

Abstract
Recent interest in the antidepressant and anti-stress effects of subanesthetic doses of ketamine, an NMDA receptor antagonist, has identified mechanisms whereby ketamine reverses the effect of stress, but little is known regarding the prophylactic effect ketamine might have on future stressors. Here we investigate the prophylactic effect of ketamine against neurochemical and behavioral changes that follow inescapable, uncontrollable tail shocks (ISs) in Sprague Dawley rats. IS induces increased anxiety, which is dependent on activation of serotonergic (5-HT) dorsal raphe nucleus (DRN) neurons that project to the basolateral amygdala (BLA). Ketamine (10 mg/kg, i.p.) administered 2 h, 1 week, or 2 weeks before IS prevented the increased extracellular levels of 5-HT in the BLA typically produced by IS. In addition, ketamine administered at these time points blocked the decreased juvenile social investigation produced by IS. Microinjection of ketamine into the prelimbic (PL) region of the medial prefrontal cortex duplicated the effects of systemic ketamine, and, conversely, systemic ketamine effects were prevented by pharmacological inhibition of the PL. Although IS does not activate DRN-projecting neurons from the PL, IS did so after ketamine, suggesting that the prophylactic effect of ketamine is a result of altered functioning of this projection.
SIGNIFICANCE STATEMENT The reported data show that systemic ketamine, given up to 2 weeks before a stressor, blunts behavioral and neurochemical effects of the stressor. The study also advances understanding of the mechanisms involved and suggests that ketamine acts at the prelimbic cortex to sensitize neurons that project to and inhibit the DRN.
Keywords: anxiety, ketamine, serotonin, stress resilience
Introduction
Low doses of the nonselective NMDA antagonist ketamine have rapid antidepressant action in the treatment resistant depressive patients (Berman et al., 2000), an effect that persists 1–2 weeks (Zarate et al., 2006; Price et al., 2009). Ketamine has also produced therapeutic effects with regard to anxiety disorders, such as posttraumatic stress disorder (Zarate et al., 2006; Price et al., 2009), suggesting action at neural processes that underlie both types of disorders. Unfortunately, ketamine has properties that limit its usefulness as a clinical agent (Duman, 2014). This has led to an effort to understand the mechanism(s) by which ketamine is effective, with the hope of optimizing pharmacological treatments with fewer negative outcomes.
Peripheral injections of low dose of ketamine reduce depressive-like behavior in a variety of paradigms, including the forced swim test (Garcia et al., 2008; Engin et al., 2009) and the tail suspension test (da Silva et al., 2010). Ketamine has also been shown to reduce anxiety-like behavior (Engin et al., 2009). In these experiments, ketamine has simply been administered 30–60 min before the behavioral testing. Given a 2.5 h plasma terminal half-life in humans (Mion and Villevieille, 2013), the animals were tested while under the acute effects of ketamine. The effectiveness of ketamine in reversing the effects of acute and chronic stressors has also been assessed. With regard to acute stressors, a number of studies have exposed mice and rats to a single session of inescapable shocks (ISs) and examined a number of behavioral consequences of the IS anywhere from 1 to 18 d later (Maeng et al., 2008; Beurel et al., 2011; Koike et al., 2011). In all of these studies, ketamine was administered shortly before the behavioral testing rather than before the IS, and the IS-induced changes were reversed. With regard to chronic or repeated stressors, a single dose of ketamine given after 21 d of stressor exposure has been found to rapidly reverse the behavioral changes that are produced by the stressor regimen (Li et al., 2011).
Recently, there has been a great deal of interest in discovering manipulations and treatments that might increase resistance/resilience to the effect of future stressors, yet there has been little work directed at whether ketamine might also blunt the effect of future stressors in addition to reversing the effects of stressors that have already occurred in the past. To our knowledge, only the recent study by Brachman et al. (2015) has dealt with possible prophylactic effects of ketamine. Here we explore this prophylactic phenomenon with a different species and stressor and begin to study the mechanism of prophylaxis. The studies determine whether ketamine delivered before IS would block or reduce the behavioral effect of IS and the duration of any such effect. We have shown that the activation of serotonin (5-HT) neurons in the dorsal raphe nucleus (DRN) is necessary and sufficient for the production of the behavioral sequelae of IS (for review, see Maier and Watkins, 2005). More specifically, increases in 5-HT in the basolateral amygdala (BLA), a DRN projection region, is critical in the production of the behavioral endpoint measured here, juvenile social investigation (JSI; Amat et al., 1998; Christianson et al., 2010). Thus, we measured extracellular levels of 5-HT in the BLA during IS. It has been suggested that peripheral ketamine injections lead to changes in the medial prefrontal cortex (mPFC) that may be key to its clinical effect (Li et al., 2010; Duman and Li, 2012; Duman et al., 2012; Dwyer and Duman, 2013; Zunszain et al., 2013; Duman, 2014). Therefore, we sought to determine whether the mPFC is a critical site of action of ketamine in a prophylactic preparation. We focused on the prelimbic (PL) region of the mPFC because the PL has proved critical to the amelioration of the effects of IS produced by behavioral manipulations (Christianson et al., 2014).
Materials and Methods
Subjects
Male Sprague Dawley rats (Harlan Laboratories), weighing 275–350 g, were housed two per cage on a 12 h light/dark cycle (on at 7:00 A.M. and off at 7:00 P.M.). Experiments were conducted between 9:00 A.M. and 4:00 P.M.. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and was approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
Overall organization
The first set of experiments examined whether systemic ketamine would modify behavioral and neurochemical effects of IS. Ketamine (10 mg/kg, i.p.; Li et al., 2010) was administered 2 weeks, 1 week, or 2 h before IS, and anxiety was evaluated by the JSI test 24 h after the stressor. BLA extracellular 5-HT levels induced by IS were assessed in a separate set of rats using in vivo microdialysis. The second set of experiments investigated whether the PL contributes to the effects of systemic ketamine administration. The experiments assessed the following: (1) whether local ketamine microinjection in the PL 90 min before stress (time before IS was reduced from 2 h above because of the more rapid action of direct brain injection) would blunt the behavioral and neurochemical effects of IS; and (2) whether systemic ketamine effects require an intact PL. This was achieved by reversibly inactivating the PL cortex with muscimol immediately before systemic ketamine, both occurring 2 h before the IS treatment. JSI was assessed 24 h later. Additionally, to test for specificity of the behavioral effect of ketamine on the PL cortex, muscimol was infused 2 mm caudal to the PL infusion, within area 2 of the cingulate (Cg2) cortex, as well as in the infralimbic (IL) cortex at the same anteroposterior (AP) site as PL infusions. These additional infusion tests for specificity were followed, as before, by systemic ketamine, IS 2 h later, and JSI the next day. Muscimol was chosen for the present purpose because Fuchikami et al. (2015) found that muscimol prevented the Fos induction in the mPFC produced by ketamine. In a final experiment, after DRN deposit of the retrograde tracer Fluorogold (FG), we examined whether ketamine would directly cause activation of DRN-projecting PL neurons or perhaps would sensitize these neurons to activation by IS. This was done because neurons projecting from PL to DRN are thought to inhibit stressor-induced 5-HT neuronal activation (Baratta et al., 2009). Neuronal activity was estimated by quantifying the early immediate gene product Fos in FG-labeled cells immediately after IS, with previous ketamine (Ket) or vehicle (Veh) treatments.
