

Abstract
Many chronic pain disorders alternate between bouts of pain and periods of remission. The latent sensitization model reproduces this in rodents by showing that the apparent recovery (“remission”) from inflammatory or neuropathic pain can be reversed by opioid antagonists. Therefore, this remission represents an opioid receptor-mediated suppression of a sustained hyperalgesic state. To identify the receptors involved, we induced latent sensitization in mice and rats by injecting complete Freund's adjuvant (CFA) in the hindpaw. In WT mice, responses to mechanical stimulation returned to baseline 3 weeks after CFA. In μ-opioid receptor (MOR) knock-out (KO) mice, responses did not return to baseline but partially recovered from peak hyperalgesia. Antagonists of α2A-adrenergic and δ-opioid receptors reinstated hyperalgesia in WT mice and abolished the partial recovery from hyperalgesia in MOR KO mice. In rats, antagonists of α2A adrenergic and μ-, δ-, and κ-opioid receptors reinstated hyperalgesia during remission from CFA-induced hyperalgesia. Therefore, these four receptors suppress hyperalgesia in latent sensitization. We further demonstrated that suppression of hyperalgesia by MORs was due to their constitutive activity because of the following: (1) CFA-induced hyperalgesia was reinstated by the MOR inverse agonist naltrexone (NTX), but not by its neutral antagonist 6β-naltrexol; (2) pro-enkephalin, pro-opiomelanocortin, and pro-dynorphin KO mice showed recovery from hyperalgesia and reinstatement by NTX; (3) there was no MOR internalization during remission; (4) MORs immunoprecipitated from the spinal cord during remission had increased Ser375 phosphorylation; and (5) electrophysiology recordings from dorsal root ganglion neurons collected during remission showed constitutive MOR inhibition of calcium channels.
SIGNIFICANCE STATEMENT Chronic pain causes extreme suffering to millions of people, but its mechanisms remain to be unraveled. Latent sensitization is a phenomenon studied in rodents that has many key features of chronic pain: it is initiated by a variety of noxious stimuli, has indefinite duration, and pain appears in episodes that can be triggered by stress. Here, we show that, during latent sensitization, there is a sustained state of pain hypersensitivity that is continuously suppressed by the activation of μ-, δ-, and κ-opioid receptors and by adrenergic α2A receptors in the spinal cord. Furthermore, we show that the activation of μ-opioid receptors is not due to the release of endogenous opioids, but rather to its ligand-independent constitutive activity.
Keywords: adrenergic receptor, constitutive activity, hyperalgesia, kappa-opioid receptor, latent sensitization, mu-opioid receptor
Introduction
Latent sensitization is a physiological phenomenon that displays key features of chronic pain disorders: indefinite duration and episodic presentation (Taylor and Corder, 2014). It can be initiated by a wide variety of noxious stimuli, including paw incision (Li et al., 2001; Richebé et al., 2005; Rivat et al., 2007; Campillo et al., 2011), paw inflammation with complete Freund's adjuvant (CFA) (Corder et al., 2013) or carrageenan (Bessière et al., 2007; Le Roy et al., 2011), and nerve injury (Solway et al., 2011). These stimuli lead to a period of hyperalgesia ranging from several days (Li et al., 2001) to months (Yalcin et al., 2011), after which the animal enters a state of susceptibility to pain (“remission”) when hyperalgesia can be reinstated by opioid receptor antagonists that normally have no effect (Campillo et al., 2011; Corder et al., 2013). This indicates that hyperalgesia is still present during remission, but is suppressed by a compensatory activation of opioid receptors. Remarkably, reinstatement by opioid antagonists can be repeated any number of times over a period of at least 5 months (Campillo et al., 2011; Corder et al., 2013), suggesting that both the hyperalgesia and the opioid compensatory response last indefinitely. A recent study indicates that latent sensitization takes place in humans (Pereira et al., 2015).
Latent sensitization may be similar to hyperalgesic priming, a phenomenon elicited by injecting interleukin-6 or carrageenan in the paw of an animal, after which injections in the same paw of inflammatory compounds such as prostaglandin E2, 5-hydroxytriptamine, or adenosine produce prolonged hyperalgesia (Aley et al., 2000; Parada et al., 2005; Wang et al., 2013; Kim et al., 2015).
The type of opioid receptor that suppresses hyperalgesia during the remission phase of latent sensitization remains unclear. One study showed that the κ-opioid receptor (KOR) antagonist nor-binaltorphimine (nor-BNI) reinstated the hyperalgesia (Campillo et al., 2011), whereas another study showed that hyperalgesia can be reinstated by the selective μ-opioid receptor (MOR) antagonist CTOP (Corder et al., 2013). The involvement of other receptors known to produce analgesia at the spinal cord, such as α2A adrenergic receptors (α2ARs) (Stone et al., 1997; Pertovaara, 2006), has not been explored. Another important issue is whether the anti-hyperalgesia produced by MORs in latent sensitization is caused by the sustained release of opioid peptides (Le Roy et al., 2011) or by agonist-independent, constitutive activity of MORs (Connor and Traynor, 2010; Corder et al., 2013), a phenomenon previously observed in β-arrestin knock-out (KO) mice (Walwyn et al., 2007; Lam et al., 2011).
In this study, we used MOR KO mice to assess the contribution of MORs to anti-hyperalgesia during the remission phase of latent sensitization. The involvement of δ-opioid receptors (DORs), KORs, and α2ARs was examined using selective antagonists in mice and rats. MOR constitutive activity was investigated by using a MOR inverse agonist and a neutral antagonist; in mice lacking pro-enkephalin (pENK), pro-opiomelanocortin (pOMC), and pro-dynorphin (pDYN); by studying its internalization and phosphorylation in the spinal cord, and with patch-clamp recordings from dorsal root ganglion (DRG) neurons.
Materials and Methods
Animals.
All animal procedures were approved by the Animal Research Committee of University of California–Los Angeles (UCLA) and the Institutional Animal Care and Use Committee of the Veteran Affairs Greater Los Angeles Healthcare System and conformed to National Institutes of Health guidelines. Efforts were made to minimize the number of animals used and their suffering. Adult male WT mice were C57BL/6J (Jackson Laboratories). Male or female mice lacking MORs (MOR KO) (Matthes et al., 1996), pre-pro-enkephalin (pENK KO) (König et al., 1996), pro-opiomelanocortin (pOMC KO) (Rubinstein et al., 1996), and pre-pro-dynorphin (pDYN KO) (Sharifi et al., 2001) and their WT littermates and were bred from heterozygotes matings of fully backcrossed C56BL/6J mice by the Animal Breeding Core of the Center for the Study of Opioid Receptors and Drugs of Abuse, UCLA. Rats were male adult (2–4 months old) Sprague Dawley (Harlan Laboratories). Additional rats and mice were used to prepare spinal cord slices and DRG neuron cultures. Rats were given an antibiotic (enrofloxacin) and an analgesic (carprofen) twice daily for 3 d after surgical procedures.
Chemicals.
BRL44408 and naltrexone (NTX) were from Tocris Bioscience. Amastatin, captopril, CFA, [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), naltrindole, nor-BNI, phosphoramidon, and common reagents were from Sigma-Aldrich. 6β-naltrexol and JDTic were from the National Institute on Drug Abuse Drug Supply Program.
CFA injections.
CFA was injected undiluted in a volume of 5 μl for mice and 50 μl for rats. One hindpaw was injected subcutaneously using a 50 μl Hamilton syringe and a 26 G needle. The needle was inserted at an oblique angle from the heel in the middle of the paw near the base of the third toe. It was held in place for 15 s and then gently withdrawn.
Mechanical hyperalgesia measures.
In mice, mechanical allodynia was assessed using the up-down method (Chaplan et al., 1994). After 7 d of habituation to the tester and the testing room, mice were habituated for 30 min over 3 consecutive days to acrylic enclosures on an elevated metal grid (10 × 10 × 12 cm W × D × H; IITC Life Sciences). A set of calibrated plastic von Frey filaments (Touch-Test; North Coast Medical) was then used to measure allodynia using the up-down method and the data were analyzed by the Dixon algorithm (Dixon, 1965) to obtain the 50% paw withdrawal threshold (PWT).
In rats, mechanical allodynia was measured by the two-out-of-three method (Kingery et al., 2000; Michot et al., 2012; Jarahi et al., 2014). Rats were habituated for periods of 30 min for 3 d to acrylic enclosures on an elevated metal grid (10 × 20 × 12 cm W × D × H; IITC Life Sciences). A series of von Frey filaments were applied in ascending order (Touch-Test, 0.8, 1.0, 1.4, 2.0, 4.0, 6.0, 8.0, 10, 15 g) to the plantar surface of the hindpaw for a maximum period of 3 s. A withdrawal response was counted only if the hindpaw was completely removed from the customized platform. Each filament was applied three times and the minimal value that caused at least two responses was recorded as the PWT. The 15 g filament was taken as cutoff threshold.
Intrathecal catheters and injections.
Rats were implanted with chronic intrathecal catheters under isoflurane (2–4%) anesthesia (Størkson et al., 1996). After cutting the skin and muscle, a 20 G needle was inserted between the L5 and L6 vertebrae to puncture the dura mater. The needle was removed and the catheter (20 mm of PE-5 tube heat fused to 150 mm of PE-10 tube) was inserted into the subdural space and pushed rostrally to terminate over L5–L6. The PE-10 end of the catheter was tunneled under the skin and externalized over the head. The skin was sutured and the catheter was flushed with 10 μl of saline and sealed. Rats were housed separately and used 5–7 d after surgery. The presence of motor weakness or signs of paresis was established as the criterion for immediate euthanasia, but this did not occur in any of the rats. Intrathecal injection volume was 10 μl of injectate plus 10 μl of saline flush. Solutions were preloaded into a PE-10 tube and delivered within 1 min. The position of the catheter was examined postmortem and the following exclusion criteria were used: (1) loss of the catheter, (2) termination of the catheter inside the spinal cord, or (3) occlusion of the catheter tip.
Immunoprecipitation and Western blots.
The L4–S1 spinal cord segments were quickly isolated and frozen on dry ice. Frozen tissues (∼70–90 mg) were thawed in ice-cold homogenization buffer containing the following (in mm): 300 sucrose, 25 Tris-HCl, pH 7.5, 10 Na+-glycerophosphate, and 1 EDTA supplemented with protease inhibitors (complete protease inhibitor mixture;, Roche Diagnostics) and phosphatase inhibitors (10 mm NaF and Phosphatase Inhibitor Cocktail 2; Sigma-Aldrich). After homogenization with a Teflon-glass homogenizer, extracts were centrifuged at 5000 × g for 5 min at 4°C. The pellet was resuspended in ice-cold immunoprecipitation buffer containing 50 mm Tris-HCl, pH 7.5, 50 mm NaCl, and 1% NP-40 supplemented with proteinase and phosphatase inhibitors. The extract was briefly sonicated and set on ice for 10 min before centrifugation at 18,000 × g for 75 min at 4°C. The supernatant was precleared by incubating for 1 h with MagnaBind Protein A/G beads (Pierce) at 4°C with mixing. The supernatant was collected, 5 μg of anti-MOR antibody (ab134054; Abcam) was added, and the mixture was incubated at 4°C overnight with continuous mixing. The next day MagnaBind Protein A/G beads were added and the extracts incubated for 2 h 4°C. The MagnaBind beads were collected, washed 4 times with immunoprecipitation buffer, and the immunoprecipitate eluted with 1× sample buffer (NuPAGE; Invitrogen) containing 10 mm DTT. Equal portions (25%) of the immunoprecipitates were electrophoresed on 3–8% NuPAGE Tris-acetate SDS gels (Invitrogen) and proteins transferred to PVDF membranes. Blots were blocked with 5% BSA in Tris-buffered saline containing 0.05% Tween 20 and probed with antibodies to MOR (RA10104; Neuromics), p-Ser375-MOR (pMOR, RA18001; Neuromics) and p-Tyr416-Src family kinase (pSFK, 2101; Cell Signaling Technology). Blots were developed using peroxidase-conjugated, light-chain-specific mouse monoclonal anti-rabbit IgG (Jackson ImmunoResearch Laboratories) and ECL detection reagents (GE Healthcare Biosciences). Bands and intensities of pMOR and pSFK were expressed relative to that of total immunoprecipitated MOR.
Immunohistochemistry.
Spinal cord sections were labeled for MORs as described previously (Song and Marvizón, 2003b; Chen et al., 2007; Chen et al., 2008; Chen and Marvizón, 2009). Rats were killed with pentobarbital (100 mg/kg) and fixed immediately by aortic perfusion of 100 ml phosphate buffer (0.1 m sodium phosphate, pH 7.4) containing 0.01% heparin, followed by 400 ml of ice-cold fixative (4% paraformaldehyde, 0.18% picric acid in phosphate buffer). Spinal segments C2, T10, and L4 (located based on root identification) were postfixed, cryoprotected in 20% sucrose, embedded in Tissue-Tek (Sakura Finetek USA), and frozen on dry ice. Free-floating transversal sections (25 μm thick) were cut with a cryostat. Sections were washed twice with PBS and twice with PBS containing 0.5% Triton X-100, 0.01% thimerosal (PBS/Triton) and 5% normal goat serum (Jackson ImmunoResearch). Sections were incubated overnight in PBS/Triton with a MOR antiserum (1:6000) raised against amino acids 384–398 of rat MOR-1 (ImmunoStar), which has been characterized previously (Arvidsson et al., 1995; Spike et al., 2002). After 3 washes with PBS, sections were incubated for 2 h with Alexa Fluor 488 goat anti-rabbit IgG secondary antibody (1:2000; Invitrogen). After four more washes, sections were mounted in Prolong Gold (Invitrogen). All incubations and washes were done at room temperature.
