

Wylie et al. show that p53 restricts retrotransposon activity and genetically interacts with components of the piRNA pathway. In gene complementation studies, normal human p53 alleles restrained these mobile elements, but mutant p53 alleles from cancer patients could not. Consistent with these observations, they also found patterns of unrestrained retrotransposons in p53-driven mouse and human cancers.
Keywords: p53, retrotransposons, Drosophila, zebrafish, mouse cancer models, human cancers, piRNAs
Abstract
Throughout the animal kingdom, p53 genes govern stress response networks by specifying adaptive transcriptional responses. The human member of this gene family is mutated in most cancers, but precisely how p53 functions to mediate tumor suppression is not well understood. Using Drosophila and zebrafish models, we show that p53 restricts retrotransposon activity and genetically interacts with components of the piRNA (piwi-interacting RNA) pathway. Furthermore, transposon eruptions occurring in the p53− germline were incited by meiotic recombination, and transcripts produced from these mobile elements accumulated in the germ plasm. In gene complementation studies, normal human p53 alleles suppressed transposons, but mutant p53 alleles from cancer patients could not. Consistent with these observations, we also found patterns of unrestrained retrotransposons in p53-driven mouse and human cancers. Furthermore, p53 status correlated with repressive chromatin marks in the 5′ sequence of a synthetic LINE-1 element. Together, these observations indicate that ancestral functions of p53 operate through conserved mechanisms to contain retrotransposons. Since human p53 mutants are disabled for this activity, our findings raise the possibility that p53 mitigates oncogenic disease in part by restricting transposon mobility.
The broadly conserved p53 family of transcription factors regulates target genes to specify distinct adaptive responses (Vousden and Lane 2007; Levine and Oren 2009; Vousden and Prives 2009). Although p53 mutations occur in most human cancers, the precise mechanisms by which p53 acts to restrict oncogenesis are not well understood. In mice, for example, p53 retained tumor suppression activity despite the combined absence of three downstream canonical effector proteins (p21, Puma, and Noxa) that arrest proliferation and engage apoptosis (Valente et al. 2013). Moreover, evolutionary analyses strongly suggest that p53 genes predate the adaptive need for tumor suppression. Thus, tumor suppression by p53 was likely co-opted from unknown ancestral functions conferred by this gene family (Lu et al. 2009). These and related observations suggest the existence of crucial, unidentified p53 effectors and highlight conspicuous gaps in our understanding of p53 function.
Results
p53 restrains transposons in Drosophila
Previously, we showed that lesions in the Drosophila piwi-interacting RNA (piRNA) pathway consistently triggered p53 activity (Wylie et al. 2014), raising the possibility that p53 might function to restrain retrotransposons that are targets for piRNA suppression. To address this possibility, we examined the expression of TAHRE elements in p53− flies, since these retrotransposons are well-documented piRNA targets (Shpiz et al. 2011). In ovaries of p53− females, TAHRE retrotransposons were highly expressed relative to wild-type counterparts, as shown by RT–PCR on bulk samples (Fig. 1A). To extend these findings and enable measurements of individual animals, we developed a droplet digital PCR (ddPCR) assay (see the Materials and Methods). As seen in Figure 1B, similar p53-dependent effects on TAHRE expression were observed using this assay. Furthermore, while TAHRE dysregulation was consistently seen in p53− individuals, the extent of derepression was variable from animal to animal. Importantly, dysregulated TAHRE expression was not observed in p53Rescue (p53 genomic rescue transgene) strains, which transgenically restore the fly p53 gene to strains mutated at the native dp53 locus (see Supplemental Fig. 1; Wylie et al. 2014). We further validated these findings by in situ detection using fluorescent in situ hybridization (FISH) probes. As seen in Figure 1, C and C′, TAHRE transcripts visibly accumulated in p53− animals but were undetectable in wild-type or p53Rescue counterparts. Derepressed TAHRE transcripts were first detectable in the early egg chambers of p53− ovaries (Supplemental Fig. 2A; Supplemental Table 1), and, like several piRNA pathway proteins, RNAs from these dysregulated retroelements distinctly accumulated in the oocyte germ plasm (Fig. 1C,C′; Supplemental Figs. 2B, 4D,G). The oocyte germ plasm induces primordial germ cells in the developing embryo (Illmensee and Mahowald 1974), and, to examine whether TAHRE transcripts are maternally loaded into the embryo, we tested for TAHRE dysregulation in staged samples resulting from reciprocal crosses. Figure 1D shows that p53− females crossed to wild-type males produced embryos exhibiting TAHRE transposon dysregulation, but wild-type females mated to p53− males did not. These results establish that TAHRE dysregulation in the early embryo is a maternal effect phenotype and indicates that retrotransposon transcripts are maternally loaded. Consistent with this, we observed elevated TAHRE transcripts in early 1- to 4-h stage p53− embryos but not late 21- to 24-h stage p53− embryos (Supplemental Fig. 3). Together, these data establish that p53 normally functions to restrict TAHRE elements in the female germline. Furthermore, observations in Figure 1, C, C′, and D, raise the intriguing possibility that TAHRE transcripts and possibly other retroelement RNAs engage mechanisms to accumulate in the oocyte germ plasm (Lehmann and Ephrussi 1994) and thereby promote germline propagation.
Figure 1.
