

Abstract
In Huntington's disease (HD), mutant Huntingtin (mHtt) protein causes striatal neuron dysfunction, synaptic loss, and eventual neurodegeneration. To understand the mechanisms responsible for synaptic loss in HD, we developed a corticostriatal coculture model that features age-dependent dendritic spine loss in striatal medium spiny neurons (MSNs) from YAC128 transgenic HD mice. Age-dependent spine loss was also observed in vivo in YAC128 MSNs. To understand the causes of spine loss in YAC128 MSNs, we performed a series of mechanistic studies. We previously discovered that mHtt protein binds to type 1 inositol (1,4,5)-trisphosphate receptor (InsP3R1) and increases its sensitivity to activation by InsP3. We now report that the resulting increase in steady-state InsP3R1 activity reduces endoplasmic reticulum (ER) Ca2+ levels. Depletion of ER Ca2+ leads to overactivation of the neuronal store-operated Ca2+ entry (nSOC) pathway in YAC128 MSN spines. The synaptic nSOC pathway is controlled by the ER resident protein STIM2. We discovered that STIM2 expression is elevated in aged YAC128 striatal cultures and in YAC128 mouse striatum. Knock-down of InsP3R1 expression by antisense oligonucleotides or knock-down or knock-out of STIM2 resulted in normalization of nSOC and rescue of spine loss in YAC128 MSNs. The selective nSOC inhibitor EVP4593 was identified in our previous studies. We now demonstrate that EVP4593 reduces synaptic nSOC and rescues spine loss in YAC128 MSNs. Intraventricular delivery of EVP4593 in YAC128 mice rescued age-dependent striatal spine loss in vivo. Our results suggest EVP4593 and other inhibitors of the STIM2-dependent nSOC pathway as promising leads for HD therapeutic development.
SIGNIFICANCE STATEMENT In Huntington's disease (HD) mutant Huntingtin (mHtt) causes early corticostriatal synaptic dysfunction and eventual neurodegeneration of medium spine neurons (MSNs) through poorly understood mechanisms. We report here that corticostriatal cocultures prepared from YAC128 HD mice feature age-dependent MSN spine loss, mirroring YAC128 MSN spine loss in vivo. This finding establishes a system for mechanistic studies of synaptic instability in HD. We use it to demonstrate that sensitization of type 1 inositol (1,4,5)-trisphosphate receptors by mHtt, which depletes endoplasmic reticulum calcium, contributes to synaptotoxic enhancement of STIM2-dependent store-operated calcium (SOC) entry. Treatment with EVP4593, a neuroprotective inhibitor of neuronal SOC channels, rescues YAC128 MSN spine loss both in vitro and in vivo. These results suggest that enhanced neuronal SOC causes synaptic loss in HD-afflicted MSNs.
Keywords: calcium, Huntingtin, imaging, synapse, transgenic
Introduction
Huntington's disease (HD) is a dominantly inherited and incurable neurodegenerative disorder involving progressive motor, psychiatric, and cognitive malfunctions, culminating in death 21.4 ± 20 years after diagnosis (Foroud et al., 1999). HD symptoms typically manifest around age 40, with significant temporal variation related to polyglutamine (polyQ) tract length in Huntingtin (Htt) protein, as well as environmental factors and genetic modifiers (Wexler, 2004; Kim, 2014). HD management involves symptomatic treatment and supportive care, but lacks disease-modifying therapeutics (Dayalu and Albin, 2015) due to an insufficient understanding of early pathogenic events. Symptoms manifest after synaptic dysfunction and changes in dendritic spine density in HD patients (Graveland et al., 1985; Ferrante et al., 1991; Sotrel et al., 1993; Paulsen et al., 2008; Miller and Bezprozvanny, 2010; Orth et al., 2010) and animal models of HD (Murphy et al., 2000; Guidetti et al., 2001; Milnerwood and Raymond, 2010). This suggests that HD symptoms may result from synaptic dysfunction rather than overt neurodegeneration; however, the early pathogenic mechanisms underlying synaptic instability are poorly understood.
HD results from expansion of the CAG repeat tract in huntingtin, which encodes mutant Htt (mHtt) with an elongated polyQ region (Huntington's Disease Collaborative Research Group, 1993; Kim, 2014). mHtt is predominantly thought to cause pathology via a toxic gain-of-function (Davies et al., 1997; Tobin and Signer, 2000); however, loss of normal Htt functions, potentially including regulation of synapse formation (McKinstry et al., 2014), may also contribute to HD initiation and progression. Htt is ubiquitously expressed in the brain and body, but striatal medium spiny neurons (MSNs) are particularly susceptible to mHtt (Eidelberg and Surmeier, 2011). The cortex innervates MSNs with glutamatergic inputs and influences striatal connectivity (Miller and Bezprozvanny, 2010; Kozorovitskiy et al., 2012). Cell-specific deletion of mHtt in BACHD mice demonstrated that a full rescue of the HD phenotype requires the absence of mHtt in both cortical and striatal neurons (Wang et al., 2014). Together, this and other evidence suggests preservation of the synaptic connection between cortical neurons and MSNs as a primary therapeutic objective (Miller and Bezprozvanny, 2010; Milnerwood and Raymond, 2010).
Accumulating evidence indicates that dysregulation of intracellular neuronal Ca2+ signaling plays a role in HD progression (Bezprozvanny, 2009; Miller and Bezprozvanny, 2010; Bezprozvanny, 2011). We discovered previously that mHtt directly and specifically binds to the carboxyterminal region of type 1 inositol-1,4,5-trisphosphate receptor (InsP3R1), a neuronal endoplasmic reticulum (ER) Ca2+ release channel (Tang et al., 2003). Association with mHtt sensitizes InsP3R1 to activation by InsP3 (Tang et al., 2003) and promotes glutamate-induced cell death of HD MSNs (Tang et al., 2005; Tang et al., 2009). Similar to mHtt, polyQ-expanded ataxin 3 and ataxin 2 proteins associate with InsP3R1 and sensitize it to InsP3, contributing to neurodegeneration in spinocerebellar ataxias type 2 and type 3 (Chen et al., 2008; Liu et al., 2009; Kasumu and Bezprozvanny, 2012; Kasumu et al., 2012). Ca2+ release from the ER stimulates neuronal store-operated calcium entry (nSOC) channels in the plasma membrane (Putney, 2003). In the previous studies, we discovered that the nSOC pathway is enhanced in HD MSNs (Wu et al., 2011). Moreover, we identified EVP4593 as a specific inhibitor of nSOC entry and found that it exerted neuroprotective effects in a glutamate toxicity assay with HD MSN cultures and normalized motor behavior in a fly model of HD (Wu et al., 2011). In the present study, we investigated the contribution of ER Ca2+ dysregulation to synaptic instability in HD MSNs. Our results suggest that excessive nSOC entry plays an important role in the loss of synaptic spines in YAC128 MSNs and identify the synaptic nSOC pathway as a potential therapeutic target for treatment of HD and potentially other polyglutamine expansion disorders.
Materials and Methods
Dendritic spine analysis of MSNs in primary corticostriatal cocultures.
All procedures involving mice were approved by the Institutional Animal Care and Use Committee of the University of Texas (UT) Southwestern Medical Center at Dallas and followed the National Institutes of Health's Guidelines for the Care and Use of Experimental Animals. WT (FVBN/NJ) and YAC128 transgenic mice [FVB-Tg(YAC128)53Hay/J; Jackson Laboratories: stock # 004938; Slow et al., 2003) were housed in a UT Southwestern Medical Center barrier facility (12 h light/dark cycle) and maintained and genotyped as in Wu et al. (2011).
Corticostriatal cocultures were established from WT and heterozygous YAC128 littermates to study the dendritic spines of MSNs. Briefly, striata and cortices were dissected from postnatal day 0–1 pups (in 1× HBSS, 16.36 mm HEPES, 10 mm NaHCO3, 1× penicillin-streptomycin) digested with papain (30 min at 37°C; Worthington), rinsed (Neurobasal-A medium with 10% FBS), dissociated (in dissection media with 5 mg/ml DNaseI), and plated on poly-d-lysine coated 12 mm coverslips in Neurobasal-A medium supplemented with 5% FBS, 2% B27 and 0.5 mm l-glutamine (Invitrogen) and maintained at 37°C in a 5% CO2 incubator, feeding weekly by addition of 400 μl of NBA, 2% B27, and 0.5 mm l-glutamine. Cortices from one hemisphere and striata from three brains were used to plate 24 wells of a 24-well plate. We estimated that, on average, the resulting plating densities were 350 cells/mm2 for cortical neurons (Ctx) and 1060 cells/mm2 for MSNs, resulting in a 3:1 MSN:Ctx ratio.
For assessment of spine morphology, 14 d in vitro (DIV14) and DIV20 corticostriatal cocultures were fixed for 30 min in 4% formaldehyde plus 4% sucrose in PBS, pH 7.4, and permeabilized for 5 min in 0.25% Triton X-100. Fixed MSNs were blocked with 5% BSA in PBS and immunostained using a rabbit anti-DARPP-32 antibody (1:500; Cell Signaling Technology, 2306s) and a goat anti-rabbit Alexa Fluor 488 secondary antibody (1:1000) or a goat anti-rabbit Alexa Fluor 594 secondary antibody (1:1000). Z-stacks were captured using a 100× oil objective [Plan-Neofluor, 1.3 numerical aperture (NA)] with a confocal miscroscope (LSM510 Meta) with the pinhole set to one airy unit and optical sections every 0.3 μm in Z. Dendritic spine density was quantified using NeuronStudio software in at least three batches of cultures for each experiment.
Dendritic spine analysis in mouse striatum.
To establish a quantitative in vivo analysis of MSN spines, we adopted a procedure developed by Dumitriu et al. (2011). WT and YAC128 mice at the age of 2, 6, and 12 months were perfused intracardially with 5 ml of ice-cold 1% paraformaldehyde (PFA) solution in 0.1 m phosphate buffer (PB), followed by 60 ml of 4% PFA plus 0.0625% glutaraldehyde in 0.1 m PB over 12 min. Brains were removed and postfixed (4% PFA, 0.0625% glutaraldehyde, 0.1 m PB) for 4 h at 4°C. Fixed brains were coronally sliced (300 μm sections) using a vibratome (Leica 1200S) and stored at 4°C in PBS with 0.05% NaN3. Slices were mounted on a patch-clamp setup (Olympus BX51 microscope with infrared camera portal). Glass electrodes for dye injection were pulled (150–300MΩ; Sutter Instruments P97) and striatal neurons were randomly injected and filled with Lucifer yellow solution (L-12926; Invitrogen). A constant negative current in the range of 1–3 nA promoted dye infusion. MSN spines were imaged using a two-photon microscope (Zeiss LSM780) with a 40× lens and a 5× zoom. MSNs were identified by their characteristic shape and abundance of spines. For each neuron, a dendritic segment spaced at least 50 μm from the soma or after the first branching point was considered for analysis. The spine density and the prevalence of different types of spines (thin, stubby, mushroom) were analyzed using NeuronStudio software as described previously (Sun et al., 2014).
Knock-down of InsP3R1 by antisense oligonucleotides (ASOs).