In all the initial experiments, IS consisted of a series of fixed-duration tail shocks. However, IS is often delivered as shocks of durations that are yoked to subjects that can terminate each of the tail shocks with an instrumental escape response. This ES versus yoked IS design is used so that the effect of behavioral control over the shocks can be studied. Although behavioral control over stressors is not an issue here, it is important for future studies to ensure that any prophylactic effects of ketamine are not specific to fixed-duration inescapable tail shocks, Thus, in one of the experiments, IS consisted of tail shocks whose durations were yoked to subjects that could terminate each of the tail shocks with a behavioral response [escapable shock (ES)]. As has been reported often, the ability to exert behavioral control over shock duration (ES) blocks the typical consequences of the tail shocks (Maier and Watkins, 2005). Thus, this experiment also allowed assessment of whether ketamine might interact with behavioral control in such a way as to reduce or eliminate the beneficial effects of being able to escape. It should be noted that a large number of experiments have found that ISs delivered in this yoked manner have the identical behavioral and neurochemical outcomes as do ISs delivered as fixed-duration shocks. This has been demonstrated specifically for both the behavioral and neurochemical endpoints measured in the present series of experiments (Amat et al., 1998, 2014).
Drug administration
Ketamine (Ketaset; Boehringer Ingelheim) was administered at 10 mg/kg (intraperitoneally) or microinjected into the PL region at 1, 10, 100, or 1000 ng or saline vehicle, in 0.5 μl volume, for a dose–response assessment. After the dose–response experiment, all other PL ketamine microinjections were 10 ng, which was found to be the effective dose. Muscimol (500 ng/side in 0.5 μl; Sigma) or saline was microinjected bilaterally into the PL, Cg2, or IL cortices, a dose found sufficient previously to inactivate the mPFC (Amat et al., 2006).
Stress procedure
Inescapable tail shock was administered as described previously (Amat et al., 2005; Christianson et al., 2010). Briefly, rats were placed in a loose Plexiglas restraint tube (8 × 18 cm, diameter × length) with a rod extending from the rear to which the tail was taped and affixed with copper electrodes. One hundred 5-s shocks were delivered on a variable-interval 60 s schedule. The shock intensity was 1 mA for the first 33 shocks, 1.3 mA for the next 33 shocks, and 1.6 mA for the final 34 shocks. Non-shocked home-cage control (HC) rats remained undisturbed in the colony.
To examine the effects of ketamine on ES as well as equated IS, we used a yoking procedure, as described previously (Amat et al., 2005). Rats were placed in Plexiglas box (14 × 11 × 17 cm) with a wheel mounted in the front. In the stressor control condition, the rat could turn the wheel to terminate shock and escape the stressor (ES). An inescapable stress (IS) subject was yoked to each ES subject and received the identical series of shocks as did the ES rat, but could not escape. The treatment consisted of 100 trials with an average inter-trial interval of 60 s. Shocks began simultaneously for both rats in a pair and terminated for both whenever the ES rat met a wheel-turn response criterion. Initially, the shock was terminated by a quarter turn of the wheel. The response requirement was increased by one-quarter turn when each of three consecutive trials was completed in fewer than 5 s. Subsequent latencies under 5 s increased the requirement by 50% up to a maximum of four full turns. If the requirement was not reached in fewer than 30 s, the shock was terminated and the requirement reduced to a single quarter turn. This procedure was used to ensure that the ES rats learned an operant response. Shock intensity was the same as described above. It should be noted that this procedure produces yoked ISs that are similar to the fixed-duration tail shocks.
JSI tests
Social investigation testing was conducted 24 h after the stress procedure, as described previously (Christianson et al., 2010). Any rats showing signs of injury after the stress session were excluded (these include irritated/inflamed hindpaws and sometimes toenails). Every effort is made to reduce the frequency of these events, but nonetheless this occurred in a few cases in the present experiments. Social interaction is particularly sensitive to inflammation and social behavior is disrupted when the test subject tends to a paw for significant periods during the test. Briefly, each experimental subject was allocated to a single plastic cage with shaved wood bedding. To begin, the test rats were placed into the test cage, and, after 45 min, a 28 ± 2-d-old juvenile was introduced to the cage for 3 min, and an observer, blind to treatment, timed exploratory behaviors (sniffing, pinning, and allogrooming) initiated by the adult. Juveniles were used for multiple tests but were never used twice for the same adult rat. The testing order was counterbalanced for stress and drug treatments.
Intracerebral cannula placement and microinjection
Under inhaled isoflurane (2–5% v/v in O2) anesthesia, rats were placed in a stereotaxic frame, and the skull was leveled by adjusting the incisor so that bregma and lambda were in the same horizontal plane. Microdialysis and microinjection guide cannulae were implanted and fixed in place with stainless steel screws and acrylic cement as described previously (Amat et al., 2006). For microdialysis, a cannula guide (SciPro) was implanted with the tip terminating just above the BLA [from bregma in mm: AP, −3.0; mediolateral (ML), 4.8; dorsoventral (DV), −6.2]. A screw cap of a 15 ml conical centrifuge tube, with the central portion removed, was also affixed to the skull so that its threads were exposed and it encircled the cannula guide. This was done so that the skull assembly could be protected during microdialysis. For microinjection, cannulae (26 g; Plastics One) were inserted bilaterally into the PL (from bregma in mm: AP, 2.7; ML, ±0.5; DV, −3.5 from the skull surface), into the Cg2 for a site specificity control (in mm: AP, 0.5; ML, ±0.5; DV, 2.5 from the skull surface) or into the IL cortex (in mm: AP, 2.7; ML, ±3.3; DV, 4.5 from the skull at a 30° angle). All coordinates were based on the atlas of Paxinos and Watson (1998). After surgery, rats were inoculated with 0.25 ml/kg (subcutaneous) penicillin (Combi-Pen; Agrilabs) and the analgesic Loxicam (0.5 mg/kg; Norbrook Laboratories). Rats were allowed to recover for 1–2 weeks after surgery before experimentation. Microinjections were made with a 25 μl Hamilton syringe in a micromanipulator (model 5000; David Kopf Instruments) connected to a 33 g injector by polyethylene-50 tubing. Injectors protruded 1 mm past the cannula end, and 0.5 μl was infused per side at a rate of ∼1 μl/min with an additional minute allowed for diffusion. Rats in the vehicle control groups received equal volume injections of saline. Rats were restrained gently in a cloth towel during injections. At the conclusion of the experiment, rats were overdosed with sodium pentobarbital (60 mg/kg), and brains were removed and frozen. Forty micrometer sections were stained with cresyl violet and inspected for cannula placement verification. Data are included only from rats with the injector tip within the PL region (Fig. 1A) and dialysis probe at least 60% within the BLA (Fig. 1B).
