytoplasmic protein preparations from C. gattii (62). Intranasal vaccination with these preparations resulted in a decreased fungal burden and prolonged survival following challenge with C. gattii.
While the full translational potential of GP-based vaccines can only be addressed with clinical studies, related β-glucan-containing preparations have been tested in humans. Notably, β-glucan preparations derived from fungi have a record of safety in both preclinical and human trials (63–65). Moreover, heat-killed S. cerevisiae genetically engineered to express antigens has undergone clinical trials in immunotherapeutic vaccines (66, 67). In phase I trials, no major toxicities, clinically significant laboratory abnormalities, or serious adverse events were noted, even at the highest doses tested (66). Importantly, in both human and animal studies, strong antigen-specific helper and cytotoxic T lymphocyte responses were elicited (66, 68–70). Due to their greater purity, including a lack of endogenous baker’s yeast proteins, we predict GPs will be even safer than heat-killed S. cerevisiae.
MATERIALS AND METHODS
Chemicals and cell culture media.
Chemical reagents were purchased from Thermo, Fisher Scientific (Pittsburg, PA), unless stated otherwise. RPMI 1640 medium was obtained from Invitrogen Life Technologies (Carlsbad, CA). R10 medium contained RPMI 1640 with 10% fetal bovine serum (FBS; Tissue Culture BioLogicals, Tulare, CA), 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM l-glutamine (Invitrogen), and 55 µM 2-mercaptoethanol (Invitrogen). Incubations with mouse cells were performed at 37°C in humidified air supplemented with 5% CO2.
Strains of Cryptococcus spp.
Three strains of C. neoformans var. grubii were used in these studies: Kn99 (wild type) (71), cap59 (acapsular) (40), and cda1Δ cda2Δ cda3Δ (cda123; a chitosan-deficient mutant strain) (39). Strains of C. gattii included R265 (wild type) (46) and cap59 (acapsular, derived from C. gattii wild-type strain NIH444) (45). R265 is the major genotype strain from the Vancouver Island outbreak. NIH444 was originally isolated in Seattle and is identical to R265 across all 30 tested loci (72). Each strain was maintained as a glycerol stock at −80°C and was initially cultured on YPD agar (yeast extract [Difco], Bacto peptone [Becton, Dickinson, Sparks, MD], and dextrose [Sigma, St. Louis, MO]), which served as inoculum for liquid cultures of YPD or YNB medium (yeast nitrogen base [Becton, Dickinson]). To prepare Cryptococcus for in vivo challenge studies, strains Kn99 and R265 were cultured in liquid YPD for 18 h at 30°C with shaking. Yeast cells were then harvested by centrifugation, washed once with phosphate-buffered saline (PBS), and finally suspended in PBS.
Alkaline extraction of cryptococcal cell wall antigens.
Strains of C. neoformans were cultured in 500 ml of 2× YNB, 1% glucose from a starting optical density at 600 nm (OD600) of 0.1. Flasks were shaken for 2 to 3 days at 30°C, followed by 1 day at 37°C. Yeast cells, typically having reached an OD600 of 9.0 to 11.0 in that time, were collected by centrifugation at 1,600 × g, 22°C, for 5 min. Cells were suspended in PBS, split between two 50-ml tubes, and then centrifuged again. The yield of 500 ml of culture was about 5 to 6 ml of packed cells in each of the 50-ml tubes; PBS-washed cell pellets were stored at −20°C. For alkaline extraction, the cell pellets were thawed at 22°C. Cells were rapidly suspended in 1.5 volumes of 0.1 M potassium hydroxide (KOH) and rotated for 10 min at 22°C, then removed by centrifugation, as described above. For each milliliter of supernatant, 25 µl 1 M sodium acetate buffer, pH 4.5, was added to neutralize the KOH, followed by mixing with 1.5 volumes of isopropanol and storage at −20°C overnight. The precipitate was collected following centrifugation at 6,000 × g for 10 min at 4°C, suspended in 15 ml 0.1 M ammonium acetate, then reprecipitated with 2 volumes of ethanol, and kept overnight at −20°C. Centrifugation was repeated, and the precipitate was dried by lyophilization. It was then suspended in 7.5 ml 0.01 M ammonium acetate and clarified by centrifugation at 6,000 × g for 10 min at 4°C. The supernatant containing soluble alkaline extracted antigens was saved and stored at −80°C. The protein concentration was measured in a bicinchoninic acid (BCA) assay, and carbohydrate was determined by the phenol-sulfuric acid method of Dubois (73). Endotoxin levels of the extracts were measured using the Limulus amebocyte lysate (LAL) pyrochrome reagent in Glucashield buffer according to the manufacturer’s instructions (Associates of Cape Cod, Falmouth, MA). SDS-PAGE was used to separate antigens in extracts. Gels were stained with either Coomassie stain (RapidStain; G-Biosciences, St. Louis, MO) for protein or PAS (Schiff’s reagent; Electron Microscopy Sciences, Hatfield, PA) for carbohydrates.