Isis Phamaceuticals generated and provided ASOs targeting InsP3R1 including the following ASOs: 536177-2, 536178-2, 536179-2, 536188-2, 536195-3, 536198-2, 536199-2, 536251-2, 536254-2, 536255-2, 536262-2, and 536263-2. Herein, we refer to ASO ID numbers by the three digits following 536. Cells were incubated with ASOs for 3-6 d to knock down InsP3R1. ASO efficacy (at 500 nm) was evaluated by qPCR (in bEnd.3 cells 3 d after ASO transfection with FuGENE 6; Promega) and Western blotting (in MEF cells after 6 d of naked ASO treatment) as described previously (Zhang et al., 2008b). ASO efficacy (at 200 nm) was also evaluated by Western blotting lysates from cortical neuron cultures (after 6 d of naked ASO treatment) with the rabbit polyclonal anti-InsP3R1 antibody T443 (Tu et al., 2005). To evaluate potential ASO-induced toxicity, mixed WT and YAC128 corticostriatal cultures were incubated with 200 nm of various ASOs starting at DIV14. After 6 d of treatment, the cells were fixed and stained with TMR red (In Situ Cell Death Detection Kit, Roche). Nuclei were counterstained with DAPI. For quantification, six randomly chosen microscopic fields containing 70-150 neurons were counted. The number of TMR red positive neuronal nuclei was calculated as a fraction of DAPI-positive neuronal nuclei in each microscopic field. The fractions of TMR red positive nuclei determined for each microscopic field were averaged and the results are presented as means ± SE.
Western blot analysis.
Striatum regions from WT and YAC128 mice at different ages were dissected, homogenized, and solubilized at 4°C for 1 h in lysis buffer containing 1% CHAPS plus the following (in mm): 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, 1.4 KH2PO4, pH 7.2, 5 EDTA, 5 EGTA, 1 PMSF, 50 NaF, and 1 Na3VO4 plus protease inhibitors. Primary pure MSN cultures of WT and YAC128 were prepared as described previously (Wu et al., 2009). MSNs were collected, homogenized, and solubilized in the lysis buffer on DIV14. The total protein lysates were separated by SDS-PAGE and analyzed by Western blotting with anti-STIM1 pAb (1:500; Cell Signaling Technology, 4961s), anti-STIM2 pAb (1:500; Cell Signaling Technology, 4917s) or anti-STIM2 pAb (1:500; AnaSpec, 54681), anti-TRPC1 mAb (1:1000; Santa Cruz Biotechnology, SC-133076) and anti-tubulin (1:1000; Developmental Studies Hybridoma Bank, E7-c). HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (115-035-146 and 111-035-144) were from Jackson ImmunoResearch. Data were analyzed using ImageJ. The integrated density of each band was normalized to tubulin signal of the same sample.
Lentivirus preparation.
We used polyethylenimine (PEI) to transfect HEK293T cells with a lentiviral expression vector (FUGW; addgene.org/14883/) to drive expression of GFP or Cherry, lenti-Cas9 (addgene.org/52962/) or lenti-Guide (addgene.org/52963/) targeting STIM2, along with the plasmids Δ8.9 and VSVG for lentiviral production and packaging. After 48 h, culture media were collected, centrifuged (2000 RPM for 5 min), filtered (0.45 μm pore size), and stored at −80°C until use. For lentiviral transfection of neurons, 100 μl of lentivirus media was added to each well. Transfection of primary cultures with lenti-GFP, followed by immunostaining for neuronal marker MAP2, revealed selective and efficient (89.9%) transfection of neurons.
Fura-2 Ca2+-imaging experiments.
Fura-2 Ca2+-imaging experiments with DIV14–20 MSNs in corticostriatal cocultures were performed as described previously (Tang et al., 2003; Zhang et al., 2010). Fura-2 340/380 ratio images were collected using a DeltaRAM-X illuminator, Evolve camera, and EasyRatioPro software (Photon Technology International). The entire cell somas were set as the regions of interest for image analysis. In preparation of the coculture, the cortical neurons were plated on DIV0 and infected with lenti-GFP. After 24 h, the medium was replaced with fresh medium and striatal neurons were plated on DIV1. GFP-negative striatal neurons were included in analyses. In DHPG-induced calcium release experiments, neurons were loaded with 5 μm Fura-2-AM (Invitrogen) in artificial CSF (ACSF) containing the following (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, pH 7.3, for 45 min at 37°C. After washing with Ca2+-free ACSF (omitting CaCl2 and supplementing with 100 μm EGTA), the MSNs were incubated in Ca2+-free medium for 1 min before application of 1 μm DHPG. The basal calcium level in Ca2+-free medium was recorded for ∼50 s before the addition of 1 μm 3,5-DHPG (Tocris Bioscience). Images from 340 and 380 nm excitation and 510 nm emissions were captured every 2 s to measure the cytosolic calcium concentration (340F/380F ratio; F = 340F/380F at peak response within 1 min of stimulus application. F0 = 340F/380F at baseline). In ionomycin (IO) experiments, the neurons were washed with Ca2+-free ACSF for 30 s before the addition of 5 μm IO. The size of the ER Ca2+ pool was calculated by integrating the area under the IO-induced Ca2+ response curve as we described previously (Tu et al., 2006).
GCaMP5.3 Ca2+-imaging experiments.
GCaMP5.3 Ca2+-imaging experiments were performed as described previously (Sun et al., 2014). The corticostriatal cocultures were prepared as described in the Fura-2 Ca2+-imaging experiment except that lenti-Cherry was used to infect cortical neurons. The WT and YAC128 neurons were transfected with a GCaMP5.3 expression plasmid using a high calcium-phosphate transfection method (Jiang and Chen, 2006) at DIV7 (CalPhos Transfection Kit; Clontech). MSNs in the coculture were identified by GCaMP5.3 expression, morphology, and the lack of Cherry expression. GCaMP5.3 fluorescent images were collected using an Olympus IX70 inverted epifluorescence microscope equipped with a 60× lens, Cascade 650 digital camera (Roper Scientific) and Prior Lumen 200 illuminator. Images from 488 nm excitation were collected at 0.5 Hz using MetaFluor (Universal Imaging). To measure synaptic nSOC, the neurons were incubated in Ca2+-free medium (supplemented with 400 μm EGTA) containing 1 μm thapsigargin (Tg) for 5 min before returning to the ACSF containing 2 mm Ca2+, 1 μm Tg and a Ca2+ channel inhibitor mixture (1 μm TTX, 50 μm AP5, 10 μm CNQX, and 50 μm nifedipine). The basal calcium level (F0) in Ca2+-free medium was recorded for ∼40 s before the addition of 2 mm Ca2+ (F = the peak response to Ca2+ readdition). Data analysis was performed using ImageJ.
For neuron-type identification in cocultures, we choose to label cortical neurons via lentiviral expression of GFP or Cherry to minimize perturbations to MSN physiology that might complicate experimental results. We determined that, in cortical cultures, 90% of neurons were transfected with lenti-GFP. Based on the transfection efficiency and the relative prevalence of MSNs and cortical neurons (3:1 MSN:Ctx ratio), we estimated that 3% of GFP-negative cells were cortical neurons and 97% were MSN neurons in our experiments. Therefore, the predicted error rate in Ca2+-imaging experiments is no more than 3% even if we ignored morphological traits of each cell type.
Dicer siRNA preparation.
Dicer-generated siRNAs were used to knock down STIM2 in corticostriatal neuronal cultures. The STIM2 siRNA was prepared as described previously (Wu et al., 2011). Full-length mouse STIM2 cDNA was obtained from Open Biosystems (clone ID: 5028346) and used as a PCR template. The T7 promoter sequence was included in the sequence of both forward and reverse primers as follows: FP = GCG TAA TAC GAC TCA CTA TAG GGA GAA GTA GTT TAT GCC GCT CTC, RP = GCG TAA TAC GAC TCA CTA TAG GGA GAT CAC TTA GAC TTC TTC TTG. The product of PCR was purified by NucleoSpin Extract II (Clontech) and dsRNA was obtained by T7 polymerase-mediated transcription for 16 h at 37°C (TurboScript T7 Transcription Kit; Genlantis) followed by DNase I digestion for 15 min at 37°C. dsRNA was purified by lithium chloride precipitation and quantified using NanoDrop 2000c (Thermo Scientific). Four micrograms of dsRNA and four units of recombinant PowerCut Dicer enzyme (Finnzymes) were combined with PowerCut Dicer Reaction Buffer in a total volume of 20 μl. After 16 h at 37°C, dsiRNA was purified on two consecutive RNA purification columns to remove salts and free nucleotides. This yielded a size-homogeneous population of dsiRNA <30 bp without contamination by long dsRNAs. The concentration of dsiRNA was determined with a NanoDrop 2000c. WT and YAC128 corticostriatal cultures were transfected on DIV7 using PEI combined with 200 ng of siRNA-STIM2. The same amount of nontargeting siRNA reagent was used in control transfections (Dicer-GFP). The efficiency of the Dicer siRNA was determined functionally by the GCaMP5.3 nSOC experiment.
CRISPR/Cas9 preparation.
To knock out neuronal STIM2 in corticostriatal cocultures, we used the CRISPR/Cas9 system. guideRNA sequences targeting mouse STIM2 were designed using a bioinformatics tool (crispr.mit.edu) and gSTIM2 plasmids were generated. The sgRNA sequence targeting exon 1 of STIM2 (GTCGGGATCGGCCGGAGCGG) was cloned into the lentiGuide-Puro plasmid (addgene.org/52963/) as described previously (Sanjana et al., 2014; Shalem et al., 2014) following the same protocol (addgene.org/static/data/plasmids/52/52963/52963-attachment_IPB7ZL_hJcbm.pdf). A gLacZ plasmid (GTGCGAATACGCCCACGCGAT) was used as a negative control (Platt et al., 2014).
The lenti-Cas9-Blast plasmid (addgene.org/52962/) was used to express Cas9. To validate these plasmids, MEF cells were cotransfected with Cas9 and gSTIM2 or gLacZ plasmids and the cells transfected with both plasmids were selected using 5 μg/ml blasticidin and 10 μg/ml puromycin. The lysates prepared from MEF cells were analyzed by Western blotting with anti-STIM2 antibodies.
Drug infusion experiments.
Mini-osmotic pumps (Alzet; model 2006) loaded with EVP4593 (0.25 mg/ml) or the vehicle (10% DMSO in PEG300) were implanted in mice with the infusion cannula targeting the ventricles. The surgical procedure followed Alzet's instructions. Briefly, after mice were anesthetized with ketamine/xylazine, the skull was exposed by a midline sagittal incision. A hole was drilled through the skull using the stereotaxic coordinates for the ventricle. The cannula was inserted and secured using dental cement. The pump was implanted subcutaneously in the back. The intraventricular delivery began when mice were 10.5 months old. After 45 d, brains were fixed, sliced, injected with Lucifer yellow to visualize MSN spine morphology, and imaged by two-photon microscopy as described above.
Statistical analyses.
