

Cheung et al. examined the opposing effects of TIGAR- and RAC1-derived ROS in a model of Apc deletion. Their results indicate that the pro- and anti-proliferative effects of ROS can be independently modulated in the same cell, with two key targets in the Wnt pathway functioning to integrate the different ROS signals for optimal cell proliferation.
Keywords: TIGAR, ROS, RAC1, APC, Wnt, proliferation
Abstract
Reactive oxygen species (ROS) participate in numerous cell responses, including proliferation, DNA damage, and cell death. Based on these disparate activities, both promotion and inhibition of ROS have been proposed for cancer therapy. However, how the ROS response is determined is not clear. We examined the activities of ROS in a model of Apc deletion, where loss of the Wnt target gene Myc both rescues APC loss and prevents ROS accumulation. Following APC loss, Myc has been shown to up-regulate RAC1 to promote proliferative ROS through NADPH oxidase (NOX). However, APC loss also increased the expression of TIGAR, which functions to limit ROS. To explore this paradox, we used three-dimensional (3D) cultures and in vivo models to show that deletion of TIGAR increased ROS damage and inhibited proliferation. These responses were suppressed by limiting damaging ROS but enhanced by lowering proproliferative NOX-derived ROS. Despite having opposing effects on ROS levels, loss of TIGAR and RAC1 cooperated to suppress intestinal proliferation following APC loss. Our results indicate that the pro- and anti-proliferative effects of ROS can be independently modulated in the same cell, with two key targets in the Wnt pathway functioning to integrate the different ROS signals for optimal cell proliferation.
Many cellular processes lead to the generation of reactive oxygen species (ROS), which can take different forms and occupy different cellular locations. The major sources of ROS production are mitochondrial oxidative respiration, the activity of NADPH oxidase (NOX) enzymes, and nitric oxide synthase (NOS). The generation of ROS can be an important component of signaling for cell proliferation as well as other responses such as angiogenesis, immortalization, metastasis, and drug resistance. However, ROS can also be damaging to the cell, resulting in DNA damage, lipid oxidation, and, ultimately, cell death. In this latter context, ROS is cytotoxic. Tight regulation of oxidative stress is therefore important to maintain cell viability, and numerous antioxidant activities exist to balance these different activities of ROS for maximal survival (Sabharwal and Schumacker 2014; Schieber and Chandel 2014). This requirement to balance ROS activity is particularly evident in cancer cells, where the oncogenic process drives increased ROS accumulation. While the pro-oncogenic activities of ROS signaling can contribute to abnormal proliferation, this must be balanced by antioxidant functions to ensure cell survival. Indeed, cancer cells frequently show enhanced expression of antioxidant defense to counteract the potentially lethal increases in ROS that accompany malignant development (D'Autreaux and Toledano 2007; Trachootham et al. 2009; Gorrini et al. 2013; Sullivan and Chandel 2014b). Given the complexity of the response to ROS signaling, it is not surprising that predicting the effect of ROS modulation is difficult. The use of antioxidants to limit the damaging effects of ROS has been suggested to promote longevity or, by preventing genotoxic damage and oncogenic signaling, limit cancer development. However, in practice, it has been extremely difficult to generate compelling evidence to support either of these proposals, leading to alternative models in which limitation of ROS may be counterproductive to therapy by helping cancer cells to survive (Dickinson and Chang 2011). The fact that many cancer cells exist in a high ROS state suggests that they will be selectively sensitive to further increases in oxidative stress or inhibition of the attendant antioxidant defense (DeNicola et al. 2011; Harris et al. 2015). In this context, it is worth noting that many standard forms of cancer therapy, such as irradiation (IR) and chemotherapy, depend on ROS generation (Myers et al. 1977; Powell and McMillan 1990).
Most models of how ROS can exert these very different effects on the cell focus primarily on the overall level of ROS, proposing that low ROS levels promote beneficial responses, such as proliferation and survival, while high ROS levels lead to damage and cell death. However, it is also possible that different ROS-generating systems lead to different responses; for example, mitochondrial ROS may be more likely to promote damage and death (Giulivi et al. 1995; Heales et al. 1999; Adam-Vizi and Chinopoulos 2006; Abramov et al. 2007), while membrane-generated ROS is more often described as contributing to signaling for cell proliferation (Vilhardt and van Deurs 2004; Choi et al. 2005; Li et al. 2006; Ushio-Fukai 2006). These distinctions are clearly not absolute; mitochondrial ROS has also been shown to contribute to proliferation, tumorigenicity, migration, and metastasis (Lee et al. 2002; Kwon et al. 2004; Weinberg et al. 2010; Porporato et al. 2014), while NOX-generated ROS at the membrane can induce cell death through ferroptosis and necrosis (Kim et al. 2007; Dixon et al. 2012). Taken together, it seems likely that the cell response to ROS reflects a complex integration of ROS type, location, and level. These are important factors to consider when developing a therapeutic strategy that involves the modulation of redox levels, where it will be critical to reconcile the beneficial and deleterious effects of ROS in cancer development. To explore more fully whether different responses to ROS simply reflect ROS levels or whether the responses are more complex, we explored the effects of combining alterations in two previously described ROS-modulating pathways. The first focuses on TIGAR, a protein that can control glucose metabolism and helps to maintain NADPH levels to regenerate glutathione (GSH), a key intracellular antioxidant. Loss of TIGAR expression leads to increased ROS, and while this is not obviously detrimental to normal growth and development, in most model systems, this results in defects in both proliferation and survival following stress (Bensaad et al. 2006, 2009; Lui et al. 2011; Wanka et al. 2012; Yin et al. 2012). In the mouse intestine, loss of TIGAR can decrease the proliferation that accompanies tissue regeneration after genotoxic stress and limit the hyperproliferation seen in Wnt-driven adenoma through the failure to limit ROS (Cheung et al. 2013). RAC1, in contrast, is a component of the NOX signaling complex that uses NADPH to signal proliferation. Similar to loss of TIGAR, loss of RAC1 also leads to defects in proliferation, but, in this case, the effect is a reflection of decreased ROS levels (Myant et al. 2013). This raises a paradox in that both decreased and increased ROS in the intestine can lead to a decrease in proliferation. Here we found that both TIGAR and RAC1 expression are induced under conditions of intestinal proliferation in response to abnormal Wnt signaling following Apc deletion. We therefore exploited the observation that the activation of Wnt results in the control of both signaling ROS and deleterious ROS, using this opportunity to modulate the endogenous amounts of these different types of ROS by manipulating these two pathways in a three-dimensional (3D) organoid culture system as well as genetically in vivo. We demonstrated that differentially modulating the different types of ROS can affect the optimal growth response.
Results
TIGAR supports intestinal proliferation through cell-autonomous mechanisms
Our previous studies found that, although whole-body constitutive deletion of TIGAR did not prevent normal growth and development, TIGAR-null mice showed defects in intestinal regeneration and decreased tumor development in a model of intestinal adenoma development following Apc deletion (Cheung et al. 2013). To determine whether the contribution of TIGAR to intestinal regeneration following damage reflected a requirement for TIGAR in the intestinal cells themselves, we generated a conditional TIGARfl/fl mouse and crossed these animals with Ahcre (Ahcre+TIGARfl/fl) to express cre in the intestine after induction by the cytochrome P450 inducer β-napthoflavone (Ireland et al. 2004). The loss of TIGAR staining by immunohistochemistry confirmed successful deletion (Fig. 1A). As seen in the TIGAR−/− mice (Cheung et al. 2013), deletion of TIGAR in the absence of stress did not cause an overt phenotype, and both wild-type and TIGAR-deleted intestines showed similar rates of proliferation, as measured by Ki67 staining (Fig. 1A). However, following genotoxic damage (cisplatin or IR) (Fig. 1B–D; Supplemental Fig. 1A), Ahcre+TIGARfl/fl mice showed a clear defect in regeneration compared with the control Ahcre+TIGAR+/+ mice, a phenotype that was manifested by both the size of the crypts and proliferation measured by Ki67 staining (Fig. 1C,D). Using malondialdehyde (MDA) staining to indicate the accumulation of damage by ROS, we found that TIGAR-deficient intestines sustained increased ROS damage following cisplatin- or IR-induced damage (Fig. 1E), correlating with a defect in regeneration after genotoxic damage.