Figure 1.

Schematic representation of rat brain sections at the levels of the PFC and amygdala (adapted from Paxinos and Watson, 1998). A, Microinjection cannula placements. The black circles represent the sites of the injection cannula tips position in PL and Cg2 cortices, and the × symbols represent those in the IL cortex. B, Black bars represent the placement of microdialysis probes. Numerals indicate distance from bregma (in millimeters).
In vivo microdialysis
The afternoon before the experiment, the rats were wrapped gently in a towel, and a Scipro microdialysis probe (MAB 6.14.2: 0.6 mm in diameter, 2 mm membrane) was introduced through the cannula guide so that the membranous tip of the probe was within the BLA. A portion of a 15 ml Eppendorf tube was screwed onto the skull-mounted screw cap, through which the dialysis tubing, protected within a metal spring, entered and attached to the probe. Each animal was placed individually in a Plexiglas bowl (Bioanalytical Systems) and infused with artificial CSF (in mm: 145 NaCl, 2.7 KCl, 1.2 CaCl, and 1.0 KCl, pH 7.2) at a rate of 0.2 μl/min overnight. At ∼9:00 A.M., the next day, the flow rate was increased to 1.5 μl/min for 90 before sampling. The infusion flow remained constant throughout the experiment, and samples were collected every 20 min. Systemic ketamine was injected 2 h before stress or microinjected into the PL 90 min before stress. Before stress, three or four baseline samples were collected. The rats were then placed in Plexiglas wheel-turn boxes that were designed to accommodate the dialysis tubing and had the wheel locked. There they received 100 ISs. Five samples were collected during the stress session. After the stress session, the rats were transferred back to the Plexiglas bowls in which three or six post-stress samples were collected. During collection of the last sample, brisk movements of the skull-mounted screw cap were performed to test for possible 5-HT increases attributable to rat head movement during the dialysis. The data from the rat were discarded if that procedure caused 5-HT increases. The dialysis flow was lowered to 0.2 μl/min overnight for a group of rats to continue dialysis the following day to collect four additional baseline samples.
5-HT quantification
5-HT concentrations were determined as described previously (Amat et al., 1998, 2005) by HPLC with electrochemical detection. The system consisted of an ESA 5600A Coularray detector with an ESA 5014B analytical cell and an ESA 5020 guard cell. The column was an ESA MD-150 (C-18, 3 μm, 150 × 3.2 mm) maintained at 40°C, and the mobile phase was the ESA buffer MD-TM. The analytical cell potentials were kept at −100 and +200 mV and the guard cell at +220 mV. Dialysate (25 μl) was injected with an ESA 542 autosampler that kept the dialysates at 6°C. External standards (Sigma) were run each day to quantify 5-HT by means of peak height, using an ESA software, with the experimenter being blind to experimental condition.
Microinjection of retrograde tracer
Under the same anesthetic and postoperative procedures as above, a small craniotomy (1.0 mm diameter) was made to allow for penetration of a needle (31 gauge, 45° bevel) attached to a 10 μl Hamilton syringe. The needle was placed stereotaxically in the DRN (AP, −8.0 mm from bregma; DV, −6.7 mm from the skull surface, at midline). Two hundred nanoliters of a 2% solution of FG (Fluorochrome) dissolved in a 0.9% saline buffer was injected over the course of 3 min and allowed to diffuse for an additional 10 min using a microinjection pump (UMP3-1; World Precision Instruments).
Tissue preparation
To quantify stressor-induced Fos, rats were given an overdose of sodium pentobarbital (60 mg/kg, i.p.), perfused transcardially with 100 ml ice-cold 0.9% saline, followed by 250 ml 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH ∼7.4. Brains were removed and postfixed overnight in the same fixative and transferred to a 30% sucrose solution in 0.1 m PB and then stored at 4°C until sectioning. Coronal brain sections containing both PL and IL, as well as separate sections containing DRN, were obtained at 40 μm. PL and IL tissue used for immunohistochemistry was stored at 4°C in cryoprotectant, and DRN tissue was mounted directly onto Superfrost Plus slides (Fisher Scientific) and coverslipped with Vectashield (Vector Laboratories) mounting medium.
Immunohistochemistry.
Staining for Fos was performed using a general immunofluorescence protocol. After a series of washes in PBS containing 0.5% Triton X-100, slices were incubated overnight in a 0.01 m PBS blocking solution containing 2% normal goat serum, 0.5% Triton X-100, and 2.5% bovine serum albumin at 4°C. Then, slices were washed in PBS and incubated for 24 h at room temperature (RT) in rabbit polyclonal primary antibody (1:2000; Santa Cruz Biotechnology) in blocking solution. After a series of PBS washes, slices were incubated for 2 h at RT in Alexa Fluor 546 goat anti-rabbit secondary antibody (1:250; Life Technologies). After a series of PBS washes, tissue was floated onto slide glass and coverslipped.
Image analysis.
Brain sections were observed using an epifluorescence microscope (Zeiss Axio Imager Z1; Zeiss), and images were captured using Axio Vision software (Zeiss). All monochromatic digital images of PL and IL were captured using a 20× objective [numerical aperture (NA) 0.8]. The precise location of the FG deposit in the DRN was observed using a 5× objective (NA 0.16). For imaging and quantification of PL and IL (taken between AP 2.5 and 2.75 mm relative to bregma), boundaries were based on those described previously (Baratta et al., 2009) with consultation to the brain atlas (Paxinos and Watson 1998). FG-positive cell bodies in the PL and IL were detected using a BFP filter set [excitation bandpass (BP), 377/28 nm; beam splitter, 403 nm; emission BP, 464/100 nm (Zeiss)] and were pseudocolored blue (FG; Fig. 2A). Fos-stained nuclei were detected using a Cy3 filter [excitation BP, 550/25 nm; beam splitter, 570 nm; emission BP, 605/70 nm (Zeiss)] and were pseudocolored red (Fos; Fig. 2A). FG-positive and Fos-positive cells in each subregion were quantified and recorded separately. Colocalization of FG and Fos was identified when a magenta cell body representing the intermixture of the two fluorophores was observed and verified to be overlapping FG and Fos (Fig. 2A, FG + Fos). For each subject, two separate counts were taken from PL and two separate counts from IL in sections between AP 2.5 and 2.75 mm relative to bregma. Both regions were quantified to determine whether neuronal activation was specific to the PL–DRN pathway or rather a more generalized activation of the mPFC. The two counts for each subregion were averaged and used for the statistical analysis. Representative fluorescent photomicrographs showing an FG deposit in the DRN are shown in Figure 2A.