MOR internalization.
MOR internalization was measured as described previously (Song and Marvizón, 2003a, 2005; Song and Marvizón, 2003b; Chen and Marvizón, 2009). MOR neurons with or without internalization were counted using an objective of 63× (numerical aperture 1.40) and a Zeiss Axio-Imager A1 microscope. Somata with >5 MOR-immunoreactive endosomes were considered to have internalization. An endosome was defined as a small region of bright MOR staining separated from the cell surface. Counting was done blind to the treatment. Four ipsilateral half-sections and four contralateral half-sections were counted for each spinal segment, counting all MOR neurons in laminas I–II.
Confocal microscopy and image processing.
Confocal images were acquired using a Zeiss LSM 710 confocal microscope with objectives of 10× and 63× oil (numerical apertures 0.3 and 1.4, respectively). The Alexa Fluor 488 fluorophore was excited by the 488 nm line of an argon laser and the emission window was 500–560 nm. The pinhole was 1.0 Airy unit: 37.7 μm for 10× and 50.7 μm for 63×. Images were acquired as stacks of sections of 1024 × 1024 pixels, spaced 5.89 μm for 10× and 0.38 μm for 63× (determined using the Nyquist formula). Images of the entire dorsal horn obtained with the 10× objective were used to show the location of the neurons imaged with the 63× objective. Imaris 6.1.5 software (Bitplane) was used to crop the 63× images and select one optical section through the center of the cell. Adobe Photoshop 5.5 was used to assemble the multipanel figures.
DRG preparation.
Constitutive MOR activity was studied in dissociated L4–L6 DRG neurons (Walwyn et al., 2007) from saline-injected or CFA-injected adult mice. DRG were collected in complete saline solution (CSS) containing the following (in mm): NaCl 137, KCl 5.3, MgCl2 1, sorbitol 25, HEPES 10, CaCl2 3) and incubated in collagenase (1.25 U of high thermolysin; Roche) with 250 nm EDTA for 20 min at 32°C, transferred to fresh CSS containing collagenase (1.25 U of medium thermolysin; Roche) with 250 nm EDTA and 0.25 U papain (Roche) and incubated for 10 min at 32°C. After 2 washes and physical trituration through a series of graded Pasteur pipettes, the cells were spun (1000 rpm, 3 min) and plated in Neurobasal/B27/Glumax/Antibiotic/Antimycotic (Life Technologies) supplemented with 10 ng/ml NGF (Life Technologies). All recordings were performed 40–48 h after plating.
Patch-clamp recordings.
Recordings were made from small- to medium-sized DRG neurons (30–70 pF) under whole-cell voltage-clamp conditions as described previously (Walwyn et al., 2007; Lam et al., 2011). The cells were perfused with an external solution containing 10 mm CaCl2, 130 mm tetraethyl ammonium chloride, 5 mm HEPES, 25 mm d-glucose, and 0.2 μm tetrodotoxin at pH 7.35 (Sigma-Aldrich). The patch electrode was filled with an internal solution composed of 105 mm CsCl, 40 mm HEPES, 5 mm d-glucose, 2.5 mm MgCl2, 10 mm EGTA, 2 mm Mg-ATP, and 0.5 mm GTP at pH 7.2 (Sigma-Aldrich). Episodic recordings were obtained using an Axopatch 200B patch-clamp amplifier set at a gain of 1.0, β = 0.1, and 2 kHz filter, and a Nidaq digitizer (NI USB-6221; National Instruments). Capacitance and series resistance were corrected; series resistance was compensated by 80–90% and included a 10 μs lag. Leak currents were subtracted using a P/6 protocol. Recorded signals were acquired and analyzed using winWCP software (University of Strathclyde, Glasgow, Scotland).
Data analysis.
Data were analyzed using Prism 6 and 7 (GraphPad Software) and are expressed as mean ± SEM. Statistical significance was set at 0.05. Statistical analyses consisted in most cases of repeated-measures two-way ANOVA followed by Holm-Sidak's post hoc tests. Experiments with one variable were analyzed by t test or one-way ANOVA, as appropriate.
Results
MOR KO mice do not fully recover from hyperalgesia in CFA-induced latent sensitization
Mice injected with CFA in the hindpaw develop mechanical hyperalgesia in the affected paw lasting for ∼3 weeks. Paw withdrawal responses then return to baseline, but the hyperalgesia can be temporarily reinstated by the MOR inverse agonist NTX (Corder et al., 2013). This suggests that this remission from hyperalgesia is not a return to normal physiological conditions, but rather a compensatory mechanism mediated by the sustained activation of MORs. This hypothesis predicts that mice lacking MORs (MOR KO) will not recover from CFA-induced hyperalgesia. We tested this by injecting MOR KO mice (n = 8) with 5 μl CFA subcutaneously in the hindpaw. Control groups were littermate WT mice injected in the hindpaw with the same volume of CFA (n = 9) or saline (n = 3). The CFA-injected WT and MOR KO mice, but not the saline-injected mice, developed mechanical hyperalgesia of the injected paw (Fig. 1A, statistical analysis in Table 1, row 1). In the WT mice, paw withdrawal responses returned to baseline by day 20. However, in the MOR KO mice, the responses recovered initially, but stayed below 50% of baseline for up to 55 d after CFA, when all mice were killed. Baseline values, measured on days −4, −1, and 0 before CFA, were not significantly different between the three groups of mice (Table 1, row 2). These results demonstrate that MOR activation contributes to the suppression of hyperalgesia during the remission phase of latent sensitization.
Figure 1.
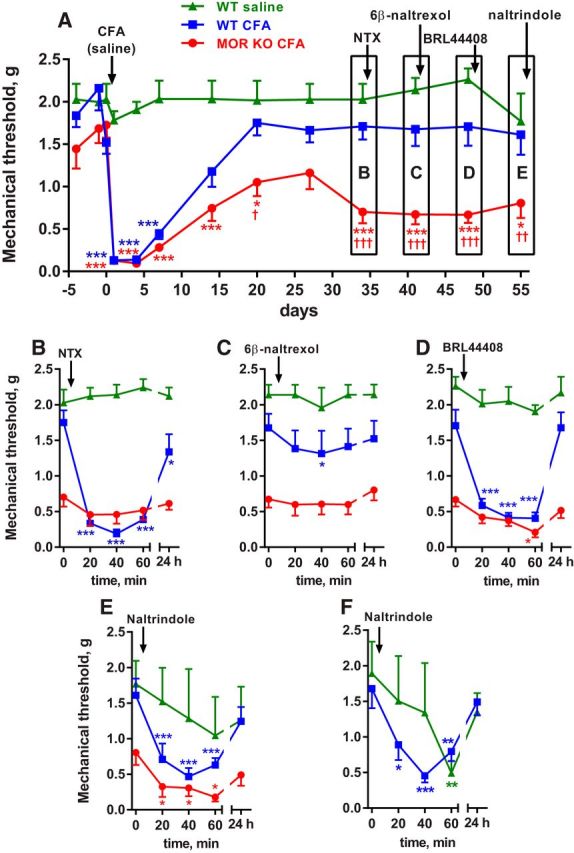
Latent sensitization in MOR KO mice and effect of receptor antagonists. MOR KO mice (MOR KO CFA, n = 8) or their WT littermates were injected with 5 μl CFA (WT CFA, n = 9) or 5 μl of saline (WT saline, n = 3) in the hindpaw. A, Responses to von Frey hairs were tested on the days indicated. Short-term responses to drugs are not shown. Holm-Sidak's post hoc tests: *p < 0.05, ***p < 0.001 compared with WT saline; †p < 0.05, ††p < 0.01, †††p < 0.001 compared with WT CFA. B, On day 34, the MOR inverse agonist NTX (3 mg/kg) was injected subcutaneously and von Frey responses tested every 20 min and at 24 h. C, On day 41, the MOR neutral antagonist 6β-naltrexol (10 mg/kg) was injected subcutaneously. D, On day 48, the α2AR antagonist BRL44408 (1 mg/kg) was injected subcutaneously. E, On day 55, the DOR antagonist naltrindole (3 mg/kg) was injected subcutaneously. F, New groups of WT mice were injected with 5 μl CFA (n = 14) or saline (n = 6) in the hindpaw and on day 29 with naltrindole (3 mg/kg, s.c.). Holm-Sidak's post hoc tests (for B–F): *p < 0.05, **p < 0.01, ***p < 0.001 compared with 0 min.
Table 1.
Two-way ANOVA of the data shown in Figure 1 (MOR KO mice)
| Row | Figure | Experiment | n | Variable 1 | Variable 2 | Interaction | Subject matching |
|---|---|---|---|---|---|---|---|
| 1 | 1A | MOR KO mice (CFA) | WT saline: 3 WT CFA: 9 KO CFA: 8 |
Time (CFA) | Group | p < 0.0001 | p = 0.001 |
| p < 0.0001 | p < 0.0001 | F(24,204) = 4.2 | F(17,204) = 2.5 | ||||
| F(12,204) = 14.4 | F(2,17) = 43.8 | ||||||
| 2 | 1A | MOR KO mice (baseline) | Time | Group | p = 0.18 | p = 0.044 | |
| p = 0.43 | p = 0.25 | F(4,34) = 1.7 | F(17,34) = 1.98 | ||||
| F(2,34) = 0.86 | F(2,17) = 1.5 | ||||||
| 3 | 1B | MOR KO mice (NTX) | Time (NTX) | Group | p < 0.0001 | p = 0.001 | |
| p < 0.0001 | p < 0.0001 | F(8,64) = 8.4 | F(16,64) = 2.58 | ||||
| F(4,64) = 8.35 | F(2,16) = 46.8 | ||||||
| 4 | 1C | MOR KO mice (6β-naltrexol) | Time (6β-naltrexol) | Group | p = 0.80 | p < 0.0001 | |
| p = 0.17 | p = 0.0023 | F(8,68) = 0.56 | F(17,68) = 21.7 | ||||
| F(4,68) = 1.65 | F(2,17) = 8.85 | ||||||
| 5 | 1D | MOR KO mice (BRL 44408) | Time (BRL44408) | Group | p < 0.0001 | p = 0.0265 | |
| p < 0.0001 | p < 0.0001 | F(8,68) = 6.2 | F(17,68) = 1.96 | ||||
| F(4,68) = 13.9 | F(2,17) = 65.0 | ||||||
| 6 | 1E | MOR KO mice (naltrindole) | Time (naltrindole) | Group | p = 0.19 | p < 0.0001 | |
| p < 0.0001 | p = 0.0069 | F(8,68) = 1.45 | F(17,68) = 0.45 | ||||
| F(4,68) = 9.28 | F(2,17) = 6.75 | ||||||
| 7 | 1F | WT mice (naltrindole) | WT saline: 6 | Time (naltrindole) | Group | p = 0.11 | p = 0.0002 |
| WT CFA: 14 | p = 0.0002 | p = 0.39 | F(4,72) = 2.0 | F(18,72) = 3.26 | |||
| F(4,72) = 14.4 | F(1,18) = 0.79 |
Each panel in Figure 1 (indicated in the second column) was analyzed independently by two-way ANOVA. In row 2, “baseline” refers to data obtained before the CFA injection. In the fourth column, n is the number of animals in the experiment. In the fifth and sixth columns, the variable is identified first: “time” refers to the time after the injection of the compound in parentheses; “group” is the animal group according to genotype and treatment. Subscript numbers in the F ratio are the degrees of freedom and the residual.
MOR constitutive activity in latent sensitization: effects of a MOR inverse agonist and a MOR neutral antagonist in mice
We investigated whether the continuous activation of MORs during the remission phase of latent sensitization is caused by sustained release of opioid peptides or by MOR constitutive activity. MOR activation by opioids should be blocked by both the MOR inverse agonist NTX and the MOR neutral antagonist 6β-naltrexol, whereas MOR constitutive activity should be blocked by NTX but not by 6β-naltrexol. In addition, the MOR specificity of NTX and 6β-naltrexol could be established by their lack of effect on MOR KO mice.
NTX (3 mg/kg, s.c.) was injected in the three groups of mice in Figure 1 on day 34 after CFA. In the CFA-injected WT mice, NTX produced a robust reinstatement of the hyperalgesia that had largely dissipated by 24 h (Fig. 1B, statistics in Table 1, row 3). In the saline-injected mice, NTX had no effect, confirming that the hyperalgesic effect of NTX requires previous sensitization by CFA. In the MOR KO mice, NTX also had no effect, showing that reinstatement by NTX is due to MOR inhibition.