Analysis of cap59 extracts by electrospray ionization LC-MS/MS.
Samples were processed for analysis by LC-MS/MS as previously described (74). Briefly, for each sample, 50 µg protein was electrophoresed into a 10% Mini-Protean Tris-glycine extended (TGX)-PAGE gel (Bio-Rad Laboratories) in Tris-glycine–SDS buffer, stained with Coomassie, destained with water, and removed as a gel slice. Gel slices were cut into 1- by 1-mm pieces, reduced in a 45 mM solution of 1,4-dithiothreitol (DTT) at 50°C for 30 min, alkylated in 100 mM iodoacetamide for 30 min, and digested with 2 ng/µl of sequencing-grade trypsin (Sigma) in 0.01% ProteaseMAX surfactant (Promega)–50 mM ammonium bicarbonate at 37°C for 18 h. The supernatant of each sample was then placed in a 0.5-ml microcentrifuge tube. The gel slices were further extracted with 80:20 acetonitrile–1% formic acid, combined with the supernatants of each sample, and completely dried in a SpeedVac.
Tryptic peptide digests were reconstituted in 25 µl 5% acetonitrile containing 0.1% (vol/vol) trifluoroacetic acid and separated using a NanoAcquity ultraperfomance liquid chromatography system (Waters Corporation, Milford, MA). In brief, a 3.0-µl injection was loaded in 5% acetonitrile containing 0.1% formic acid at 4.0 µl/min for 4.0 min onto a 100-µm-inner-diameter (i.d.) fused-silica precolumn packed with 2 cm of 5-µm (200-Å) Magic C18AQ resin (Bruker-Michrom, Auburn, CA) and eluted using a gradient at 300 nl/min onto a 75-µm-i.d. analytical column packed with 25 cm of 3-µm (100-Å) Magic C18AQ particles to a gravity-pulled tip. The solvents were A (water–0.1% formic acid) and B (acetonitrile–0.1% formic acid). A linear gradient was developed from 5% solvent A to 35% solvent B in 60 min. Ions were introduced by positive electrospray ionization via liquid junction into a Q Exactive hybrid mass spectrometer (Thermo Scientific). Mass spectra were acquired over m/z 300 to 1750 at 70,000 resolution (m/z 200), and data-dependent acquisition selected the 10 most abundant precursor ions for tandem mass spectrometry by higher-energy collisional dissociation fragmentation using an isolation width of 1.6 Da, collision energy of 27, and a resolution of 17,500.
Raw data files were peak processed with Proteome Discoverer (version 1.3; Thermo Scientific) or Mascot Distiller (version 2.4; Matrix Science, Inc., Boston, MA) prior to database searching with the Mascot server (version 2.4) against the C. neoformans var. grubii database (National Center for Biotechnology Information). Search parameters included both trypsin specificity with two missed cleavages and no enzymatic specificity. The variable modifications of oxidized methionine, pyroglutamic acid for N-terminal glutamine, deamidation of aspargine and glutamine, N-terminal acetylation of the protein, and a fixed modification for carbamidomethyl cysteine were considered. The mass tolerances were 10 ppm for the precursor and 0.05 Da for the fragments. Search results were then loaded into the Scaffold viewer (Proteome Software, Inc., Portland, OR) for peptide/protein validation. MS data were analyzed with Mascot software using two search criteria: a conventional search based on tryptic digestion sites and an expanded search in which no digestion enzyme was specified. Proteins were identified with Scaffold software (Proteome Software, Inc.), and probabilities were assigned by using the Protein Prophet algorithm; identifications were accepted if they could be established at >95% probability for at least two peptides. The Mascot Distiller average quantitation method was used, in which the precursor intensities of the three most abundant peptides are used to compute a protein’s relative abundance in a sample.
GP vaccines.