Data are presented as mean ± SE and were analyzed statistically with GraphPad Prism 6 or OriginPro 8 using one-way or two-way ANOVAs followed by a multiple-comparisons test. Bonferroni's post hoc test was used when only comparing WT versus YAC128 for each condition. Tukey's post hoc test was used to make all comparisons. Dunnett's post hoc test was used when comparing against the control condition. The multiplicity adjusted p value is reported (p > 0.05 = n.s., *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Results
Dendritic spine loss in aged YAC128 MSNs in vitro and in vivo
Mixed cortical/striatal cultures are necessary for the formation of functional MSN dendritic spines (Segal et al., 2003). To study synaptic defects in HD MSNs, we set up mixed corticostriatal cultures from WT and YAC128 (YAC) mice. In control electrophysiological experiments, we confirmed that functional synaptic contacts are established between cortical neurons and MSNs in these cultures (Artamonov et al., 2013), in agreement with previously published observations (Segal et al., 2003; Tian et al., 2010; Fishbein and Segal, 2011; Milnerwood et al., 2012). To examine MSN spines, we plated MSNs and infected them on DIV0 with lenti-GFP. On DIV1, culture media was replaced and cortical cells were plated. On DIV20, cultures were fixed and stained for DARPP32, a protein abundantly expressed in striatal neurons. In MSNs expressing GFP at high enough levels, spines could be visualized by both GFP and DARPP32 in confocal imaging experiments (Fig. 1A). Immunostained cultures were imaged by confocal microscopy and the density and shape of synaptic spines was determined by automated analysis (Rodriguez et al., 2008). Both labeling methods revealed a substantial reduction in the density of DIV20 MSN spines from 9.6 ± 0.67 spines/10 μm of dendritic length in WT cocultures to 4.9 ± 0.46 spines/10 μm of dendritic length in YAC128 cocultures (p < 0.001; n = 7 for all conditions; Fig. 1B). Although both methods labeled spines identically (Fig. 1A), DARPP32 staining was brighter, produced less background fluorescence, and enabled the analysis of spines for a greater number of MSNs (the GFP signal was too dim in most MSNs for spine analysis). Therefore, for the remaining experiments, we used DARPP32 staining as a simplified method to analyze MSN spines. To estimate potential error associated with misidentifying MSNs in spine analysis experiments, we examined the density of DARPP32-positive neurons in isolated cortical and striatal cultures. We observed occasional DARPP32-positive neurons in cortical cultures, but the density of these cells was much lower than in striatal cultures. From these control studies, we estimated that, on average, one of six DARPP32-positive cells in the mixed cultures may correspond to a cortical neuron. In addition, cortical neurons had different shape and much weaker DARPP32 staining than striatal neurons, further facilitating the confirmation of MSN identity. MSNs were included in the analysis if they had a small soma size, multiple dendrites originating from the soma, and dendrites that were highly spiny in addition to intense DARPP32 staining.
Figure 1.

Age-dependent dendritic spine loss in YAC128 MSNs. A, WT and YAC128 MSNs were plated on DIV0 and transfected with lenti-GFP. Medium was replaced on DIV1 and cortical cells of the same genotype were plated. On DIV20, the corticostriatal cocultures were immunolabeled for DARPP32. Confocal images of GFP and DARPP32 signals are shown as indicated. A merged image is also shown (GFP, green; DARPP32, red). Scale bar, 5 μm. B. Average MSN spine density was calculated for DIV20 WT and YAC128 MSNs using GFP images and DARPP32 staining images as indicated. An average density is shown as mean ± SE (n = 7). ***p < 0.01. C. WT and YAC128 corticostriatal cocultures were fixed at DIV14 or DIV20 and stained for DARPP32. Volumetric reconstructions of confocal images are shown. Scale bar, 5 μm. D. An average MSN spine density is shown for DIV14 and DIV20 WT and YAC128 MSNs cultures as mean ± SE (DIV14, n = 37–39 neurons; DIV20, n = 25–30 neurons). ****p < 0.0001. E. Striatal slices from WT and YAC128 mice were obtained at 2, 6, and 12 months of age. Two-photon images of MSNs filled with Lucifer yellow are shown. Scale bar, 5 μm. F. An average MSN spine density in striatal slices is shown as mean ± SE (2 months, WT, n = 34 neurons; YAC128, n = 30 neurons; 6 months, WT, n = 39 neurons, YAC128, n = 52 neurons; 12 months, WT, n = 36 neurons, YAC128, n = 42 neurons). The data were pooled together from 3–4 mice for each age group and genotype. **p < 0.01.
Using DARPP32 staining alone, we determined that, on DIV14, spine density for WT MSNs was equal to 10.0 ± 0.5 spines/10 μm of dendritic length (n = 37 neurons) and, for YAC128 MSNs, it was equal to 8.9 ± 0.5 spines/10 μm of dendritic length (n = 39), not significantly different from each other (Fig. 1C,D). On DIV20, the spine density of WT MSNs remained at 10.3 ± 0.5 spines/10 μm of dendritic length (n = 25; Fig. 1C, 1D). In contrast, spine density of YAC128 MSNs was decreased to 5.1 ± 0.3 (n = 30) spines/10 μm of dendritic length (Fig. 1C,D), significantly (p < 0.0001) lower than for WT cultures and for YAC128 cultures at DIV14. Analysis of spine shapes revealed that all spine types (thin, stubby, and mushroom) were equally affected in YAC128 MSNs (data not shown).
To validate these findings in vivo, we analyzed spine density in striatal slices from WT and YAC128 mice at different ages. In these experiments, striatal neurons in fixed slices were loaded by Lucifer yellow fluorescent dye using glass electrodes (Dumitriu et al., 2011) and the spine shape and density was evaluated by two-photon imaging (Fig. 1E). In these experiments, we discovered that MSN spine density was similar in WT and YAC128 mice at 2 and 6 months of age, on the order of 17–20 spines/10 μm of dendritic length (Fig. 1E,F). The spine density was reduced in WT MSNs at 12 months of age to 15.3 ± 1.1 spines/10 μm of dendritic length (n = 36 from 3 mice; Fig. 1E,F). At the same age, the spine density of YAC128 MSNs was reduced even further to 10.3 ± 0.5 spines/10 μm of dendritic length (n = 42 from 4 mice; Fig. 1E,F), significantly (p = 0.0012) lower than for the WT. Similar to in vitro results (Fig. 1A–D), all spine types (thin, stubby, and mushroom) were uniformly lost in 12-month-old YAC128 MSNs (data not shown).
Supranormal InsP3R1 activity, reduced ER Ca2+ levels, and increased spine SOC in YAC128 MSNs
What causes spine loss in YAC128 MSNs? In previous studies, we demonstrated that mHtt causes supranormal activation of InsP3R1 and excessive Ca2+ release from the ER (Tang et al., 2003; Tang et al., 2005). Therefore, we set out to determine whether dysregulation of InsP3R1 function and ER Ca2+ homeostasis contributes to spine loss in YAC128 MSNs. In these experiments, we took an advantage of ASOs targeting mouse InsP3R1. In control experiments, we established that all of the tested ASOs (500 nm) reduced InsP3R1 mRNA levels in bEnd.3 cells 3 d after transfection, but ASOs 178 (p = 0.0007) and 195 (p = 0.0017) were the most effective (Fig. 2A). ASOs 178 (p = 0.0029), 195 (p < 0.0001), and 255 (p < 0.0001) were also the most effective at reducing InsP3R1 protein levels in MEF cells after 6 d of 500 nm ASO treatment (Fig. 2B,C). Several ASOs (200 nm) also reduced InsP3R1 protein levels in cortical neuron cultures (Fig. 2D,E). For example, ASO178 reduced InsP3R1 expression in cortical neuron cultures to 0.67305 ± 0.06041 (n = 6) relative to the control (n = 6; p < 0.001), whereas ASO177 had no effect (0.91277 ± 0.05412; n = 6; Fig. 2E). Because ASO178 was the most effective across several cell lines and assays, we used it as the primary ASO to suppress InsP3R1 function selectively in the corticostriatal coculture HD model.
Figure 2.
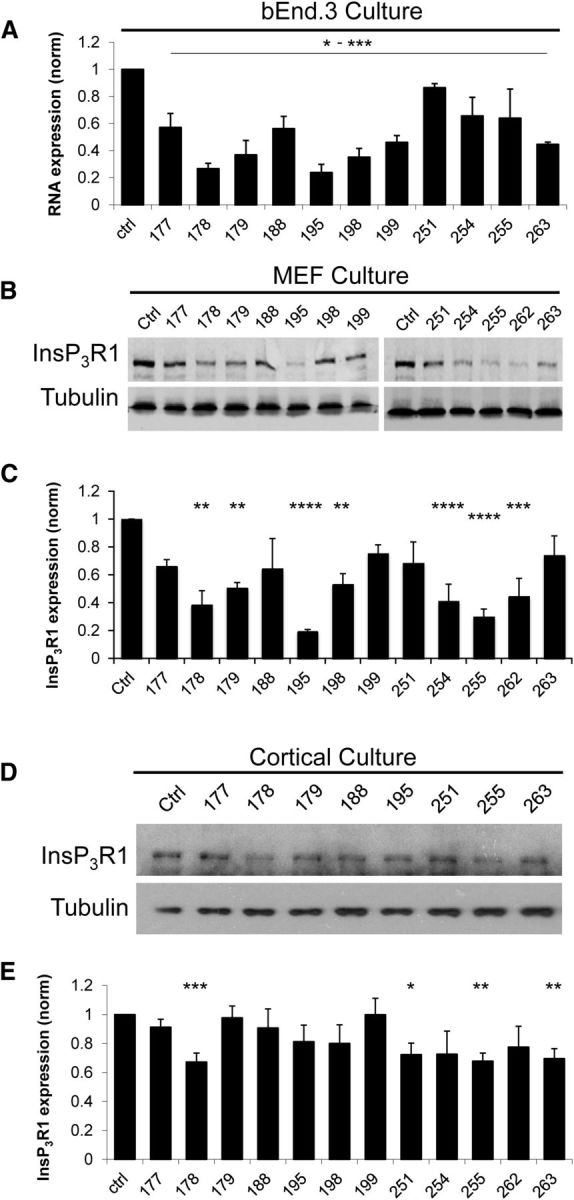
Validation of InsP3R1 knock-down ASOs. A, bEnd.3 cells were transfected with 500 nm of different ASOs or the vehicle control. Three days after transfection, InsP3R1 mRNA levels were quantified by qPCR. The levels of InsP3R1 mRNA for each ASO were normalized to the levels of InsP3R1 mRNA in control cultures. p = 0.0488–0.0005; n = 3 experiments. B, Western blot analysis of InsP3R1 levels in lysates from MEF cells incubated for 6 d with 500 nm of different ASOs or the vehicle control (culture medium). C, The mean InsP3R1 expression levels in MEF cells determined in Western blotting experiments are shown as mean ± SE for each ASO (n = 2–5). **p < 0.01; ***p < 0.001; ****p < 0.0001. D, Western blot analysis of InsP3R1 levels in lysates from cortical cultures incubated for 6 d with 200 nm of different ASOs or the vehicle control (culture media). E, The mean InsP3R1 expression levels in cortical neurons cells determined in Western blotting experiments are shown as mean ± SE for each ASO (n = 3–7). *p < 0.05; **p < 0.01; ***p < 0.001.