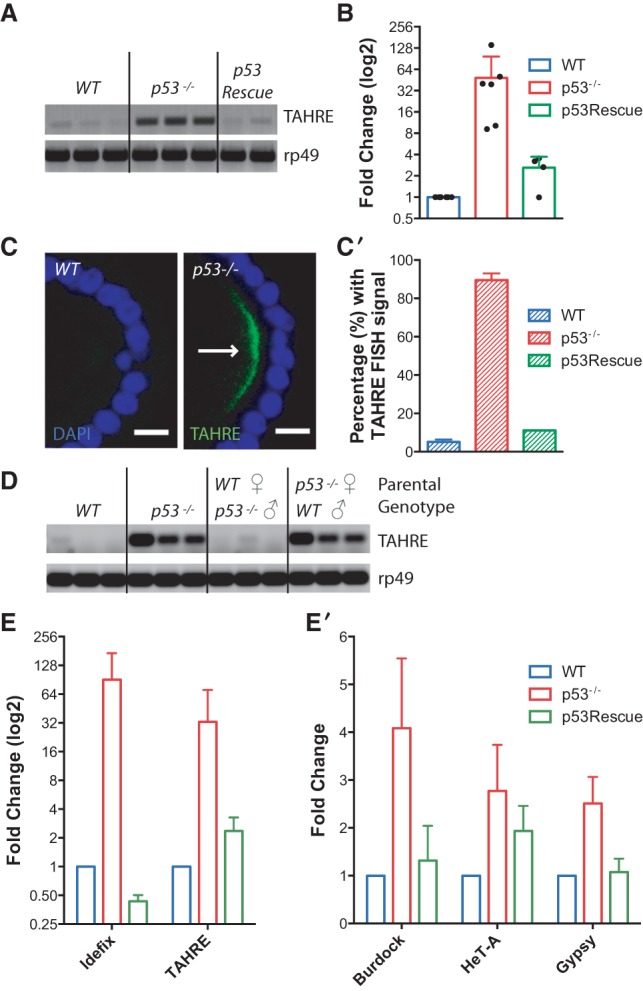
p53 restrains transposon activity in the Drosophila germline. (A) TAHRE retrotransposons, measured by RT–PCR, are highly expressed in dp53− ovaries but minimally expressed in parental wild-type or dp53− flies carrying p53Rescue. The control reference gene ribosomal protein L32 (rp49) is present at similar levels among all genotypes. (B) Derepression of TAHRE transcripts in ovaries of single animals was quantified using ddPCR standardized to the housekeeping gene rp49. Each dot represents measurements from an ovary pair from a single female. TAHRE retrotransposons were consistently dysregulated in dp53− animals (red bar). Normal repression, comparable with wild type (blue bar), occurred when the p53Rescue transgene was present in these mutants (green bar). p53− was significantly different from wild type (P-value = 0.0172) and p53Rescue (P = 0.0347) (see the Materials and Methods). (C,C′) TAHRE expression was assayed by FISH. In C, TAHRE RNAs (arrow) accumulate in the germ plasm of p53− oocytes (stage 9) of stage 9 and 10 egg chambers but not in wild-type egg chambers. (Green) TAHRE signal; (blue) DAPI counterstain. These data are quantified in C′, illustrating TAHRE derepression in p53− ovaries (red bar) Bars, 10 μm. Wild type was significantly different from p53− (P = 0.001) (see the Materials and Methods). (D) TAHRE transcripts, measured by RT–PCR, are maternally loaded into the 1- to 4-h-old embryo. TAHRE elements are derepressed in the p53− embryo (parental genotypes were p53−/−) but undetectable in the wild-type embryo (parental genotypes were wild type). Robust TAHRE expression was also observed in embryos from p53− mothers mated to wild-type fathers (parental genotypes are p53−/− female; wild-type male) but not in embryos from the reciprocal cross (wild-type female; p53−/− male). The control reference transcript rp49 is present at similar levels among all genotypes. Three independent biological replicates are shown for all genotypes. (E,E′) Expression from the indicated retroelements was measured by quantitative RT–PCR. In E the Idefix and TAHRE elements were highly derepressed in dp53− ovaries (red bars) relative to wild-type (blue bars) or p53Rescue (green bars) samples. In E′, retroelements from the Burdock, Gypsy, and HeT-A families (red bars) were similarly but more modestly derepressed in p53− ovaries. Note that in E, the fold change is plotted on a log2 scale to better appreciate differences in transcript levels between wild-type and p53Rescue flies. The error bars represent standard deviations. p53− samples were significantly different from wild type (P-value < 0.05) for Idefix, TAHRE, Burdock, HeT-A, and Gypsy. p53− samples were significantly different from p53Rescue (P -value < 0.05) for TAHRE, Burdock, and Gypsy.
To determine whether p53 generally suppresses retroelements, we tested whether other retrotransposons were active in p53− ovaries. Figure 1, E and E′, shows quantitative RT–PCR (qRT–PCR) indicating that multiple retrotransposon classes are derepressed in p53− ovaries. Notably, the expression of telomeric non-long terminal repeat (non-LTR) retrotransposons (TAHRE and HeT-A) as well as nontelomeric LTR retrotransposons (Idefix, Burdock, and Gypsy) was elevated in p53− ovaries, and, in all cases, this effect was reversed in p53Rescue strains. Since complete or partial sterility is commonly observed in fly mutants defective for retroelement silencing (Czech et al. 2013), we also assessed fertility in p53− and p53Rescue adults. Consistent with previous reports (Hu et al. 2011), we observed partial infertility phenotypes in p53− female flies that were rescued in p53Rescue strains (see the Materials and Methods; Supplemental Table 2). Thus, in Drosophila, p53 loss permits widespread dysregulation of retroelements and associated phenotypes.
Retrotransposition could occur in meiotic progenitors, since DNA breaks needed for recombination may facilitate de novo integration events (Brouha et al. 2002). Furthermore, we previously showed that p53 is transiently activated in these same cells during the process of meiotic recombination (Lu et al. 2010). These observations, together with outcomes seen in Figure 1, raise the possibility that p53 contains retrotransposon activity during meiosis. To test this possibility, we generated animals deficient for both p53 and Spo11 (McKim and Hayashi-Hagihara 1998), a universally conserved enzyme that forms dsDNA breaks to initiate meiotic recombination. Using these double mutants, we found that spo11 is epistatic to p53 when transposon derepression was assessed (Fig. 2). Specifically, transposon derepression was completely reversed for Idefix, Gypsy, and Burdock elements (which were present at or below wild-type levels) and partially reversed for the TAHRE and HeT-A retroelements (Fig. 2) in the ovaries of spo11−;p53− animals. Hence, programmed DNA breaks during meiotic recombination are required for transposon eruptions that accompany the p53− state. Furthermore, since the action of Spo11 is cell-autonomous and evidently confined to meiotic cells, these data strongly suggest that p53 normally restrains mobile elements within meiotic cells of the germline. The fact that TAHRE and HeT-A retroelements were only partially affected by loss of spo11 is consistent with this interpretation, since these elements are also involved in telomere elongation (Shpiz et al. 2007).
Figure 2.
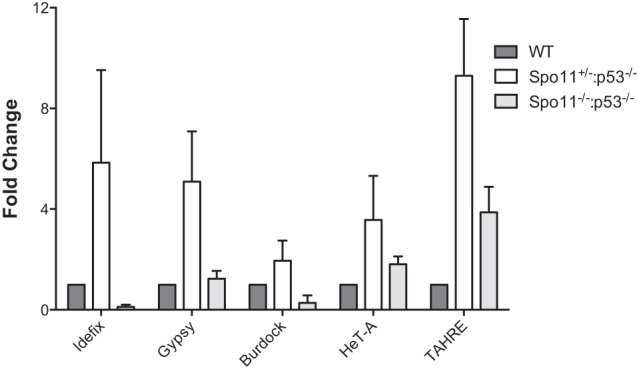
Retrotransposon derepression in p53− animals requires Spo11 function. Expression from the indicated retroelements was measured in Drosophila ovaries of the indicated genotypes by qRT–PCR. Idefix, Gypsy, Burdock, HeT-A, and TAHRE elements are derepressed in spo11+/−; p53−/− ovaries (white bars), unlike wild-type (dark-gray bars) or spo11−/−;p53−/− (light-gray bars) samples. Error bars represent standard deviations from three biological replicates. For HeT-A, spo11+/−;p53−/− samples were significantly different from spo11−/−; p53−/−at the 90% confidence interval (P-value = 0.0803). For all other samples, spo11+/−;p53−/− was statistically significant from wild-type (P-value < 0.05) and spo11−/−;p53−/− (P-value < 0.05) samples (see the Materials and Methods).