Figure 1.
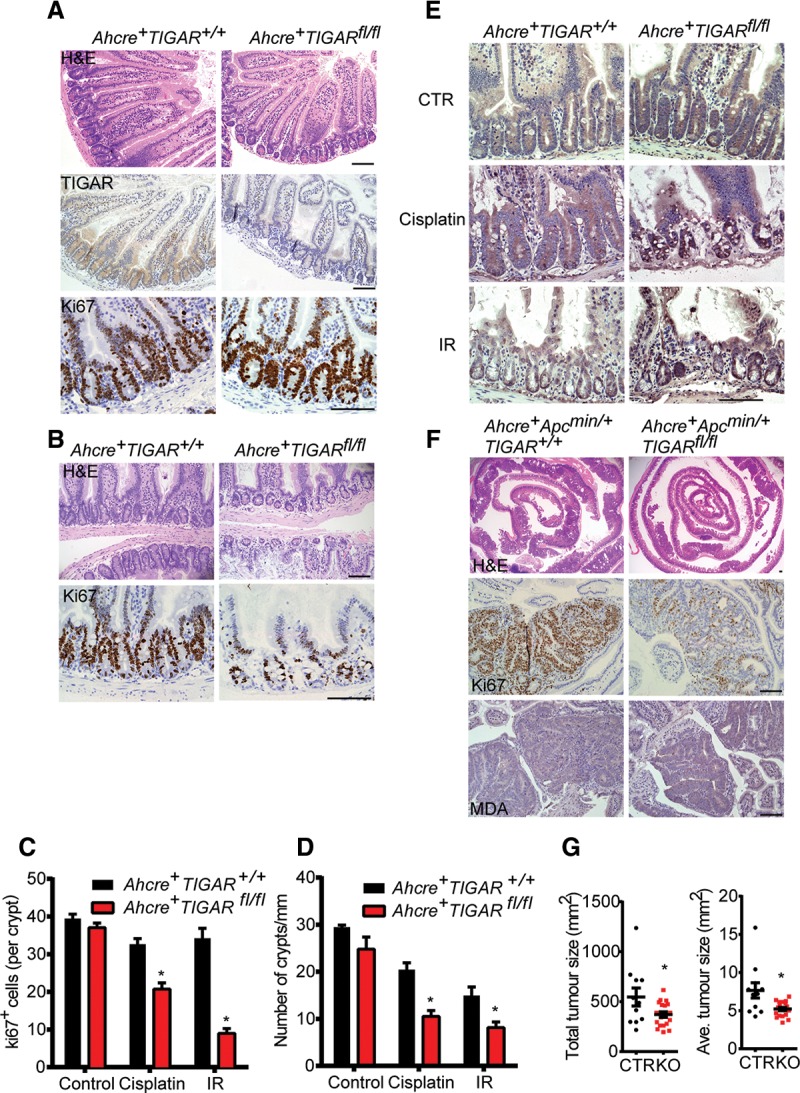
TIGAR supports intestinal proliferation through cell-autonomous mechanisms. (A) H&E staining (top), TIGAR staining (middle), and Ki67 staining (bottom) of small intestines from Ahcre+ TIGAR+/+ and Ahcre+TIGARfl/fl animals 3 d after induction of cre by β-napthoflavone. (B) Small intestines from induced Ahcre+TIGAR+/+ and Ahcre+TIGARfl/fl animals 3 d after CDDP (cisplatin; 10 mg/kg) treatment. (Top) H&E staining. (Bottom) Ki67 staining. (C) Quantification of Ki67+ cells per intestinal crypt of induced Ahcre+TIGAR+/+ and Ahcre+TIGARfl/fl 3 d after CDDP or 10 Gy of γ-IR. (*) P < 0.05 compared with Ahcre+TIGAR+/+. Control Ahcre+TIGAR+/+, n = 11; control Ahcre+TIGARfl/fl, n = 8; cisplatin Ahcre+ TIGAR+/+, n = 6; cisplatin Ahcre+TIGARfl/fl, n = 7; IR Ahcre+TIGAR+/+, n = 4; cisplatin Ahcre+TIGARfl/fl, n = 6. (D) Quantification of number of crypts per millimeter of intestine of induced Ahcre+TIGAR+/+ and Ahcre+TIGARfl/fl 3 d after CDDP (cisplatin) or 10 Gy of γ-IR. (*) P < 0.05 compared with Ahcre+TIGAR+/+. Control Ahcre+TIGAR+/+, n = 3; control Ahcre+TIGARfl/fl, n = 3; cisplatin Ahcre+TIGAR+/+, n = 6; cisplatin Ahcre+TIGARfl/fl, n = 7; IR Ahcre+TIGAR+/+, n = 4; cisplatin Ahcre+TIGARfl/fl, n = 6. (E) MDA staining of small intestines from induced Ahcre+TIGAR+/+ and Ahcre+TIGARfl/fl animals after CDDP or γ-IR. (F) H&E staining (top), Ki67 staining (middle), and MDA staining (bottom) of the small intestines from induced Ahcre+Apcmin/+TIGAR+/+ and Ahcre+Apcmin/+TIGARfl/fl at 80 d of age. (G) Quantification of total tumor size and average tumor size of induced Ahcre+Apcmin/+TIGAR+/+ and Ahcre+Apcmin/+TIGARfl/fl at 80 d of age. (*) P < 0.05 compared with control. Control (CTR), n = 11; knockout (KO), n = 19. Bars, 100 µm.
Our previous work showed that systemic loss of TIGAR decreased intestinal adenoma development following APC loss and enhanced the survival of these mice (Cheung et al. 2013). Using Apcmin/+ mice, which develop adenomas in response to a reduced expression of APC (Moser et al. 1990), we found that loss of TIGAR specifically in intestinal cells also resulted in a significant increase in ROS damage, as measured by MDA staining, and reduction in tumor burden and tumor proliferation, as measured by Ki67 staining (Fig. 1F,G; Supplemental Fig. 1B), although, in this model, no significant survival advantage of loss of TIGAR could be detected. These results support a cell-autonomous function for TIGAR in limiting ROS and supporting proliferation in the small intestine after injury or in adenomas.
TIGAR expression is regulated by Wnt signaling through Myc
Since TIGAR plays an important role in regulating and supporting proliferation of Apc-deficient intestinal cells, we examined the effect of activation of Wnt signaling on TIGAR expression. β-Napthoflavone treatment of Ahcre+Apcfl/fl mice resulted in loss of APC in both the intestine and liver (Sansom et al. 2004) and was accompanied by a clear increase in expression of the Wnt target gene Axin2 (Fig. 2C; Supplemental Fig. 2A). APC loss also led to an increase in TIGAR expression in both organs (Fig. 2A–C). Although we were unable to consistently recapitulate this Wnt-induced increase in TIGAR expression in two-dimensional (2D) tissue culture systems, a similar increase in TIGAR expression at both the protein and mRNA level was detected in crypt organoid cultures from Apcmin/+ mice (Supplemental Fig. 2B,C) and following acute Apc deletion in Ahcre+Apcfl/fl organoid cultures (Fig. 2D). Interestingly, while an increase in the known Wnt target gene Ccnd1 (cycline D1) and Myc was detected in each of these crypt cultures in response to APC loss, there was no clear induction of p53 or the p53-dependent target gene Cdkn1a (p21) (Fig. 2D,E). These results are consistent with previous reports (Reed et al. 2008) and our recent observations that the expression of TIGAR in the mouse intestine is not dependent on p53 (Lee et al. 2015).
Figure 2.