Figure 2.
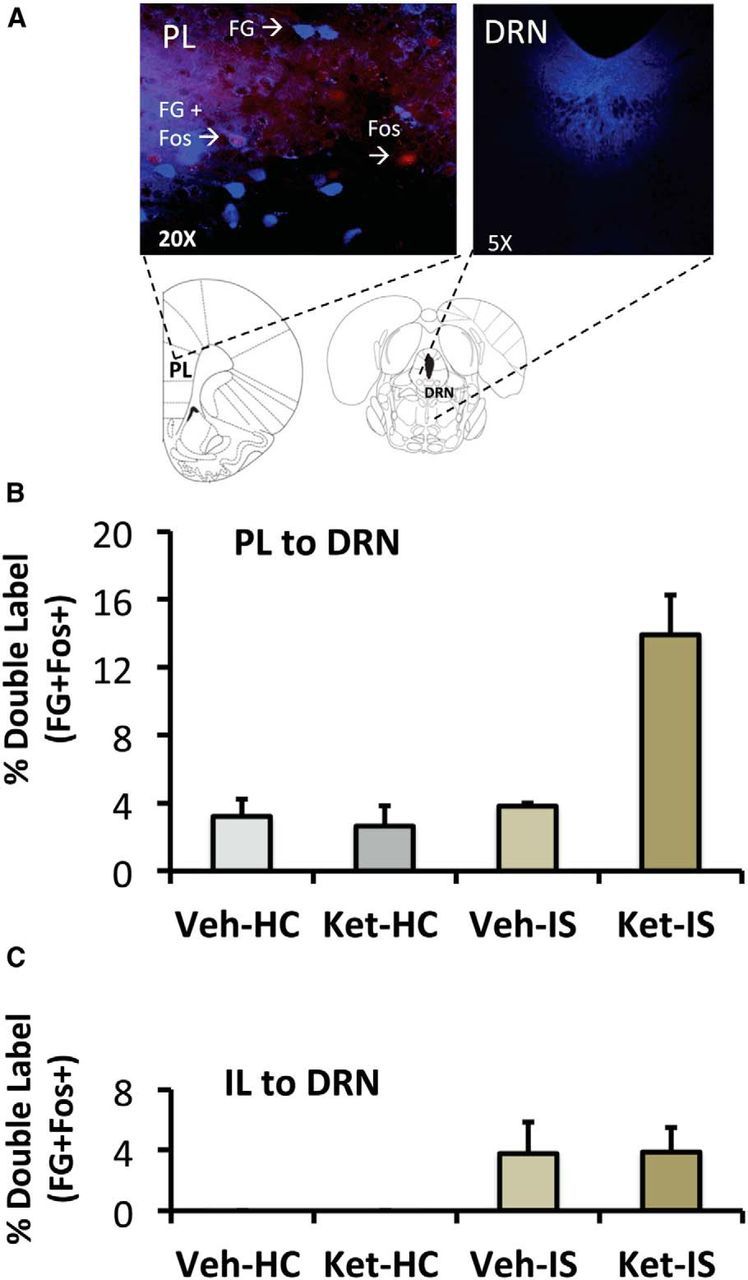
Effect of systemic ketamine on mPFC neurons projecting to the DRN in IS-treated rats. A, Representative fluorescence photomicrograph showing an FG-immunoreactive cell, a Fos-immunoreactive (Fos) cell, and a double-labeled (FG + Fos) PL neuron after intraperitoneal ketamine and IS and a representative fluorescent photomicrographs showing an FG deposit in the DRN. B, Effect of intraperitoneal ketamine (10 mg/kg) or saline vehicle, followed by HC or IS treatments on Fos activation in the PL–DRN pathway. The groups are vehicle injection followed 2 h later by home cage control (Veh-HC), ketamine injection followed 2 h later by HC (Ket-HC), vehicle injection followed by IS (Veh-IS), or ketamine injection followed by IS (Ket-IS). n = 7 for Veh-HC, Ket-HC, and Veh-IS, n = 8 for Ket-IS. C, Effect of intraperitoneal ketamine (10 mg/kg) or saline vehicle followed by HC or IS treatments on Fos activation in the IL–DRN pathway. Same groups as in B. n = 6 for Veh-HC and Ket-HC, n = 7 for Veh-IS, and n = 8 for Ket-IS.
Statistical analysis
Data were analyzed by two-way ANOVA or repeated-measures ANOVA, followed by Fisher's protected least significant difference test (PLSD) post hoc comparison (α set at 0.05).
Results
Effect of systemic ketamine
Behavior
Rats received either ketamine (10 mg/kg) or vehicle (intraperitoneal). Then, 2 h, 1 week, or 2 weeks later, they received IS or home-cage control treatment. JSI was assessed 24 h later. Thus, the design was a 2 (ketamine or vehicle) × 2 (IS or home-cage control) × 3 (2 h, 1 week, or 2 week) factorial. Figure 3 shows the time spent in social investigation for the 12 groups. As is typical, exposure to IS reduced JSI. Ketamine alone did not influence social investigation at any of the time intervals between ketamine and testing. Importantly, previous ketamine administration completely blocked the behavioral effect of IS and did so at all intervals between ketamine and IS; the proactive effects of ketamine had not diminished even when ketamine was given 2 weeks before IS. ANOVA yielded significant effects of IS (F(1,84) = 10.88, p < 0.001), ketamine (F(1,84) = 7.16, p < 0.01), and the interaction between IS and ketamine (F(1,84) = 14.15, p < 0.001). Post hoc multiple comparisons indicate that the three groups given vehicle–IS differed from all the others (p values <0.05) but did not differ among themselves. That is, all three ketamine groups given IS differed from the IS groups not given ketamine, and none of the IS groups given ketamine differed from nonshocked controls.
Figure 3.