In metazoans, the highly conserved piRNA network acts through protein components, P-element-induced wimpy testis (PIWIs), in concert with small RNAs (piRNAs) to silence the expression of retrotransposons at the transcriptional and post-transcriptional levels (Aravin et al. 2007; Khurana and Theurkauf 2010; Siomi et al. 2011; Ross et al. 2014). To examine whether p53 might collaborate with the piRNA system to suppress retrotransposons, we tested for possible genetic interactions between p53 and core elements of the piRNA pathway encoded by aubergine (aub) and cutoff (cuff). For these assays, we scored stunted egg morphology as a maternal effect readout and, despite normal localization of piRNA pathway proteins in p53− ovaries (Supplemental Fig. 4D–G′), found that aub−;p53− and cuff−;p53− double mutants produced significantly stunted eggs (Supplemental Fig 4A,B). Consistent with these data, we also found that piRNA biogenesis was altered in p53 mutants. Mature piRNAs are processed from large precursor RNAs that map to transposon “graveyards,” representing fossilized templates of previous exposures (Aravin et al. 2007; Khurana and Theurkauf 2010; Siomi et al. 2011; Ross et al. 2014). In Drosophila, precursor RNAs encoded by the flamenco locus generate piRNAs that silence retroelements (Aravin et al. 2007; Khurana and Theurkauf 2010; Siomi et al. 2011; Ross et al. 2014), and, like other mutants defective for piRNA biogenesis (Siomi et al. 2011), the flamenco piRNA precursor abnormally accumulated in p53− animals (Supplemental Fig. 4C). Together, these observations suggest that p53 collaborates with the piRNA pathway to repress retrotransposons.
p53 limits retrotransposition in fish
To test whether vertebrate p53 genes similarly restrain mobile elements, we developed assays to measure de novo retrotransposition in zebrafish using a well-characterized retrotransposition indicator that takes advantage of an engineered reporter construct, designated pLRE3 (Brouha et al. 2002). As illustrated in Figure 3A, this construct is a retrotransposition-competent human LINE-1 (L1) element containing a reporter cassette in its 3′ untranslated region (UTR) (Fig. 3A) that encodes a backward copy of a CMV-driven enhanced green fluorescence protein (EGFP) gene. A key feature of this integration reporter is that EGFP is interrupted by an artificial intron oriented in the same transcriptional direction as L1 expression (Ostertag et al. 2000). This arrangement ensures that EGFP-positive (EGFP+) cells will arise only when pLRE3 transcripts undergo successful retrotransposition (Ostertag et al. 2000; Garcia-Perez et al. 2010). Specifically, LRE3 RNAs must be expressed, properly spliced, reverse-transcribed, and ultimately integrated in order to produce a CMV-driven GFP signal (Ostertag et al. 2000). To assay LINE-1 expression levels, we injected the pLRE3 reporter into wild-type and p53− zebrafish embryos and performed immunohistochemistry to detect the human LINE-1 ORF1 protein (ORF1p) at 11 h post-fertilization (hpf) (see the Materials and Methods). As shown in Figure 3, B and B′, we observed abundant ORF1p expression in p53− embryos, but this signal was undetectable in the wild-type embryos. Thus, like the Drosophila p53 counterpart, zebrafish p53 similarly acts to restrain retroelement expression. To determine whether derepressed LINE-1 activity in p53− gametes predicts extensive de novo integration events, we injected the pLRE3 reporter into wild-type and p53− zebrafish embryos and performed immunohistochemistry for EGFP at 48 hpf (see the Materials and Methods). EGFP+ cells were detected in both wild-type and p53− zebrafish (Fig. 3C,C′; Supplemental Table 3), but the frequency of retrotransposition within individual animals was dramatically elevated in p53− mutants (Fig. 3C,C′). Consistent with our previous data (Fig. 1B), the number of EGFP+ cells seen in p53− individuals varied from animal to animal (Fig. 3C′). To confirm that EGFP+ cells reflect authentic LINE-1 retrotransposition events, we injected a control LINE-1 indicator, designated pLRE3H230A-mEGFPI. This synthetic retroelement is unable to integrate by virtue of a mutation in the ORF2 endonuclease but is otherwise identical to LRE3 (Coufal et al. 2011). As expected, the pLRE3H230A-mEGFPI reporter failed to produce EGFP+ cells in both wild-type and p53− zebrafish (Supplemental Table 3) despite elevated LINE-1 expression in the p53− embryo (Supplemental Fig. 5). These data indicate that p53 repression of retroelements lies upstream of the integration event. Moreover, as in human cells, L1 retrotransposition depended on ORF2 endonuclease function (Feng et al. 1996) and exhibited a preference for cis transcripts (Wei et al. 2001). Hence, using assays for expression (Figs. 1, 3B,B′) or movement (Fig. 3C,C′), we conclude that zebrafish p53 and Drosophila p53 similarly restrict retrotransposon activity.
Figure 3.