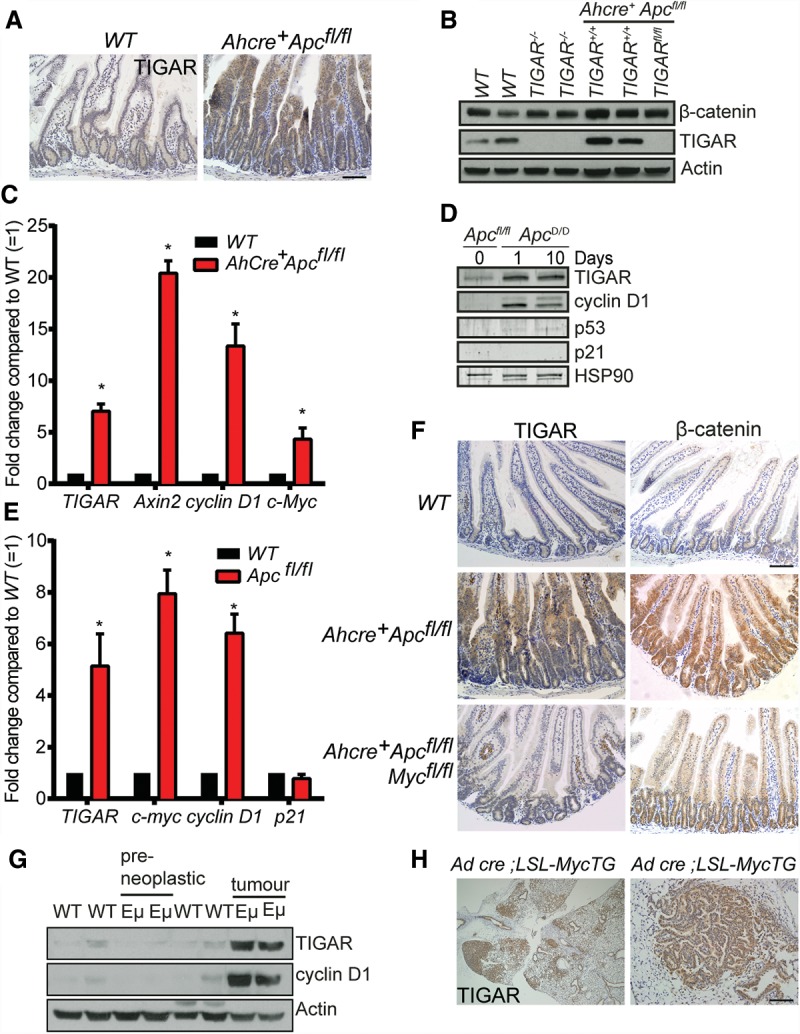
TIGAR expression is regulated by Wnt signaling through Myc. (A) TIGAR staining of small intestines from wild-type (WT) and Ahcre+Apcfl/fl animals 3 d after cre induction by β-napthoflavone. (B) Western blot analysis of liver tissues from wild-type, TIGAR−/−, Ahcre+Apcfl/flTIGAR+/+, and Ahcre+Apcfl/flTIGARfl/fl 3 d after cre induction. (C) mRNA expression of TIGAR, Axin2, cyclin D1, and c-Myc from wild-type and Ahcre+Apcfl/fl liver tissues 3 d after cre induction. (*) P < 0.05 compared with wild type. n = 3. (D) Western blot analysis of Ahcre+Apcfl/fl crypt cultures before (0 d) and after (1 and 10 d) cre induction. (E) mRNA expression of TIGAR, c-Myc, cyclin D1, and p21 from wild-type and Apcfl/fl crypts. (*) P < 0.05 compared with wild type. n = 3. (F) TIGAR staining (left) and β-catenin staining (right) of intestines from wild-type, Ahcre+Apcfl/fl, and Ahcre+Apcfl/flMycfl/fl 3 d after cre induction. (G) Western blot analysis of tissues from wild-type thymus, preneoplastic thymus, and thymic lymphoma from Eµ-Myc animals. (H) TIGAR staining of lung tissues from inhaled Adeno-cre; LSL-MycTG; LSL-KRasG12D animals. Bars, 100 µm.
Induction of Wnt signaling can promote transcriptional activation through several mechanisms. Loss of APC results in the stabilization and nuclear translocation of β-catenin, which interacts with TCF to directly control gene expression (MacDonald et al. 2009). A key transcriptional target of β-catenin/TCF is Myc, which has been shown to mediate many of the cellular responses to Wnt signaling (Sansom et al. 2007). To determine whether Myc is important for the induction of TIGAR expression, we examined the effect of simultaneous deletion of Apc and Myc on TIGAR expression in vivo (Fig. 2F). As seen previously (Sansom et al. 2007), loss of Myc significantly decreased the enhanced crypt proliferation seen in response to Apc deletion. As shown previously (Sansom et al. 2007), the absence of Myc did not prevent the activation of Wnt, as shown by the accumulation of nuclear β-catenin in these smaller crypts. However, deletion of Myc led to a failure to induce both cyclin D1—a direct Myc target gene (Supplemental Fig. 2D)—and TIGAR in Apc-deleted crypts (Fig. 2F). Taken together, the results suggest that the induction of TIGAR is not a direct response to β-catenin/TCF activity but is a response to the activation of Myc. Further support for a role of Myc in the regulation of TIGAR expression was seen in transgenic mice expressing deregulated Myc. In the Eμ-Myc lymphoma model, transgenic Myc is expressed from the IgH enhancer, leading to deregulated Myc expression in B cells and the development of lymphomas within the first 5 mo of age (Adams et al. 1985). Thymus from tumor-bearing mice showed clear evidence of TIGAR overexpression compared with normal tissue from wild-type mice (Fig. 2G). However, no evidence of increased TIGAR levels was seen in the preneoplastic tissue of these mice, suggesting that Myc-mediated expression of TIGAR may be indirect or require additional signals. A lung cancer model driven by Adeno-cre inhalation in an LSL-KRasG12D, LSL-Hu-Myc mouse (Murphy et al. 2008) showed evidence of premalignant lesions within 6 wk of KRas and Myc activation. Substantial up-regulation of TIGAR was seen in these lesions compared with the surrounding normal lung tissue (Fig. 2H), again suggesting that Myc deregulation contributes to the up-regulation of TIGAR expression.
TIGAR induction is ROS-dependent
Previous studies have shown that both inhibition of APC and treatment with IR increased ROS in the mouse intestine (Cheung et al. 2013; Myant et al. 2013). Importantly, the induction of ROS (as measured by MDA staining) in the intestine following Apc deletion was dependent on Myc (Fig. 3A). We therefore considered that the activation of TIGAR may in part reflect increased levels of ROS. To test this directly, we examined the effect of ROS-inducing treatments on TIGAR expression in the normal and tumor organoid cultures. Induction of oxidative stress following treatment of the crypts with hydrogen peroxide (H2O2) or rotenone led to an increase in the expression of the known ROS-responsive gene heme oxygenase-1 (HO-1) and TIGAR, although expression of the Myc target cyclin D1 was not affected (Fig. 3B,C). The activation of TIGAR expression was not accompanied by an increase in the p53 target p21 (Fig. 3B), consistent with our recent report that TIGAR expression is not dependent on p53 activity in the mouse intestine (Lee et al. 2015). Conversely, treatment of the organoids with antioxidants such as allopurinol (a xanthine oxidase inhibitor), L-NAME (L-NG-nitroarginine methyl ester; an NOS inhibitor) resulted in decreased TIGAR expression (Fig. 3D). These results indicate that TIGAR expression can be regulated by the levels of oxidative stress in the cell.
Figure 3.