Effect of intraperitoneal ketamine (10 mg/kg) or vehicle injection 2 h, 1 week, or 2 weeks before IS or control treatment on JSI measured 24 h after stress. ket-HC, HC animals given intraperitoneal ketamine; veh-HC, control animals given intraperitoneal saline vehicle; veh-IS, animals given intraperitoneal saline vehicle before IS; ket-IS, subjects give intraperitoneal ketamine before IS. The intraperitoneal injections were given 2 h, 1 week, or 2 weeks before IS (n = 8, for all groups).
BLA 5-HT release
Ketamine or vehicle was given 2 h before IS or home-cage control treatment and BLA 5-HT was quantified. There were no differences in absolute 5-HT baseline concentrations between the groups (all F values <1). The results are shown in Figure 4A, which depicts the extracellular levels of 5-HT as a percentage of baseline. IS produced a large increase in 5-HT that persisted after the IS ended and the subjects were removed from the tail shock apparatus (Veh–IS). Ketamine or vehicle by themselves had no effect on 5-HT (Ket–HC, Veh–HC), but ketamine almost completely blocked the increase produced by IS (Ket–IS). Repeated-measures ANOVA identified a main effect of stress in the vehicle-injected group (F(3,22) = 46.4, p < 0.001). Fisher's PLSD revealed significant differences between the vehicle-stressed group and the ketamine-stressed group, and the nonstressed vehicle or ketamine groups (p < 0.001), which did not differ among themselves. In a second experiment, rats were given ketamine or vehicle either 1 or 2 weeks before IS. Groups receiving home-cage control treatment during dialysis 1 or 2 weeks after ketamine were not included, because ketamine did not by itself alter BLA 5-HT during control treatment even when given 2 h before, in the experiment above. In addition, previous ketamine did not influence basal 5-HT concentrations measured before the onset of IS in the present experiment. Figure 4B shows 5-HT levels in animals given ketamine (1 week Ket and 2 week Ket) or vehicle (1 and 2 week Veh), either 1 or 2 weeks before IS. The two groups given vehicle were pooled because they did not differ, and the typical 5-HT increase during and after IS is shown in the figure. Ketamine given either 1 or 2 weeks before IS almost completely blocked the 5-HT increase produced by the stressor. Repeated-measures ANOVA on 5-HT during and after the stressor for the groups given ketamine 1 and 2 weeks before IS identified a main effect of ketamine (F(2,18) = 14.8, p < 0.0002) and a main effect of stress (F(7,14) = 6.1, p < 0.0001). Fisher's PLSD indicates that ketamine injected either 1 or 2 weeks before stress produced an effect different from vehicle, but there was no difference between them.
Figure 4.

A, Effect of intraperitoneal ketamine (10 mg/kg) or vehicle injection given 2 h before IS (Ket-IS and Veh-IS, n = 8 for both groups) or before HC treatment (Ket-HC and Veh-HC, n = 5 for each group), on extracellular levels of 5-HT in the BLA. B, Effect of intraperitoneal ketamine 1 week (1 w ket) or 2 weeks (2 w ket) before IS, and vehicle 1 or 2 weeks before IS (1 and 2 w Veh), on extracellular levels of 5-HT in the BLA. The 1 week (n = 4) and 2 week (n = 3) vehicle groups were pooled because there was no difference between them. IS indicated by gray rectangle in both A and B.
PFC involvement in ketamine effects
Behavior after PL ketamine
In this experiment, the main goal was to determine whether ketamine microinjected in the PL would mimic the systemic effect and block the effects of IS. A second goal was to determine whether, as in clinical practice, ketamine would lose its effectiveness at higher doses. Finally, we sought to verify that ketamine would not alter the behavior of subjects given ES as opposed to yoked IS. Thus, this experiment used the ES, yoked IS, home-cage controls design. Zero, 1, 10, 100, or 1000 ng of ketamine was microinjected in the PL (Fig. 1A) 90 min before these treatments. Social investigation testing occurred 24 h later. However, we microinjected only 0, 10, or 1000 ng of ketamine in HCs and before ES to minimize the number of animals. As shown in Figure 5, IS, but not ES, reduced social investigation. Thus, the yoked ISs duplicated the effect of the fixed-duration ISs. Ketamine had no effects on the behavior of ES or control animals at any dose. Ten nanograms of ketamine blocked the effects of IS on social investigation, but neither higher or lower doses did so. One-way ANOVA for Veh–HC and the five ketamine doses found a significant main effect of ketamine infusion (F(5,48) = 8.1, p < 0.0001). Fisher's PLSD indicated that the Veh–HC and 10 ng–IS groups differed significantly from 1, 100, and 1000 ng of ketamine, which did not differ among themselves. Also, two-way ANOVAs for the 3 ketamine × 3 stress groups revealed significant main effects of ketamine infusion (F(2,65) = 12.7, p < 0.0001) and stress condition (F(2,65) = 12.4, p < 0.0001). Importantly, Fisher's PLSD indicated that the 10 ng Ket–IS group differed from the 0, 1, 100, and 1000 ng Ket–IS group, which did not differ among themselves.
Figure 5.

Effect of intra-PL ketamine microinjection (0, 1, 10, 100, and 1000 ng, in 0.5 μl bilaterally) 90 min before IS (black squares) on social investigation tested 24 h later. The HC (white triangles) and ES (white circles) groups did not include all the ketamine doses to minimize animal use. n = 10 for 10 and 1000 ng ketamine–IS, n = 8 for all other groups.
Effect of PL ketamine on BLA 5-HT release
This experiment sought to determine whether the effective intra-PL dose of 10 ng ketamine would block the increase in BLA 5-HT produced by IS. Thus, either 10 ng of ketamine or vehicle was microinjected 90 min before IS, and 5-HT was measured. There were no significant differences in absolute 5-HT baseline concentrations between groups after the 10 ng ketamine injection (all F values <1). Figure 6 shows the effect of the microinjection of 10 ng of ketamine into the PL region 90 min before IS on extracellular 5-HT concentrations in the BLA during and after IS. Clearly, intra-PL ketamine blocked the IS-induced increase in BLA extracellular 5-HT. Repeated-measures ANOVA on the IS post-stress period (day 1) indicated a main effect of ketamine (F(1,12) = 22.2, p = 0.0005). Ketamine did not have a significant effect on the day 2 basal levels (F(1,12) = 2.93, p = 0.1125).
Figure 6.

Effect on the BLA 5-HT of PL microinjections of ketamine [10 ng in 0.5 μl bilaterally (Ket, n = 6)] or saline vehicle (Veh, n = 8) 90 min before IS or control treatment. Also shown are baseline 5-HT levels 24 h later (day 2).