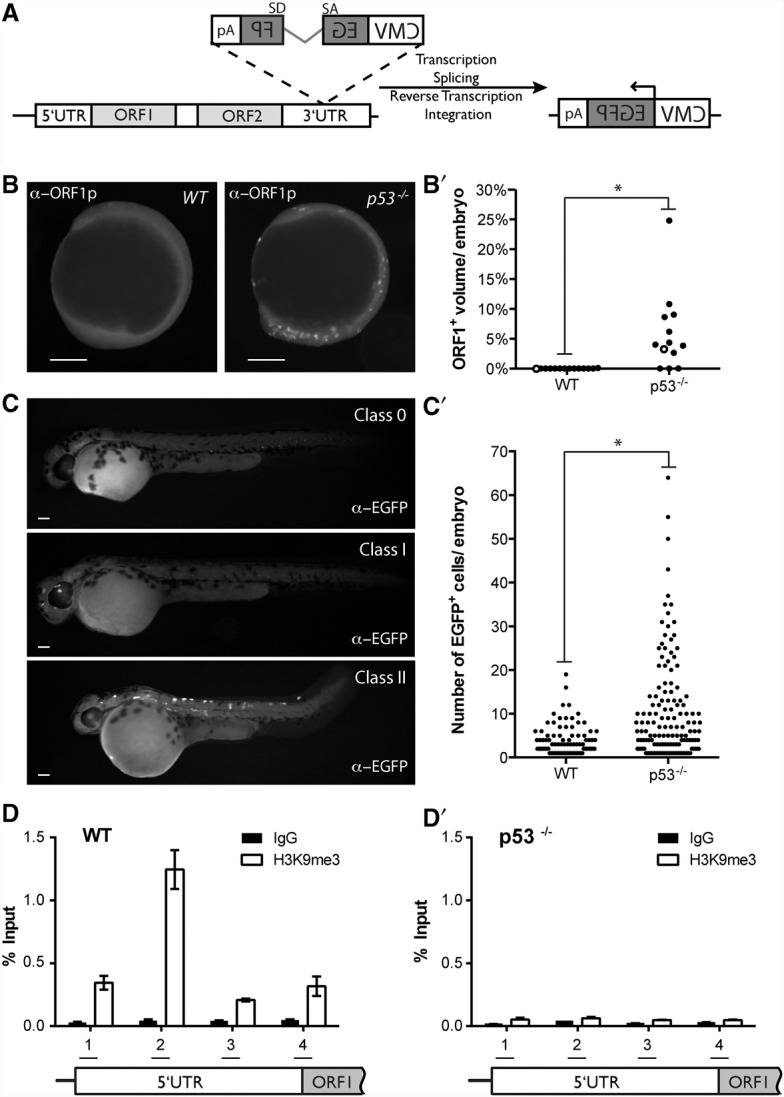
Unrestrained retroelements in p53− fish are integration-competent and lack repressive chromatin marks. (A) pLRE3-mEGFPI is a widely used integration reporter (Coufal et al. 2009, 2011; Garcia-Perez et al. 2010) schematized here. It consists of a retrotransposition-competent human LINE-1 (LRE3) (Brouha et al. 2002) containing an internal RNA polymerase II promoter in its 5′ UTR (light-gray box), two ORFs (ORF1 [light-gray box] and ORF2 [light-gray box]), and the mEGFPI retrotransposition indicator cassette in its 3′ UTR (Ostertag et al. 2000; Garcia-Perez et al. 2010). The mEGFPI retrotransposition indicator cassette encodes a backward copy of a CMV-driven EGFP (dark-gray box) that is interrupted by an intron ([SD] splice donor; [SA] splice acceptor) that is in the same transcriptional orientation as LRE3 (Ostertag et al. 2000). The arrangement of the indicator cassette ensures that EGFP+ cells will arise only if the LRE3 transcript undergoes a successful round of retrotransposition. LRE3 expression levels were assayed using a previously described antibody that detects the human ORF1-encoded protein (α-ORF1p) (Rodic et al. 2014). LRE3 integration events were visualized using an antibody against EGFP (α-EGFP). (B,B′) Human LRE3 ORF1p expression in 11-hpf embryos injected with the pLRE3-mEGFPI expression construct. In B, ORF1p immunoreactivity is undetectable in wild-type embryos (left panel) but is abundant in p53− embryos (right panel). In B′, quantification of ORF1p expression in wild-type and p53− embryos is plotted. The X-axis indicates genotypes injected. The Y-axis plots the volume of ORF1 expression normalized to total embryonic volume (see the Materials and Methods) for individual animals (black dots). The two embryos shown in B are each represented as an open circle on the graph in B′. Note that prominent ORF1p expression is frequently observed in p53− embryos but is absent in wild-type animals. (*) P-value < 0.0025. (C,C′) Retrotransposition events derived from pLRE3-mEGFPI can be stratified into three classes in 48-hpf embryos, as indicated in C. Class 0 consists of embryos with no EGFP+ cells. Class I consists of embryos that have <13 EGFP+ cells. Class II consists of embryos that have ≥13 EGFP+ cells. Note that all animals in C are p53−. In C′, the number of EGFP+ cells in class I and class II embryos is plotted for the indicated genotypes (X-axis). The Y-axis indicates the number of EGFP+ cells per embryo. Each dot represents an individual animal. Class II embryos were frequently observed in p53− embryos (27.3%) but were only rarely observed in wild-type animals (2.1%). (*) P-value < 0.0001. The pLRE3H230A-mEGFPI expression plasmid contains a missense mutation in the endonuclease domain of the LRE3 ORF2-encoded protein (ORF2p) (Coufal et al. 2011) and serves as a negative control (Supplemental Table 3). The pLRE3H230A-mEGFPI control plasmid produced only class 0 embryos when injected into wild-type and p53− animals (wild type, n = 149; p53−/−, n = 49) (Supplemental Table 3). Similarly, uninjected controls only produced class 0 animals (wild type, n = 209; p53−/−, n = 178) (Supplemental Table 3). Bars, 200 μm. (D,D′) H3K9 trimethylation across the LRE3 5′ UTR in wild-type (D) and p53− (D′) zebrafish. Chromatin immunoprecipitation (ChIP) analysis was performed in 4-hpf zebrafish embryos injected with the pLRE3-mEGFPI reporter construct using a H3K9 trimethyl (H3K9me3) antibody (open bars) and control IgG (closed bars). H3K9me3 levels were determined at four sites (1–4) spanning the L1 5′ UTR by ddPCR (see the schematic in the bottom panel). H3K9me3 marks were enriched in wild-type embryos, notably at primer pair 2 (D), but the signal for H3K9me3 was similar to background IgG controls in p53− embryos (D′). H3K9me3 levels were normalized to input, and mean values with 95% confidence intervals are presented. Total H3 levels were similar across the 5′ UTR for each genotype (Supplemental Fig. 7). See Supplemental Figure 7 for H3K9me3 levels normalized to total H3 and Supplemental Figure 6 for a second biological replicate.
To test how p53 might influence LRE3 activity, we examined these synthetic elements for p53-dependent H3K9 trimethyl (H3K9me3) marks after injection into zebrafish embryos by chromatin immunoprecipitation (ChIP). This histone modification reflects a prominent mechanism of piRNA-mediated repression and is generally associated with silenced genes (for review, see Malone and Hannon 2009; Castaneda et al. 2011; Holoch and Moazed 2015). Consistent with this, H3K9me3 marks were clearly enriched at the L1 enhancer region of wild-type embryos injected with the LRE3 reporter (Fig. 3D; Supplemental Fig. 6). However, when LRE3 molecules were injected into p53− embryos, the signal for H3K9me3 was similar to background IgG controls (Fig. 3D′; Supplemental Fig. 6). These starkly contrasting results suggest that p53 regulates transcription of retrotransposons by impacting chromatin marks at these mobile elements. Furthermore, since little or no cell deaths occur during the stages examined in these assays (Cole and Ross 2001), p53-dependent restraint of LINE-1 activity is separable from apoptotic functions associated with p53 (Lu et al. 2009).
Human p53 corrects dysregulated transposon activity, but cancer-associated variants do not
To test whether suppression of transposons might be a conserved property also encoded by human p53 (hp53) genes, we engineered a collection of fly strains that are, in effect, “humanized” for p53 function. In these lines, the Drosophila p53 gene is replaced by human wild-type or cancer-associated p53 variants regulated by flanking sequences of the native fly locus (see the Materials and Methods; D'Brot 2014). The majority of TP53 mutations in human cancers are missense and cause single amino acid changes that correspond to the DNA-binding domain (Soussi 2007). We generated cancer-associated alleles that comprise five of the most prevalent hp53 mutations in cancer. These five hot spot mutations all map to the DNA-binding domain and are thought to directly contact DNA (R248 and R273) or support the structure of the DNA-binding surface (R175 and G245) (Soussi 2007). As seen in Figure 4, the normal human p53 gene complemented the fly counterpart and effectively restrained retroelements. However, despite comparable expression (Fig. 4, inset), all five cancer-associated p53 alleles failed to rescue this defect. Together, these observations establish that retrotransposon suppression by p53 genes is a broadly conserved property shared by the human counterpart. Moreover, since p53 mutant alleles commonly seen in cancer patients were disabled for this function, our results raise the possibility that suppression of retrotransposon activity may contribute to p53-mediated tumor suppression.