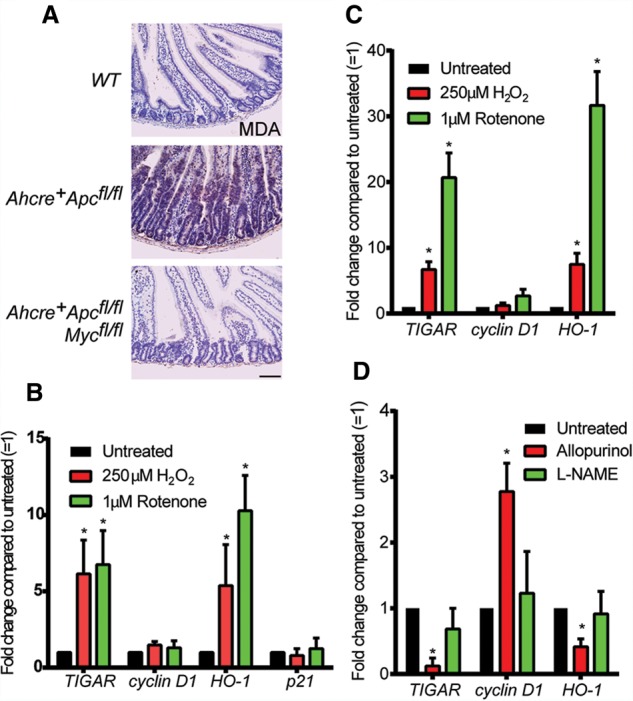
TIGAR expression is regulated by ROS. (A) MDA staining of small intestines from wild-type (WT), Ahcre+Apcfl/fl, and Ahcre+Apcfl/flMycfl/fl 3 d after cre induction. (B) mRNA expression of TIGAR, cyclin D1, HO-1, and p21 from normal wild-type intestinal crypt cultures after 24 h of the indicated ROS treatments. (*) P < 0.05 compared with untreated. n = 3. (C) mRNA expression of TIGAR, cyclin D1, and HO-1 from Apcmin/+ organoid cultures after 24 h of the indicated ROS treatments. (*) P < 0.05 compared with untreated. n = 3. (D) mRNA expression of TIGAR, cyclin D1, and HO-1 from Apcmin/+ organoid cultures after 24 h of the indicated anti-ROS treatments. Bars, 100 µm.
TIGAR supports intestinal cell growth by limiting damaging ROS
Taken together, our data suggest that TIGAR is induced in response to ROS, and previous work has shown that TIGAR can function to provide antioxidant defense (Bensaad et al. 2006, 2009; Lui et al. 2011; Wanka et al. 2012; Yin et al. 2012; Cheung et al. 2013). Normal and tumor intestinal crypts derived from constitutive TIGAR-null mice grew less well in 3D cultures, and these could be rescued with antioxidants such as N-acetyl L-cysteine (NAC) (Cheung et al. 2013). To examine the effects of ROS regulation in more detail, we established 3D organoid cultures from Rosa-creERT2Apcmin/+TIGARfl/fl mice. This system allowed us to acutely delete TIGAR in the organoids by treating the cultures with 4-hydroxytamoxifen (Fig. 4A). Acute deletion of TIGAR in vitro led to an inhibition of growth of these Apcmin/+ organoids that could be rescued following treatment with the antioxidants NAC, Trolox, and EUK134 (a catalase mimetic) (Baker et al. 1998) at concentrations that did not affect the growth of the control Apcmin/+ organoids (Fig. 4B,C; Supplemental Fig. 3A,B). Using MDA staining to detect lipid damage by oxidation, we detected increased oxidative damage in the TIGAR-deleted organoids compared with TIGAR-expressing controls. This increased ROS damage was prevented by treatment with NAC or Trolox (Fig. 4D; Supplemental Fig. 3C).
Figure 4.
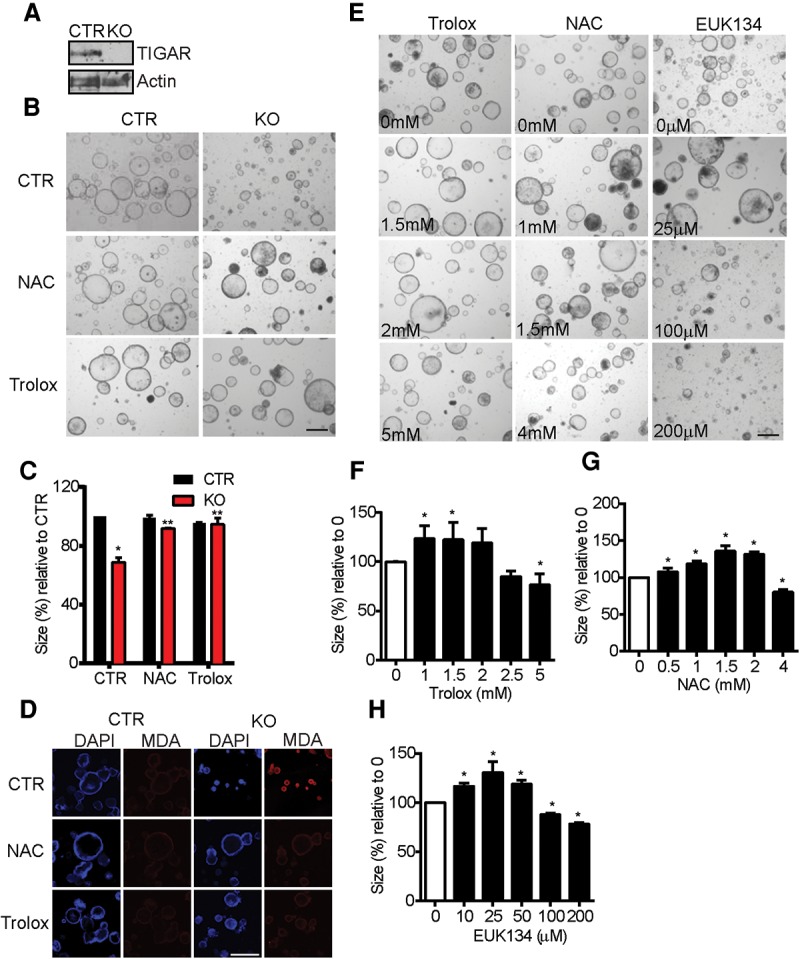
General ROS inhibitors can rescue the growth defects of TIGAR-deficient Apcmin/+ intestinal organoids. (A) Western blot analysis from Rosa-creERT2Apcmin/+TIGARfl/fl intestinal organoid cultures that were not induced (control [CTR]) or were induced by 4-hydroxytamoxifen (knockout [KO]) for 2 d. (B) Organoid cultures from noninduced (CTR) and induced (knockout) organoid cultures 2 d after 0.5 mM NAC or 1 mM Trolox treatments. (C) Measurement of relative crypt size (relative to CTR with CTR treatment) after the indicated induction and antioxidant treatments. (*) P < 0.05 compared with control treatment of control organoids; (**) P < 0.05 compared with knockout crypt without drugs. (D) MDA staining of noninduced (CTR) and induced (knockout) organoids treated with 0.5 mM NAC or 1 mM Trolox for 2 d. (E) Apcmin/+ organoid cultures 2 d after various concentrations of Trolox, NAC, or EUK134. (F) Measurements of relative organoid size after increasing concentrations of Trolox. (*) P < 0.05 compared with untreated. (G) Measurements of relative organoid size after increasing concentrations of NAC. (*) P < 0.05 compared with untreated. (H) Measurements of relative organoid size after increasing concentrations of EUK134. (*) P < 0.05 compared with untreated. Bars, 100 µm.
While our data support a role for TIGAR in limiting oxidative damage and thus supporting cell proliferation and survival, it is clear that some ROS signaling is important for cell proliferation. Indeed, using the Apcmin/+ organoids, we were able to demonstrate this effect of ROS limitation. While our previous studies were carried out with concentrations of NAC, Trolox, and EUK134 that did not inhibit the growth of these cells, we found that treating the cultures with even higher concentrations of these antioxidants, which inhibit ROS levels in a dose-dependent manner (Supplemental Fig. 4), led to a clear inhibition of organoid growth (Fig. 4E–H). These data therefore indicate that ROS levels must be kept within tight boundaries and that too little ROS may be as inhibitory to cell growth as too much ROS.