Saccharomyces cerevisiae cells were converted into GPs following a series of hot alkali, organic, and aqueous extraction steps, as described previously (23, 24, 27, 31). The final product consisted of a highly purified 3- to 4-μm-diameter yeast cell wall preparation devoid of cytoplasmic content and bounded by a porous, insoluble shell of β-glucans. The final GP vaccine consisted of Cryptococcus alkaline extract, mouse serum albumin (Equitech-Bio, Kerrville, TX), and yeast RNA (yRNA; Sigma) complexed within the glucan shells. Samples to be loaded into GPs were concentrated by lyophilization and dissolved in water at 5 to 10 mg/ml by protein content. Antigens were loaded into GPs and complexed with MSA and yRNA as described elsewhere (23, 24, 27). Vaccines were diluted in sterile 0.9% saline for injection to deliver the indicated amount (ranging from 0.01 to 10 µg) of antigen in 200-µg GPs (approximately 108 particles) per 0.1-ml dose. A control vaccine consisted of GPs loaded with MSA and yRNA but without cryptococcal alkaline extract. Vaccines were stored in 0.6-ml aliquots at −80°C and briefly vortexed prior to use. Vaccine formulations were quality controlled by manual counting of the number of GPs per milliliter by using a hematocytometer and microscopically assessing for intact GPs with a phase-distinct protein-yRNA complex. Vaccines were also extracted and analyzed by 10% SDS-PAGE to quantify antigen/MSA loading.
GP antigen fraction vaccines.
Cryptococcus extracts were size fractionated by Sephadex G-100 gel filtration. One milligram (based on protein content) in 0.5 ml of 0.1 M ammonium acetate, 20 mM Tris-HCl, pH 7.4, was separated on a 1.5- by 7.0-cm column equilibrated with the same buffer as the sample. One-milliliter fractions were collected and analyzed for protein and carbohydrate. A 260-µl portion from each fraction was lyophilized and suspended in 26 µl water, and then 5-µl aliquots of each 10× concentrated fraction were loaded into 1-mg GPs and complexed with MSA and yRNA, as described above. The loaded GPs were each suspended in 0.5 ml 0.9% saline and frozen for later use as stimuli for LP assays. The fractions were also tested in GP-based vaccines. Pools of four sequential fractions containing 0.6 ml of one fraction were lyophilized, suspended in 20 µl of water, and loaded into 4-mg GPs, as described above. Vaccines were diluted to 2 mg/ml in sterile 0.9% saline for injection. Aliquots containing 0.6 ml of vaccine were frozen until use in vaccination studies.
Mice.
Six-week-old female C57BL/6 mice were obtained from either Charles River Laboratories (Kingston, NY) or The Jackson Laboratory (Bar Harbor, ME). Mice were housed in a pathogen-free environment in the animal facility at the University of Massachusetts Medical School, All animal procedures were carried out under a protocol approved by the University of Massachusetts Medical School Institutional Use and Care of Animals Committee.
Vaccination and challenge studies.
The GP-based vaccines and the live Cryptococcus vaccines (0.1 ml dose) were administered three times at 2-week intervals as a subcutaneous injection at the midline of the abdomen, as described elsewhere (23, 24). Two weeks after the third vaccination, the mice were challenged with C. neoformans strain Kn99 or C. gattii strain R265. Mice were anesthetized with 2% isoflurane (Piramal Health Care, Andrah Pradesh, India) in a laboratory animal anesthesia system (VetEquip, Livermore, CA) and inoculated intranasally with 50 µl of fungal suspension, so as to deliver 103, 104, or 105 CFU of Cryptococcus. For survival studies, mice were monitored twice daily and euthanized if they developed advanced signs of disease, including ataxia, listlessness, and failure to groom. At the termination of the study, surviving mice were euthanized, and lungs were removed and homogenized in 4 ml PBS containing penicillin and streptomycin. Undiluted and diluted homogenates were plated on Sabouraud dextrose agar, Emmons, and incubated at 30°C for 2 to 3 days, at which time CFU of Cryptococcus were enumerated. For other studies, mice were sacrificed 2 weeks after fungal challenge, at which time lung and brain CFU were counted.
Lymphoproliferation.
An LP assay was performed essentially as described elsewhere (23, 24). Briefly, 2 or more weeks after the third vaccination, mice were euthanized and spleens and inguinal lymph nodes were collected. Single-cell suspensions were prepared, and CD4+ T cells were isolated over magnetic bead columns by negative selection according to the manufacturer’s protocol (Miltenyi Biotec, San Diego, CA). Purified CD4+ T cells (105/well) were incubated with the indicated stimuli in round-bottom 96-well plates containing 200 µl R10 medium. Mitomycin C-treated murine bone marrow-derived dendritic cells (BMDCs; 104/well, generated as described elsewhere [23]) served as antigen-presenting cells. After a 3-day incubation, [3H]thymidine (1 µCi/well; PerkinElmer, Boston, MA) was added, and the plates were incubated for an additional 24 h. Cells were then collected on filter paper (Wallac, Turku, Finland) by using a harvester (Brandel, Gaithersburg, MD), and [3H]thymidine incorporation was measured with a beta counter (1450 MicroBeta; Wallac). Each condition was analyzed in triplicate.