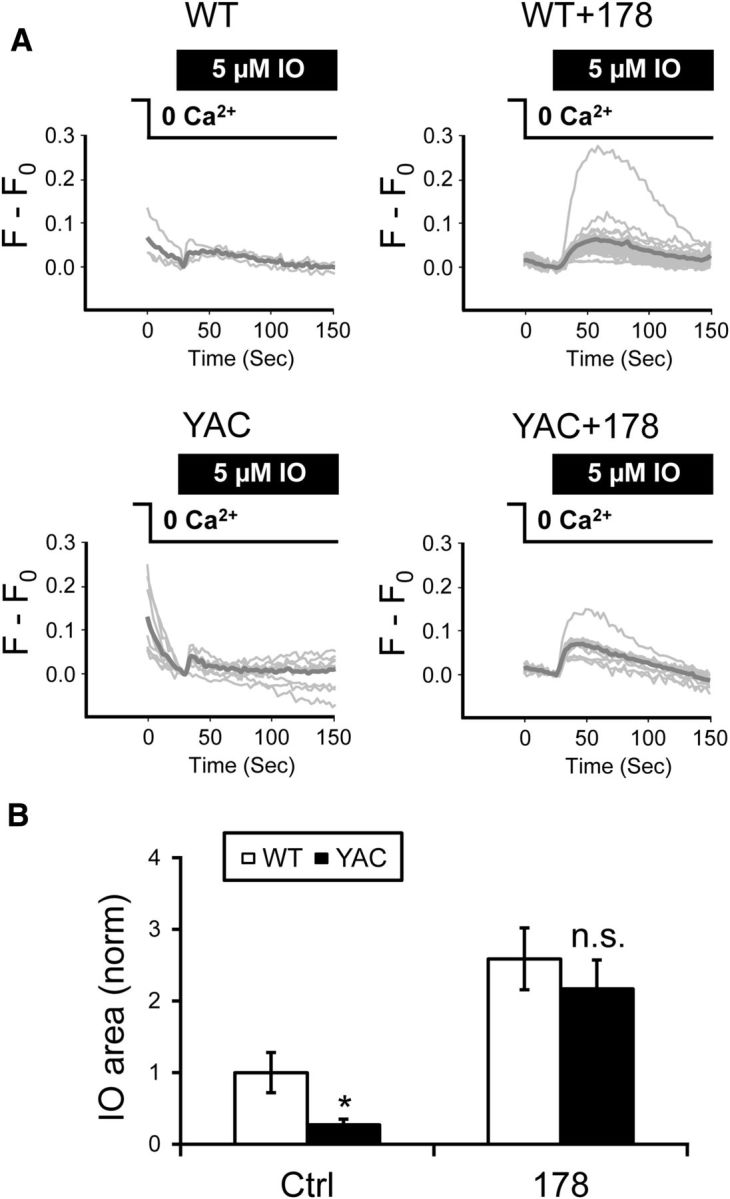
To evaluate InsP3R1-mediated ER Ca2+ release, neurons in WT and YAC128 DIV14–20 corticostriatal cocultures were loaded with Fura-2 AM. Cortical neurons in these experiments were infected with lenti-EGFP virus at DIV0 before plating of MSNs at DIV1. In Fura-2 Ca2+-imaging experiments, MSNs were identified as GFP-negative cells with characteristic morphology. In these experiments, application of the selective mGluR1/5 agonist DHPG (1 μm) evoked InsP3R1-mediated Ca2+ release from the ER (Fig. 3A). The response was enhanced in YAC128 MSNs compared with WT MSNs (Fig. 3A). On average, the amplitude of response to DHPG was 1 ± 0.1346 (n = 20) in WT MSNs and 3.02075 ± 0.25483 (n = 25; p < 0.0001) in YAC128 MSNs (Fig. 3B). These results confirmed that InsP3R1 is indeed hyperactive in YAC128 mixed MSN cultures, in agreement with our previous findings (Tang et al., 2003; Tang et al., 2005). Interestingly, the increase in DHPG-induced Ca2+ release was significantly more dramatic in the YAC128 MSN corticostriatal culture preparation (Fig. 3A,B) than in our previous studies with “naked” YAC128 MSNs (Tang et al., 2003; Tang et al., 2005).
Figure 3.

InsP3R1-mediated Ca2+ release is enhanced in YAC128 MSNs. A, Fura-2 340/380 fluorescence ratio traces are shown for WT and YAC128 MSN cell bodies in response to stimulation by 1 μm DHPG. Individual cell traces are shown by thin gray lines, and average traces are shown by thick gray lines. The traces are shown for control cultures and for the cultures treated with ASO178. B, C, The mean amplitude (B) and slope (C) of DHPG-evoked Fura-2 responses is shown as mean ± SE (WT, n = 20; YAC128, n = 25; WT + ASO178, n = 34, YAC128 + ASO178, n = 27). Comparisons were made to the WT control condition; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To further test the involvement of InsP3R1 in the corticostriatal coculture HD model, we used ASO178 to knock down InsP3R1 expression levels. ASO178 treatment of cocultures before Ca2+ imaging normalized MSN ER Ca2+ release to WT levels (Fig. 3A,B). ASO255 comparably suppressed DHPG-induced Ca2+ release in WT and YAC128 cocultures (data not shown). The incomplete suppression of the DHPG response after ASO treatment most likely reflects partial knock-down of InsP3R1 expression and calcium-induced Ca2+ release from ryanodine receptors. Quantification of the Ca2+ release rate (by measuring slope of the initial Fura-2 response) provides a more direct readout of InsP3R1 function in cells. The Fura-2 signal slope measurements confirmed enhanced InsP3R1 function in YAC128 MSNs and efficient knock-down of InsP3R1 by ASO178 in both WT and YAC128 MSNs (Fig. 3C).
Some basal levels of InsP3 are present in most cells, including MSNs, due to steady-state activation of PLC-coupled receptors. This causes basal activity of InsP3R1, contributing to ER Ca2+ leakage. Therefore, we reasoned that sensitization of InsP3R1 may lead to enhanced ER Ca2+ leakage and reduced steady-state ER Ca2+ levels in YAC128 MSNs. To test this hypothesis, we measured the ionomycin-sensitive ER Ca2+ pool in WT and YAC128 corticostriatal cocultures. In these experiments, mixed cultures were loaded by Fura-2 and MSNs were identified as GFP-negative cells as described above. The cells were transferred to Ca2+-free media for 30 s and challenged with 5 μm IO. The content of the IO-sensitive ER Ca2+ pool was determined by integrating the area under the IO-induced curve as we described previously (Tu et al., 2006). In these experiments, we found that ER Ca2+ content was significantly lower in YAC128 MSNs than in WT MSNs (Fig. 4A). On average, the IO-sensitive pool size was equal to 1 ± 0.28065 (n = 34) in WT MSNs and 0.2715 ± 0.40065 (n = 30) in YAC128 MSNs, significantly (p < 0.05) lower (Fig. 4B). Pretreatment with ASO178 suppressed the ER Ca2+ leak in WT MSNs, resulting in a 2.5-fold increase in the ER Ca2+ pool size (Fig. 4A,B). Pretreatment with ASO178 also suppressed the ER Ca2+ leak in YAC128 MSNs, increasing the ER Ca2+ pool size to the same levels as in WT MSNs (Fig. 4A,B). These data support the hypothesis that supranormal steady-state activity of InsP3R1 is a major cause of elevated ER Ca2+ leakage and reduced ER Ca2+ levels in YAC128 MSNs.
Figure 4.
ER Ca2+ content is reduced in YAC128 MSNs. A, Fura-2 340/380 fluorescence ratio traces are shown for WT and YAC128 MSN cell bodies in response to application to 5 μm IO. The cells were moved to Ca2+-free media 30 s before application of IO as indicated. Individual cell traces are shown by thin gray lines and average traces are shown by thick gray lines. The traces are shown for control cultures and for the cultures treated with ASO178. B, The average size of the IO-sensitive Ca2+ pool was calculated by integrating an area under Fura-2 traces. The results were normalized to IO pool size in WT control cultures and are shown as mean ± SE (WT, n = 34; YAC128, n = 30; WT + ASO178, n = 20, YAC128 + ASO178, n = 19). Comparisons were made between genotypes for each condition; *p < 0.05.
Given that ER Ca2+ depletion activates the SOC entry pathway, persistently reduced ER levels should trigger enhanced SOC in YAC128 MSNs. In agreement with this prediction, we previously observed elevated nSOC in mHtt expressing MSN somata (Wu et al., 2011). Here, we investigated whether nSOC is also upregulated in YAC128 MSN spines. To measure synaptic MSN nSOC, we used lenti-Cherry to infect cortical neurons at DIV0 and plated MSNs at DIV1. The mixed cultures were transfected with a GCaMP5.3 expression plasmid at DIV7 as we described previously (Sun et al., 2014). During imaging experiments, GCaMP5.3-transfected MSNs were identified by morphology and the lack of Cherry expression (Fig. 5A). To evaluate synaptic nSOC, we transferred the DIV14 cultures to Ca2+-free medium in the presence of 1 μm Tg, an inhibitor of the SERCA Ca2+ pump. Increasing extracellular Ca2+ to 2 mm resulted in Ca2+ influx into the spines, which was measured by GCaMP5.3 fluorescence (Fig. 5A). We determined that nSOC was enhanced in YAC128 spines compared with WT spines (Fig. 5B). On average, the nSOC amplitude was 1.00 ± 0.08 (n = 72) in WT MSN spines and 2.5 ± 0.2 (n = 54; p < 0.0001) in YAC128 MSN spines (Fig. 5C). The 2.5-fold increase in spine nSOC was much greater than the 50% increase in somatic nSOC observed in our previous studies with YAC128 MSNs (Wu et al., 2011). Pretreatment with ASO178 reduced MSN spine nSOC in WT cultures 4-fold (Fig. 5B) to the average level of 0.25 ± 0.02 (n = 29; Fig. 5C). Pretreatment with ASO178 also reduced nSOC in YAC128 MSN spines the same levels as in WT MSNs (Fig. 5B,C). ASO255 similarly reduced nSOC in both WT and YAC128 MSNs (data not shown). These results suggest that the InsP3R1-mediated ER Ca2+ leak plays a critical role in control of MSN spine nSOC activity and that hyperactivation of InsP3R1 leads to elevated spine nSOC in YAC128 MSNs.
Figure 5.
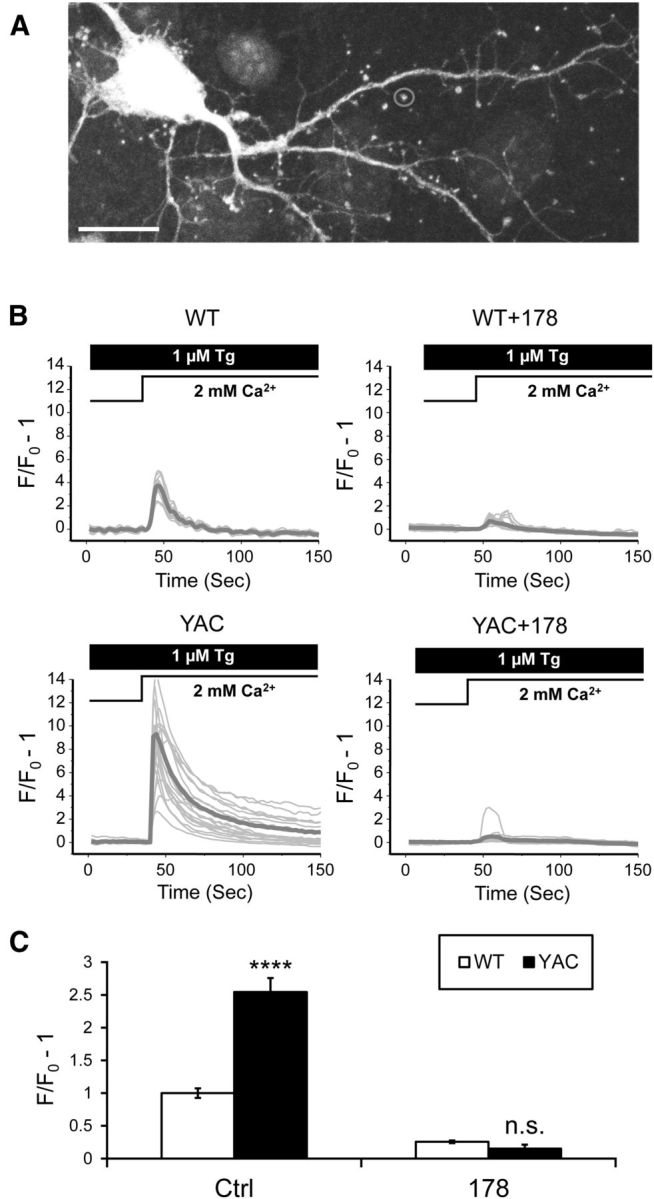
Spine SOC is enhanced in YAC128 MSNs. A, A GCaMP5.3 fluorescent image is shown for a DIV14 MSN in a WT corticostriatal culture. A MSN spine is indicated by a circle. Scale bar, 10 μm. B, GCaMP5.3 fluorescence traces in WT and YAC128 MSN spines after Ca2+ “add-back” protocol. Activity of SERCA pump in these experiments is blocked by 1 μm Tg. Individual cell traces are shown by thin gray lines and average traces are shown by thick gray lines. The traces are shown for control cultures and for the cultures treated with ASO178. C, The average peak of spine SOC signals. The results were normalized to SOC peak size in WT control cultures and shown as mean ± SE (WT, n = 72, YAC128, n = 54; WT + ASO178, n = 29, YAC128 + ASO178, n = 14). Comparisons were made between genotypes for each condition; ****p < 0.0001.