Qualitative differences between signaling and damaging ROS
Previous work has suggested that signaling and damaging ROS may be generated through different mechanisms as well as in different cell compartments (Trachootham et al. 2009). ROS generated in mitochondria as a consequence of oxidative phosphorylation or nitric oxide generated by nitric oxide synthase can contribute to proliferation but are generally considered to be damaging to the cell and, if produced at excessive levels, lead to cell death (Porasuphatana et al. 2003; Abramov et al. 2007). On the other hand, the membrane-associated NOXs are induced by growth factors and produce ROS that is required for cell proliferation signaling (Bedard and Krause 2007). Clearly, ROS can be supportive or detrimental to cell growth and survival, although whether this reflects only differences in ROS levels or whether different forms of ROS can have different functions based on source or location within the cell is not clear. We therefore sought to test whether the ROS signal induced by NOX is qualitatively different from the ROS induced by loss of TIGAR.
To explore the effects of ROS from different sources on organoid proliferation, we treated Apcmin/+ organoid cultures with different antioxidants, each of which reduces ROS levels in the organoid cultures in a dose-dependent manner, as indicated by the reduction of DHE (dihydroethidium) staining (Supplemental Fig. 4). As shown above, NAC, Trolox, and EUK134 are general antioxidants that lower both proliferative and damaging ROS. Using Mito tempo to inhibit mitochondrial ROS (Murphy and Smith 2007), L-NAME to inhibit nitric oxide synthase (Atlante et al. 1997), and allopurinol to inhibit xanthine oxidase that generate deleterious ROS (Cheng and Sun 1994), we found that the growth of the organoids was enhanced and could not be inhibited even at high concentrations of the antioxidants, suggesting that the ROS targeted by these compounds was only growth inhibitory and not required for the growth of these cells (Fig. 5A–D). In contrast, diphenyleneiodonium (DPI) and ML171, two inhibitors of NOX1 (Morre 2002; Gianni et al. 2010a,b), resulted in a dose-dependent reduction in organoid growth with no evidence of enhanced proliferation, indicating that NOX1-derived ROS only promotes, but does not inhibit, cell growth in this system (Fig. 5A,E,F).
Figure 5.
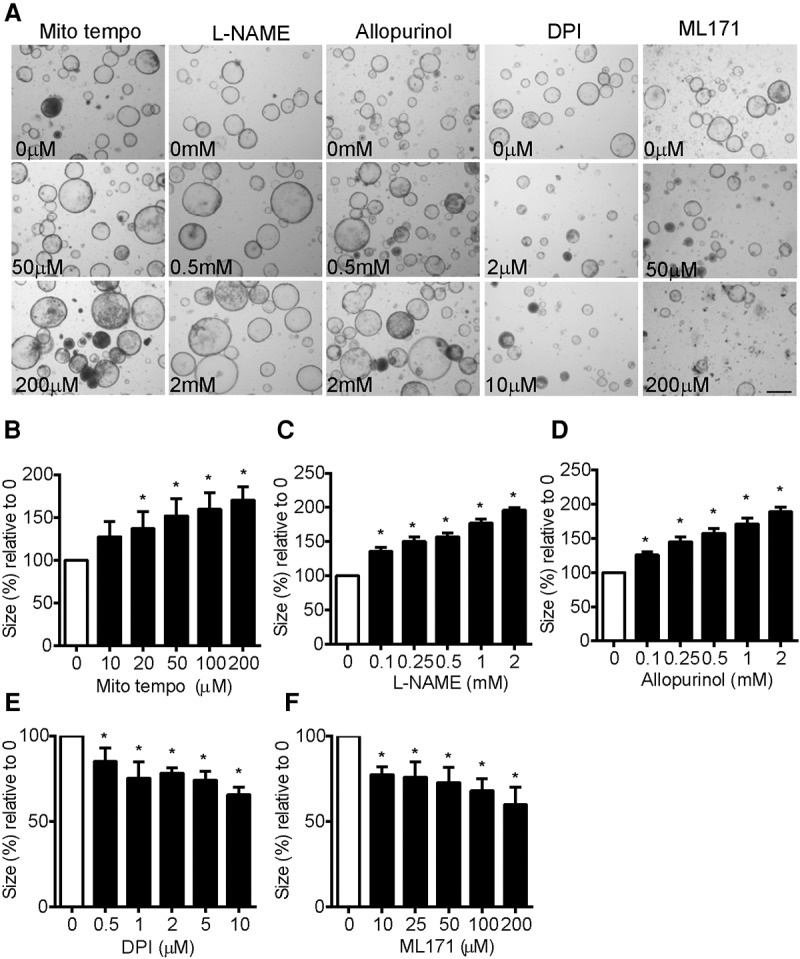
Qualitative differences between signaling and damaging ROS in Apcmin/+ organoids. (A) Uninduced Apcmin/+ organoid cultures 2 d after addition of the indicated concentrations of Mito tempo, L-NAME, allopurinol, DPI, and ML171. (B) Measurements of relative organoid size after increasing concentrations of Mito tempo. (*) P < 0.05 compared with untreated. (C) Measurements of relative organoid size after increasing concentrations of L-NAME. (*) P < 0.05 compared with untreated. (D) Measurements of relative organoid size after increasing concentrations of allopurinol. (*) P < 0.05 compared with untreated. (E) Measurements of relative organoid size after increasing concentrations of DPI. (*) P < 0.05 compared with untreated. (F) Measurements of relative organoid size after increasing concentrations of ML171. (*) P < 0.05 compared with untreated. Bars, 100 µm.
To determine the interplay of ROS increases in response to loss of TIGAR expression and ROS limitation by selective antioxidants, we tested the effects of Mito tempo, L-NAME, allopurinol, DPI, and ML171 on the growth of Rosa-creERT2Apcmin/+TIGARfl/fl organoids with and without acute deletion of TIGAR (Fig. 6A,B; Supplemental Fig. 3). In each case, inhibition of deleterious ROS (using Mito tempo, L-NAME, or allopurinol) rescued the growth of TIGAR-deleted organoids. On the other hand, inhibition of NOX1-derived ROS using DPI and ML171 inhibited the growth of the control organoids and led to a further reduction in the growth of TIGAR-null Apcmin/+ organoids (Fig. 6A,B; Supplemental Fig. 3A,B). Inhibition of deleterious ROS by Mito tempo, L-NAME, and allopurinol decreased ROS damage in TIGAR knockout organoids compared with control, as indicated by MDA staining (Fig. 6C). In contrast, inhibition of NOX-derived ROS by DPI and ML171 did not decrease ROS damage in the TIGAR knockout organoids compared with control (Fig. 6C; Supplemental Fig. 3C) despite a reduction in total ROS level, as indicated by DCFDA (2′,7′–dichlorofluorescin diacetate) staining in both control and knockout organoids (Fig. 6D; Supplemental Fig. 3D). These data therefore suggest a qualitative difference between damaging ROS produced following loss of TIGAR and signaling ROS generated through NOX.
Figure 6.