6β-Naltrexol (10 mg/kg, s.c.) was injected to these three groups of mice on day 42 after CFA. 6β-Naltrexol did not reinstate hyperalgesia in the MOR KO mice or in the saline-injected controls (Fig. 1C, statistics in Table 1, row 4). In the CFA-injected WT mice, 6β-naltrexol produced a slight hyperalgesia at 40 min, but this effect was negligible compared with the reinstatement produced by NTX (Fig. 1B).
Adrenergic α2ARs suppress hyperalgesia in latent sensitization in mice
The fact that MOR KO mice showed a partial recovery from hyperalgesia (Fig. 1A) suggests that receptors other that MORs contribute to the suppression of hyperalgesia during the remission phase of latent sensitization. Descending noradrenergic pathways produce analgesia by acting on α2 receptors in the dorsal horn (Yaksh, 1985; Pertovaara, 2006), which are mainly α2ARs located in primary afferent terminals (Stone et al., 1998; Chen et al., 2008). Therefore, we hypothesized that α2ARs contribute to suppressing hyperalgesia in latent sensitization and predicted that an α2AR antagonist would produce reinstatement during the remission phase.
To test this hypothesis in mice, the three groups of mice in the experiment shown in Figure 1 were injected with the selective α2AR antagonist BRL44408 (1 mg/kg, s.c.) (Li and Eisenach, 2001; Nazarian et al., 2008) on day 48 after CFA. In the CFA-injected WT mice, BRL44408 produced robust reinstatement that lasted for <24 h (Fig. 1D, Table 1, row 5). In the MOR KO mice, BRL44408 also produced reinstatement, inducing a degree of hyperalgesia similar to that measured immediately after CFA. BRL44408 had no effect in saline-injected mice, showing that its pronociceptive effect did not occur in the absence of the sensitizing effect of CFA.
Effect of the DOR antagonist naltrindole in mice
Because DORs are present in the spinal cord and produce analgesia (Marvizón, 2009), it is possible that they contribute to the suppression of hyperalgesia in latent sensitization. We investigated the involvement of DORs by determining whether the DOR antagonist naltrindole produces reinstatement of hyperalgesia in mice with CFA-induced latent sensitization. The three groups of mice in the experiment shown in Figure 1 were injected with naltrindole (3 mg/kg, s.c.) on day 55 after CFA. In the CFA-injected WT and MOR KO mice, naltrindole reinstated hyperalgesia (Fig. 1E, Table 1, row 6). However, in the saline-injected mice, we observed a highly variable effect and a trend toward hyperalgesia. The effect of naltrindole was studied after sequential injections of three other drugs, NTX, 6β-naltrexol, and BRL44408, and multiple testing with von Frey filaments over a period of almost 2 months, which could have sensitized the mice. To rule out interference from other drugs and prolonged testing, we studied additional groups of WT mice injected in the hindpaw with CFA (n = 14) or saline (n = 6), and with naltrindole (3 mg/kg, s.c.) 29 d later. Naltrindole induced substantial hyperalgesia in the CFA-injected mice (Fig. 1F, Table 1, row 7). In the saline-injected mice, naltrindole again produced a variable effect, with clear hyperalgesia at the 60 min time point (Fig. 1F).
Effects of the MOR inverse agonist and MOR neutral antagonist in rats
The effects of NTX and 6β-naltrexol were further investigated in rats. We administered these compounds intrathecally to assess whether the MORs that suppress hyperalgesia during latent sensitization are in the spinal cord. Rats (n = 6) were implanted with intrathecal catheters terminating at the lumbar spinal cord and, 5 d later, were injected with 50 μl of CFA in 1 hindpaw. Robust mechanical hyperalgesia occurred in the ipsilateral hindpaw and lasted 21 d (Fig. 2A, statistics in Table 2, row 1).
Figure 2.
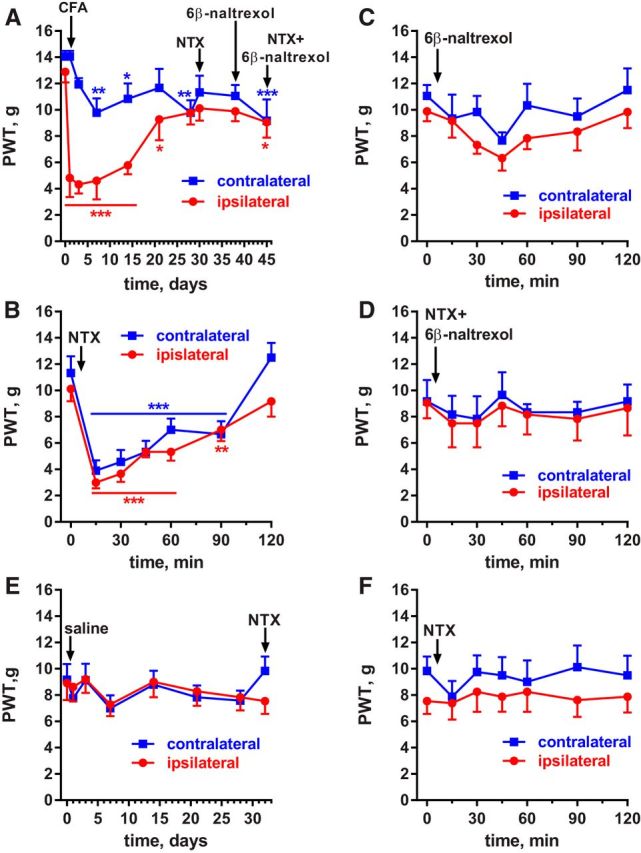
Effect of a MOR inverse agonist and neutral antagonist in rats with CFA-induced latent sensitization. Saline-injected controls. A, PWTs of rats (n = 6) implanted with intrathecal catheters and injected in the hindpaw with 50 μl of CFA subcutaneously. B, On day 30, the rats received intrathecal NTX (2.6 nmol), a MOR inverse agonist. C, On day 38, the MOR neutral antagonist 6β-naltrexol (8.7 nmol) was injected intrathecally. D, On day 45, NTX (2.6 nmol) plus 6β-naltrexol (8.7 nmol) were injected intrathecally. Holm-Sidak's post hoc tests of two-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline (0 d or 0 min). E, PWTs of rats (n = 8) with intrathecal catheters that were injected in the hindpaw with 50 μl of saline subcutaneously. F, On day 32, the saline-injected rats (n = 8) received intrathecal NTX (2.6 nmol).
Table 2.
Two-way ANOVA of the data shown in Figure 2 (NTX and 6β-naltrexol)
| Row | Figure | Experiment | n | Variable 1 | Variable 2 | Interaction | Subject matching |
|---|---|---|---|---|---|---|---|
| 1 | 2A | CFA | 6 | Time (CFA) | Side | p < 0.0001 | p = 0.001 |
| p < 0.0001 | p = 0.0096 | F(9,90) = 7.8 | F(10,90) = 7.88 | ||||
| F(9,90) = 8.98 | F(1,10) = 10.21 | ||||||
| 2 | 2B | CFA (NTX) | 6 | Time (NTX) | Side | p = 0.15 | p < 0.0001 |
| p < 0.0001 | p = 0.25 | F(6,60) = 1.6 | F(10,60) = 6.45 | ||||
| F(6,60) = 40 | F(1,10) = 1.5 | ||||||
| 3 | 2C | CFA (6β-naltrexol) | 6 | Time (6β-naltrexol) | Side | p = 0.89 | p < 0.0001 |
| p = 0063 | p = 0.24 | F(6,60) = 0.38 | F(10,60) = 5.68 | ||||
| F(9,90) = 3.37 | F(1,10) = 1.5 | ||||||
| 4 | 2D | CFA (NTX + 6β-naltrexol) | 6 | Time (NTX + 6β-naltrexol) | Side | p = 0.999 | p < 0.0001 |
| p = 0.42 | p = 0.81 | F(6,60) = 0.04 | F(10,60) = 14.6 | ||||
| F(9,90) = 1.02 | F(1,10) = 0.06 | ||||||
| 5 | 2E | Saline | 8 | Time (saline) | Side | p = 0.50 | p < 0.0001 |
| p = 0.066 | p = 0.97 | F(7,98) = 0.91 | F(14,98) = 10.3 | ||||
| F(7,98) = 1.97 | F(1,14) = 0.002 | ||||||
| 6 | 2F | Saline (NTX) | 8 | Time (NTX) | Side | p = 0.95 | p < 0.0001 |
| p = 0.87 | p = 0.29 | F(6,84) = 0.28 | F(14,84) = 7.08 | ||||
| F(6,84) = 0.42 | F(1,14) = 1.2 |
Each panel in Figure 2 (indicated in the second column) was analyzed independently by two-way ANOVA. In the fourth column, n is the number of animals in the experiment. In the fifth and sixth columns, the variable is identified first: “time” refers to the time after the injection of the compound in parentheses; “side” is ipsilateral versus contralateral paw. Subscript numbers in the F ratio are the degrees of freedom and the residual.
NTX (1 μg or 2.6 nmol) injected intrathecally on day 30 after CFA reinstated hyperalgesia for 2 h in both the ipsilateral and contralateral hindpaws (Fig. 2B, statistics in Table 2, row 2).
6β-Naltrexol (3 μg or 8.7 nmol) injected intrathecally on day 38 after CFA produced little hyperalgesia (Fig. 2C); a two-way ANOVA revealed a significant effect of time (Table 2, row 3), but the Holm-Sidak's post hoc test did not reveal significant effects at any of the time points.
A stringent test for MOR constitutive activity is that the neutral antagonist 6β-naltrexol should block the effect of the inverse agonist NTX. Accordingly, NTX and 6β-naltrexol (2.6 and 8.7 nmol, respectively) were coinjected intrathecally to these rats on day 45 after CFA and no reinstatement of hyperalgesia was observed (Fig. 2D, Table 2, row 4).
Implanting chronic intrathecal catheters involves surgery, which could induce latent sensitization (Rivat et al., 2007; Campillo et al., 2011). To control for this possibility and to confirm that intrathecal NTX does not reinstate hyperalgesia in the absence of CFA-induced inflammation, we studied a control group of rats (n = 8) implanted with intrathecal catheters and injected in one hindpaw with saline (50 μl) instead of CFA. These rats did not develop mechanical hyperalgesia in the ipsilateral or contralateral hindpaws after the saline injection (Fig. 2E, Table 2, row 5). NTX (2.6 nmol intrathecally) did not produce any effect in these rats (Fig. 2F, Table 2, row 6).
Adrenergic α2ARs suppress hyperalgesia in latent sensitization in rats
The hypothesis that α2ARs suppress hyperalgesia during latent sensitization was also tested in rats. In addition, BRL44408 was administered intrathecally to determine whether the targeted α2ARs are in the spinal cord. Rats (n = 9) were implanted with intrathecal catheters terminating at the lumbar spinal cord (L4–L5) and injected with CFA (50 μl, s.c.) in 1 hindpaw. This resulted in hyperalgesia in the ipsilateral but not the contralateral paw (Fig. 3A, Table 3, row 1). On day 30, 6 of the rats received an intrathecal injection of 0.3 nmol BRL44408 (0.1 μg), which produced hyperalgesia in both paws at 15 min (Fig. 3B, Table 3, row 2). On day 40, all 9 rats received intrathecal BRL44408 at the higher dose of 3 nmol (1 μg), which produced robust hyperalgesia in both paws for >2 h (Fig. 3C, Table 3, row 3). No differences were noted between the rats that received 2 injections of BRL44408 (0.3 nmol followed by 3 nmol) and the other rats.
Figure 3.

Effect of the α2AR antagonist BRL44408 in rats with CFA-induced latent sensitization. A, PWTs of rats (n = 9) implanted with intrathecal catheters and injected in the hindpaw with 50 μl of CFA subcutaneously. B, On day 30, 6 of the rats received 0.3 nmol BRL44408 (α2AR antagonist) intrathecally. C, On day 40, all rats (n = 9) received 3 nmol BRL44408 intrathecally. Holm-Sidak's post hoc tests of two-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline (0 d or 0 min). D, PWTs of rats (n = 8) with intrathecal catheters that were injected in the hindpaw with 50 μl of saline subcutaneously. E, On day 31, the saline-injected rats (n = 8) received intrathecal BRL-44408 (3 nmol).