Effect of systemic ketamine with concomitant PL inhibition
The purpose of this experiment was to determine whether inactivation of the PL with muscimol would abrogate the protective effects of systemic ketamine. Five hundred nanograms of muscimol or saline was microinjected bilaterally into the PL, followed immediately by intraperitoneal ketamine or vehicle. IS or HC treatment was administered 2 h later. Thus, the design was a 2 (muscimol or vehicle) × 2 (ketamine or vehicle) × 2 (IS or home-cage control) factorial. In addition, a site specificity control was added as a ninth group. This group received muscimol, ketamine, and IS, but the muscimol was microinjected 2 mm caudal to PL in the Cg2 cortex. The data are shown in Figure 7A. The group given only saline vehicle injections followed by HC treatment (V/V–HC) indicates the control level of social investigation measured 24 h later. In the HC groups, neither systemic ketamine (V/K–HC), intra-PL muscimol (M/V–HC), or their combination (M/K–HC) altered the level of social investigation. As in previous data, exposure to IS reduced social investigation (V/V–IS), and muscimol in the PL (M/V–IS) did not influence the effect of IS. As before, systemic ketamine blocked the effects of IS (V/K–IS) so that now IS subjects interacted at control levels. The important new finding is that intra-PL muscimol completely eliminated the protective effect of systemic ketamine (M/K–IS). ANOVA indicated a main effect of stress (F(1,56) = 54.1, p < 0.0001), PL muscimol infusion (F(1,56) = 5.9, p < 0.05), and a stress × PFC infusion interaction (F(1,56) = 7.698, p < 0.01). Fisher's PLSD demonstrated that V/V–IS, M/V–IS, and M/K–IS differed from all other groups (p values < 0.05) but do not differ among themselves. Importantly, intra-PL muscimol before intraperitoneal ketamine (M/K–IS) prevented the stress-protective effect of ketamine (V/K–IS; p < 0.001). Importantly, muscimol microinjected 2 mm caudal to PL (Cg2 cortex) did not prevent the stress-protective effect of systemic ketamine, providing evidence of site specificity.
Figure 7.

Effect on JSI of mPFC microinjections of muscimol (0.5 μl bilaterally, 500 ng or saline vehicle) immediately followed by intraperitoneal injections of ketamine, 2 h before IS. JSI was measured 24 h after IS. In A, the groups include the following: PL saline and intraperitoneal saline in HC (V/V–HC), PL saline and intraperitoneal ketamine in HC (V/K–HC), PL muscimol and intraperitoneal ketamine in HC (M/K–HC), PL saline and intraperitoneal saline before IS (V/V–IS), PL saline and intraperitoneal ketamine before IS (V/K–IS), PL muscimol and intraperitoneal ketamine before IS (M/K–IS), and PL muscimol and intraperitoneal saline before IS (M/V–IS). For a site specificity control, muscimol was microinjected 2 mm caudal to the PL (into Cg2 cortex, site control), or into the IL cortex (B) at the same AP level as PL, followed by the same treatments as above. n = 7 for the site control group, n = 8 for all other groups.
Effect of systemic ketamine with concomitant IL inhibition
Given the close anatomical and functional relationship of IL to PL, IL inactivation with muscimol, followed by ketamine and IS treatments, as above, was performed. The data shown in Figure 7B demonstrates that IL cortex inactivation does not alter the stress-blunting effect of ketamine.
Effect of systemic ketamine on activity in the PL–DRN and IL–DRN pathways
As described above, FG was microinjected in the DRN before the experiment, which involved systemic ketamine or vehicle 2 h before IS or control treatment. We will describe results for the PL–DRN and IL–DRN pathways separately. To verify that the size of the FG deposits made in the DRN did not differ across groups, total FG-labeled cells were analyzed in the PL. There was no significant differences in the total number of PL–DRN FG-labeled cells between any of the groups. Fos was assessed immediately after IS (4 h after ketamine), and IS increased the total number of Fos-positive cells in the PL (F(1,25) = 51.50, p < 0.0001). Post hoc analyses revealed that IS groups had significantly greater Fos-immunoreactive cells in the PL relative to those that received HC (p < 0.001). The IS groups given ketamine or vehicle did not differ from one another, nor did the HC groups given ketamine versus vehicle (data not shown). Importantly, ketamine and IS interacted to increase Fos specifically in FG-labeled cells in the PL (Fig. 2B). The percentage of PL cells expressing Fos plus FG revealed a significant effect of stress treatment (F(1,25) = 17.97, p < 0.001), drug treatment (F(1,25) = 7.49, p < 0.05), and a stress × drug interaction (F(1,25) = 16.08, p < 0.05). Post hoc analyses found that the group that received ketamine and IS had a significantly higher percentage of PL–DRN double-labeled cells (p < 0.0001) compared with the group that received vehicle and HC, ketamine and HC, as well as the group that received vehicle and IS.
With regard to the IL, there were no significant differences between groups in the number of FG-labeled cells. As for the PL, IS increased the total number of Fos-positive cells in the IL (F(1,23) = 15.74, p < 0.001; data not shown). Post hoc analyses revealed that IS groups had a significantly greater number Fos-immunoreactive cells relative to those that received HC (p < 0.05). There was no effect of ketamine and no interaction between IS and ketamine. Interestingly, the pattern for double-labeled cells was different from that for the PL. Here, IS led to a significant increase in the number of double-labeled Fos-positive cells in both groups that received ketamine or vehicle (F(1,23) = 7.30, p < 0.05; Fig. 2C). That is, unlike the PL–DRN pathway, ketamine and IS did not interact to increase Fos specifically in FG-labeled cells.
Discussion
A subanaesthetic systemic dose of ketamine (10 mg/kg) administered 2 h, 1 week, and 2 weeks before IS blocked the behavioral effects of IS. These results support the findings of Brachman et al. (2015) using a different species, different stressor paradigm, and different behavioral endpoint, suggesting that the finding has generality and replicability.
It is known that that the reduced social investigation produced by IS is mediated by increased 5-HT release in the BLA produced by IS (Amat et al., 1998; Christianson et al., 2010). Thus, the present experiments assessed whether ketamine administered before IS would block or reduce the 5-HT increase in the BLA produced by IS. Ketamine given 2 h, 1 week, and 2 weeks before IS did indeed strongly reduce the increased levels of extracellular 5-HT in the BLA produced by IS. Finally, the data clearly implicate the PL region of the mPFC as a site of action for systemic ketamine. Microinjection of ketamine into the PL before IS blocked both the reduction in JSI and the increase in BLA 5-HT that are normally produced by IS. In addition, the effect of intra-PL ketamine revealed an inverted U-shaped dose–response curve, with both lower and higher doses being ineffective, as is the case with clinical efficacy. A PL site of action is consistent with the finding that intra-PL muscimol eliminated the efficacy of systemic ketamine in blunting the effects of IS. Strengthening the conclusion that the PL is here the effective site, microinjection of muscimol in neither the Cg2 nor the immediately adjacent IL had any effect at all in reducing the effect of systemic ketamine.