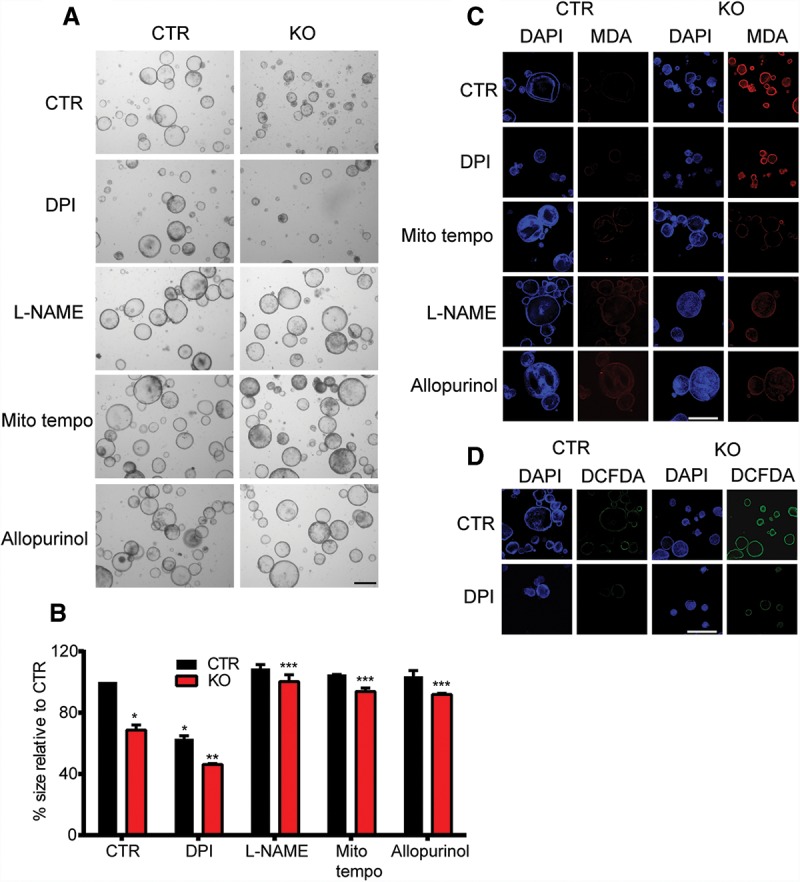
Only ROS inhibitors that decrease damaging ROS can rescue the defects of TIGAR-deficient organoids. (A) Rosa-creERT2Apcmin/+TIGARfl/fl organoid cultures were induced (knockout [KO]) or not induced (control [CTR]) by 4-hydroxytamoxifen for 2 d and then treated with the indicated drugs (2 µM DPI, 20 µM Mito tempo, 100 µM L-NAME, and 100 µM allopurinol) for another 2 d. (B) Measurements of the relative size of organoids from A. (*) P < 0.05 compared with untreated CTR organoids; (**) P < 0.05 compared with untreated (CTR) knockout organoids (KO) or CTR organoids with DPI; (***) P < 0.05 compared with untreated knockout organoids. (C) MDA staining of organoids from A. (D) DCFDA live imaging of CTR and knockout organoids treated with DPI or without DPI (CTR). Bars, 100 µm.
To test whether it is possible to distinguish the two categories of ROS that affect crypt proliferation in vivo, we examined the consequences of deletion of Rac1 and TIGAR. The RAC1 GTPase has been shown to play an important role in controlling cell proliferation and is activated in several human cancers (Sanz-Moreno et al. 2008; Krauthammer et al. 2012; Yang et al. 2012). The deletion of Apc in the mouse intestine leads to the induction of RAC1, with deletion of Rac1 inhibiting the crypt hyperproliferation seen in this model. Importantly, RAC1 is a mediator of ROS generation by NOX, and a key contribution of RAC1 to support proliferation in the intestine is through the production of ROS by NOX (Myant et al. 2013).
To investigate the relative importance of endogenous ROS regulated by different mechanisms, we examined the effects of intestine-specific deletion of TIGAR and/or Rac1 (Fig. 7). As shown previously, Apc deletion resulted in the hyperproliferation of intestinal crypts, which is accompanied by an increased expression of TIGAR. Deletion of either TIGAR or Rac1 attenuated this proliferation to similar extents (Fig. 7A,B; Supplemental Fig. 5). Importantly, Rac1 deletion (which leads to a decrease in ROS) did not prevent induction of TIGAR expression, and the deletion of TIGAR does not affect RAC1 expression (Fig. 7A), demonstrating that the ROS produced by NOX through RAC1 is not necessary to induce TIGAR expression. Simultaneous deletion of both TIGAR (which increases ROS) and RAC1 (which decreases ROS) resulted in an even more severe inhibition of crypt proliferation in response to APC loss, supporting the suggestion that these pools of ROS have separate functions in these cells (Fig. 7A,B; Supplemental Fig. 5). This is further supported by the observation that MDA staining (which indicates ROS damage) is only increased in TIGAR-deleted but not Rac1-deleted intestines, and this increase in ROS damage is not decreased by the loss of RAC1 in the TIGAR and RAC1 double mutants (Fig. 7A). Taken together, our results suggest that, during rapid proliferation induced by the loss of APC, there is a selective modulation of two pools of ROS that have different and opposing properties on cell growth, with RAC1 promoting signaling ROS, and TIGAR limiting damaging ROS.
Figure 7.
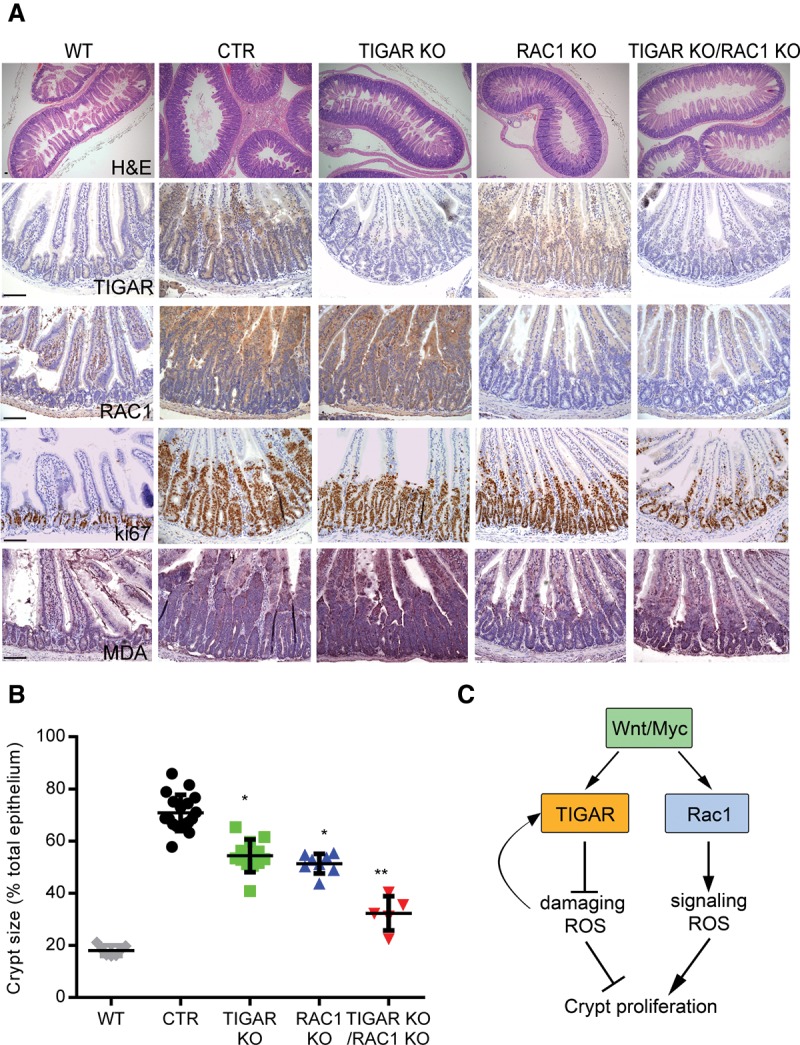
In vivo deletion of both TIGAR and RAC1 can synergistically decrease hyperproliferation of Apc-deficient intestinal crypts. (A) H&E staining (top row), TIGAR staining (second row), RAC1 staining (third row), Ki67 staining (fourth row), and MDA staining (bottom row) of small intestines from wild-type (WT), Ahcre+Apcfl/fl (CTR), Ahcre+Apcfl/flTIGARfl/fl (TIGAR knockout [KO]), Ahcre+Apcfl/flRac1fl/fl (RAC1 knockout), and Ahcre+Apcfl/flTIGARfl/flRac1fl/fl (TIGAR/RAC1 knockout) animals 3 d after β-napthoflavone induction of Ahcre. Bars, 100 µm. (B) Measurements of crypt thickness from A as a percentage of total thickness of epithelium. (*) P < 0.05 compared with CTR; (**) P < 0.05 compared with TIGAR knockout or RAC1 knockout. CTR, n = 18; TIGAR, n = 12, RAC1 knockout, n = 7; TIGAR knockout RAC1 knockout, n = 5. (C) Proposed model of the role of ROS after Wnt signaling activation.