In the previous studies, we demonstrated that the nSOC pathway plays an important role in the maintenance of postsynaptic mushroom spines in hippocampal neurons (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015). Is it possible that ER Ca2+ depletion and enhanced synaptic nSOC contributes to spine loss in YAC128 MSNs? To answer this question, starting at DIV14, we treated WT and YAC128 corticostriatal cocultures with ASOs (dissolved in Neurobasal-A media) that effectively reduce InsP3R1 levels in neuron cultures (Fig. 2E) or less effective control ASOs (Fig. 2E). Additional control cultures were treated with Neurobasal-A medium alone. In control experiments, we established that incubation with these ASOs do not result in significant neurotoxic effects in WT and YAC128 corticostriatal cultures (Fig. 6A). After ASO treatments, the cultures were fixed at DIV20, stained with DARPP32 antibodies, and analyzed by confocal imaging (Fig. 6B). Consistent with the previous results (Fig. 1), the spine density in YAC128 MSNs was reduced compared with WT MSNs in control experiments (Fig. 6B). On average, the density of WT MSN spines in these experiments was equal to 10.6 ± 0.7 spines/10 μm (n = 10) and the density of YAC128 MSN spines was equal to 6.5 ± 0.2 spines/10 μm (n = 12; p < 0.0001; Fig. 6C). Treatment with ASOs 177, 178, 195, or 255 had no effect on the spine density of WT MSNs, whereas ASO 263 reduced the density of WT spines (Fig. 6B,C). Treatment with effective ASOs (178, 255, or 263) rescued YAC128 MSN spines, increasing them to the same level as in WT cultures (Fig. 6B,C). In contrast, ASOs 177 and 195 did not fully rescue YAC128 MSN spines (Fig. 6B,C), consistent with their lesser efficacy in neuronal InsP3R1 knock-down (Fig. 2D,E). Therefore, ASO-mediated suppression of InsP3R1 expression was sufficient to prevent spine loss in YAC128 MSNs, presumably due to normalization of ER Ca2+ levels (Fig. 4A,B) and spine nSOC (Fig. 5B,C) in these neurons.
Figure 6.

InsP3R1 knock-down rescues spine loss in YAC128 MSNs. A, ASO neuronal toxicity assay. After 6 d of exposure of WT and YAC128 corticostriatal cultures to ASOs, the fraction of dead cells was quantified by TMR assay. The fraction of TMR Red+ nuclei is shown for control conditions (Ctrl) and each ASO as mean ± SE (n = 3 cultures). B, WT and YAC128 corticostriatal cultures were exposed to control solution (culture media) or different ASOs, fixed at DIV20, and stained for DARPP32. Spine reconstructions are shown. Scale bar, 5 μm. C, Average spine density for WT and YAC128 MSNs exposed to different ASOs. The spine density for each genotype and ASO is shown as mean ± SE (n = 10 for WT, n = 12 for YAC128). Comparisons were made between genotypes for each condition; ***p < 0.001; ****p < 0.0001.
STIM2 controls spine nSOC in YAC128 MSNs
Upon ER Ca2+ depletion, SOC channels are activated by STIM1 and/or STIM2 ER resident proteins (Liou et al., 2005). We previously identified a key role for the SOC-regulated channel TRPC1 (Cheng et al., 2011) in MSN soma nSOC (Wu et al., 2011). We also previously demonstrated that STIM2, but not STIM1, plays a major role in control of synaptic nSOC in hippocampal neurons (Sun et al., 2014). To better understand the molecular mechanism of synaptic nSOC upregulation in YAC128 MSNs, we compared expression levels of TRPC1, STIM1, and STIM2 proteins in WT and YAC128 MSNs by Western blotting. We found that the expression levels of STIM2 are significantly elevated in YAC128 MSN cultures compared with WT cultures (Figs. 7A,B). The levels of TRPC1 and STIM1 were not significantly different between WT and YAC128 cultures (Figs. 7A,B). We also evaluated expression levels of TRPC1, STIM1, and STIM2 proteins in striatal lysates prepared from WT and YAC128 mice at 2, 6, and 12 months of age (Fig. 7C). In these experiments, we did not observe significant differences in 2- and 6-month-old samples (Fig. 7D). However, expression levels of STIM2 protein were elevated in striatal samples from YAC128 mice at 12 months of age (Fig. 7D). There was no difference in expression levels of TRPC1 and STIM1 proteins at 12 months (Fig. 7D). From these results, we concluded that the increase in STIM2 expression is likely to contribute to enhanced spine nSOC in YAC128 MSNs.
Figure 7.

STIM2 protein is upregulated in YAC128 MSNs. A, Western blot analysis of lysates from DIV14 WT and YAC128 MSN cultures. Levels of TRPC1, STIM1 and STIM2 were evaluated. Tubulin was used as a loading control. B, Expression levels of TRPC1, STIM1, and STIM2 in WT and YAC128 MSN cultures. The data for each protein were normalized to WT and are shown as mean ± SE (n = 3 cultures). Comparisons were made between genotypes for each protein; *p < 0.05. C. Western blot analysis of striatal lysates from 2-, 6-, and 12-month-old WT and YAC128 mice. Levels of TRPC1, STIM1, and STIM2 were evaluated. Tubulin was used as a loading control. D, Expression levels of TRPC1, STIM1, and STIM2 in striatal lysates from 2-, 6-, and 12-month-old WT and YAC128 mice. The data for each protein were normalized to WT and are shown as mean ± SE (n = 3 independent experiments). Comparisons were made between genotypes for each protein; *p < 0.05.
To test the potential role of STIM2 protein more directly, we used Dicer-generated STIM2-siRNA (dSTIM2) to knock down STIM2 expression levels. In these experiments, DIV7 WT and YAC128 corticostriatal cultures were transfected with dSTIM2 or with control Dicer-generated EGFP-siRNA (dGFP) and spine nSOC measurements were performed with DIV14 cultures using GCaMP5.3 as described above. Consistent with previous experiments (Fig. 5), in control cultures (treated with dGFP), spine nSOC was significantly higher in YAC128 MSNs than in WT MSNs (Fig. 8A). On average, for dGFP-treated cultures, the amplitude of spine nSOC was 1.00 ± 0.04 (n = 30) in WT MSN spines and 1.9 ± 0.1 (n = 24; p < 0.0001) in YAC128 MSN spines (Fig. 8B). Treatment of WT cocultures with dSTIM2 decreased nSOC amplitude to 0.67 ± 0.03 (n = 18; Fig. 8A,B), indicating that STIM2 plays a significant role in control of spine nSOC in MSNs. Remarkably, treatment of YAC128 cultures with dSTIM2 nearly completely abolished spine nSOC (Fig. 8A). On average, the MSN spine nSOC amplitude in dSTIM2-treated YAC128 cocultures was reduced to 0.23 ± 0.09 (n = 28; p < 0.0001; Fig. 8B). These results indicated that STIM2 plays a predominant role in supporting supranormal nSOC in YAC128 MSNs.
Figure 8.
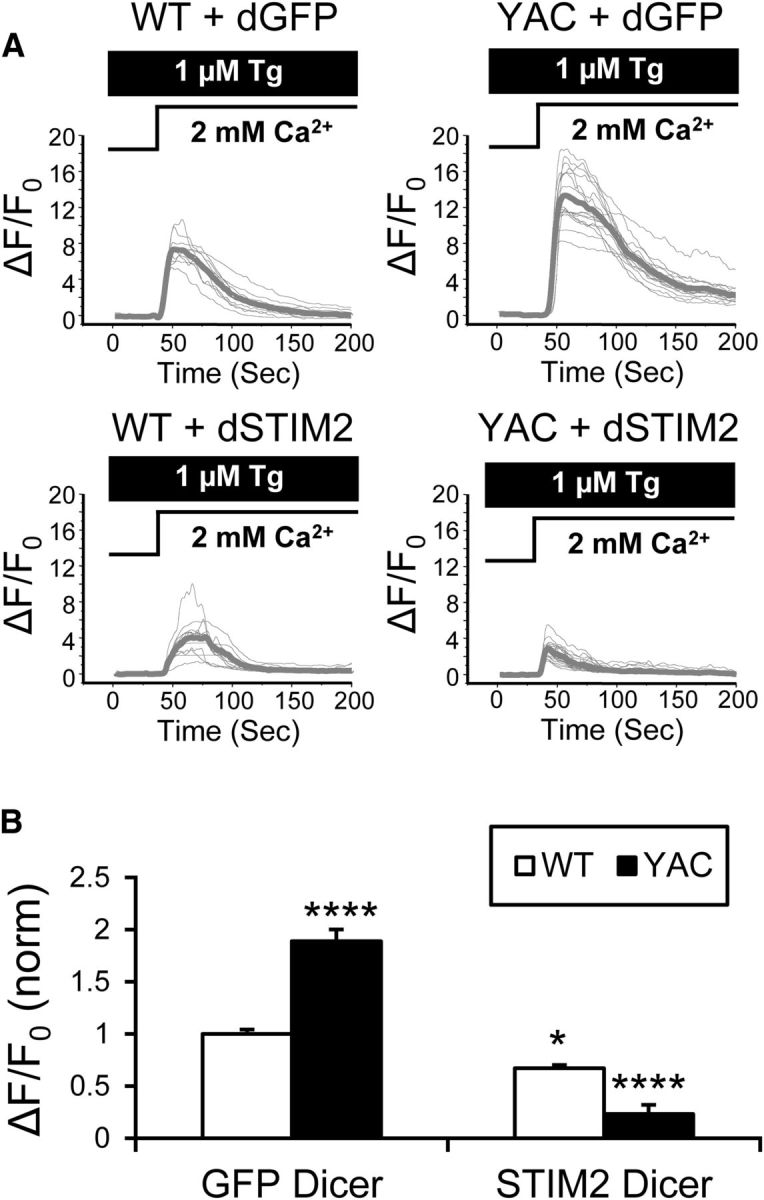
STIM2 mediates enhanced spine nSOC in YAC128 MSNs. A, GCaMP5.3 fluorescence traces in WT and YAC128 MSN spines after Ca2+ “add-back” protocol. Activity of SERCA pump in these experiments is blocked by 1 μm Tg. Individual cell traces are shown by thin gray lines and average traces are shown by thick gray lines. The traces are shown for cultures treated with Dicer-generated siRNAs targeting STIM2 (dSTIM2) or control GFP (dGFP). B, Average peak of spine SOC signals. The results were normalized to SOC peak size in WT control cultures and are shown as mean ± SE (WT + dGFP, n = 30, YAC128 + dGFP, n = 24; WT + dSTIM2, n = 18, YAC128 + dSTIM2, n = 28). Statistical comparisons were made with WT + dGFP data; *p < 0.05; ****p < 0.0001.