Table 3.
| Row | Figure | Experiment | n | Variable 1 | Variable 2 | Interaction | Subject matching |
|---|---|---|---|---|---|---|---|
| 1 | 3A | CFA | 9 | Time (CFA) | Side | p < 0.0001 | p < 0.0001 |
| p < 0.0001 | p < 0.0001 | F(7,112) = 18.10 | F(16,112) = 7.30 | ||||
| F(7,112) = 18.66 | F(1,16) = 30.49 | ||||||
| 2 | 3B | CFA, BRL44408 0.3 nmol | 6 | Time (BRL44408) | Side | p = 0.75 | p < 0.0001 |
| p = 0.0064 | p = 0.93 | F(5,50) = 0.53 | F(10,50) = 6.25 | ||||
| F(5,50) = 3.69 | F(1,10) = 0.007 | ||||||
| 3 | 3C | CFA, BRL44408 3 nmol | 9 | Time (BRL44408) | Side | p = 0.17 | p < 0.0001 |
| p < 0.0001 | p = 0.25 | F(5,80) = 1.60 | F(16,80) = 3.88 | ||||
| F(5,80) = 19.34 | F(1,16) = 1.43 | ||||||
| 4 | 3D | saline | 8 | Time (saline) | Side | p = 0.92 | p < 0.0001 |
| p = 0.018 | p = 0.78 | F(7,98) = 0.36 | F(14,98) = 13.1 | ||||
| F(7,98) = 2.56 | F(1,14) = 0.08 | ||||||
| 5 | 3E | saline (BRL 44408) | 8 | Time (BRL44408) | Side | p = 0.73 | p < 0.0001 |
| p = 0.033 | p = 0.72 | F(6,84) = 0.60 | F(14,84) = 12.1 | ||||
| F(6,84) = 2.42 | F(1,14) = 0.14 | ||||||
| 6 | 4A | CFA | 11 | Time (CFA) | Side | p < 0.0001 | p < 0.0001 |
| p < 0.0001 | p = 0.0001 | F(7,140) = 21.1 | F(20,140) = 12.8 | ||||
| F(7,140) = 23.15 | F(1,20) = 22.94 | ||||||
| 7 | 4B | CFA, naltrindole i.th. | 11 | Time (naltrindole) | Side | p = 0.87 | p = 0.0007 |
| p = 0.0088 | p = 0.22 | F(5,100 = 0.36 | F(20,100 = 2.66 | ||||
| F(5,100) = 3.28 | F(1,20) = 1.60 | ||||||
| 8 | 4C | CFA, naltrindole s.c. | 8 | Time (naltrindole) | Side | p = 0.14 | p < 0.0001 |
| p = 0.0033 | p = 0.97 | F(5,70) = 1.72 | F(14,70) = 8.34 | ||||
| F(5,70) = 3.93 | F(1,14) = 0.0017 | ||||||
| 9 | 4D | saline | 8 | Time (saline) | Side | p = 0.30 | p < 0.0001 |
| p = 0.0001 | p = 0.71 | F(7,98) = 1.22 | F(14,98) = 7.66 | ||||
| F(7,98) = 4.7 | F(1,14) = 0.15 | ||||||
| 10 | 4E | saline, naltrindole s.c. | 8 | Time (naltrindole) | Side | p = 0.75 | p = 0.006 |
| p = 0.49 | p = 0.022 | F(5,70) = 0.53 | F(14,70) = 2.51 | ||||
| F(5,70) = 0.89 | F(1,14) = 6.6 |
Each panel in Figure 3 or 4 (indicated in the second column) was analyzed independently by two-way ANOVA. In the fourth column, n is the number of animals in the experiment. In the fifth and sixth columns, the variable is identified first: “time” refers to the time after the injection of the compound in parentheses; “side” is ipsilateral versus contralateral paw. Subscript numbers in the F ratio are the degrees of freedom and the residual.
To confirm that BRL44408 does not reinstate hyperalgesia in the absence of CFA-induced inflammation, we studied rats (n = 8) injected in 1 hindpaw with saline (50 μl). These rats did not develop mechanical hyperalgesia in the ipsilateral or contralateral hindpaws (Fig. 3D), although atypical hypoalgesia at day 28 resulted in a statistically significant effect of the variable “time” (p = 0.018; Table 3, row 4). In these saline-injected rats, BR44408 (3 nmol intrathecal; Fig. 3E) did not produce the marked hyperalgesic effect found in the CFA-injected rats. However, there was a small but statistically significant (p = 0.033, Table 3, row 5) trend toward hyperalgesia.
Therefore, intrathecal BRL44408 had a potent, dose-dependent effect in CFA-injected rats, marginal at 0.3 nmol and robust at 3 nmol. By comparison, Nazarian et al. (2008) reported that the analgesic effects of the α2AR agonists dexmedetomidine and ST-91 in rats were blocked by intrathecal BRL44408 at 30 and 300 nmol, but not at 3 nmol. This 10-fold difference in dose was likely due to the presence of agonists in the experiments by Nazarian et al. (2008).
Effect of the DOR antagonist naltrindole in rats
To determine whether DORs contribute to the suppression of hyperalgesia in latent sensitization in rats, we studied the effect of the DOR antagonist naltrindole. Rats (n = 11) were implanted with intrathecal catheters terminating at the lumbar spinal cord (L4–L5) and injected with CFA (50 μl, s.c.) in one hindpaw. This induced hyperalgesia in the ipsilateral but not the contralateral paw (Fig. 4A, Table 3, row 6). On day 30, the rats received an intrathecal injection of naltrindole (3 μg or 2.2 nmol), which produced a small and short reinstatement of hyperalgesia that was more noticeable in the ipsilateral paw at 15 min (Fig. 4B, Table 3, row 7).
Figure 4.

Effect of the DOR antagonist naltrindole in rats with CFA-induced latent sensitization. A, PWTs of rats (n = 11) injected in the hindpaw with 50 μl of CFA subcutaneously. B, On day 30, these rats received 2.2 nmol naltrindole (a DOR antagonist) intrathecally. C, On day 48, some of the rats (n = 8) received 1 mg/kg naltrindole subcutaneously. Holm-Sidak's post hoc tests of two-way ANOVA: *p < 0.05, ***p < 0.001 compared with baseline (0 d or 0 min). D, PWTs of rats (n = 8) without intrathecal catheters that were injected in the hindpaw with 50 μl of saline subcutaneously. E, On day 30, the saline-injected rats (n = 8) received naltrindole subcutaneously (1 mg/kg).
Because DORs in supraspinal areas could also contribute to suppression of hyperalgesia, we studied the effect of systemic naltrindole in a subgroup of these rats (n = 8). Naltrindole was injected subcutaneously at 1 mg/kg on day 48 after CFA. Again, naltrindole produced a short-lived reinstatement of the hyperalgesia (Fig. 4C, Table 3, row 8).
As control, we used rats injected in the hindpaw with saline instead of CFA. In these rats, the strong hyperalgesia induced by CFA during the first week was completely absent (Fig. 4D), but an atypical slight hyperalgesia developed after 20 d, resulting in a significant effect of the variable “time” (p = 0.0001; Table 3, row 9). Naltrindole injected (1 mg/kg, s.c.) in these control rats did not produce hyperalgesia (Fig. 4E, Table 3, row 10).
KORs suppress hyperalgesia in rats and mice with latent sensitization
In rats, the effect of KORs was studied by injecting the KOR antagonist nor-BNI intrathecally. It is known that the KOR antagonists nor-BNI (Endoh et al., 1992) and JDTic (Carroll et al., 2004) produce effects in vivo with a slow onset (taking several hours) and persisting for many days (Munro et al., 2012). Therefore, we studied the effect of nor-BNI 12 h after injecting it in rats. As usual, these rats (n = 5) were injected with CFA in the hindpaw, which induced hypersensitivity ipsilaterally but not contralaterally (Fig. 5A, Table 4, row 1). On day 30, the rats received an intrathecal injection of 1.3 nmol nor-BNI and its effect was assessed 12 h later. At that time and for the next 2 h, there was a robust reinstatement of hyperalgesia in both hindpaws, which largely dissipated 36 h after the injection. (Fig. 5B, Table 4, row 2). In saline-injected rats that did not develop hyperalgesia (Fig. 5C, Table 4, row 3), nor-BNI (1.3 nmol intrathecal) did not induce hyperalgesia after 12 h (Fig. 5D, Table 4, row 4).
Figure 5.
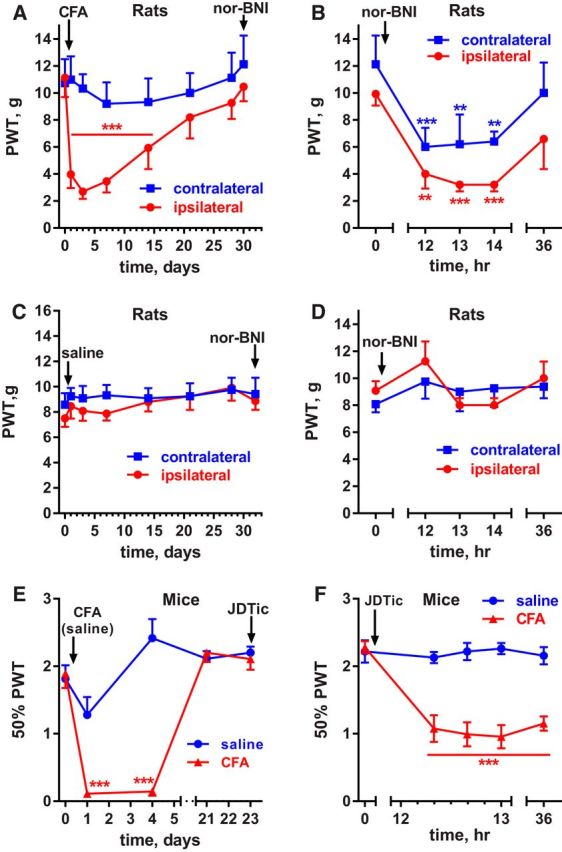
Effect of the KOR antagonists nor-BNI and JDTic in rats and mice with CFA-induced latent sensitization. A, PWTs of rats (n = 5) injected in the hindpaw with 50 μl of CFA subcutaneously. B, On day 30, these rats received 1.3 nmol nor-BNI (a KOR antagonist) intrathecally, which reinstated hyperalgesia 12 h after the injection. C, PWTs of rats (n = 8) with intrathecal catheters that were injected in the hindpaw with 50 μl of saline subcutaneously. D, On day 32, the saline-injected rats (n = 8) received intrathecal nor-BNI (1.3 nmol). E, Mice received 5 μl of CFA (n = 6) or 5 μl of saline (n = 6) subcutaneously in the hindpaw. F, On day 43, the mice received an injection of the KOR antagonist JDTic (10 mg/kg, s.c.), which reinstated hyperalgesia after 12 h. Holm-Sidak's post hoc tests of two-way ANOVA: **p < 0.01, ***p < 0.001 compared with baseline (0 d or 0 min).
Table 4.
Two-way ANOVA of the data shown in Figure 5 (nor-BNI and JDTic)
| Row | Figure | Experiment | n | Variable 1 | Variable 2 | Interaction | Subject matching |
|---|---|---|---|---|---|---|---|
| 1 | 5A | Rats CFA | 5 | Time (CFA) | Side | p < 0.0001 | p < 0.0001 |
| p < 0.0001 | p = 0.092 | F(7,56) = 5.7 | F(8,56) = 16.59 | ||||
| F(7,56) = 10.56 | F(1,8) = 4.23 | ||||||
| 2 | 5B | Rats CFA, nor-BNI 1.3 nmol (12 h) | 5 | Time (nor-BNI) | Side | p = 0.95 | p < 0.0001 |
| p < 0.0001 | p = 0.16 | F(4,32) = 0.18 | F(8,32) = 7.26 | ||||
| F(4,32) = 14.66 | F(1,8) = 2.42 | ||||||
| 3 | 5C | Saline | 8 | Time (saline) | Side | p = 0.96 | p < 0.0001 |
| p = 0.46 | p = 0.42 | F(7,98) = 0.26 | F(14,98) = 3.82 | ||||
| F(7,98) = 0.96 | F(1,14) = 0.69 | ||||||
| 4 | 5D | Saline (nor-BNI) | 8 | Time (nor-BNI) | Side | p = 0.37 | p < 0.0001 |
| p = 0.073 | p = 0.88 | F(4,56) = 1.1 | F(14,56) = 4.42 | ||||
| F(4,56) = 2.27 | F(1,14) = 0.025 | ||||||
| 5 | 5E | Mice CFA / saline | 6/6 | Time (CFA) | CFA | p < 0.0001 | p = 0.011 |
| p < 0.0001 | p = 0.021 | F(6,60) = 11.2 | F(10,60) = 2.60 | ||||
| F(6,60) = 15.09 | F(1,10) = 7.48 | ||||||
| 6 | 5F | Mice CFA / saline, JDTic | 6/6 | Time (JDTic) | CFA | p < 0.0001 | p < 0.0001 |
| p < 0.0001 | p = 0.0001 | F(4,40) = 15.3 | F(10,40) = 5.67 | ||||
| F(4,40) = 16.08 | F(1,10) = 36.03 |
Each panel in Figure 5 (indicated in the second column) was analyzed independently by two-way ANOVA. In the fourth column, n is the number of animals in the experiment. In the fifth and sixth columns, the variable is identified first: “time” refers to the time after the injection of the compound in parentheses; “side” is ipsilateral versus contralateral paw, and “CFA” refers to CFA versus saline. Subscript numbers in the F ratio are the degrees of freedom and the residual.