The FG/Fos experiment deserves special comment. FG was first injected into the DRN, and later ketamine or vehicle was given intraperitoneally 2 h before IS (2 h session) or control treatment. The purpose of this experiment was not to determine whether ketamine would induce Fos in PL or IL neurons. It is already known that systemic intraperitoneal ketamine does indeed do so. Fuchikami et al. (2015) reported increases in Fos in both the PL and IL 1 h after intraperitoneal ketamine at the same dose used here. Fos protein peaks at ∼1 h after Fos induction and returns to basal levels after 4 h (Reddy et al., 2013). In the experiment here, there was a 4 h interval between ketamine and when the animals were killed, and so it is not surprising that ketamine by itself did not increase Fos. The purpose was also not to determine IS effects, because it is already known that IS exactly as used here induces Fos in the PL and IL in unlabeled neurons. However, retrogradely labeled neurons from the DRN are activated by IS in the IL but not in the PL cortices (Baratta et al., 2009). This is exactly the pattern found here. The PL-to DRN pathway is critical because activation of this pathway inhibits the DRN (Hajós et al., 1998), and indeed, pharmacological activation of this pathway during IS blocks the behavioral and neurochemical effects of IS (for review, see Maier, 2015). The question here was whether previous ketamine would alter the pattern of IS effects, and we found that previous ketamine produced only one change: now IS activated neurons that project from the PL to the DRN. This is consistent with the idea that ketamine acts by altering PL pyramidal cells that can inhibit DRN 5-HT cells, so that now these PL cells become activated by IS. The mechanisms suggested here could be readily applied to the findings by Brachman et al. (2015).
It might be noted that Fuchikami et al. (2015) have very recently provided data showing that the direct effects of systemic ketamine on a number of depression and anxiety-related behaviors are mediated by action at the IL region of mPFC. By direct effect is meant that their studies assessed the effect of ketamine on behaviors such as forced swim test rather than stress-induced alterations in the behaviors, as was done here. It may be that ketamine has action at multiple sites, with the site that is critical for a particular paradigm varying with the paradigm. Here the proactive effects of systemic ketamine on stress-induced changes in a behavior known to be DRN 5-HT dependent was studied, and almost all of the mPFC input to the DRN derives from the PL, not the IL (Gabbott et al., 2005). Consistent with this conclusion, very few retrogradely labeled cells were found here in the IL. Had another paradigm been chosen, another region might have played the dominant role. However, it should also be noted that the PL and IL are interconnected intrinsically, with PL layer 5/6 projecting to IL layer 5/6, and IL layers 1–6 projecting to primarily PL layers 1, 3, and 5 (Jones et al., 2005; Hoover and Vertes, 2007). Moreover, the projections between regions synapse primarily on spines on pyramidal projecting neurons (Gabbott et al., 2003), and so anatomy exists to allow effects that begin at the PL to use the IL as an output pathway to other structures, and conversely.
Ketamine blunted the effect of IS occurring both shortly after drug administration and at a delay of 1 and 2 weeks. The only other manipulation of which we are aware that has this sort of effect is the presence of behavioral control over the stressor. Control is typically studied with a paradigm in which each of a series of tail shocks terminates whenever the subject turns a small wheel (ES). Thus, the subjects in this condition are given behavioral control over when each of the tail shocks terminates. A comparison group is yoked to the ES group and simply given the same shocks; here the subjects do not have control over shock durations (IS), but the durations are matched to the ES group. The presence of control blocks the 5-HT activation and behavioral effect of the tail shocks being experienced and, in addition, blocks the 5-HT activation and behavioral effects of IS occurring 7 d later (Amat et al., 2006). Importantly, the mechanism by which the experience of ES blocks the effects of the immediate stressor as well as the IS occurring later has been explored extensively. As noted above, IS produces behavioral effects such as reduced social investigation because it potently activates DRN 5-HT neurons. The presence of control blocks the effect of the stressor being experienced by inhibiting this excitation of DRN 5-HT neurons (Maier and Watkins, 2005). Importantly, control does so by activating mPFC pyramidal projections to the DRN that synapse preferentially on GABAergic neurons within the DRN that inhibit the 5-HT cells (Amat et al., 2005). Thus, for example, intra-mPFC muscimol microinjection blocks the protective effects of control (Amat et al., 2005). Previous experience with control blocks the effects of subsequent IS because it alters the PL–DRN projecting neurons so that now IS activates these cells, which it would not do without this previous experience (Baratta et al., 2009). A variety of findings suggest that the experience of control alters the PL–DRN projection so that it becomes responsive to uncontrollable stressors by inducing a plasticity process (for review, see Maier, 2015). Thus, mPFC microinjection of protein synthesis inhibitors (Amat et al., 2006), NMDA antagonists (Amat et al., 2014), and ERK inhibitors (Christianson et al., 2014), before the ES experience, all prevent ES-induced blockade of the neurochemical and behavioral effects of IS occurring 7 d later. Furthermore, the PL rather than the IL appears to be the critical site of action of these effects (Christianson et al., 2014). In addition, the experience of ES leads to the production of “plasticity proteins” in the PL (Christianson et al., 2014) and increases the excitability of layer 5/6 pyramidal neurons in the PL as measured in patch-clamp experiments (Varela et al., 2012). The outcome of this plasticity process is that now IS activates the glutamatergic PL-to-DRN pathway, an effect that IS does not have without previous ES, as assessed by Fos expression in cells retrogradely labeled from the DRN by FG (Baratta et al., 2009).
The data reported here suggest that ketamine might mimic the neural effects of behavioral control. As with behavioral control, ketamine blunted the 5-HT activation produced by IS and required action at the mPFC to do so. Moreover, as with control, ketamine led the stressor to activate PL neurons that project to the DRN. Perhaps ketamine also induces plasticity in this pathway as does the experience of control, a matter for future investigation. It has been argued that behavioral control, by inducing plasticity in mPFC projections, increases the connectivity from mPFC to limbic and brainstem stress-responsive structures, thereby increasing top-down inhibitory control over the activity of these structures during exposure to aversive stimuli (Maier, 2015). Duman (2014) has recently made a quite similar argument for the operation of ketamine, and the data reported here are fully in accord with this idea. Behavioral control and ketamine might well converge on the same resiliency circuitry.
Footnotes
This work was supported by National Institutes of Health Grant MH050479 (S.F.M.) and National Institute of Child Health and Human Development Grant T32 HD7289-30 (S.D.D.).