Figure 4.
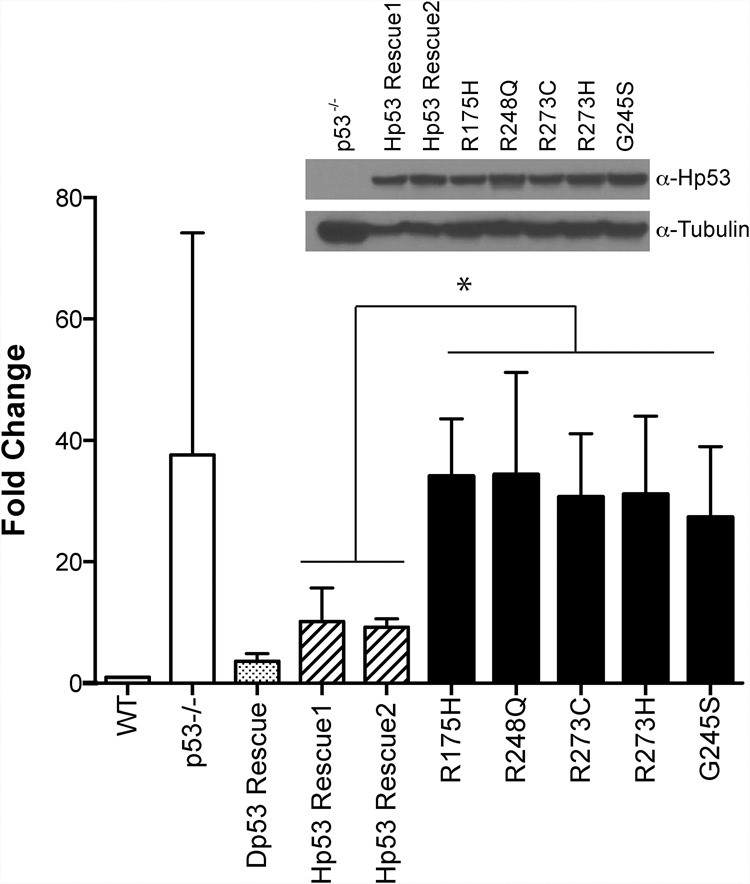
Human p53 corrects dysregulated transposon activity in p53− flies, but variants commonly seen in patients do not. TAHRE retrotransposon expression was quantified in ovaries from humanized p53 Drosophila strains (see the text) using ddPCR (standardized to the housekeeping gene rp49). Note that dysregulation seen in p53− flies (white bar) is effectively corrected in rescue lines encoding either the fly p53 gene (Dp53 Rescue; dotted bar) or the wild-type human p53 gene (striped bars). Lines humanized with distinct p53 mutant alleles commonly seen in cancers (black bars) were not corrected for transposon dysregulation despite comparable expression from these alleles, as verified by Western blot shown using Drosophila Tubulin as a loading control (inset). Note that each cancer-associated allele (black bars) differs from wild-type human p53 (striped bars) by the single amino acid indicated, and all human transgenes are positioned at the same “landing site” in the fly genome (see the Materials and Methods). Hp53 Rescue1 and Hp53 Rescue2 are independently generated lines. All p53 cancer-associated alleles are significantly different from wild-type human p53 strains, denoted by the asterisk (P-value < 0.05) (see the Materials and Methods). Error bars represent standard deviations.
Elevated retrotransposon activity in p53− human and mouse cancers
Recent studies have documented elevated retrotransposon activity in cancer tissues (Doucet-O'Hare et al. 2015; Ewing et al. 2015; Rodic et al. 2015), but this oncogenic trait has not been directly coupled to p53 status. To empirically assess whether elevated retrotransposon activity might be coupled to p53 loss in human cancers, we profiled LINE-1 expression in wild-type and p53 mutant Wilms tumors (Rakheja et al. 2014) using an α-ORF1p antibody (Rodic et al. 2014). Figure 5A shows that ORF1p expression was consistently detected in p53 mutant tumors, but, in stark contrast, little or no ORF1p expression was detected in Wilms tumors that were normal for p53 (Fig. 5A′; Supplemental Fig. 9). Likewise, little or no ORF1p expression was seen in matched normal tissue controls (Fig. 5A′′). We extended this analysis to all remaining Wilms tumors that could be curated for p53 status from this same archive (Rakheja et al. 2014). These additional samples (seven total) were wild type for p53 and also clearly negative for ORF1p expression (Fig. 5B; Supplemental Fig. 10). Together, these results exposed a highly significant association between p53 mutations and dysregulated LINE-1 expression in Wilms tumors.
Figure 5.