In mice, we used the KOR antagonist JDTic (Carroll and Dolle, 2014). One hindpaw was injected with 5 μl of either saline (n = 6) or CFA (n = 6). As usual, the CFA-injected but not the saline-injected mice developed hypersensitivity in the paw that was gone by day 21 (Fig. 5E, Table 4, row 5). On day 43, the mice received an injection of JDTic (10 mg/kg, s.c.). In the CFA-injected mice, this resulted in reinstatement of hyperalgesia that was observed 12 h after injecting JDTic and was still present at 36 h afterward (Fig. 5F, Table 4, row 6). JDTic had no effect in the control mice injected with saline.
pENK, pOMC (β-endorphin), and pDYN KO mice display normal latent sensitization
To determine whether the suppression of hyperalgesia produced by MORs was caused by endogenous opioids, we studied CFA-induced latent sensitization in mice lacking opioid peptides: pENK KO, pOMC KO, or pDYN KO. Enkephalin and dynorphins are abundant in the dorsal horn, whereas endorphins are largely absent (Marvizón et al., 2009).
Unlike the MOR KO mice, pENK KO mice (König et al., 1996) injected with CFA (5 μl) in one hindpaw developed mechanical hyperalgesia that fully subsided after 21 d (Fig. 6A, Table 5, row 1). There were no differences in baseline responses between WT and pENK KO mice over the 4 d before the CFA injection (Table 5, row 2). CFA induced hyperalgesia in WT and KOs. After day 4, the pENK KO mice showed less hyperalgesia than their WT littermates, suggesting that enkephalins delay the recovery. However, this effect was inconsistent and not seen in a previous experiment. On day 28, the pENK and WT mice received NTX (3 mg/kg, s.c.), which reinstated hyperalgesia in both groups of mice (Fig. 6B, Table 5, row 3). Therefore, in the absence of enkephalins, there was full recovery from hyperalgesia and activation of MORs.
Figure 6.
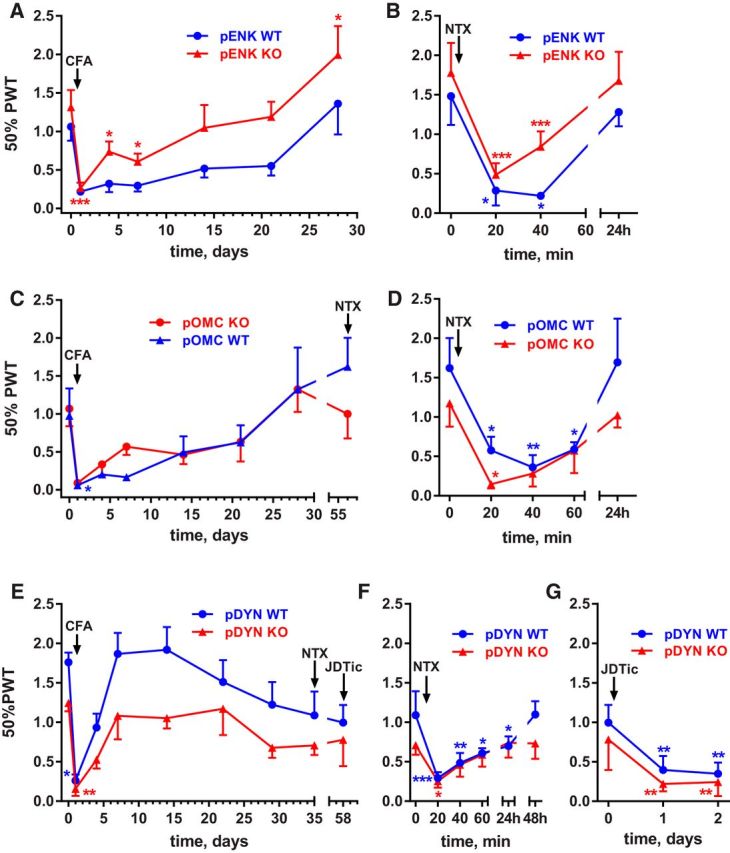
Latent sensitization in pENK, pOMC, and pDYN KO mice. Mice were injected in the hindpaw with 5 μl of CFA and, upon return to baseline, they received NTX (3 mg/kg, s.c.). PWTs to von Frey filaments were obtained on the days indicated. A, pENK KO mice (n = 10) and their WT littermates (n = 5). B, NTX injected on day 28 to pENK KO and WT mice reinstated hyperalgesia. C, pOMC KO mice (n = 5) and their WT littermates (n = 4). D, NTX injected on day 56 to pOMC KO and WT mice reinstated hyperalgesia. E, pDYN KO mice (n = 8) and their WT littermates (n = 8). F, NTX injected on day 35 to pDYN KO and WT mice reinstated hyperalgesia. G, JDTic (10 mg/kg, s.c.) injected on day 58 to pDYN KO and WT mice reinstated hyperalgesia after 1 d. Holm-Sidak's post hoc tests of two-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline (time 0 d or 0 min).
Table 5.
Two-way ANOVA of the data shown in Figure 6 (pENK, pOMC, and PDYN KO mice) and Figure 7 (MOR internalization)
| Row | Figure | Experiment | n | Variable 1 | Variable 2 | Interaction | Subject matching |
|---|---|---|---|---|---|---|---|
| 1 | 6A | pENK KO, CFA | KO: 10 | Time (CFA) | Genotype | p = 0.83 | p = 0.0093 |
| WT: 5 | p < 0.0001 | p = 0.048 | F(6,78) = 0.47 | F(13,78) = 2.39 | |||
| F(6,78) = 9.52 | F(1,13) = 4.78 | ||||||
| 2 | — | pENK KO, baseline | KO: 10 | Time | Genotype | p = 0.18 | p = 0.0006 |
| WT: 5 | p = 0.49 | p = 0.48 | F(3,39) = 1.70 | F(13,39) = 3.77 | |||
| F(3,39) = 0.81 | F(1,13) = 0.53 | ||||||
| 3 | 6B | pENK KO, NTX | KO: 10 | Time (NTX) | Genotype | p = 0.88 | p = 0.014 |
| WT: 5 | p < 0.0001 | p = 0.229 | F(3,39) = 0.23 | F(13,39) = 2.48 | |||
| F(3,39) = 0.23 | F(1,13) = 1.59 | ||||||
| 4 | 6C | pOMC KO, CFA | KO: 4 | Time (CFA) | Genotype | p = 0.68 | p = 0.016 |
| WT: 5 | p < 0.0001 | p = 0.99 | F(7,49) = 0.69 | F(7,49) = 2.79 | |||
| F(7,49) = 7.7 | F(1,7) = 0.00001 | ||||||
| 5 | — | pOMC KO, baseline | KO: 4 | Time | Genotype | p = 0.06 | p < 0.0001 |
| WT: 5 | p = 0.58 | p = 0.91 | F(9,36) = 2.0 | F(12,36) = 4.91 | |||
| F(3,36) = 0.66 | F(3,12) = 0.18 | ||||||
| 6 | 6D | pOMC KO, NTX | KO: 4 | Time (NTX) | Genotype | p = 0.67 | p = 0.04 |
| WT: 5 | p < 0.0001 | p = 0.23 | F(4,28) = 0.59 | F(7,28) = 2.48 | |||
| F(4,28) = 8.8 | F(1,7) = 1.69 | ||||||
| 7 | 6E | pDYN KO, CFA | KO: 8 | Time (CFA) | Genotype | p = 0.51 | p < 0.0001 |
| WT: 8 | p < 0.0001 | p = 0.037 | F(8,112) = 0.91 | F(14,112) = 5.38 | |||
| F(8,112) = 11.38 | F(1,14) = 5.34 | ||||||
| 8 | — | pDYN, baseline | KO: 8 | Time | Genotype | p = 0.51 | p = 0.79 |
| WT: 8 | p = 0.16 | p = 0.0069 | F(2,28) = 0.68 | F(14,28) = 0.66 | |||
| F(2,28) = 1.94 | F(1,14) = 10.0 | ||||||
| 9 | 6F | pDYN KO, NTX | KO: 8 | Time (NTX) | Genotype | p = 0.25 | p < 0.0001 |
| WT: 8 | p < 0.0001 | p = 0.45 | F(5,70) = 1.37 | F(14,70) = 6.40 | |||
| F(5,70) = 9.47 | F(1,14) = 0.61 | ||||||
| 10 | 6G | pDYN KO, JDTic | KO: 8 | Time (JDTic) | Genotype | p = 0.91 | p < 0.0001 |
| WT: 8 | p < 0.0001 | p = 0.55 | F(2,26) = 0.096 | F(13,26) = 6.90 | |||
| F(2,26) = 14.60 | F(1,13) = 0.38 | ||||||
| 11 | 7A | Rats CFA | 6 | Time (CFA) | Side | p < 0.0001 | p < 0.0001 |
| p < 0.0001 | p = 0.0007 | F(7,70) = 17.4 | F(10,70) = 5.98 | ||||
| F(7,70) = 20.2 | F(1,10) = 23.4 | ||||||
| 12 | 7B | Rats NTX | 6 | Time (NTX) | Side | p = 0.39 | p < 0.0001 |
| p < 0.0001 | p = 0.92 | F(8,80) = 14.9 | F(10,80) = 4.7 | ||||
| F(8,80) = 14.9 | F(1,10) = 0.01 |
Each panel in Figure 6 or 7 (indicated in the second column) was analyzed independently by two-way ANOVA except for rows 2, 5, and 8 which are baseline data obtained before the CFA injection. In the fourth column, n is the number of animals in the experiment (KO or their WT littermates). In the fifth and sixth columns, the variable is identified first: “time” refers to the time after the injection of the compound in parentheses; “genotype” is KO versus WT, and “side” is ipsilateral versus contralateral paw. Subscript numbers in the F ratio are the degrees of freedom and the residual.
To study the effect of deleting endorphins on latent sensitization, we used mice that were KO in the pOMC gene (Rubinstein et al., 1996), which encodes these peptides. pOMC KO and WT mice injected with CFA (5 μl) in one hindpaw developed mechanical hyperalgesia that fully subsided by day 28 (Fig. 6C, Table 5, row 4). There were no differences in baseline responses between pOMC KO and WT mice over the 4 d before the CFA injection (Table 5, row 5). On day 56, the pOMC and WT mice received NTX (3 mg/kg, s.c.), which reinstated hyperalgesia in both groups of mice (Fig. 6D, Table 5, row 6). Therefore, in the absence of endorphins, there was full recovery from hyperalgesia and activation of MORs.
Likewise, pDYN KO mice completely recovered from hyperalgesia compared with their own baseline responses (Fig. 6E, Table 5, row 7). In contrast to other groups of mice, both the pDYN mice, whether WT or KO, recovered more rapidly from CFA-induced hyperalgesia. These mice were tested in the same environment and by the same tester as all other groups of mice. However, this line was raised in a different room with a greater number of mice and daily interruptions. These factors could have altered their genetic or epigenetic background to facilitate a more rapid recovery from CFA regardless of genotype. In addition, the baseline for the pDYN KO mice was significantly lower than the baseline for their WT littermates (Table 5, row 8). Therefore, the differences between pDYN and WT observed in Figure 6E should be attributed to this difference in baseline. In both the pDYN and WT mice, NTX (3 mg/kg, s.c.) given on day 35 reinstated hyperalgesia (Fig. 6F, Table 5, row 9), showing that hyperalgesia was being suppressed by MOR activation at this time. Furthermore, hyperalgesia was also reinstated by the KOR antagonist JDTic (10 mg/kg, s.c.) given on day 58 (Fig. 7G, Table 5, row 10). This shows that KORs contributed to suppression of hyperalgesia in latent sensitization, even in the absence of dynorphin.
Figure 7.
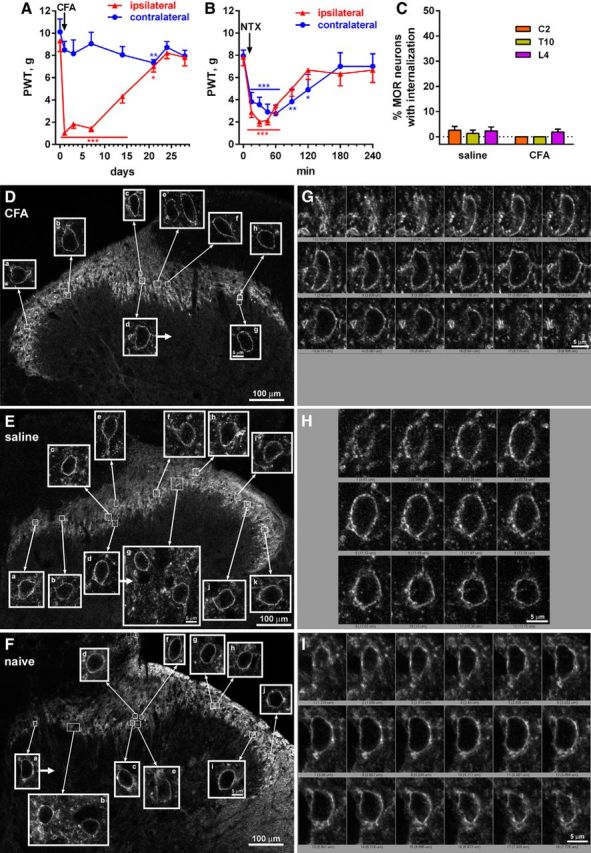
Absence of MOR internalization in the spinal cord during the remission phase of latent sensitization. A, Rats (n = 6) implanted with intrathecal catheters were injected in the hindpaw with 50 μl of CFA subcutaneously and responses to von Frey hairs were tested. B, On day 28, the rats received NTX (1 mg/kg, s.c.) and PWTs were tested. Holm-Sidak's post hoc tests: *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline (time 0 d or 0 min). C, On day 30, rats injected with CFA (n = 4) or saline-injected controls (n = 4) received intrathecal injections of peptidase inhibitors (amastatin, phosphoramidon, and captopril, 100 nmol) and fixed 15 min later. MOR immunohistochemistry was performed in spinal segments C2, T10, and L4 to measure MOR internalization. D, Images taken from the L4 segment of a rat 30 d after injecting CFA in the hindpaw; MORs are not internalized. E, Images taken from the L4 segment of a rat 30 d after injecting saline in the hindpaw; MORs are not internalized. F, Images taken from the L4 segment of a naive rat; MORs are not internalized. Images are single optical sections taken with a 10× objective (main panels, voxel size 830 × 830 × 5983 nm, scale bar 100 μm) or a 63× objective (insets, voxel size 132 × 132 × 383 nm, scale bar 5 μm). G, Confocal stack of cell d (arrow) in D. H, Confocal stack of cell d (arrow) in E. I, Confocal stack of cell a (arrow) in F.