Discussion
It is clear that ROS can both function in support of proliferation and cell survival and induce damage and cell death (D'Autreaux and Toledano 2007; Trachootham et al. 2009; Gorrini et al. 2013; Sullivan and Chandel 2014a). Our work indicates that ROS can be differentiated into those supporting and those inhibiting proliferation and that these different pools of ROS can be separately controlled to maximize cell growth in vitro and in vivo. We used ROS inhibitors and genetic mechanisms to modulate levels of endogenous ROS produced by Apc deletion in the small intestinal cells in 3D cultures and in vivo. These models allowed us to determine whether the growth response reflects only the levels of ROS (with low ROS signaling proliferation and high ROS driving cell death) or whether there is a qualitative difference between signaling and damaging ROS. APC loss leads to the activation of both RAC1, which promotes the generation of signaling ROS via NOX1 (Myant et al. 2013), and TIGAR, which limits ROS through the maintenance of reduced glutathione for antioxidant defense (Cheung et al. 2013). If the role of TIGAR is to limit the levels of RAC1-induced ROS to maintain proliferation but prevent death, deletion of either Rac1 or TIGAR would lower proliferation, but deletion of TIGAR in a Rac1-null tissue would have no further effect, since there would be no excessive ROS for TIGAR to balance. However, this was not the outcome that we observed. Rather, our data are consistent with a model in which Wnt signaling leads to the activation of both RAC1 and TIGAR, each of which regulates independent ROS pools that have different functions (Fig. 7C). RAC1 induces expression of signaling ROS to drive proliferation, while TIGAR provides antioxidant defense to prevent the accumulation of damaging ROS. As predicted by this model, we observed that loss of either Rac1 or TIGAR resulted in less proliferation in Apc-null crypts, and a codeletion of both Rac1 and TIGAR even further reduced proliferation compared with single deletions. The cooperation of TIGAR deletion and the NOX-specific inhibitors DPI and ML171 in retarding the growth of Apcmin/+ organoid cultures further supports this model.
Malignant development is associated with increased ROS, which can be induced by oncogene activation and the loss of the normal cell environment. While ROS can help support cancer cell proliferation, several studies have shown that the ability of cancer cells to limit excessive ROS is important for their survival and rapid growth (Diehn et al. 2009; DeNicola et al. 2011; Harris et al. 2015). The variable responses to ROS have made predicting the response to ROS limitation in cancer growth difficult. Inhibition of ROS can prevent proliferation or limit the accumulation of damage and thus suppress tumor development. On the other hand, inhibition of the antioxidant defense mechanisms that protect tumors would increase ROS and promote cancer cell death (D'Autreaux and Toledano 2007; Trachootham et al. 2009; Gorrini et al. 2013; Sullivan and Chandel 2014a). Our results suggest that the ROS involved in driving these different responses can be differentially targeted, potentially allowing the development of therapeutics that inhibit proliferating ROS while promoting death-inducing ROS. Understanding the details of how each pool of ROS is generated and regulated in cancer cells may be key for developing an effective strategy for therapy.
Materials and methods
Animals
Apcmin/+ (Moser et al. 1990), Ahcre+ (Ireland et al. 2004), Apcfl/fl (Sansom et al. 2004), Rac1fl/fl (Walmsley et al. 2003), Mycfl/fl (Baena et al. 2005), and TIGARfl/fl (Cheung et al. 2013) mice were used as previously described. Ahcre was induced by β-napthoflavone (three 80 mg/kg i.p. injections). Cisplatin-induced (CDDP; 10 mg/kg i.p. injection) and γ-IR-induced intestinal damage was performed as previously described (Cheung et al. 2013) 3 d after cre induction. Ahcre+Apcmin/+TIGAR+/+ and Ahcre+Apcmin/+TIGARfl/fl animals were induced at 30 d old, and animals were sacrificed at 80 d old. All animal work was carried out in line with the Animals (Scientific Procedures) Act 1986 and the EU Directive 2010 and was sanctioned by the local ethical review process (University of Glasgow).
Small intestinal crypt culture
Small intestinal crypt cultures from normal intestinal tissues and adenoma were prepared as previously described (Barker et al. 2009; Sato et al. 2009, 2011). Crypts were embedded in Matrigel (BD) and cultured in Advanced DMEM/F-12 (ADF, Life Technologies) supplemented with 1% glutamine, 1% penicillin/streptomycin, 0.1% AlbuMAX I (Life Technologies), 10 mM HEPES (Life Technologies), 0.05 μg/mL EGF (Peprotech), 0.1 μg/mL Noggin (Peprotech), and 0.5 μg/mL mR-spondin (R&D Systems). For RosacreERT2Apcmin/+TIGARfl/fl crypts, adenomas were suspended in Matrigel with ADF supplemented with 1% glutamine, 1% penicillin/streptomycin, 0.1% AlbuMAX I, 10 mM HEPES, 0.05 μg/mL EGF, and 0.1 μg/mL Noggin. The cultures were passaged every 7–10 d. 4-hydroxytamoxifen (500 nM; Sigma) was used to induce cre after 2 d in culture. Trolox (Sigma), NAC (Sigma), Mito tempo (Cayman), L-NAME (Sigma), DPI (Sigma), allopurinol (Sigma), ML171 (Tocris), and EUK134 (Cayman) were used at the indicated concentrations for 2 d after cre induction.
Western blot
Protein lysates were prepared in RIPA buffer with complete protease inhibitors (Roche), resolved via PAGE, and transferred to nitrocellulose membranes. The following primary antibodies were used: Actin I-19-R (Santa Cruz Biotechnology), β-catenin 6B3 (Cell Signaling Technology), cyclin D1 (Cell Signaling Technology), HSP90 (Cell Signaling Technology), p21 C-19 (Santa Cruz Biotechnology), p53 1C12 (Cell Signaling Technology), and TIGAR M-209 (Santa Cruz Biotechnology). Secondary antibodies were IRDye800CW-conjugated (LiCor Biosciences), and detection was performed using an Odyssey infrared scanner (LiCor Biosciences).
Gene expression analyses
RNA was extracted from organoids or mouse tissue using the RNeasy kit (Qiagen) following the manufacturer's instructions. cDNA was synthesized from 1 μg of RNA using the High-Capacity RNA-to-cDNA kit (Applied Biosystems) according to the manufacturer's instructions using a PTC-200 Peltier Thermal Cycler (MJ Research). The quantitative real-time PCR (qRT-PCR) reaction was performed with 2 μL of cDNA with the Fast SYBR Green Master mix (Applied Biosystems) using a 7500 Fast real-time PCR system (Applied Biosystems).
Gene expression was quantified relative to the housekeeping gene Gapdh (Primer Design) according to the comparative ΔΔCt method. Mouse TIGAR primers were purchased from Qiagen.
The mRNA primer sequences (5′–3′) used were as follows: Axin2 for (GCTCCAGAAGATCACAAAGAGC), Axin2 rev (AGCTTTGAGCCTTCAGCATC), c-Myc for (TGAAGAAGAGCAAGAAGATGAG), c-Myc rev (CTGGATAGTCCTTCCTTGTG), Cyclin D1 for (GAGAAGTTGTGCATCTACACTG), Cyclin D1 rev (AAATGAACTTCACATCTGTGGC), HO-1 for (CAGGAGCTGCTGACCCATGA), HO-1 rev (AGCAACTGTCGCCACCAGAA), p21 for (GGCCCGGAACATCTCAGG), and p21 rev (AAATCTGTCAGGCTGGTCTGC).
Immunohistochemistry
Immunohistochemistry and H&E staining were performed as previously described (Sansom et al. 2004; Myant et al. 2013). The primary antibodies used were β-catenin 6B3 (Cell Signaling Technology), cyclin D1 (DAKO), Ki67 (Thermo Scientific), MDA (Abcam), and TIGAR (Millipore).
RNAscope assay protocol
RNAscope assays for detecting mRNA expression in tissue sections were done on formalin-fixed paraffin-embedded slides and performed by the Cancer Research UK Beatson Institute Histology Services according to the protocol from Advanced Cell Diagnostics. All reagents and probes to detect murine TIGAR (Mm-Tigar targeting 619–1574 of NM_177003.5) and Axin2 (Mm-Axin2 NM_015732.4) were purchased from Advanced Cell Diagnostics.