The authors declare no competing financial interests.
References
- Amat J, Matus-Amat P, Watkins LR, Maier SF. Escapable and inescapable stress differentially alter extracellular levels of 5-HT in the basolateral amygdala of the rat. Brain Res. 1998;812:113–120. doi: 10.1016/S0006-8993(98)00960-3. [DOI] [PubMed] [Google Scholar]
- Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci. 2005;8:365–371. doi: 10.1038/nn1399. [DOI] [PubMed] [Google Scholar]
- Amat J, Paul E, Zarza C, Watkins LR, Maier SF. Prior experience with behavioral control over stress blocks the behavioral effects of later uncontrollable stress: role of the ventral medial prefrontal cortex. J Neurosci. 2006;26:13264–13272. doi: 10.1523/JNEUROSCI.3630-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat J, Christianson JP, Aleksejev RM, Kim J, Richeson KR, Watkins LR, Maier SF. Control over a stressor involves the posterior dorsal striatum and the act/outcome circuit. Eur J Neurosci. 2014;40:2352–2358. doi: 10.1111/ejn.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratta MV, Zarza CM, Gomez DM, Campeau S, Watkins LR, Maier SF. Selective activation of dorsal raphe nucleus-projecting neurons in the ventral medial prefrontal cortex by controllable stress. Eur J Neurosci. 2009;30:1111–1116. doi: 10.1111/j.1460-9568.2009.06867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16:1068–1070. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachman RA, McGowan JC, Perusini JN, Lim SC, Pham TH, Faye C, Gardier AM, Mendez-David I, David DJ, Hen R, Denny CA. Ketamine as a prophylactic against stress-induced depressive-like behavior. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.04.022. doi: 10.1016/j.biopsych.2015.04.022. Advance online publication. Retrieved November 21, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JP, Ragole T, Amat J, Greenwood BN, Strong PV, Paul ED, Fleshner M, Watkins LR, Maier SF. 5-hydroxytryptamine 2C receptors in the basolateral amygdala are involved in the expression of anxiety after uncontrollable traumatic stress. Biol Psychiatry. 2010;67:339–345. doi: 10.1016/j.biopsych.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JP, Flyer-Adams JG, Drugan RC, Amat J, Daut RA, Foilb AR, Watkins LR, Maier SF. Learned stressor resistance requires extracellular signal-regulated kinase signaling in the prefrontal cortex. Front Behav Neurosci. 2014;8:348. doi: 10.3389/fnbeh.2014.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva FC, do Carmo de Oliveira Cito M, da Silva MI, Moura BA, de Aquino Neto MR, Feitosa ML, de Castro Chaves R, Macedo DS, de Vasconcelos SM, de França Fonteles MM, de Sousa FC. Behavioral alterations and pro-oxidant effect of a single ketamine administration to mice. Brain Res Bull. 2010;83:9–15. doi: 10.1016/j.brainresbull.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Duman RS. Pathophysiology of depression and innovative treatments: remodeling glutamatergic synaptic connections. Dialogues Clin Neurosci. 2014;16:11–27. doi: 10.31887/DCNS.2014.16.1/rduman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond B Biol Sci. 2012;367:2475–2484. doi: 10.1098/rstb.2011.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer JM, Duman RS. Activation of mammalian target of rapamycin and synaptogenesis: role in the actions of rapid-acting antidepressants. Biol Psychiatry. 2013;73:1189–1198. doi: 10.1016/j.biopsych.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin E, Treit D, Dickson CT. Anxiolytic- and antidepressant-like properties of ketamine in behavioral and neurophysiological animal models. Neuroscience. 2009;161:359–369. doi: 10.1016/j.neuroscience.2009.03.038. [DOI] [PubMed] [Google Scholar]
- Fuchikami M, Thomas A, Liu R, Wohleb ES, Land BB, DiLeone RJ, Aghajanian GK, Duman RS. Optogenetic stimulation of infralimbic PFC reproduces ketamine's rapid and sustained antidepressant actions. Proc Natl Acad Sci U S A. 2015;112:8106–8111. doi: 10.1073/pnas.1414728112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Bacon SJ. Areal and synaptic interconnectivity of prelimbic (area 32), infralimbic (area 25) and insular cortices in the rat. Brain Res. 2003;993:59–71. doi: 10.1016/j.brainres.2003.08.056. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Garcia LS, Comim CM, Valvassori SS, Réus GZ, Andreazza AC, Stertz L, Fries GR, Gavioli EC, Kapczinski F, Quevedo J. Chronic administration of ketamine elicits antidepressant-like effects in rats without affecting hippocampal brain-derived neurotrophic factor protein levels. Basic Clin Pharmacol Toxicol. 2008;103:502–506. doi: 10.1111/j.1742-7843.2008.00210.x. [DOI] [PubMed] [Google Scholar]
- Hajós M, Richards CD, Székely AD, Sharp T. An electrophysiological and anatomical study of the medial prefrontal cortical projection to the midbrain raphe nuclei in the rat. Neuroscience. 1998;87:95–108. doi: 10.1016/S0306-4522(98)00157-2. [DOI] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- Jones BF, Groenewegen HJ, Witter MP. Intrinsic connections of the cingulate cortex in the rat suggest the existence of multiple functionally segregated networks. Neuroscience. 2005;133:193–207. doi: 10.1016/j.neuroscience.2005.01.063. [DOI] [PubMed] [Google Scholar]
- Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224:107–111. doi: 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS. Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Maier SF. Behavioral control blunts reactions to contemporaneous and future adverse events: medial prefrontal cortex plasticity and a corticostriatal network. Neurobiol Stress. 2015;1:12–22. doi: 10.1016/j.ynstr.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci Biobehav Rev. 2005;29:829–841. doi: 10.1016/j.neubiorev.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings) CNS Neurosci Ther. 2013;19:370–380. doi: 10.1111/cns.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GR, Xie C, Lindaman LL, Coss D. GnRH increases c-fos half-life contributing to higher FSHβ induction. Mol Endocrinol. 2013;27:253–265. doi: 10.1210/me.2012-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela JA, Wang J, Christianson JP, Maier SF, Cooper DC. Control over stress, but not stress per se increases prefrontal cortical pyramidal neuron excitability. J Neurosci. 2012;32:12848–12853. doi: 10.1523/JNEUROSCI.2669-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zunszain PA, Horowitz MA, Cattaneo A, Lupi MM, Pariante CM. Ketamine: synaptogenesis, immunomodulation and glycogen synthase kinase-3 as underlying mechanisms of its antidepressant properties. Mol Psychiatry. 2013;18:1236–1241. doi: 10.1038/mp.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]