MORs do not become internalized during latent sensitization
The fact that NTX reinstates hyperalgesia during the remission phase of latent sensitization (Figs. 1, 2; Corder et al., 2013) shows that there is sustained activation of MORs. It is possible that this activation would be accompanied by MOR internalization either as the result of endogenous opioid release (Song and Marvizón, 2003b) or of MOR constitutive activity leading to MOR internalization (Walwyn et al., 2007). Therefore, we measured MOR internalization during the remission phase of latent sensitization by immunofluorescence, an approach that we have used and described extensively (Song and Marvizón, 2003a; Song and Marvizón, 2003b; Chen et al., 2007; Chen et al., 2008; Chen and Marvizón, 2009). To maximize the detection of MOR internalization due to the release of opioid peptides, we prevented peptide cleavage by an intrathecal injection of a mixture of peptidase inhibitors (Song and Marvizón, 2003a; Song and Marvizón, 2003b; Chen et al., 2007; Lao et al., 2008; Chen and Marvizón, 2009).
Rats (n = 6) with intrathecal catheters terminating at the L4–L5 segments were injected with CFA (50 μl, s.c.) in one hindpaw. CFA produced hyperalgesia ipsilaterally that subsided after 3 weeks (Fig. 7A, Table 5, row 11). To confirm the presence of MOR-mediated suppression of hyperalgesia, the rats received NTX (1 mg/kg, s.c.) on day 28, which reinstated the hyperalgesia for 2 h in both the ipsilateral and the contralateral hindpaws (Fig. 7B, Table 5, row 12). As control, another group of rats (n = 6) fitted with intrathecal catheters received 50 μl of saline in the hindpaw. These rats did not develop hyperalgesia after the saline injection in the paw (Fig. 2E) or after intrathecal NTX (2.6 nmol on day 32; Fig. 2F). On day 30, a subset of the CFA-injected (n = 4) and the saline-injected (n = 4) rats received an intrathecal injection of peptidase inhibitors (amastatin, captopril, and phosphoramidon, all 100 nmol) and were killed and fixed 10–15 min later. MOR internalization was assessed in spinal segments C2, T10, and L4, ipsilaterally to the injected paw and was found to be negligible (Fig. 7C). Representative confocal microscope images of MOR neurons in the ipsilateral L4 dorsal horn show the absence of MOR-immunoreactive endosomes in a CFA-injected rat (Fig. 7D) and a saline-injected rat (Fig. 7E). However, in both cases, we observed clusters of MOR immunoreactivity at the cell surface (Fig. 7G,H, respectively) that were largely absent in naive rats that did not receive intrathecal peptidase inhibitors (Fig. 7F,I, respectively). Because these MOR clusters were found in saline-injected rats that had no latent sensitization, we attributed them to the presence of small amounts of endogenous opioids after the injection of peptidase inhibitors.
Increased phosphorylation of MORs and Src during latent sensitization
Rats were injected in the left hindpaw with 50 μl of CFA (n = 8) or saline (n = 8) and developed hyperalgesia ipsilaterally. After 32 d, the presence of latent sensitization in the CFA group was verified by measuring hyperalgesia after NTX (1 mg/kg, s.c.). Results were similar to those in Figure 7B. After 2 more days, the L4–S1 spinal cord segments were collected, snap frozen and stored at −80°C. Tissues were homogenized and membranes solubilized under nondenaturing conditions that permit retention of protein–protein interactions. MOR and associated proteins were coimmunoprecipitated with a MOR antibody (ab13405; Abcam) and separated by SDS-gel electrophoresis. Blots were probed with antibodies to serine-phosphorylated MOR (p-Ser375-MOR, RA18001; Neuromics) and a different MOR antibody (RA10104; Neuromics). All three antibodies to the MOR used in this study, including the antibody used for MOR immunofluorescence (ImmunoStar; Fig. 7), consistently labeled two protein bands with apparent molecular weights of 57 and 60 kDa (Fig. 8A). The anti-p-Ser375-MOR antibody labeled both bands, although the lower band was always more intense than the upper band. We measured the densities of both bands to evaluate the relative level of phosphorylation of MORs. Rats in the CFA group had a significantly higher level of MOR phosphorylation than rats in the saline group (Fig. 8B; p = 0.0009, t = 4.92, unpaired t tests with Welch's correction). We also determined whether activated Src family kinases (SFKs) were coimmunoprecipitated with MOR using an antibody to p-Tyr416-Src, which recognizes the equivalent site in the SFKs Lyn, Fyn, Lck, Yes, and Hck. We found that the relative level of activated SFKs in the immunoprecipitates was significantly higher in the CFA group than in the saline group (Fig. 8C; p = 0.0141, t = 3.14, unpaired t tests with Welch's correction).
Figure 8.

Increased phosphorylation of MORs at Ser375 in latent sensitization. Rats were injected in one hindpaw with 50 μl of saline (n = 8) or CFA (n = 8). After 28 d, the rats were killed and the spinal cords collected. MORs were immunoprecipitated using a MOR antibody from Abcam (ab134054). A, Western blots were probed with antibodies to MOR (Neuromics, RA10104), p-Ser375-MOR (Neuromics RA18001, pMOR), and p-Tyr416-Src (Cell Signaling Technology, pSFK). Two bands at ∼57 and 64 kDa appear in the MOR and pMOR blots and one band at 60 kDa in the pSFK blot. Data are representative from four of eight rats in each group. B, The ratio of the optical density of the pSer375-MOR blot and the MOR blot was obtained and normalized to saline. C, The ratio of the optical density of the pSFK blot and the MOR blot was obtained and normalized to saline. *p = 0.0141, ***p = 0.0009, unpaired t tests with Welch's correction.
MOR constitutive inhibition of voltage-gated Ca2+ (Cav) channels in DRG neurons from mice with latent sensitization
An established method to assess constitutive inhibition by opioid receptors is to measure Cav channel currents in the absence and presence of a strong depolarizing pulse able to dissociate the Gβγ subunits responsible for their voltage-dependent inhibition (Walwyn et al., 2007; Connor and Traynor, 2010; Lam et al., 2011). We applied this method to DRG neurons (L4–L5 ipsilateral to the injected hindpaw) dissociated from mice >21 d after injection in the hindpaw of either saline or CFA (5 μl). The presence of Cav channels constitutively inhibited by MORs was assessed by a 2-pulse protocol (Fig. 9A): protocol P1consisted of a 100 ms hold at −80 mV, followed by a 10 ms test pulse from −80 mV to +10 mV; protocol P2 consisted of a 100 ms depolarizing prepulse from −80 mV to +80 mV, followed by the 10 ms test pulse from −80 mV to +10 mV. Constitutive inhibition of the Cav channels was determined by comparing the peak amplitude of the test pulse, averaged over 5 ms, in the absence (P1) or presence (P2) of the prepulse. A P2/P1 ratio >1.0 indicates Cav channel inhibition.
Figure 9.

Ligand-independent and ligand-dependent inhibition of Cav channels in DRG neurons from mice with latent sensitization. Mice were injected in one hindpaw with 5 μl of saline or CFA. DRG neurons (L4–L5) were acutely isolated from the saline-injected or the CFA-injected mice after day 21. A, Whole-cell patch-clamp recordings were used to measure Cav currents evoked by a two-pulse protocol (bottom traces). P2 includes a high voltage prepulse from −80 mV to +80 mV to dissociate Gβγ subunits from the Cav channels, whereas P1 consists only of a test pulse from −80 mV to +10 mV. Constitutive inhibition of the Cav channels was indicated by a larger current with the P2 protocol (red traces) resulting in an increase in the P2/P1 ratio. B, P1/P2 ratio obtained from DRG of saline-injected and CFA-injected mice in the absence and presence of NTX (1 μm). Two-way ANOVA: NTX p = 0.023, CFA p = 0.15, interaction p = 0.0497. Holm-Sidak's post hoc tests: **p < 0.01 compared with baseline, †p < 0.05 as shown. C, P1/P2 ratio obtained from DRG of CFA-injected mice untreated (baseline) or in presence of NTX (1 μm), 6β-naltrexol (10 μm), and NTX plus 6β-naltrexol. One-way ANOVA: p < 0.0001. D, DAMGO was used to assess MOR ligand inhibition of Cav currents in saline-injected mice and mice with CFA-induced latent sensitization. A single depolarizing 100 ms pulse from −70 mV to +10 mV was used to evoke Cav currents. An exemplar recording shows the following: (1) the basal Cav current (initial), (2) DAMGO (1 μm) inhibition of Cav current, and (3) the current after DAMGO had been washed off. E, DAMGO inhibition of Cav currents expressed as a percentage of the total current in neurons from the ipsilateral L4–L6 DRG from CFA- and saline-injected mice; there was no effect of CFA. Holm-Sidak's post hoc tests: *p < 0.05 compared with baseline; ††p < 0.01, †††p < 0.001 compared with NTX. Numbers indicate the number of neurons recorded in each group (n).
In DRG neurons from saline-injected mice, a P2/P1 ratio of 1.0 was obtained both in the absence and presence of the MOR inverse agonist NTX (one-sample t test to compare with 1.0: p = 0.80, t = 0.27, for baseline; p = 0.90, t = 0.13, for NTX; Fig. 9B). In DRG neurons from CFA-injected mice, the P2/P1 ratio was significantly higher than 1.0 (one-sample t test to compare with 1.0: p < 0.0001, t = 6.5). This was reduced to 1.0 in the presence of 1 μm NTX (one-sample t test to compare with 1.0: p = 0.39, t = 0.89, for NTX; Fig. 9B). Two-way ANOVA of the data in Figure 9B yielded p = 0.15, F(1,50) = 2.09 for CFA; p = 0.023, F(1,50) = 5.48 for NTX, and p = 0.0497, F(1,50) = 4.04 for interaction.
The P2/P1 ratio was assessed in DRG neurons from CFA-injected mice in the absence of ligands or in the presence of NTX (1 μm), the MOR neutral antagonist 6β-naltrexol (10 μm), or NTX + 6β-naltrexol (Fig. 9C). Whereas NTX decreased the P2/P1 ratio from a baseline value of 1.1 to 1.0, 6β-naltrexol (10 μm) yielded a P2/P1 ratio similar to baseline and blocked the effect of NTX. One-way ANOVA of the data in Figure 9C yielded p < 0.0001, F(3,57) = 9.02. These results demonstrate the presence of MOR-mediated constitutive inhibition of Cav channels in DRG neurons from mice with CFA-induced latent sensitization.
We found previously that an increase in constitutively active MORs is associated with a decrease in Cav channel inhibition induced by MOR agonists (Walwyn et al., 2007). To determine whether a similar inverse relationship exists between ligand-dependent and ligand-independent receptor pools, we examined Cav channel inhibition elicited by DAMGO, a selective MOR agonist, in mice with CFA-induced latent sensitization. Recordings were made from L4–L6 DRG ipsilateral to the injection and collected 28 d after the CFA or saline injection. A single 100 ms depolarizing pulse from −70 mV to +10 mV was used to evoke Ca2+ currents. Once three to four stable basal recordings were obtained, external solution containing DAMGO was applied to the cells until maximum inhibition was obtained (20–40 s) and then washed off by the extracellular solution (Fig. 9D). Mean Cav amplitudes were measured 5–10 ms after initiating the depolarizing step and plotted against time. Stable recordings were fitted by a linear function to compare, by extrapolation, control current amplitude with the current amplitude recorded in the presence of DAMGO. DAMGO (1 μm) inhibition was assessed as the percentage inhibition of the Ca2+ current amplitude before and after DAMGO application. We found no effect of CFA on Cav channel inhibition by DAMGO (Fig. 9E): an unpaired t test yielded p = 0.0796, t14 = 1.89. Therefore, the ability of DAMGO-activated MORs to inhibit Cav channels in DRG neurons does not decrease in mice with CFA-induced latent sensitization.