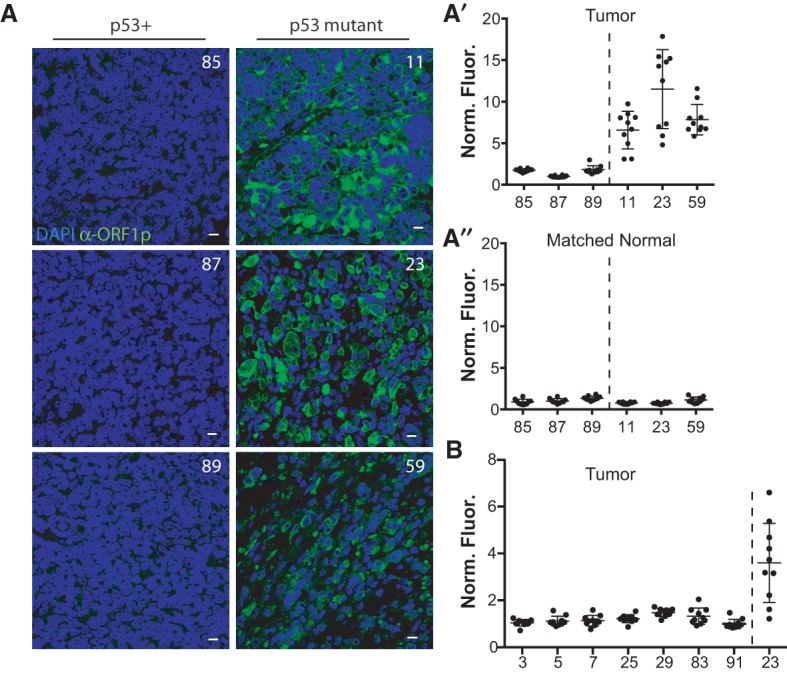
Deregulated retroelements stratify with p53 mutations in Wilms tumors. (A) Compared with Wilms tumors that are wild type for p53 (left panels: 85, 87, and 89), Wilms tumors that are mutant for p53 (right panels: 11, 23, and 59) show dramatically elevated human LINE-1 ORF1p expression (α-ORF1p [green]; counterstained with DAPI [blue]) (Rodic et al. 2014). Bars, 10 μm. (A′) Quantification of results in A was measured here using automated image analyses (see the Materials and Methods). On the X-axis, tumors wild type for p53 (tumors 85, 87, and 89) are separated by a dotted line from tumors mutant for p53 (tumors 11, 23, and 59). The Y-axis plots the normalized fluorescence intensity (Norm. Fluor.), where the fluorescence intensity of ORF1p expression is normalized to the DAPI volume (see the Materials and Methods) for individual fields of view, each represented as a dot. Ten fields of view were taken per tumor (shown in Supplemental Fig. 9). The normalized fluorescence intensity of tumors mutant for p53 (tumors 11, 23, and 59) is significantly different from the tumors wild type for p53 (tumors 85, 87, and 59) (P-value < 0.0001) (see the Materials and Methods). In A′′ note that fluorescence intensities were similar across all matched normal tissue. (B) Seven additional Wilms tumors wild type for p53 were quantified. On the X-axis, tumors wild type for p53 (tumors 3, 5, 7, 25, 29, 83, and 91) are separated by a dotted line from the tumor mutant for p53 (tumor 23, used as a positive control). The Y-axis plots the normalized fluorescence intensity (Norm. Fluor.), where the fluorescence intensity of ORF1p expression is normalized to the DAPI volume (see the Materials and Methods) for individual fields of view, each represented as a dot. Ten fields of view were taken per tumor. The normalized fluorescence intensity of the tumor mutant for p53 (tumor 23) is significantly different from the tumors wild type for p53 (tumors 3, 5, 7, 25, 29, 83, and 91) (P < 0.0001) (see the Materials and Methods). Note that p53 mutations appearing in these Wilms tumors are listed in Supplemental Table 5. Specificity controls for the human L1 ORF1p antibody are shown in Supplemental Figure 8.
To determine whether this association extended to other cancer types, we examined colon cancer RNA sequencing (RNA-seq) data sets (see the Materials and Methods) in The Cancer Genome Atlas (TCGA). In colon cancers, p53 mutations are prominent. Furthermore, raw RNA-seq reads from tumor and matched normal tissue were accessible for an anonymous cohort of 18 patients, eight of which were wild type for p53, and 10 of which were mutant for p53 (see Supplemental Table 5). As shown in Figure 6, loss of p53 was associated with a statistically significant elevation of transcripts corresponding to the human-specific LINE-1 (L1Hs) subfamily. These represent the most recent LINE-1 lineage and contain the majority of retrotransposition-competent elements (Beck et al. 2010; Carreira et al. 2014). Furthermore, as the evolutionary distance increased through the LINE-1 family, the P-value for this relationship also increased (Supplemental Fig. 11), suggesting that p53 preferentially impacts expression of elements within the L1Hs lineage. This effect was specific to retrotransposons, since no association emerged between p53− status and the expression of either simple repeats or pseudogenes in these same data sets (Supplemental Fig. 11).
Figure 6.
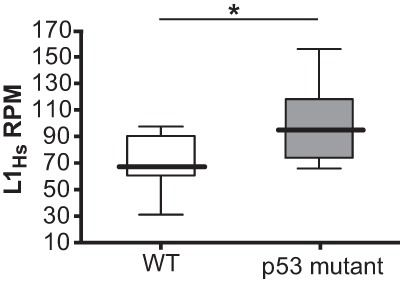
Deregulated retroelements stratify with p53 mutations in colon cancers. L1Hs expression was analyzed in colon cancer patients corresponding to all TCGA single-end RNA-seq data sets for which matched normals and raw sequences were available. Eight patient samples were wild type for p53 (open box plot), and 10 patient samples were mutant for p53 (gray box plot). The X-axis indicates p53 status. The Y-axis indicates L1Hs reads per million (RPM). Medians are represented by thick lines, and, for each box, the top edge is the 75th percentile, and the bottom edge is the 25th percentile. The top and bottom whiskers are maximum and minimum values, respectively. Note that cancers bearing p53 mutations (gray) are elevated for L1Hs expression relative to cancers that are wild type for p53 (open). (*) P-value = 0.03. In contrast, RNA-seq reads corresponding to simple repeats or pseudogenes are similar for p53 wild-type and p53 mutant genotypes (see Supplemental Fig. 11). See Supplemental Table 5 and Petitjean et al. (2007) for p53 mutations in colon cancer samples.
To empirically extend these findings, we also assessed this association in mouse cancer models. Here, retrotansposons were examined in myc-driven liver tumors that were either wild type or null for p53 (Fig. 7). Using antibodies to detect expression of mouse IAP retroelements, αIAP gag, (Dewannieux et al. 2004), or mouse LINE-1 retroelements (αL1 ORF1p) (Soper et al. 2008), we identified clear expression in testes as expected (Supplemental Fig. 12; Ma et al. 2009) but found little or no expression in wild-type livers (Fig. 7A,B). In contrast, expression from both retrolement classes was present and heterogenous in almost all tumors (Fig. 7C–F), but, notably, tumors lacking p53 were significantly elevated for IAP and L1 activity (Fig. 7G,H).
Figure 7.

Elevated retrotransposon expression in mouse tumors lacking p53. (A–F) Photomicrographs of livers from a wild-type (A,B), a c-Myc-expressing (C,D), and a p53-null c-Myc-expressing (E,F) mouse depicting elevated expression of mouse IAP gag (red staining; left panels) (Dewannieux et al. 2004) and mouse L1 ORF1p (red staining; right panels) (Soper et al. 2008) in tumors (C–F). The dotted line in F denotes the boundary between a tumor with modest expression of L1 ORF1p (left) and one expressing high levels of L1 ORF1p (right). Original magnifications, ×125. Specificity controls for antibody stainings are shown in Supplemental Figure 12 for IAP gag and L1 ORF1p. (G,H) Graphs showing quantitation of relative levels of IAP gag and L1 ORF1p in the wild-type liver, adjacent liver (AL), and liver tumors (T) in p53+/+:Myc and p53−/−:Myc mice. Quantitation was performed as described in the Materials and Methods and in Comerford et al. (2014). Red dots in G and H correspond to immunohistochemistry images in C–F. In F, two distinct tumors are shown, and the matching red dot in H corresponds to the highest-expressing tumor at the right. The intensity of staining of IAP gag (G) and L1 ORF1p (H) in Myc-driven tumors in p53-null mice is significantly different from Myc-driven tumors in p53 wild-type mice, denoted by asterisks (P-value < 0.0001) (see the Materials and Methods).