In the next series of experiments, we aimed to evaluate the importance of STIM2-mediated nSOC for stability of MSN synaptic spines. In these experiments, WT and YAC128 cocultures were transfected by dSTIM2 or control dGFP on DIV7. At DIV21, the cultures were fixed, stained for DARPP32 and analyzed by confocal imaging (Fig. 9A). In agreement with previous results (Fig. 1), in cultures treated with dGFP, the average MSN spine density was 10.0 ± 0.5 spines/10 μm in WT cultures (n = 20) and 6.3 ± 0.1 spines/10 μm in YAC128 cultures (n = 20, p < 0.01; Fig. 9B). Transfection with dSTIM2 reduced the WT MSN spine density to 7.69 ± 0.03 spines/10 μm (n = 20, p < 0.01), indicating a potential role for basal STIM2-dependent nSOC in WT MSN spine maintenance. In contrast, transfection with dSTIM2 completely rescued the YAC128 MSN spine density (Fig. 9A). On average, the MSN spine density in dSTIM2-treated YAC128 cultures was 10.7 ± 0.5 spines/10 μm (n = 20; Fig. 9B), not significantly different from dGFP-treated WT MSNs.
Figure 9.
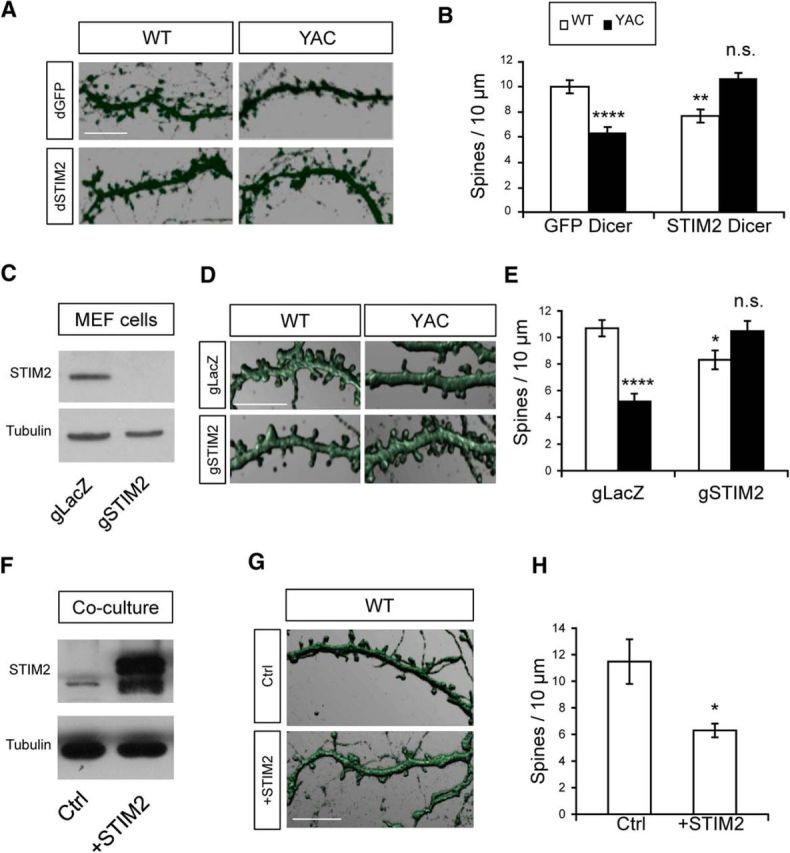
STIM2 expression levels affect MSN spines. A, WT and YAC128 corticostriatal cultures were transfected with dSTIM2 or control dGFP Dicer RNAi preparations on DIV7. The cultures were fixed at DIV20 and stained for DARPP32. Spine reconstructions are shown. Scale bar, 10 μm. B, Average spine density for DIV20 WT and YAC128 MSNs transfected with dSTIM2 or dGFP. Spine densities for each experimental condition are shown as mean ± SE (n = 20 for all conditions). Statistical comparisons were made with WT + dGFP data; *p < 0.05; **p < 0.01; ****p < 0.0001. C. Western blot analysis of STIM2 expression in MEF cells transfected with Cas9 and gSTIM2 or Cas9 and gLacZ plasmids. Transfected cells were selected using a mixture of blasticidin and puromycin. Tubulin was used as a loading control. D. WT and YAC128 corticostriatal cultures were infected with lenti-Cas9 and lenti-gSTIM2 (gSTIM2) or lenti-Cas9 and lenti-gLacZ (gLacZ) on DIV7. The cultures were fixed at DIV20 and stained for DARPP32. Spine reconstructions are shown. Scale bar, 10 μm. E, Average spine density for DIV20 WT and YAC128 MSNs coinfected with lenti-Cas9 and lenti-gSTIM2 (gSTIM2) or lenti-Cas9 and lenti-gLacZ (gLacZ) viruses. The spine density for each experimental condition is shown as mean ± SE (gLacZ experiments, n = 11, gSTIM2 experiments, n = 13). Statistical comparisons were made with WT + gLacZ data; *p < 0.05; ****p < 0.0001. F, WT corticostriatal cultures were infected with lenti-STIM2 or lenti-RFP (Ctrl) viruses. Expression of STIM2 was analyzed by Western blotting of culture lysates. G, WT corticostriatal cultures were infected with lenti-STIM2 or lenti-RFP (Ctrl) viruses on DIV7. The cultures were fixed at DIV20 and stained for DARPP32. Spine reconstructions are shown. Scale bar, 10 μm. H, Average spine density for WT MSNs infected with lenti-STIM2 or lenti-RFP (Ctrl) viruses. The spine density is shown as mean ± SE (n = 10); *p < 0.05.
To confirm these findings, the CRISPR/Cas9 system was used as an alternate approach to knock out neuronal STIM2. To validate CRISPR plasmids, MEF cells were cotransfected with pCas9-Blast and gSTIM2 plasmids. The gSTIM2 plasmid was developed using pGuide-Puro as a backbone (see Materials and Methods for details). As a control, MEF cells were cotransfected with pCas9-Blast and gLacZ plasmids (targeting bacterial β-galactosidase gene). Cotransfected MEF cells were selected using blasticidin and puromycin. Western blot analysis confirmed efficient knock-out of STIM2 in gSTIM2-transfected but not in gLacZ-transfected MEF cells (Fig. 9C). These results confirmed efficient targeting of the mouse STIM2 gene by the gSTIM2 plasmid. Cas9 and gSTIM2 plasmids were packaged into lentiviruses and used to coinfect corticostriatal cocultures on DIV7. Control cultures were coinfected with Cas9 and gLacZ lentiviruses. At DIV20, spine density in gLacZ-expressing YAC128 MSNs was 5.2 ± 0.58 spines/10 μm (n = 11), significantly lower than 10.7 ± 0.61 spines/10 μm in gLacZ-expressing WT MSNs (n = 11; p < 0.0001; Fig. 9D,E). STIM2 knock-out by gSTIM2 expression elevated the density of YAC128 MSN spines to 10.5 ± 0.76 spines/10 μm (n = 13), which was comparable to the levels observed in gLacZ-treated WT cultures (p > 0.05), confirming a role for STIM2 in YAC128 MSN spine loss (Fig. 9D,E). Expression of gSTIM2 in WT neurons resulted in a decreased spine density with 8.3 ± 0.71 spines/10 μm (n = 13; p < 0.05), confirming a role for STIM2-mediated nSOC in WT MSN spine maintenance (Fig. 9D,E).
Because excessive STIM2-dependent nSOC activity appears to lead to spine loss in YAC128 MSN spines (Fig. 9A–E), we tested whether STIM2 overexpression would be sufficient to destabilize WT MSN spines (Fig. 9F–H). STIM2 cDNA was cloned into the lentivirus expression plasmid and packaged into lentiviruses. Resulting lentiviruses were used to infect WT corticostriatal cocultures on DIV7. STIM2 overexpression was validated by Western blotting of coculture lysates using lenti-RFP as the negative control (Fig. 9F). We discovered that STIM2 overexpression was indeed sufficient to cause spine loss in cocultured WT MSNs at DIV20, reducing the density of MSN spines from 11.5 ± 1.7 spines/10 μm (n = 10) in lenti-RFP-infected cocultures to 6.3 ± 0.52 spines/10 μm (n = 10) in lenti-STIM2-infected cocultures (p < 0.05; Fig. 9G,H). Results obtained with WT cocultures suggest that either a reduction (Fig. 9B,E) or an increase (Fig. 9H) in synaptic nSOC can lead to destabilization of MSN spines.
nSOC inhibitor EVP4593 prevents spine loss in YAC128 MSNs
In the previous study, we demonstrated that EVP4593 (6-amino-4-(4-phenoxyphenethyl-amino)quinazoline; Fig. 10A) is an effective inhibitor of nSOC at nanomolar concentrations (Wu et al., 2011). We also demonstrated that EVP4593 improves motor symptoms in a Drosophila HD model and protects YAC128 MSNs in a glutamate toxicity assay (Wu et al., 2011). In previous studies, we demonstrated that EVP4593 blocks SOC in the soma of YAC128 MSNs (Wu et al., 2011). To determine whether EVP4593 can also block synaptic nSOC, we performed synaptic Ca2+-imaging experiments with WT and YAC128 mixed cultures at DIV14–15. These experiments were performed using the GCaMP5.3 Ca2+ indicator, as described above. In agreement with previous results (Fig. 5), we observed that synaptic nSOC is significantly potentiated in control (vehicle-treated) YAC128 MSNs compared with WT MSNs (Fig. 10B). On average, synaptic nSOC amplitude in vehicle-treated cultures was equal to 1.00 ± 0.07 (n = 45) in WT MSNs and 3.4 ± 0.3 (n = 21, p < 0.001) in YAC128 MSNs (Fig. 10C). Incubation with 30 nm EVP4593 reduced synaptic nSOC in WT MSNs twofold (Fig. 10B) and dramatically inhibited synaptic nSOC in YAC128 MSNs (Fig. 10B). On average, the synaptic nSOC amplitude in the presence of EVP4593 was equal to 0.53 ± 0.06 (n = 19) in WT MSNs and 0.51 ± 0.06 (n = 23) in YAC128 MSNs (Fig. 10C), not significantly different from each other. These results indicated that EVP4593 efficiently blocks supranormal nSOC in YAC128 MSN spines, in agreement with our previous results with somatic SOC measurements (Wu et al., 2011).
Figure 10.