Discussion
This study shows that inflammation produces a state of latent sensitization to pain that is suppressed by the activation of spinal MORs, DORs, KORs, and α2ARs. Furthermore, the suppression of hyperalgesia is not due to sustained opioid release, but rather to receptor constitutive activity.
MORs, DORs, KORs, and α2ARs suppress hyperalgesia in latent sensitization
Our results show that MORs suppress hyperalgesia in animals with latent sensitization. Thus, in MOR KO mice, which have normal responses to acute noxious stimuli (Matthes et al., 1996), responses to mechanical stimulation during latent sensitization remained below baseline (although this contrasts with results reported by Gavériaux-Ruff et al., 2008). However, in these MOR KO mice there was a partial recovery from the peak hyperalgesia induced by CFA, indicating that receptors other than MORs contribute to the suppression of hyperalgesia.
Adrenergic α2ARs mediate the analgesic effects of norepinephrine (Yaksh, 1985; Stone et al., 1997; Pertovaara, 2006; De Felice et al., 2011) and are located in primary afferent terminals (Zeng and Lynch, 1991; Stone et al., 1997; Stone et al., 1998; Nazarian et al., 2008). The α2AR antagonist BRL44408 reinstated hyperalgesia during latent sensitization and eliminated the partial recovery from hyperalgesia in MOR KO mice, showing that α2ARs are one of the receptors that suppress hyperalgesia in latent sensitization.
Another of those receptors are DORs, because naltrindole reinstated hyperalgesia in rats and mice with latent sensitization. A role for DORs in the recovery from CFA hyperalgesia has been shown in mice lacking DORs in Nav1.8-expresing DRG neurons (Gavériaux-Ruff et al., 2011). Naltrindole induced hyperalgesia in saline-injected mice, but not saline-injected rats, suggesting that DORs may have an analgesic effect in mice.
Two KOR antagonists, nor-BNI and JDTic, reinstated hyperalgesia during latent sensitization, but only 12 h after their injection, consistent with the slow onset of their effects in vivo (Endoh et al., 1992; Munro et al., 2012). In agreement, Campillo et al. (2011) found that nor-BNI produced reinstatement in mice with latent sensitization induced by plantar incision. KORs suppress hyperalgesia in the absence of their natural agonist dynorphin, because JDTic produced reinstatement in pDYN KO mice. This suggests that KORs are constitutively active during latent sensitization.
In addition, neuropeptide Y receptors have been found to mediate anti-hyperalgesia in latent sensitization (Solway et al., 2011).
Receptor constitutive activity in latent sensitization
Although studies from overexpressed or mutated G-protein-coupled receptors show that they can become constitutively active (Connor and Traynor, 2010), there are few examples of constitutively active endogenous receptors. Constitutive activity is defined by the negative effect of an inverse agonist that can be blocked by a neutral antagonist (Kenakin, 2001). Here, we demonstrate that the suppression of hyperalgesia by MORs is not due to continuous opioid release, but rather to MOR constitutive activity (Kenakin, 2001; Connor and Traynor, 2010), as proposed by Corder et al. (2013). Unfortunately, the lack of well defined inverse agonists and neutral antagonists for KORs, DORs, and α2ARs prevents us from determining whether they were also constitutively active.
First, the MOR inverse agonist NTX, but not its neutral antagonist 6β-naltrexol, reinstated hyperalgesia in mice and rats. Importantly, 6β-naltrexol prevented the reinstatement induced by NTX. NTX acted on MORs, since it had no effect in MOR KO mice.
Second, unlike MOR KO mice, pENK, pOMC, and pDYN KO mice had normal latent sensitization: they recovered fully from hyperalgesia, which was reinstated by NTX. Therefore, suppression of hyperalgesia during latent sensitization can occur in the absence of opioid peptides. The fact that pDYN KO mice had higher baseline responses to noxious stimulation suggests that they are in an analgesic state. This is consistent with the fact that dynorphin has pronociceptive effects (Lai et al., 2001; Lai et al., 2006) in addition to its anti-nociceptive effects (Auh and Ro, 2012; Taylor et al., 2015). Indeed, pDYN KO mice exhibit reduced pain after nerve injury (Gardell et al., 2004; Xu et al., 2004).
Third, the suppression of hyperalgesia by MORs was not accompanied by MOR internalization. We have shown previously that all endogenous opioids produce MOR internalization when their degradation is suppressed with peptidase inhibitors (Song and Marvizón, 2003b) and that opioid release induces MOR internalization in slices (Song and Marvizón, 2003a; Chen et al., 2008) and in vivo (Lao et al., 2008; Chen and Marvizón, 2009). Therefore, the absence of MOR internalization in the presence of peptidase inhibitors argues against sustained opioid release in latent sensitization.
Fourth, in DRG neurons collected from mice with latent sensitization (but not from control mice), Cav channels were constitutively inhibited. This inhibition was suppressed by the MOR inverse agonist NTX, but was not affected by its neutral antagonist 6β-naltrexol, which prevented the effect of NTX.
Mechanisms for MOR constitutive activity
There are several possible mechanisms for receptor constitutive activity. The first is based on a competitive balance between constitutively active receptors and receptors available for ligand activation, so that an increase in one population of receptors occurs at the expense of the other. This happens in DRG neurons lacking β-arrestin 2 (Walwyn et al., 2007), which show increased constitutively active MORs and reduced Cav channel inhibition by MOR agonists. In contrast, in latent sensitization there was an increase in constitutively active MORs, but not a decrease in the ability of the MOR agonist DAMGO to inhibit Cav channels. This suggests that the constitutively active MORs are not extracted from the pool of MORs available for ligand activation.
Second, the increase in constitutively active MORs could be a result of a change in the intrinsic energy constraint that limits constitutive activity (Vezzi et al., 2013). This new model is an extension to the extended ternary complex model (De Léan et al., 1980; Wreggett and De Léan, 1984; Weiss et al., 1996) and proposes that an intrinsic, intramolecular energy constraint limits constitutive activity. This explains the low levels of basal constitutive MOR activity yet high levels of constitutive DOR activity. It is possible that, by activating MORs, agonists could lessen this constraint and shift the balance between constitutive- and ligand-activated receptors. This relationship may explain the increase in constitutive activity after chronic morphine (Liu and Prather, 2001; Sadée et al., 2005; Shoblock and Maidment, 2006; Meye et al., 2012; Meye et al., 2014) that occurs as a result of morphine-induced enkephalin release (Shoblock and Maidment, 2007). However, our results show that deleting each of the opioid peptides did not affect MOR suppression of hyperalgesia in latent sensitization, indicating that MOR constitutive activity was not initiated by an agonist.
Third, the increase in MOR constitutive activity could have resulted from changes in their phosphorylation. Chronic pain induces MOR constitutive phosphorylation (Illing et al., 2014) and we found that MORs immunoprecipitated from the spinal cord of rats with latent sensitization had increased Ser375 phosphorylation. However, this residue is typically associated with agonist-induced receptor activation and is not part of the phosphorylation “barcode” typical of constitutively active MORs (Williams et al., 2013; Illing et al., 2014). We also observed increased phosphorylation of SFKs at a site equivalent to Tyr416 of c-Src, indicative of SFK activation (Ohnishi et al., 2011). Targeting of c-Src to MORs by β-arrestin 2 is involved in MOR constitutive activation (Walwyn et al., 2007; Williams et al., 2013).
Fourth, the increase in constitutive activity could be a result of an increase in the expression levels of MORs after CFA. There are reports of changes in MOR expression after inflammatory and neuropathic pain (Nũnéz et al., 2007; Lee et al., 2011; Aoki et al., 2014). It is possible that, with an increase in the total amount of MORs, there is also an increase in the number of receptors that are constitutively active.
Physiological mechanisms
The phenomenon of latent sensitization indicates that chronic pain consists of an ongoing hyperalgesic state suppressed by analgesic mechanisms in the spinal cord. In addition, there is indirect evidence that latent sensitization involves descending pathways. Therefore, stress is able to interrupt the suppression of hyperalgesia during latent sensitization (Rivat et al., 2007; Le Roy et al., 2011), a phenomenon that would require either triggering descending pain facilitation or stopping ongoing descending pain inhibition. The fact that antagonists of these four receptors reinstated hyperalgesia contralaterally shows that both pain sensitization and its suppression occur bilaterally in the spinal cord, which supports the involvement of descending signals.
Latent sensitization may represent the same phenomenon as hyperalgesic priming, a model of chronic pain elicited by the injection in the paw of a sensitizing agent (interleukin-6 or carrageenan) followed by prostaglandin E2 or other inflammatory compounds (Aley et al., 2000; Parada et al., 2005; Reichling and Levine, 2009; Kim et al., 2015). Hyperalgesic priming also occurs bilaterally and involves a descending serotonergic signal for its initiation and a descending dopaminergic signal for its maintenance (Kim et al., 2015). It would be interesting to determine whether these descending pathways are similarly involved in latent sensitization. In a small trial, latent sensitization was shown to occur in 4 of 12 human subjects (Pereira et al., 2015), making these mechanisms promising targets to treat chronic pain.
Footnotes
This work was supported by the Rehabilitation Research and Development Service, Department of Veterans Affairs (Grant 1I01RX000378 to J.C.G.M.), the National Institutes of Health–National Institute of Drug Abuse (Grant R01-DA033059 to J.C.G.M. and J.A.M. and Grant DA005010 to W.M.W and A.M.), and by the Shirley and Stefas Hatos Center for the Study of Opioid Receptors. This study was under the venue of the following UCLA institutes: the Brain Research Institute, the Center for the Study of Opioid Receptors and Drugs of Abuse, the CURE: Digestive Diseases Research Center, and the Oppenheimer Family Center for Neurobiology of Stress. We thank Bradley K. Taylor for critical discussion.
The authors declare no competing financial interests.