Discussion
We establish here that p53 genes from flies, zebrafish, and humans act to restrain retrotransposons (Figs. 1–4). Moreover, in at least two p53-driven human cancers and a comparable mouse model, p53 loss was strongly associated with elevated retrotransposon activity (Figs. 5–7). Furthermore, we showed previously that p53 is acutely responsive when these transposons are dysregulated (Wylie et al. 2014). Combined, these findings suggest that ancestral functions of the p53 gene family were linked to transposon control, raising the possibility that p53 restricts oncogenesis in part by suppressing the movement of mobile elements. Powerful support for this concept emerged from our complementation studies (Fig. 4), where human p53 corrected retroelement dysregulation seen in fly p53 mutants, but all five cancer-associated p53 alleles were clearly disabled for this activity. Since p53 mutant alleles arising in cancers are typically compromised for DNA binding, repression could occur through direct action at putative p53-binding sites in these retrotransposons (Harris et al. 2009). Indeed, our observations suggest that p53 contains these mobile elements by impacting chromatin marks (Fig. 3D,D′ on H3K9me3) and associated production of transcripts (Fig. 1; Supplemental Fig. 5).
Retrotransposition in somatic tissues is relevant for human disease (Carreira et al. 2014; Richardson et al. 2015), but retroelements also have the potential to impact subsequent generations and, as illustrated in Figure 1, C and D, their RNAs clearly target the presumptive germline during oogenesis. Long-standing questions have focused on stimuli that might instigate transposon movement, and, in the germline, DNA breaks formed during meiotic recombination are thought to provoke retrotransposition (Beauregard et al. 2008). Consistent with this, we found that loss of p53 did not incite transposon derepression if spo11 was also defective (Fig. 2). Hence, Spo11-dependent p53 activity occurring in the meiotic precursors of flies and in mice (Lu et al. 2010) reflects functions that contain movement in the germline. Moreover, these functions must be distinct from apoptosis, since p53− germline cells are normal for programmed cell death (Peterson et al. 2003; Wylie et al. 2014). Viewed from this perspective, transposon eruptions in the germline are consistent with infertility phenotypes seen in p53− flies, zebrafish, and mice (see the Materials and Methods; Supplemental Tables 2,4) (Hu et al. 2011). Combined, these observations suggest that p53-mediated tumor suppression was evolutionarily co-opted from ancestral meiotic functions that restricted mobile elements to insure germline integrity. This model provides an attractive framework for understanding why p53 loss provokes destabilized genomes and could also explain why p53 is universally coupled to genotoxic stressors. Viewed more broadly, this framework facilitates efforts to interrogate roles for p53 as a guardian against “transposopathy” in human health and disease.
Materials and methods
Fly stocks and genetics
All fly stocks were maintained at 22°C–25°C on standard food medium. We obtained aubergine and cutoff mutants aubHN, aubQC, cuffWM, and cuffQQ from T. Schupbach (Princeton University, Princeton, NJ). All other stocks were obtained from Bloomington Stock Center (Indiana University, Bloomington, IN). Unless otherwise noted, two p53-null alleles, 238H (ns) and 5A-1-4 (k1), were used in transcombination to reduce genetic background influences. Two wild-type strains, yw and w1118, were used in transcombination for comparison. The Spo11 mei-W68(1) allele was obtained from the Bloomington Stock Center and was crossed into the p53−/− 238H (ns) and 5A-1-4 (k1) strains. To test genetic interactions between Spo11 and p53, we generated flies that placed these p53 alleles in trans: Spo11−/−;p53−/−(ns)/(k1). As previously described in Wylie et al. (2014) the Dp53 rescue strain was engineered by φC31 integration of a 20-kb genomic fragment BAC containing the Dp53 locus into an attP site on the X chromosome of the PBac{y+-attP-9A}VK00006 line (Bloomington Stock Center, no. 9726). The parent BAC CH322-15D03 was obtained from the P[acman] resource library (Venken et al. 2009) and Rainbow Transgenic Flies performed the injection and screening for recombinants. The Dp53 rescue strain was crossed into two p53-null alleles, 238H (ns) and 5A-1-4 (k1), and used in transcombination to reduce genetic background influences unless otherwise noted. Similarly, as described in D'Brot (2014), the humanized p53 lines were generated by replacing the Dp53 ORF of BAC CH322-15D03 with either wild-type or mutant human p53 cDNA via recombineering. These constructs were integrated into the attP site on the X chromosome of the PBac{y+-attP-9A}VK00006 line (Bloomington Stock Center, no. 9726). Templates for the p53 mutants alleles were generated by site-directed mutagenesis of the p53 cDNA before recombineering into the BAC. For Figure 4, the p53−, Dp53Rescue, and all humanized lines were homozygous for fly p53 5A-1-4 (k1)-null allele and were compared with the yw wild-type strain.
RT–PCR
Retrotransposon expression was measured using RT–PCR assays (Figs. 1,2,4). Wild-type, p53−, and p53Rescue fly ovaries were dissected in PBS, and total RNA was isolated using TRIzol reagent (Life Technologies). RNA was isolated from single ovary pairs for Figure 1B. Five ovary pairs per RNA preparation were used for all other RT–PCR data (Figs. 1A,D,E,E′, 2, 4; Supplemental Figs. 2, 3C). cDNA was generated using iScript cDNA synthesis kit (Bio-Rad). Semi-qRT–PCR (Fig. 1A,D; Supplemental Fig. 2) was performed using GoTaq Green master mix (Promega). Samples were run on a 1.2% ethidium bromide gel and visualized on the Typhoon Trio Imager. qRT–PCR (Figs. 1E,E′, 2; Supplemental Fig. 3C) was performed using the iQ SYBR Green supermix (Bio-Rad) on the CFX96 real-time PCR machine (Bio-Rad). Primer efficiency was taken into account for all reactions. rp49 was used for normalization. Controls for DNA contamination included reactions with and without reverse transcriptase. Primers are listed in Supplemental Table 6. Droplet digital RT–PCR reactions (Figs. 1B, 4) were previously described (Link et al. 2013). Primers and fluorescent probes specific for the TAHRE transcript are listed in Supplemental Table 6. rp49 was used for normalization.
FISH
Custom Stellaris FISH probes were designed against TAHRE transcripts by using the Stellaris RNA FISH probe designer version 4.1 (Biosearch Technologies, Inc.; available online at http://www.biosearchtech.com/stellarisdesigner). The wild-type, p53−, and p53Rescue ovaries were hybridized with the TAHRE Stellaris RNA FISH probe set labeled with Quasar 570 (Biosearch Technologies, Inc.) following the manufacturer's instructions (available online at http://www.biosearchtech.com/stellarisprotocols). Briefly, ovaries were dissected into PBS and fixed for 45 min at room temperature with 4% formaldehyde solution in PBS. After fixation, ovaries were placed in 70% EtOH overnight at 4°C. The following day, the EtOH was aspirated, and wash buffer (2× SSC, 10% deionized formamide in nuclease-free water) was added for 5 min. The probe was diluted at a concentration of 50 nM in hybridization buffer (2× SSC, 10% dextran sulfate [Sigma, D8906], 1 mg/mL tRNA [Sigma, R8759], 2 mM vanadyl robonucleoside complex [New England Biolabs], 10% deionized formamide in nuclease free water). The wash buffer was aspirated, and the hybridization + probe solution was added to each sample and placed for 24 h at 37°C. The samples were then washed with wash buffer twice for 30 min each at 37°C. VectaShield (Vector Laboratories) with DAPI was added before mounting and imaging.