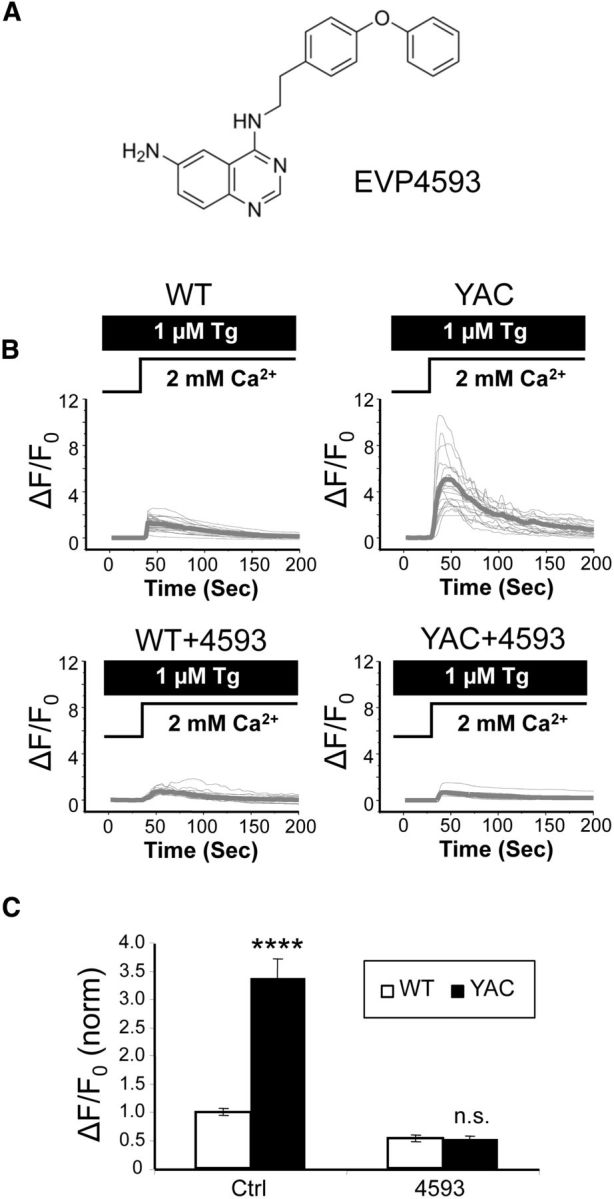
The nSOC inhibitor EVP4593 normalizes YAC128 MSN spine SOC entry. A, Chemical structure of EVP4593 (adapted from Wu et al., 2011). B, GCaMP5.3 fluorescence traces in WT and YAC128 MSN spines after Ca2+ “add-back” protocol. The activity of the SERCA pump in these experiments is blocked by 1 μm Tg. Individual cell traces are shown by thin gray lines and average traces are shown by thick gray lines. The traces are shown for control cultures and cultures exposed to 30 nm EVP4593. C, Average peak of spine SOC signals. The results were normalized to SOC peak size in WT control cultures and are shown as mean ± SE (WT, n = 45, YAC128, n = 21; WT + EVP4593, n = 19, YAC128 + EVP4593, n = 23). Comparisons were made between genotypes for each condition; ****p < 0.0001.
In the next series of experiments, we evaluated neuroprotective effects of EVP4593. In these experiments, WT and YAC128 mixed cultures were treated with 30 nm EVP4593 at DIV20. After 16 h of incubation with EVP4593, the cultures were fixed at DIV21, stained for DARPP32, and analyzed by confocal imaging (Fig. 11A). Control cultures were treated with the vehicle alone. In agreement with previous findings (Fig. 1), we observed significant loss of MSN spines in control YAC128 cultures (Fig. 11A). On average, MSN spine density in control cultures in these experiments was 9.1 ± 0.6 spines/10 μm (n = 15) for WT cultures and 3.50 ± 0.24 (n = 15, p < 0.001) for YAC128 cultures (Fig. 11B). Treatment with EVP4593 significantly reduced MSN spine density in WT cultures to 6.6 ± 0.9 spines/10 μm (n = 15, p = 0.027), further supporting an essential role of nSOC in MSN spine maintenance. In contrast, treatment with EVP4593 rescued spine loss in YAC128 MSNs (Fig. 11A). On average, MSN spine density in EVP4593-treated YAC128 cultures was equal to 9.0 ± 0.6 (n = 15; Fig. 11B), not significantly different from spine density in control WT MSNs. These results are consistent with dSTIM2 data and gSTIM2 data (Fig. 9) and further suggest that inhibition of nSOC may help to prevent synaptic spine loss in HD MSNs.
Figure 11.

The SOC inhibitor EVP4593 rescues YAC128 MSN spines both in vitro and in vivo. A. WT and YAC128 corticostriatal cultures were incubated with 30 nm EVP4593 for 16 h starting at DIV20, fixed, and stained for DARPP32. Spine reconstructions are shown. Scale bar, 5 μm. B, Average spine density of WT and YAC128 MSNs in control conditions and after exposure to 30 nm EVP4593. The spine density for each experimental condition is shown as mean ± SE (n = 15 for all conditions). Statistical comparisons were made with the WT control group; *p < 0.05; ****p < 0.0001. C, Striatal slices were obtained from WT and YAC128 mice at 12 months of age. The mice were infused with EVP4593 (0.25 mg/ml) or the vehicle solution (10% DMSO in PEG300) into the ventricles over 6 weeks starting at 10.5 months of age. Two-photon images of MSNs filled with Lucifer yellow are shown. Scale bar, 5 μm. D, An average MSN spine density in striatal slices is shown as mean ± SE (WT Ctrl, n = 52 neurons; YAC128 Ctrl, n = 64 neurons; WT + EVP4593, n = 32 neurons, YAC128 + EVP4593, n = 36 neurons). The data were pooled together from 3–6 mice for each experimental group. Statistical comparisons were made with the WT control group; ***p < 0.001; ****p < 0.0001.
To further evaluate potential neuroprotective effects of EVP4593, we tested it in vivo with YAC128 mice and WT controls. In pilot studies, we determined that EVP4593 does not efficiently pass blood–brain barrier in mice (data not shown). This precluded us from evaluating EVP4593 in behavioral studies. Instead, we used an osmotic minipump loaded with 200 μl of EVP4593 (0.25 mg/ml) to infuse EVP4593 into the ventricles of 10.5-month-old WT and YAC128 mice over 6 weeks. In control experiments, mice were infused with the same volume of vehicle solution (10% DMSO in PEG300). At the conclusion of drug treatment, mice were perfused, brains extracted, and MSNs were injected with the fluorescent dye Lucifer yellow (Dumitriu et al., 2011). MSN spine density was evaluated by two-photon imaging (Fig. 11C). In agreement with previous results (Fig. 1), we observed a significant reduction in the MSN spine density in vehicle-treated YAC128 mice (Fig. 11C). On average, the MSN spine density was reduced from 15.7 ± 0.47 spines/10 μm dendritic length (n = 52 neurons from 4 mice) in vehicle-treated WT mice to 9.76 ± 0.26 spines/10 μm (n = 64 neurons from 4 mice; p < 0.0001) in vehicle-treated YAC128 mice (Fig. 11D). Consistent with previous experiments indicating a role for tonic STIM2-dependent nSOC in normal MSN spine maintenance (Figs. 9, 11), the MSN spine density in EVP4593-treated WT mice was 13.04 ± 0.57 spines/10 μm (n = 32 neurons from 3 mice), significantly lower than in vehicle-treated WT mice (p = 0.0007; Fig. 11D). Also consistent with Figures 9 and 11, nSOC inhibition restored the YAC128 MSN spine density to WT levels (Fig. 11C). The spine density in EVP4593-treated YAC128 mice was 14.64 ± 0.57 spines/10 μm (n = 36 neurons from 6 mice), not significantly different from vehicle-treated WT mice (p = 0.3795; Fig. 11D). These data support an important role for nSOC in MSN spine maintenance and suggest that supranomal nSOC leads to spine destabilization in YAC128 MSNs in vivo.
Discussion
In vitro model of synaptic loss in HD MSNs
Previously, we used “naked” MSN cultures to investigate the increased sensitivity of YAC128 MSNs to glutamate-induced apoptotic cell death (Tang et al., 2005; Wu et al., 2006; Tang et al., 2007; Wu et al., 2008; Zhang et al., 2008a; Tang et al., 2009; Wu et al., 2011). A similar approach was taken by others in studies of NMDA-induced apoptosis of YAC128 MSNs (Zeron et al., 2002; Shehadeh et al., 2006). This approach provided important insights into mechanisms of excitotoxic cell death in HD. However, this approach has significant limitations and the naked MSN model is not appropriate for studies of synaptic dysfunction. Mixed cortical/striatal cultures are necessary for the proper formation of functional MSN dendritic spines (Segal et al., 2003; Tian et al., 2010; Fishbein and Segal, 2011; Milnerwood et al., 2012; Parsons et al., 2014). Synapse physiology in these cultures was shown to be modulated by Htt and mHtt (Milnerwood et al., 2012; Parsons et al., 2014), but the impact of mHtt on synaptic stability was not previously investigated in corticostriatal cocultures. Preliminary characterization of the mixed corticostriatal coculture system used here was reported in our earlier study (Artamonov et al., 2013). We report here that this model enabled us to quantify age-dependent spine loss in YAC128 MSNs (Fig. 1). Importantly, this HD “synaptic loss” model does not rely on any additional stressors (such as glutamate or NMDA), with the exception of mHtt expression and the time spent in culture. YAC128 MSN spine loss in the coculture mirrored age-dependent YAC128 MSN spine loss in vivo except that the extent of spine loss was more mild in vivo, presumably due to stresses associated with culturing and/or differences in the rate of aging in vitro versus in vivo. In both YAC128 corticostriatal cocultures and in YAC128 mouse brains, the most predominant aging phenotype that we observed was spine loss (Fig. 1). In the human brain, as some neurons lose connections, others might compensate by forming an overabundance of connections (Graveland et al., 1985). Although cocultures may model only one aspect of spine pathology in HD, we consider the developed model to be useful for mechanistic studies of MSN spine instability in HD and for validating HD therapeutic targets and compounds.
Role of synaptic nSOC pathway in stability of synaptic spines
Our recent results suggested that the STIM2-gated nSOC pathway is essential for the stability of mushroom spines in hippocampal neurons (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015). Our present results suggest that the STIM2-gated nSOC pathway also plays an important role in supporting the stability of MSN spines (Fig. 12A). Knock-down of STIM2 with Dicer or knock-out of the STIM2 gene with Cas9/CRISPR reduced the density of WT MSN spines (Fig. 9B,E). Reduction of WT MSN spine density was also observed after exposure to the nSOC inhibitor EVP4593 in vitro and in vivo (Fig. 11B,D). Although nSOC homeostasis is important for both hippocampal and striatal neurons for spine stability, there are some important differences in their reliance on nSOC. Striatal spines in WT neurons are only partially affected when SOC is blocked by STIM2 knock-down or knock-out (Fig. 9) and by EVP4593 (Fig. 11). In addition, striatal neurons are sensitive to supranormal SOC, as observed when STIM2 is overexpressed in WT MSN neurons (Fig. 9). In contrast, mushroom hippocampal spines are very sensitive to genetic deletion of STIM2 or pharmacological inhibition of SOC, but are not affected by STIM2 overexpression (Sun et al., 2014). Therefore, the signaling mechanisms that connect spine SOC and spine stability are likely to be different in hippocampal and striatal neurons. In hippocampal neurons, nSOC drives tonic synaptic CaMKII activity, which is necessary for maintenance of mushroom spines (Sun et al., 2014). CaMKII is abundant in the striatum (Erondu and Kennedy, 1985) and is highly enriched in MSN spines (Fukunaga et al., 1988). Therefore, it is possible that CaMKII is also involved in the control of MSN spine maintenance. MSN targets downstream of synaptic nSOC need to be identified in future studies.
Figure 12.