References
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Mizoguchi H, Watanabe C, Takeda K, Sakurada T, Sakurada S. Potential involvement of mu-opioid receptor dysregulation on the reduced antinociception of morphine in the inflammatory pain state in mice. J Pharmacol Sci. 2014;124:258–266. doi: 10.1254/jphs.13242FP. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, Dado RJ, Loh HH, Law PY, Wessendorf MW, Elde R. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J Neurosci. 1995;15:3328–3341. doi: 10.1523/JNEUROSCI.15-05-03328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auh QS, Ro JY. Effects of peripheral kappa opioid receptor activation on inflammatory mechanical hyperalgesia in male and female rats. Neurosci Lett. 2012;524:111–115. doi: 10.1016/j.neulet.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessière B, Richebé P, Laboureyras E, Laulin JP, Contarino A, Simonnet G. Nitrous oxide (N2O) prevents latent pain sensitization and long-term anxiety-like behavior in pain and opioid-experienced rats. Neuropharmacology. 2007;53:733–740. doi: 10.1016/j.neuropharm.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Campillo A, Cabañero D, Romero A, García-Nogales P, Puig MM. Delayed postoperative latent pain sensitization revealed by the systemic administration of opioid antagonists in mice. Eur J Pharmacol. 2011;657:89–96. doi: 10.1016/j.ejphar.2011.01.059. [DOI] [PubMed] [Google Scholar]
- Carroll FI, Dolle RE. The discovery and development of the N-Substituted trans-3,4-Dimethyl-4-(3′-hydroxyphenyl)piperidine class of pure opioid receptor antagonists. ChemMedChem. 2014;9:1638–1654. doi: 10.1002/cmdc.201402142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll I, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: a novel kappa-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen W, Marvizón JC. Acute inflammation induces segmental, bilateral, supraspinally mediated opioid release in the rat spinal cord, as measured by μ-opioid receptor internalization. Neuroscience. 2009;161:157–172. doi: 10.1016/j.neuroscience.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Song B, Lao L, Pérez OA, Kim W, Marvizón JCG. Comparing analgesia and μ-opioid receptor internalization produced by intrathecal enkephalin: requirement for peptidase inhibition. Neuropharmacology. 2007;53:664–667. doi: 10.1016/j.neuropharm.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Song B, Marvizón JC. Inhibition of opioid release in the rat spinal cord by α2C adrenergic receptors. Neuropharmacology. 2008;54:944–953. doi: 10.1016/j.neuropharm.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Traynor J. Constitutively active mu-opioid receptors. Methods Enzymol. 2010;484:445–469. doi: 10.1016/B978-0-12-381298-8.00022-8. [DOI] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science. 2013;341:1394–1399. doi: 10.1126/science.1239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice M, Sanoja R, Wang R, Vera-Portocarrero L, Oyarzo J, King T, Ossipov MH, Vanderah TW, Lai J, Dussor GO, Fields HL, Price TJ, Porreca F. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain. 2011;152:2701–2709. doi: 10.1016/j.pain.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Léan A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Dixon WJ. The up-and-down method for small samples. J Am Stat Assoc. 1965;60:967–978. [Google Scholar]
- Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: a potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Arch Int Pharmacodyn Ther. 1992;316:30–42. [PubMed] [Google Scholar]
- Gardell LR, Ibrahim M, Wang R, Wang Z, Ossipov MH, Malan TP, Jr, Porreca F, Lai J. Mouse strains that lack spinal dynorphin upregulation after peripheral nerve injury do not develop neuropathic pain. Neuroscience. 2004;123:43–52. doi: 10.1016/j.neuroscience.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Gavériaux-Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci. 2008;27:2558–2567. doi: 10.1111/j.1460-9568.2008.06223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavériaux-Ruff C, Nozaki C, Nadal X, Hever XC, Weibel R, Matifas A, Reiss D, Filliol D, Nassar MA, Wood JN, Maldonado R, Kieffer BL. Genetic ablation of delta opioid receptors in nociceptive sensory neurons increases chronic pain and abolishes opioid analgesia. Pain. 2011;152:1238–1248. doi: 10.1016/j.pain.2010.12.031. [DOI] [PubMed] [Google Scholar]
- Illing S, Mann A, Schulz S. Heterologous regulation of agonist-independent μ-opioid receptor phosphorylation by protein kinase C. Br J Pharmacol. 2014;171:1330–1340. doi: 10.1111/bph.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarahi M, Sheibani V, Safakhah HA, Torkmandi H, Rashidy-Pour A. Effects of progesterone on neuropathic pain responses in an experimental animal model for peripheral neuropathy in the rat: a behavioral and electrophysiological study. Neuroscience. 2014;256:403–411. doi: 10.1016/j.neuroscience.2013.10.043. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Kim JY, Tillu DV, Quinn TL, Mejia GL, Shy A, Asiedu MN, Murad E, Schumann AP, Totsch SK, Sorge RE, Mantyh PW, Dussor G, Price TJ. Spinal dopaminergic projections control the transition to pathological pain plasticity via a D1/D5-mediated mechanism. J Neurosci. 2015;35:6307–6317. doi: 10.1523/JNEUROSCI.3481-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingery WS, Guo TZ, Davies MF, Limbird L, Maze M. The alpha(2A) adrenoceptor and the sympathetic postganglionic neuron contribute to the development of neuropathic heat hyperalgesia in mice. Pain. 2000;85:345–358. doi: 10.1016/S0304-3959(99)00286-9. [DOI] [PubMed] [Google Scholar]
- König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- Lai J, Ossipov MH, Vanderah TW, Malan TP, Jr, Porreca F. Neuropathic pain: the paradox of dynorphin. Mol Interv. 2001;1:160–167. [PubMed] [Google Scholar]
- Lai J, Luo MC, Chen Q, Ma S, Gardell LR, Ossipov MH, Porreca F. Dynorphin A activates bradykinin receptors to maintain neuropathic pain. Nat Neurosci. 2006;9:1534–1540. doi: 10.1038/nn1804. [DOI] [PubMed] [Google Scholar]
- Lam H, Maga M, Pradhan A, Evans CJ, Maidment NT, Hales TG, Walwyn W. Analgesic tone conferred by constitutively active mu opioid receptors in mice lacking beta-arrestin 2. Mol Pain. 2011;7:24. doi: 10.1186/1744-8069-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao L, Song B, Chen W, Marvizón JC. Noxious mechanical stimulation evokes the segmental release of opioid peptides that induce μ-opioid receptor internalization in the presence of peptidase inhibitors. Brain Res. 2008;1197:85–93. doi: 10.1016/j.brainres.2007.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Pérez FM, Wang W, Guan X, Zhao X, Fisher JL, Guan Y, Sweitzer SM, Raja SN, Tao YX. Dynamic temporal and spatial regulation of mu opioid receptor expression in primary afferent neurons following spinal nerve injury. Eur J Pain. 2011;15:669–675. doi: 10.1016/j.ejpain.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy C, Laboureyras E, Gavello-Baudy S, Chateauraynaud J, Laulin JP, Simonnet G. Endogenous opioids released during non-nociceptive environmental stress induce latent pain sensitization via a NMDA-dependent process. J Pain. 2011;12:1069–1079. doi: 10.1016/j.jpain.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Li X, Eisenach JC. alpha2A-adrenoceptor stimulation reduces capsaicin-induced glutamate release from spinal cord synaptosomes. J Pharmacol Exp Ther. 2001;299:939–944. [PubMed] [Google Scholar]
- Li X, Angst MS, Clark JD. Opioid-induced hyperalgesia and incisional pain. Anesth Analg. 2001;93:204–209. doi: 10.1097/00000539-200107000-00040. [DOI] [PubMed] [Google Scholar]
- Liu JG, Prather PL. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol Pharmacol. 2001;60:53–62. doi: 10.1124/mol.60.1.53. [DOI] [PubMed] [Google Scholar]
- Marvizón JCG. Opioidergic transmission in the dorsal horn. In: Malcangio M, editor. Synaptic plasticity in pain. New York: Springer; 2009. [Google Scholar]
- Marvizón JC, Chen W, Murphy N. Enkephalins, dynorphins and β-endorphin in the rat dorsal horn: an immunofluorescence colocalization study. J Comp Neurol. 2009;517:51–68. doi: 10.1002/cne.22130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dollé P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Meye FJ, van Zessen R, Smidt MP, Adan RA, Ramakers GM. Morphine withdrawal enhances constitutive mu-opioid receptor activity in the ventral tegmental area. J Neurosci. 2012;32:16120–16128. doi: 10.1523/JNEUROSCI.1572-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meye FJ, Ramakers GM, Adan RA. The vital role of constitutive GPCR activity in the mesolimbic dopamine system. Transl Psychiatry. 2014;4:e361. doi: 10.1038/tp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot B, Bourgoin S, Viguier F, Hamon M, Kayser V. Differential effects of calcitonin gene-related peptide receptor blockade by olcegepant on mechanical allodynia induced by ligation of the infraorbital nerve vs the sciatic nerve in the rat. Pain. 2012;153:1939–1948. doi: 10.1016/j.pain.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Munro TA, Berry LM, Van't Veer A, Béguin C, Carroll FI, Zhao Z, Carlezon WA, Jr, Cohen BM. Long-acting kappa opioid antagonists nor-BNI, GNTI and JDTic: pharmacokinetics in mice and lipophilicity. BMC Pharmacol. 2012;12:5. doi: 10.1186/1471-2210-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian A, Christianson CA, Hua XY, Yaksh TL. Dexmedetomidine and ST-91 analgesia in the formalin model is mediated by alpha2A-adrenoceptors: a mechanism of action distinct from morphine. Br J Pharmacol. 2008;155:1117–1126. doi: 10.1038/bjp.2008.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nũnéz S, Lee JS, Zhang Y, Bai G, Ro JY. Role of peripheral mu-opioid receptors in inflammatory orofacial muscle pain. Neuroscience. 2007;146:1346–1354. doi: 10.1016/j.neuroscience.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Ohnishi H, Murata Y, Okazawa H, Matozaki T. Src family kinases: modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 2011;34:629–637. doi: 10.1016/j.tins.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113:185–190. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Pereira MP, Donahue RR, Dahl JB, Werner M, Taylor BK, Werner MU. Endogenous opioid-masked latent pain sensitization: studies from mouse to human. PLoS One. 2015;10:e0134441. doi: 10.1371/journal.pone.0134441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara A. Noradrenergic pain modulation. Prog Neurobiol. 2006;80:53–83. doi: 10.1016/j.pneurobio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richebé P, Rivat C, Laulin JP, Maurette P, Simonnet G. Ketamine improves the management of exaggerated postoperative pain observed in perioperative fentanyl-treated rats. Anesthesiology. 2005;102:421–428. doi: 10.1097/00000542-200502000-00028. [DOI] [PubMed] [Google Scholar]
- Rivat C, Laboureyras E, Laulin JP, Le Roy C, Richebé P, Simonnet G. Non-nociceptive environmental stress induces hyperalgesia, not analgesia, in pain and opioid-experienced rats. Neuropsychopharmacology. 2007;32:2217–2228. doi: 10.1038/sj.npp.1301340. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japón M, Chan EC, Allen RG, Low MJ. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci U S A. 1996;93:3995–4000. doi: 10.1073/pnas.93.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadée W, Wang D, Bilsky EJ. Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sci. 2005;76:1427–1437. doi: 10.1016/j.lfs.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knockout mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/S0169-328X(00)00264-3. [DOI] [PubMed] [Google Scholar]
- Shoblock JR, Maidment NT. Constitutively active mu opioid receptors mediate the enhanced conditioned aversive effect of naloxone in morphine-dependent mice. Neuropsychopharmacology. 2006;31:171–177. doi: 10.1038/sj.npp.1300782. [DOI] [PubMed] [Google Scholar]
- Shoblock JR, Maidment NT. Enkephalin release promotes homeostatic increases in constitutively active mu opioid receptors during morphine withdrawal. Neuroscience. 2007;149:642–649. doi: 10.1016/j.neuroscience.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Solway B, Bose SC, Corder G, Donahue RR, Taylor BK. Tonic inhibition of chronic pain by neuropeptide Y. Proc Natl Acad Sci U S A. 2011;108:7224–7229. doi: 10.1073/pnas.1017719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Marvizón JC. Peptidases prevent μ-opioid receptor internalization in dorsal horn neurons by endogenously released opioids. J Neurosci. 2003b;23:1847–1858. doi: 10.1523/JNEUROSCI.23-05-01847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Marvizón JC. Dorsal horn neurons firing at high frequency, but not primary afferents, release opioid peptides that produce μ-opioid receptor internalization in the rat spinal cord. J Neurosci. 2003a;23:9171–9184. doi: 10.1523/JNEUROSCI.23-27-09171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Marvizón JC. NMDA receptors and large conductance calcium-sensitive potassium channels inhibit the release of opioid peptides that induce μ-opioid receptor internalization in the rat spinal cord. Neuroscience. 2005;136:549–562. doi: 10.1016/j.neuroscience.2005.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike RC, Puskar Z, Sakamoto H, Stewart W, Watt C, Todd AJ. MOR-1-immunoreactive neurons in the dorsal horn of the rat spinal cord: evidence for nonsynaptic innervation by substance P-containing primary afferents and for selective activation by noxious thermal stimuli. Eur J Neurosci. 2002;15:1306–1316. doi: 10.1046/j.1460-9568.2002.01969.x. [DOI] [PubMed] [Google Scholar]
- Stone LS, MacMillan LB, Kitto KF, Limbird LE, Wilcox GL. The alpha2a adrenergic receptor subtype mediates spinal analgesia evoked by alpha2 agonists and is necessary for spinal adrenergic-opioid synergy. J Neurosci. 1997;17:7157–7165. doi: 10.1523/JNEUROSCI.17-18-07157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone LS, Broberger C, Vulchanova L, Wilcox GL, Hökfelt T, Riedl MS, Elde R. Differential distribution of α2A and α2C adrenergic receptor immunoreactivity in the rat spinal cord. J Neurosci. 1998;18:5928–5937. doi: 10.1523/JNEUROSCI.18-15-05928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Størkson RV, Kjørsvik A, Tjølsen A, Hole K. Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Roberts KW, Pradhan AA, Akbari HA, Walwyn W, Lutfy K, Carroll FI, Cahill CM, Evans CJ. Anti-nociception mediated by a kappa opioid receptor agonist is blocked by a delta receptor agonist. Br J Pharmacol. 2015;172:691–703. doi: 10.1111/bph.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BK, Corder G. Endogenous analgesia, dependence, and latent pain sensitization. Curr Top Behav Neurosci. 2014;20:283–325. doi: 10.1007/7854_2014_351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzi V, Onaran HO, Molinari P, Guerrini R, Balboni G, Calò G, Costa T. Ligands raise the constraint that limits constitutive activation in G-protein-coupled opioid receptors. J Biol Chem. 2013;288:23964–23978. doi: 10.1074/jbc.M113.474452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5104. doi: 10.1523/JNEUROSCI.1157-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Heijnen CJ, van Velthoven CT, Willemen HL, Ishikawa Y, Zhang X, Sood AK, Vroon A, Eijkelkamp N, Kavelaars A. Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J Clin Invest. 2013;123:5023–5034. doi: 10.1172/JCI66241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. III. Resurrecting efficacy. J Theor Biol. 1996;181:381–397. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreggett KA, De Léan A. The ternary complex model–its properties and application to ligand interactions with the D2-dopamine receptor of the anterior pituitary gland. Mol Pharmacol. 1984;26:214–227. [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL. Pharmacology of spinal adrenergic systems which modulate spinal nociceptive processing. Pharmacol Biochem Behav. 1985;22:845–858. doi: 10.1016/0091-3057(85)90537-4. [DOI] [PubMed] [Google Scholar]
- Yalcin I, Bohren Y, Waltisperger E, Sage-Ciocca D, Yin JC, Freund-Mercier MJ, Barrot M. A time-dependent history of mood disorders in a murine model of neuropathic pain. Biol Psychiatry. 2011;70:946–953. doi: 10.1016/j.biopsych.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Zeng DW, Lynch KR. Distribution of alpha 2-adrenergic receptor mRNAs in the rat CNS. Brain Res Mol Brain Res. 1991;10:219–225. doi: 10.1016/0169-328X(91)90064-5. [DOI] [PubMed] [Google Scholar]