Immunofluorescence
Organoids were collected in cold PBS in Eppendorf tubes (precoated with 1% BSA/PBS for 20 min), incubated for 15 min on ice, and washed three times before fixing with 4% PFA in PBS. Organoids were then permeablized with cold 0.2% Triton X-100 in PBS and blocked with 3% BSA and 0.1% Tween-20 in PBS. After blocking, the organoids were incubated overnight with primary antibody (prepared in 1% BSA in PBS), washed three times, and then incubated with secondary antibody for 1 h. After washing, the organoids were transferred onto microscope slides and mounted with mounting medium (Vector Laboratories) under a coverslip with DAPI to visualize nuclei. Live imaging with DCFDA and DHE was performed according to the manufacturer's instructions (Life Technologies). The primary antibody used was MDA (Abcam). The secondary antibody used was anti-rabbit Alexa fluor 594 (Life Technologies). Samples were examined using an Olympus FV1000 inverted laser scanning confocal microscope.
Quantification and statistical analysis
The number of Ki67-positive cells was measured in at least 50 crypts per animal per treatment. The rate of growth of the tumor organoid cultures was measured by the average size (diameter) of at least 100 crypts in each treatment. The data represent mean values ± SEM from three independent experiments (n = 3) unless otherwise noted. All P-values were obtained using a t-test.
Supplementary Material
Acknowledgments
We thank the Beatson Institute facilities at the Cancer Research UK Beatson Institute (C596/A17196), the Cancer Research UK Glasgow Centre (C596/A18076), and the Beatson Institute Histology Service. We also thank Daniel Murphy for providing access to the Myc-dependent lung cancer model. We are grateful for funding from Cancer Research UK grant C596/A10419, European Research Council grant 322842-METABOp53, and a Medical Research Council studentship.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.271130.115.
Freely available online through the Genes & Development Open Access option.
References
- Abramov AY, Scorziello A, Duchen MR. 2007. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 27: 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. 1985. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538. [DOI] [PubMed] [Google Scholar]
- Adam-Vizi V, Chinopoulos C. 2006. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 27: 639–645. [DOI] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Minervini GM, Ciotti MT, Marra E, Calissano P. 1997. Glutamate neurotoxicity in rat cerebellar granule cells: a major role for xanthine oxidase in oxygen radical formation. J Neurochem 68: 2038–2045. [DOI] [PubMed] [Google Scholar]
- Baena E, Gandarillas A, Vallespinos M, Zanet J, Bachs O, Redondo C, Fabregat I, Martinez AC, de Alboran IM. 2005. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci 102: 7286–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Marcus CB, Huffman K, Kruk H, Malfroy B, Doctrow SR. 1998. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J Pharmacol Exp Ther 284: 215–221. [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457: 608–611. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. 2006. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126: 107–120. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH. 2009. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28: 3015–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Sun AY. 1994. Oxidative mechanisms involved in kainate-induced cytotoxicity in cortical neurons. Neurochem Res 19: 1557–1564. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH. 2013. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell 25: 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, et al. 2005. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 435: 347–353. [DOI] [PubMed] [Google Scholar]
- D'Autreaux B, Toledano MB. 2007. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. 2011. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. 2011. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7: 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. 2009. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458: 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni D, Nicolas N, Zhang H, Der Mardirossian C, Kister J, Martinez L, Ferguson J, Roush WR, Brown SJ, Bokoch GM, et al. 2010a. Optimization and characterization of an inhibitor for NADPH oxidase 1 (NOX-1). In Probe Reports from the NIH Molecular Libraries Program (Internet), National Center for Biotechnology Information, NIH, Bethesda, MD: http://www.ncbi.nlm.nih.gov/books/NBK98925. [PubMed] [Google Scholar]
- Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, Rosen H. 2010b. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol 5: 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C, Boveris A, Cadenas E. 1995. Hydroxyl radical generation during mitochondrial electron transfer and the formation of 8-hydroxydesoxyguanosine in mitochondrial DNA. Archives Biochem Biophys 316: 909–916. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. 2013. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. 2015. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27: 211–222. [DOI] [PubMed] [Google Scholar]
- Heales SJ, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB. 1999. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta 1410: 215–228. [DOI] [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. 2004. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of β-catenin. Gastroenterology 126: 1236–1246. [DOI] [PubMed] [Google Scholar]
- Kim YS, Morgan MJ, Choksi S, Liu ZG. 2007. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 26: 675–687. [DOI] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. 2012. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44: 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. 2004. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci 101: 16419–16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. 2002. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277: 20336–20342. [DOI] [PubMed] [Google Scholar]
- Lee P, Hock AK, Vousden KH, Cheung EC. 2015. p53- and p73-independent activation of TIGAR expression in vivo. Cell Death Dis 6: e1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. 2006. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol 26: 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui VW, Wong EY, Ho K, Ng PK, Lau CP, Tsui SK, Tsang CM, Tsao SW, Cheng SH, Ng MH, et al. 2011. Inhibition of c-Met downregulates TIGAR expression and reduces NADPH production leading to cell death. Oncogene 30: 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. 2009. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morre DJ. 2002. Preferential inhibition of the plasma membrane NADH oxidase (NOX) activity by diphenyleneiodonium chloride with NADPH as donor. Antioxid Redox Signal 4: 207–212. [DOI] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247: 322–324. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Smith RA. 2007. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 47: 629–656. [DOI] [PubMed] [Google Scholar]
- Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. 2008. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell 14: 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, et al. 2013. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 12: 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CE, McGuire WP, Liss RH, Ifrim I, Grotzinger K, Young RC. 1977. Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science 197: 165–167. [DOI] [PubMed] [Google Scholar]
- Porasuphatana S, Tsai P, Rosen GM. 2003. The generation of free radicals by nitric oxide synthase. Comp Biochem Physiol Toxicol Pharmacol 134: 281–289. [DOI] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, et al. 2014. A mitochondrial switch promotes tumor metastasis. Cell Rep 8: 754–766. [DOI] [PubMed] [Google Scholar]
- Powell S, McMillan TJ. 1990. DNA damage and repair following treatment with ionizing radiation. Radiother Oncol 19: 95–108. [DOI] [PubMed] [Google Scholar]
- Reed KR, Meniel VS, Marsh V, Cole A, Sansom OJ, Clarke AR. 2008. A limited role for p53 in modulating the immediate phenotype of Apc loss in the intestine. BMC Cancer 8: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharwal SS, Schumacker PT. 2014. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14: 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, et al. 2004. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev 18: 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. 2007. Myc deletion rescues Apc deficiency in the small intestine. Nature 446: 676–679. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135: 510–523. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265. [DOI] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. 2011. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, Chandel NS. 2014. ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Chandel NS. 2014a. Mitochondrial metabolism in TCA cycle mutant cancer cells. Cell Cycle 13: 347–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Chandel NS. 2014b. Mitochondrial reactive oxygen species and cancer. Cancer Metab 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. 2009. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8: 579–591. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M. 2006. Localizing NADPH oxidase-derived ROS. Sci STKE 2006: re8. [DOI] [PubMed] [Google Scholar]
- Vilhardt F, van Deurs B. 2004. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J 23: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, Mathiot A, Vanes L, Williams DA, Cancro MP, Tybulewicz VL. 2003. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science 302: 459–462. [DOI] [PubMed] [Google Scholar]
- Wanka C, Steinbach JP, Rieger J. 2012. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J Biol Chem 287: 33436–33446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. 2010. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natil Acad Sci 107: 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Lan HY, Huang CH, Tai SK, Tzeng CH, Kao SY, Wu KJ, Hung MC, Yang MH. 2012. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol 14: 366–374. [DOI] [PubMed] [Google Scholar]
- Yin L, Kosugi M, Kufe D. 2012. Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH. Blood 119: 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.