Synaptic SOC and maintenance of MSN spines. A. Long-term maintenance of spines in MSNs depends on synaptic nSOC. Synaptic nSOC is gated by STIM2, which is regulated by synaptic ER Ca2+ levels. The filling state of ER Ca2+ stores is regulated by a balance between activity of SERCA Ca2+ pump and InsP3R1-mediated Ca2+ leak from the ER. When synpatic ER Ca2+ stores are appropriately filled, tonic nSOC supports MSN spine stability by promoting activity of SOC-dependent Ca2+ effectors in the spines. B, Supranormal synaptic nSOC causes spine loss in HD MSNs. mHtt sensitizes InsP3R1 to basal levels of InsP3, causing excessive Ca2+ leakage from the chronic ER depletion. Depletion of ER Ca2+ causes compensatory upregulation of STIM2 expression and supranormal synaptic nSOC. Enhanced synaptic nSOC overactivates SOC-dependent synaptic Ca2+ effectors, leading to destabilization and loss of spines in HD MSNs.
nSOC and synaptic loss in Alzheimer's disease (AD) and HD
Our results provide information that is useful for understanding selective neuronal vulnerability in neurodegenerative disorders. Our results suggest that synapses are very sensitive to magnitude of SOC and deviation in either direction is detrimental for synaptic stability. In the previous studies, we demonstrated that impaired nSOC causes loss of mushroom hippocampal spines in PS1KI and APPKI mouse models of FAD and in response to amyloid toxicity (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015). In the present study, we demonstrated that association of mHtt with InsP3R1 causes supranormal activation of InsP3R1 and depletion of ER Ca2+ levels (Figs. 3, 4). We further demonstrated that persistently depleted ER Ca2+ levels result in compensatory upregulation of STIM2 expression and supranormal levels of nSOC in YAC128 MSNs (Figs. 5, 7). Based on these results, we concluded that supranormal levels of nSOC contribute to loss of spines in YAC128 MSNs (Fig. 12B). In support of this hypothesis, we demonstrated that knock-down of InsP3R1 by ASOs, knock-down of STIM2 by Dicer, knock-out of STIM2 by Cas9/CRISPR, and treatment with the nSOC inhibitor EVP4593 reduced synaptic nSOC and rescued spine loss in YAC128 MSNs (Figs. 5, 6, 8, 9, 10, 11). Dysregulation of nSOC in HD MSN spines (Fig. 12B) is a “mirror image” of the model that we previously suggested to explain the loss of mushroom hippocampal spines in aging and AD (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015). Consistent with this dichotomy, we observed reduced expression of STIM2 in hippocampal neurons from AD mouse models and in aging mice (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015), but increased expression of STIM2 in YAC128 HD MSNs (Fig. 7). As discussed in the previous section, our results indicated that mushroom spines in hippocampal neurons are particularly vulnerable to insufficient nSOC and striatal MSN spines are sensitive to supranormal nSOC. This may explain why deviation of nSOC in either direction affects synaptic spines in different types of neurons (hippocampal and striatal) and leads to different disease phenotypes (AD or HD).
Synaptic nSOC as drug target
Our results suggest that synaptic nSOC is a promising drug target for AD, HD, and possibly for other neurodegenerative disorders. In previous studies, we demonstrated that overexpression of STIM2 resulted in rescue of mushroom spine loss in hippocampal neurons from the PS1KI and APPKI mouse models of AD and protected mushroom spines from synaptotoxic insult by amyloid (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015). In the present study, we demonstrated that knock-down or knock-out of STIM2 rescued spine loss in YAC128 MSNs (Fig. 9). These results suggest that pharmacological modulators of the nSOC pathway may exert neuroprotective effects in AD and HD. More specifically, our results suggest that positive modulators and activators of the nSOC pathway may have beneficial effects in AD and negative modulators and inhibitors may have beneficial effects in HD. Indeed, we demonstrated that the novel nSOC inhibitor EVP4593 (Wu et al., 2011; Fig. 10A) rescued spine loss in YAC128 MSNs both in vitro and in vivo (Fig. 11). These results suggest that EVP4593 and derivatives may be considered as lead compounds for HD therapeutic development. However, the therapeutic window of these compounds may be limited by their effects in other neuronal populations. For example, nSOC inhibitors such as EVP4593 lead to mushroom spine loss in hippocampal neurons (data not shown). These results suggest that more selective compounds that target the striatal nSOC pathway more specifically need to be developed to achieve a wider therapeutic window and minimize potential side effects in the clinical setting.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke–National Institutes of Health (Grants R01NS074376 and R01NS056224 to I.B.; results shown in Figures 1-10; and Grant F32NS093786 to D.A.R.) and the Russian Scientific Fund (Grant 14-25-00024 to I.B.; results shown in Figure 11). I.B. holds the Carl J. and Hortense M. Thomsen Chair in Alzheimer's Disease Research. P.E. holds Presidential Stipend 2354.2013.4. We thank members of I.B.'s laboratory for advice and suggestions; Leah Taylor and Polina Plotnikova for administrative assistance; and Dmitry Artamonov, Victoria Korzhova, Chris Im, and Olha Nazarko for technical help with developing the MSN spine loss assay.
I.B. is a paid consultant to Ataxion and TEVA in the field of neurodegeneration. The remaining authors declare no competing financial interests.
References
- Artamonov D, Korzhova V, Wu J, Rybalchenko P, Im K, Krasnoborova V, Vlasova O, Bezprozvanny I. Characterization of synaptic dysfunction in an in vitro corticostriatal model system of Huntington's disease. Biochemistry (Moscow) Supplement Series A: Membrane and Cell Biology. 2013;7:192–202. doi: 10.1134/S1990747813040028. [DOI] [Google Scholar]
- Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I. Role of inositol 1, 4, 5-trishosphate receptors in pathogenesis of Huntington's disease and spinocerebellar ataxias. Neurochem Res. 2011;36:1186–1197. doi: 10.1007/s11064-010-0393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, Nukina N, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Ong HL, Liu X, Ambudkar IS. Transient receptor potential channels. New York: Springer; 2011. Contribution of TRPC1 and Orai1 to Ca2+ entry activated by store depletion; pp. 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Dayalu P, Albin RL. Huntington disease: pathogenesis and treatment. Neurol Clin. 2015;33:101–114. doi: 10.1016/j.ncl.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Dumitriu D, Rodriguez A, Morrison JH. High-throughput, detailed, cell-specific neuroanatomy of dendritic spines using microinjection and confocal microscopy. Nat Protoc. 2011;6:1391–1411. doi: 10.1038/nprot.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidelberg D, Surmeier DJ. Brain networks in Huntington disease. J Clin Invest. 2011;121:484–492. doi: 10.1172/JCI45646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein I, Segal M. Active cortical innervation protects striatal neurons from slow degeneration in culture. J Neural Transm. 2011;118:445–451. doi: 10.1007/s00702-010-0505-5. [DOI] [PubMed] [Google Scholar]
- Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington's disease based on age at onset. J Neurol Neurosurg Psychiatry. 1999;66:52–56. doi: 10.1136/jnnp.66.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Goto S, Miyamoto E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J Neurochem. 1988;51:1070–1078. doi: 10.1111/j.1471-4159.1988.tb03070.x. [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- Jiang M, Chen G. High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
- Kasumu A, Bezprozvanny I. Deranged calcium signaling in purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum. 2012;11:630–639. doi: 10.1007/s12311-010-0182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumu AW, Liang X, Egorova P, Vorontsova D, Bezprozvanny I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J Neurosci. 2012;32:12786–12796. doi: 10.1523/JNEUROSCI.1643-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. Pathogenic polyglutamine expansion length correlates with polarity of the flanking sequences. Mol Neurodegener. 2014;9:45. doi: 10.1186/1750-1326-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry SU, Karadeniz YB, Worthington AK, Hayrapetyan VY, Ozlu MI, Serafin-Molina K, Risher WC, Ustunkaya T, Dragatsis I, Zeitlin S, Yin HH, Eroglu C. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci. 2014;34:9455–9472. doi: 10.1523/JNEUROSCI.4699-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Bezprozvanny I. Corticostriatal circuit dysfunction in Huntington's disease: intersection of glutamate, dopamine, and calcium. Future Neurology. 2010;5:735–756. doi: 10.2217/fnl.10.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Kaufman AM, Sepers MD, Gladding CM, Zhang L, Wang L, Fan J, Coquinco A, Qiao JY, Lee H, Wang YT, Cynader M, Raymond LA. Mitigation of augmented extrasynaptic NMDAR signaling and apoptosis in cortico-striatal co-cultures from Huntington's disease mice. Neurobiol Dis. 2012;48:40–51. doi: 10.1016/j.nbd.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci. 2000;20:5115–5123. doi: 10.1523/JNEUROSCI.20-13-05115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth M, Schippling S, Schneider SA, Bhatia KP, Talelli P, Tabrizi SJ, Rothwell JC. Abnormal motor cortex plasticity in premanifest and very early manifest Huntington disease. J Neurol Neurosurg Psychiatry. 2010;81:267–270. doi: 10.1136/jnnp.2009.171926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, Kang R, Buren C, Dau A, Southwell AL, Doty CN, Sanders SS, Hayden MR, Raymond LA. Bidirectional control of postsynaptic density-95 (PSD-95) clustering by Huntingtin. J Biol Chem. 2014;289:3518–3528. doi: 10.1074/jbc.M113.513945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M, Hayden M. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E, Pchitskaya E, Speshilova A, Alexandrov S, Zhang H, Vlasova O, Bezprozvanny I. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol Neurodegener. 2015;10:37. doi: 10.1186/s13024-015-0034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry in the nervous system. Cell Calcium. 2003;34:339–344. doi: 10.1016/S0143-4160(03)00143-X. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Greenberger V, Korkotian E. Formation of dendritic spines in cultured striatal neurons depends on excitatory afferent activity. Eur J Neurosci. 2003;17:2573–2585. doi: 10.1046/j.1460-9568.2003.02696.x. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington's disease: a quantitative Golgi study. Neurology. 1993;43:2088–2096. doi: 10.1212/WNL.43.10.2088. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL, 3rd, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014;82:79–93. doi: 10.1016/j.neuron.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/S0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinás R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci. 2007;27:7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Guo C, Wang H, Chen X, Bezprozvanny I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J Neurosci. 2009;29:1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Kai L, Hockberger PE, Wokosin DL, Surmeier DJ. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol Cell Neurosci. 2010;44:94–108. doi: 10.1016/j.mcn.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AJ, Signer ER. Huntington's disease: the challenge for cell biologists. Trends Cell Biol. 2000;10:531–536. doi: 10.1016/S0962-8924(00)01853-5. [DOI] [PubMed] [Google Scholar]
- Tu H, Wang Z, Nosyreva E, De Smedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1, 4, 5-trisphosphate receptor isoforms. Biophys J. 2005;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Gray M, Lu XH, Cantle JP, Holley SM, Greiner E, Gu X, Shirasaki D, Cepeda C, Li Y, Dong H, Levine MS, Yang XW. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat Med. 2014;20:536–541. doi: 10.1038/nm.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayán J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, et al. U.S.-Venezuela Collaborative Research Project Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Tang T, Bezprozvanny I. Evaluation of clinically relevant glutamate pathway inhibitors in in vitro model of Huntington's disease. Neurosci Lett. 2006;407:219–223. doi: 10.1016/j.neulet.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington's disease. Mol Neurodegener. 2008;3:15. doi: 10.1186/1750-1326-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Jeong HK, Bulin SE, Kwon SW, Park JH, Bezprozvanny I. Ginsenosides protect striatal neurons in a cellular model of Huntington's disease. J Neurosci Res. 2009;87:1904–1912. doi: 10.1002/jnr.22017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem Biol. 2011;18:777–793. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/S0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li Q, Graham RK, Slow E, Hayden MR, Bezprozvanny I. Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease. Neurobiol Dis. 2008a;31:80–88. doi: 10.1016/j.nbd.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Das S, Li QZ, Dragatsis I, Repa J, Zeitlin S, Hajnóczky G, Bezprozvanny I. Elucidating a normal function of huntingtin by functional and microarray analysis of huntingtin-null mouse embryonic fibroblasts. BMC Neurosci. 2008b;9:38. doi: 10.1186/1471-2202-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010;30:8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, Bezprozvanny I. Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer's disease. J Neurosci. 2015;35:13275–13286. doi: 10.1523/JNEUROSCI.1034-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]