Polychromatic fluorescence-activated flow cytometry.
Fluorescence-activated cell sorting (FACS) flow cytometry was performed as previously described with slight modifications (24). Briefly, single-cell suspensions were prepared from mouse lungs by using a lung dissociation kit (Miltenyi Biotec) according to the manufacturer’s protocol. The cell pellets were resuspended in 40% Percoll in PBS, layered on top of 67% Percoll in RPMI, and centrifuged at 600 × g for 20 min. The lung leukocytes at the interface were collected and washed twice with PBS. Cells were then counted on a hemocytometer and incubated with the indicated stimuli for 24 to 28 h. Brefeldin A (10 µg/ml) was added for the last 5 h of incubation. Following incubation with Fc-blocking antibodies (BD Biosciences, San Jose, CA), the cells were stained for surface antigens (CD3 conjugated to fluorescein isothiocyanate, CD4 conjugated to V500), and CD8 conjufated to V450) and intracellular cytokines IFN-γ (Alexa 700 conjugate), IL-4 (allophycocyanin conjugate), and IL-17A (phycoerythin conjugate) after fixation and permeabilization using the Cytofix/Cytoperm fixation/permeabilization solution kit (BD Biosciences). Single-color controls for CD3, CD4, CD8, IFN-γ, IL-4, and IL-17A were prepared using OneComp ebeads according to the manufacturer’s protocol (eBiosciences). FACS data were collected using an LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR). Briefly, singlets were selected while debris and SSChi cells were removed. Then, the CD4+ CD8− and CD4− CD8+ cell populations were selected from the CD3+ population. Finally, the expression levels of IFN-γ, IL-4, and IL-17A on the CD3+ CD4+ CD8− and CD3+ CD4− CD8+ gated populations were examined.
Statistics.
Data were analyzed using GraphPad Prism, version 6.0 (GraphPad Software, La Jolla, CA). Kaplan-Meier survival curves were analyzed for significance using the log rank test. For the ex vivo experiments, comparisons were made using a one-way analysis of variance (ANOVA) with the Tukey multiple correction.
ACKNOWLEDGMENTS
We thank Lorina Baker, Joseph Heitman, Vishnu Chaturvedi, and Sudha Chaturvedi for generously providing strains used in this study.
This work was supported by Public Health Service grants AI025780 (to S.M.L.), AI102618 (to S.M.L.), AI072196 (to J.K.L. and C.A.S.), and HL112671 (to S.M.L.) from the National Institute of Allergy and Infectious Diseases and the National Heart Lung and Blood Institute.
The funding agencies had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Specht CA, Lee CK, Huang H, Tipper DJ, Shen ZT, Lodge JK, Leszyk J, Ostroff GR, Levitz SM. 2015. Protection against experimental cryptococcosis following vaccination with glucan particles containing Cryptococcus alkaline extracts. mBio 6(6):e01905-15. doi:10.1128/mBio.01905-15.
REFERENCES
- 1.Heitman J, Kozel TR, Kwon-Chung KJ, Perfect JR, Casadevall A (ed). 2011. Cryptococcus: from human pathogen to model yeast. ASM Press, Washington, DC. [Google Scholar]
- 2.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv113. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 3.Jesus M, Nicola A, Chow S, Lee IR, Nong S, Specht CA, Levitz SM, Casadevall A. 2010. Glucuronoxylomannan, galactoxylomannan, and mannoprotein occupy spatially separate and discrete regions in the capsule of Cryptococcus neoformans. Virulence 1:500–508. doi: 10.4161/viru.1.6.13451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 5.Singh N, Dromer F, Perfect J, Lortholary O. 2008. Cryptococcosis in solid organ transplant recipients: current state of the science. Clin Infect Dis 47:1321–1327. doi: 10.1086/592690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes EJ III, Li W, Lewit Y, Ma H, Voelz K, Ren P, Carter DA, Chaturvedi V, Bildfell RJ, May RC, Heitman J. 2010. Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog 6:e1000850. doi: 10.1371/journal.ppat.1000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrnes EJ III, Li W, Ren P, Lewit Y, Voelz K, Fraser JA, Dietrich FS, May RC, Chatuverdi S, Chatuverdi V, Heitman J. 2011. A diverse population of Cryptococcus gattii molecular type VGIII in southern Californian HIV/AIDS patients. PLoS Pathog 7:e1002205. doi: 10.1371/journal.ppat.1002205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benedict K, Park BJ. 2014. Invasive fungal infections after natural disasters. Emerg Infect Dis 20:349–355. doi: 10.3201/eid2003.131230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devi SJN, Schneerson R, Egan W, Ulrich TJ, Bryla D, Robbins JB, Bennett JE. 1991. Cryptococcus neoformans serotype A glucuronoxylomannan-protein conjugate vaccines: synthesis, characterization, and immunogenicity. Infect Immun 59:3700–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleuridor R, Lees A, Pirofski L. 2001. A cryptococcal capsular polysaccharide mimotope prolongs the survival of mice with Cryptococcus neoformans infection. J Immunol 166:1087–1096. doi: 10.4049/jimmunol.166.2.1087. [DOI] [PubMed] [Google Scholar]
- 11.Fromtling RA, Kaplan AM, Shadomy HJ. 1983. Immunization of mice with stable, acapsular, yeast-like mutants of Cryptococcus neoformans. Sabouraudia 21:113–119. doi: 10.1080/00362178385380181. [DOI] [PubMed] [Google Scholar]
- 12.Anderson DM, Dykstra MA. 1984. Resistance to challenge and macrophage activity in mice previously vaccinated with formalin-killed Cryptococcus neoformans. Mycopathologia 86:169–177. doi: 10.1007/BF00441128. [DOI] [PubMed] [Google Scholar]
- 13.Wormley FL Jr, Perfect JR, Steele C, Cox GM. 2007. Protection against cryptococcosis using a murine interferon-gamma producing Cryptococcus neoformans strain. Infect Immun 75:1453–1462. doi: 10.1128/IAI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louria DB, Kaminski T, Finkel G. 1963. Further studies on immunity in experimental cryptococcosis. J Exp Med 117:509–520. doi: 10.1084/jem.117.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leopold Wager CM, Wormley FL Jr. 2015. Is development of a vaccine against Cryptococcus neoformans feasible? PLoS Pathog 11:e1004843. doi: 10.1371/journal.ppat.1004843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy JW, Mosley RL, Cherniak R, Reyes GH, Kozel TR, Reiss E. 1988. Serological, electrophoretic, and biological properties of Cryptococcus neoformans antigens. Infect Immun 56:424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levitz SM, North EA. 1997. Lymphoproliferation and cytokines profiles in human peripheral blood mononuclear cells stimulated by Cryptococcus neoformans. J Med Vet Mycol 35:229–236. doi: 10.1080/02681219780001201. [DOI] [PubMed] [Google Scholar]
- 18.Levitz SM, Specht CA. 2006. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res 6:513–524. doi: 10.1111/j.1567-1364.2006.00071.x. [DOI] [PubMed] [Google Scholar]
- 19.Mansour MK, Latz E, Levitz SM. 2006. Cryptococcus neoformans glycoantigens are captured by multiple lectin receptors and presented by dendritic cells. J Immunol 176:3053–3061. doi: 10.4049/jimmunol.176.5.3053. [DOI] [PubMed] [Google Scholar]
- 20.Mansour MK, Schlesinger LS, Levitz SM. 2002. Optimal T-cell responses to Cryptococcus neoformans mannoprotein are dependent on recognition of conjugated carbohydrates by mannose receptors. J Immunol 168:2872–2879. doi: 10.4049/jimmunol.168.6.2872. [DOI] [PubMed] [Google Scholar]
- 21.Specht C, Nong S, Dan J, Lee C, Levitz S. 2007. Contribution of glycosylation to T cell responses stimulated by recombinant Cryptococcus neoformans mannoprotein. J Infect Dis 196:796–800. doi: 10.1086/520536. [DOI] [PubMed] [Google Scholar]
- 22.Mansour MK, Yauch LE, Rottman JB, Levitz SM. 2004. Protective efficacy of antigenic fractions in mouse models of cryptococcosis. Infect Immun 72:1746–1754. doi: 10.1128/IAI.72.3.1746-1754.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H, Ostroff GR, Lee CK, Specht CA, Levitz SM. 2010. Robust stimulation of humoral and cellular immune responses following vaccination with antigen-loaded beta-glucan particles. mBio 1:e00164-10. doi: 10.1128/mBio.00164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Ostroff GR, Lee CK, Specht CA, Levitz SM. 2013. Characterization and optimization of the glucan particle-based vaccine platform. Clin Vaccine Immunol 20:1585–1591. doi: 10.1128/CVI.00463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levitz SM, Huang H, Ostroff GR, Specht CA. 2015. Exploiting fungal cell wall components in vaccines. Semin Immunopathol 37:199–207. doi: 10.1007/s00281-014-0460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soto ER, Caras AC, Kut LC, Castle MK, Ostroff GR. 2012. Glucan particles for macrophage targeted delivery of nanoparticles. J Drug Deliv 2012:143524. doi: 10.1155/2012/143524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H, Ostroff GR, Lee CK, Wang JP, Specht CA, Levitz SM. 2009. Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and Toll-like receptor agonists. Infect Immun 77:1774–1781. doi: 10.1128/IAI.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agarwal S, Specht CA, Haibin H, Ostroff GR, Ram S, Rice PA, Levitz SM. 2011. Linkage specificity and role of properdin in activation of the alternative complement pathway by fungal glycans. mBio 2:e00178-11. doi: 10.1128/mBio.00178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H, Ostroff GR, Lee CK, Agarwal S, Ram S, Rice PA, Specht CA, Levitz SM. 2012. Relative contributions of dectin-1 and complement to immune responses to particulate beta-glucans. J Immunol 189:312–317. doi: 10.4049/jimmunol.1200603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, Czech MP. 2009. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 458:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soto ER, Ostroff GR. 2008. Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery. Bioconjug Chem 19:840–848. doi: 10.1021/bc700329p. [DOI] [PubMed] [Google Scholar]
- 32.Hurtgen BJ, Hung C-, Ostroff GR, Levitz SM, Cole GT. 2012. Construction and evaluation of a novel recombinant T cell epitope-based vaccine against coccidioidomycosis. Infect Immun 80:3960–3974. doi: 10.1128/IAI.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wüthrich M, Brandhorst T, Sullivan T, Filutowicz H, Sterkel A, Stewart D, Li M, Lerksuthirat T, LeBert V, Shen Z, Ostroff G, Deepe G Jr., Hung C, Cole G, Walter J, Jenkins M, Klein B. 2015. Calnexin induces expansion of antigen-specific CD4+ T cells that confer immunity to fungal ascomycetes via conserved epitopes. Cell Host Microbe 17:452–465. doi: 10.1016/j.chom.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shourian M, Flaczyk A, Angers I, Mindt BC, Fritz JH, Qureshi ST. 2015. The Cnes2 locus on mouse chromosome 17 regulates host defense against cryptococcal infection through pleiotropic effects on host immunity. Infect Immun 83:4541–4554. doi: 10.1128/IAI.00697-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huffnagle GB, Boyd MB, Street NE, Lipscomb MF. 1998. IL-5 is required for eosinophil recruitment, crystal deposition, and mononuclear cell recruitment during a pulmonary Cryptococcus neoformans infection in genetically susceptible mice (C57BL/6). J Immunol 160:2393–2400. [PubMed] [Google Scholar]
- 36.Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect Immun 71:4831–4841. doi: 10.1128/IAI.71.9.4831-4841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker LG, Specht CA, Lodge JK. 2011. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot Cell 10:1264–1268. doi: 10.1128/EC.05138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banks IR, Specht CA, Donlin MJ, Gerik KJ, Levitz SM, Lodge JK. 2005. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell 4:1902–1912. doi: 10.1128/EC.4.11.1902-1912.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker LG, Specht CA, Donlin MJ, Lodge JK. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 6:855–867. doi: 10.1128/EC.00399-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang RJ, Breger J, Idnurm A, Gerik KJ, Lodge JK, Heitman J, Calderwood SB, Mylonakis E. 2005. Cryptococcus neoformans gene involved in mammalian pathogenesis identified by a Caenorhabditis elegans progeny-based approach. Infect Immun 73:8219–8225. doi: 10.1128/IAI.73.12.8219-8225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Toffaletti DL, Tenor JL, Litvintseva AP, Fang C, Mitchell TG, McDonald TR, Nielsen K, Boulware DR, Bicanic T, Perfect JR. 2014. The Cryptococcus neoformans transcriptome at the site of human meningitis. mBio 5:e01087-13. doi: 10.1128/mBio.01087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levitz SM, Nong S, Mansour MK, Huang C, Specht CA. 2001. Molecular characterization of a mannoprotein with homology to chitin deacetylases that stimulates T cell responses to Cryptococcus neoformans. Proc Natl Acad Sci U S A 98:10422–10427. doi: 10.1073/pnas.181331398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiesner DL, Specht CA, Lee CK, Smith KD, Mukaremera L, Lee ST, Lee CG, Elias JA, Nielsen JN, Boulware DR, Bohjanen PR, Jenkins MK, Levitz SM, Nielsen K. 2015. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. PLoS Pathog 11:e1004701. doi: 10.1371/journal.ppat.1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yauch LE, Lam JS, Levitz SM. 2006. Direct inhibition of T-cell responses by the Cryptococcus capsular polysaccharide glucuronoxylomannan. PLoS Pathog 2:e120. doi: 10.1371/journal.ppat.0020120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Springer DJ, Ren P, Raina R, Dong Y, Behr MJ, McEwen BF, Bowser SS, Samsonoff WA, Chaturvedi S, Chaturvedi V. 2010. Extracellular fibrils of pathogenic yeast Cryptococcus gattii are important for ecological niche, murine virulence and human neutrophil interactions. PLoS One 5:e10978. doi: 10.1371/journal.pone.0010978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kidd SE, Hagen F, Tscharke RL, Huynh M, Bartlett KH, Fyfe M, Macdougall L, Boekhout T, Kwon-Chung KJ, Meyer W. 2004. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc Natl Acad Sci U S A 101:17258–17263. doi: 10.1073/pnas.0402981101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levitz S, Golenbock D. 2012. Beyond empiricism: informing vaccine development through innate immunity research. Cell 148:1284–1292. doi: 10.1016/j.cell.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JHA, Rao NA, Aghajanirefah A, Manjeri GR, Li Y, Ifrim DC, Arts RJW, van der Veer BMJW, Deen PMT, Logie C, O’Neill LA, Willems P, van de Veerdonk FL, van der Meer JW, Ng A, Joosten LA, Wijmenga C, Stunnenberg HG, Xavier RJ, Netea MG. 2014. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torello C, Souza-Queiroz J, Queiroz M. 2012. β-1,3-Glucan given orally modulates immunomyelopoietic activity and enhances the resistance of tumour-bearing mice. Clin Exp Pharmacol Physiol 39:209–217. doi: 10.1111/j.1440-1681.2011.05655.x. [DOI] [PubMed] [Google Scholar]
- 50.Hoag KA, Street NE, Huffnagle GB, Lipscomb MF. 1995. Early cytokine production in pulmonary Cryptococcus neoformans infections distinguishes susceptible and resistant mice. Am J Respir Cell Mol Biol 13:487–495. doi: 10.1165/ajrcmb.13.4.7546779. [DOI] [PubMed] [Google Scholar]
- 51.Brown L, Wolf JM, Prados-Rosales R, Casadevall A. 2015. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol 13:620–630. doi: 10.1038/nrmicro3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandel MA, Grace GG, Orsborn KI, Schafer F, Murphy JW, Orbach MJ, Galgiani JN. 2000. The Cryptococcus neoformans gene DHA1 encodes an antigen that elicits a delayed-type hypersensitivity reaction in immune mice. Infect Immun 68:6196–6201. doi: 10.1128/IAI.68.11.6196-6201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biondo C, Beninati C, Bombaci M, Messina L, Mancuso G, Midiri A, Galbo R, Teti G. 2003. Induction of T helper type 1 responses by a polysaccharide deacetylase from Cryptococcus neoformans. Infect Immun 71:5412–5417. doi: 10.1128/IAI.71.9.5412-5417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang C, Nong S-, Mansour MK, Specht CA, Levitz SM. 2002. Purification and characterization of a second immunoreactive mannoprotein from Cryptococcus neoformans that stimulates T-cell responses. Infect Immun 70:5485–5493. doi: 10.1128/IAI.70.10.5485-5493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eigenheer RA, Lee YJ, Blumwald E, Phinney BS, Gelli A. 2007. Extracellular glycosylphosphatidylinositol-anchored mannoproteins and proteases of Cryptococcus neoformans. FEMS Yeast Res 7:499–510. doi: 10.1111/j.1567-1364.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- 56.Chaturvedi AK, Weintraub ST, Lopez-Ribot JL, Wormley FL Jr. 2013. Identification and characterization of Cryptococcus neoformans protein fractions that induce protective immune responses. Proteomics 13:3429–3441. doi: 10.1002/pmic.201300213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wozniak KL, Levitz SM. 2011. T cell and dendritic cell immune responses to Cryptococcus, p 387–396. In Heitman J, Kozel TR, Kwon-Chung JK, Perfect JR, Casadevall A (ed), Cryptococcus: from human pathogen to model yeast. ASM Press, Washington, DC. [Google Scholar]
- 58.Hardison SE, Wozniak KL, Kolls JK, Wormley FL Jr. 2010. Interleukin-17 is not required for classical macrophage activation in a pulmonary mouse model of Cryptococcus neoformans infection. Infect Immun 78:5341–5351. doi: 10.1128/IAI.00845-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wuthrich M, Filutowicz HI, Warner T, Deepe GS Jr, Klein BS. 2003. Vaccine immunity to pathogenic fungi overcomes the requirement for CD4 help in exogenous antigen presentation to CD8+ T cells: implications for vaccine development in immune-deficient hosts. J Exp Med 197:1405–1416. doi: 10.1084/jem.20030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nanjappa SG, Heninger E, Wüthrich M, Gasper DJ, Klein BS. 2012. Tc17 cells mediate vaccine immunity against lethal fungal pneumonia in immune deficient hosts lacking CD4+ T cells. PLoS Pathog 8:e1002771. doi: 10.1371/journal.ppat.1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen S-CA, Meyer W, Sorrell TC. 2014. Cryptococcus gattii infections. Clin Microbiol Rev 27:980–1024. doi: 10.1128/CMR.00126-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaturvedi AK, Hameed RS, Wozniak KL, Hole CR, Leopold Wager CM, Weintraub ST, Lopez-Ribot JL, Wormley FL Jr. 2014. Vaccine-mediated immune responses to experimental pulmonary Cryptococcus gattii infection in mice. PLoS One 9:e104316. doi: 10.1371/journal.pone.0104316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Novak M, Vetvicka V. 2008. Beta-glucans, history, and the present: immunomodulatory aspects and mechanisms of action. J Immunotoxicol 5:47–57. doi: 10.1080/15476910802019045. [DOI] [PubMed] [Google Scholar]
- 64.Weitberg AB. 2008. A phase I/II trial of beta-(1,3)/(1,6)-d-glucan in the treatment of patients with advanced malignancies receiving chemotherapy. J Exp Clin Cancer Res 27:40. doi: 10.1186/1756-9966-27-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams DL, Sherwood ER, Browder IW, McNamee RB, Jones EL, Di Luzio NR. 1988. Pre-clinical safety evaluation of soluble glucan. Int J Immunopharmacol 10:405–414. doi: 10.1016/0192-0561(88)90127-0. [DOI] [PubMed] [Google Scholar]
- 66.Ardiani A, Higgins JP, Hodge JW. 2010. Vaccines based on whole recombinant Saccharomyces cerevisiae cells. FEMS Yeast Res 10:1060–1069. doi: 10.1111/j.1567-1364.2010.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schiff ER, Everson GT, Tsai N. 2007. HCV-specific cellular immunity, RNA reductions, and normalization of ALT in chronic HCV subjects after treatment with GI-5005, a yeast-based immunotherapy targeting NS3 and core: a randomized, double-blind, placebo controlled phase 1b study. Hepatol Int 46(Suppl):816A. [Google Scholar]
- 68.Lu Y, Bellgrau D, Dwyer-Nield LD, Malkinson AM, Duke RC, Rodell TC, Franzusoff A. 2004. Mutation-selective tumor remission with Ras-targeted, whole yeast-based immunotherapy. Cancer Res 64:5084–5088. doi: 10.1158/0008-5472.CAN-04-1487. [DOI] [PubMed] [Google Scholar]
- 69.Stubbs AC, Martin KS, Coeshott C, Skaates SV, Kuritzkes DR, Bellgrau D, Franzusoff A, Duke RC, Wilson CC. 2001. Whole recombinant yeast vaccine activates dendritic cells and elicits protective cell-mediated immunity. Nat Med 7:625–629. doi: 10.1038/87974. [DOI] [PubMed] [Google Scholar]
- 70.Wansley EK, Chakraborty M, Hance KW, Bernstein MB, Boehm AL, Guo Z, Quick D, Franzusoff A, Greiner JW, Schlom J, Hodge JW. 2008. Vaccination with a recombinant Saccharomyces cerevisiae expressing a tumor antigen breaks immune tolerance and elicits therapeutic antitumor responses. Clin Cancer Res 14:4316–4325. doi: 10.1158/1078-0432.CCR-08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect Immun 71:4831–4841. doi: 10.1128/IAI.71.9.4831-4841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fraser JA, Giles SS, Wenink EC, Geunes-Boyer SG, Wright JR, Diezmann S, Allen A, Stajich JE, Dietrich FS, Perfect JR, Heitman J. 2005. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437:1360–1364. doi: 10.1038/nature04220. [DOI] [PubMed] [Google Scholar]
- 73.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. 1956. Colorimetric method for determination of sugars and related substances. Anal Chem 28:350–356. doi: 10.1021/ac60111a017. [DOI] [Google Scholar]
- 74.Smith TC, Fridy PC, Li Y, Basil S, Arjun S, Friesen RM, Leszyk J, Chait BT, Rout MP, Luna EJ. 2013. Supervillin binding to myosin II and synergism with anillin are required for cytokinesis. Mol Biol Cell 24:3603–3619. doi: 10.1091/mbc.E12-10-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]