Embryo collections
Embryos from wild-type (yw), p53− (K1), and p53Rescue;p53− (k1) strains were collected on standard juice agar plates for 3 h and aged 1 h for early stage embryos or aged 21 h for late stage embryos. Embryos were collected and dechorionated in 50% bleach, washed, and transferred to Trizol for RNA extraction. For maternal loading assays, wild-type virgin females were crossed to p53− males, and p53− virgin females were crossed to wild-type males. Embryos from these parental genotypes were collected for 3 h, aged 1 h, and processed for RNA extraction.
Drosophila fertility studies
In fertility assays, the p53-null allele 238H (ns) was compared with the p53 rescue transgene in the p53ns background (p53Rescue). To reduce genetic background influences, the p53ns allele and the p53 rescue transgene were backcrossed into the yw wild-type background for 17 and 10 generations, respectively. Female virgins were collected for 5 d. To assess fertility at the single-animal level, one female virgin and three yw wild-type males were placed in a vial. Females were allowed to lay eggs for 4 d, and fertility was scored by the presence of larvae 10 d after the parents were removed. The number of single-pair matings and the percent infertility are indicated in Supplemental Table 2.
Egg phenotypes
As previously described (Lu et al. 2010) eggs were collected on standard juice agar plates and manually orientated horizontally for imaging. Images were taken on the Zeiss SteREO Discovery version 12 and processed with ImageJ using the following script: “Enhance Contrast (saturated = 0.5), RGB color, set scale (distance = 0; known = 1; pixel = 1; unit = pixel).” Sample sizes were n = 419 for aubHN/QC; p53ns/k1, n = 298 for aubHN/+; p53ns/k1, n = 312 for aubHN/+, n = 469 for aubHN/QC, n = 248 for cuffQQ/WM; p53ns/k1, n = 891 for cuffQQ/+; p53ns/k1, n = 153 for cuffQQ/+, and n = 564 for cuffQQ/WM. Prism 6 software (GraphPad) was used to perform statistics.
Immunostaining of fly tissue
Three-day-old to 5-d-old well-fed females were dissected in PBS and fixed in 4% EM-grade formaldehyde (Polysciences) diluted in PBS–0.1% Triton X-100 (PBST) with three times the volume of heptane. After washing, tissues were blocked in 1.5% BSA and then incubated with primary antibodies overnight at 4°C. The antibodies used were as follows: α-Aubergine and α-armitage were gifts from Mikiko Siomi (Nishida et al. 2007; Saito et al. 2010), α-rhino was a gift from William Theurkoff (Klattenhoff et al. 2009), and α-vasa was from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). For fluorescence visualization, Alexa-488 (Invitrogen) secondary antibody was used, and 0.1 μg/mL DAPI (Invitrogen) for DNA staining was added in the first wash step. After three washes, ovaries were further hand-dissected and mounted in VectaShield (Vector Laboratories) for microscopy imaging.
Zebrafish maintenance, strains, and injections
Zebrafish were maintained according to standard procedures (Westerfield 2000). All work with zebrafish was carried out under protocols approved by the Institutional Animal Care and Use Committees at University of Texas Southwestern Medical Center (Dallas, TX), an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited institution. The pLRE3-mEGFPI and pLRE3H230A-mEGFPI constructs have been previously described (Coufal et al. 2009, 2011; Garcia-Perez et al. 2010). These constructs were injected into one- to two-cell zebrafish embryos from the parental AB (wild-type) or p53− (tp53M214K/M214K) strains (Berghmans et al. 2005), 2–4 nL of each construct was injected at a concentration of 125 ng/µL, and the injection mixture included phenol red and 0.3× Danieau's solution [1740 mM NaCl, 21 mM KCl, 12 mM MgSO4•7H2O, 18 mM Ca(NO3)2, 150 mM HEPES buffer]. Uninjected controls were carried alongside for both genotypes.
Immunostaining of zebrafish tissue
Forty-eight-hour-old embryos were dechorionated, euthanized with tricaine, and fixed in 4% EM-grade formaldehyde (Polysciences) diluted in PBS–0.1% Triton X-100 for 24 h at 4°C. Eleven-hour-old embryos were fixed in 4% EM-grade formaldehyde (Polysciences) diluted in PBS–0.1% Triton X-100 for 24 h at 4°C or 4 h at room temperature and then hand-dechorionated. After washing, tissues were blocked in 1.5% BSA and then incubated with anti-GFP (1:1000; Thermo Fisher Scientific) or anti-human ORF1 (1:500) primary antibody overnight at 4°C. The anti-human ORF1 monoclonal antibody was a gift from Kathleen Burns (Johns Hopkins University) (Rodic et al. 2014) For fluorescence visualization, Alexa-488 (Invitrogen) or Alexa-568 (Invitrogen) secondary antibody was used. After three washes, zebrafish were placed in PBS or mounted in 1% agar in PBS for microscopy imaging.
ChIP methods
ChIP was performed as described in Lindeman et al. (2009) and Bogdanovic et al. (2013) with modifications. As for all LRE3 studies, parental AB wild-type and p53− zebrafish eggs were injected with the LRE3 indicator at the single-cell stage. At 4 hpf, embryos were dechorionated using pronase and cross-linked for 10 min with 1% formaldehyde (final concentration). Cross-linking was quenched for 5 min on ice by addition of glycine to 125 mM. Embryos were washed three times with PBS and resuspended in 300 µL of ChIP lysis buffer (50 mM Tris-HCl at pH 7.5, 0.5% SDS, 10 mM EDTA, protease inhibitors) per 100 embryos. Embryos were mechanically lysed and incubated for an additional 10 min on ice. A Diagenode Bioruptor was used to sonicate chromatin to between 200 and 700 base pairs (two 15-min cycles, 30 sec on/30 sec off, high output). Following sonication, chromatin was diluted to a concentration of 400 ng/µL with ChIP lysis buffer before being diluted to a final concentration of 100 ng/µL using ChIP dilution buffer (10 mM Tris at pH 7.5, 1 mM EDTA, 100 mM NaCL, 1% Triton X-100, protease inhibitors). Antibodies were preincubated with protein A/G beads (Santa Cruz Biotechnology) for 3 h at 4°C. Immunoprecipitations were performed overnight at 4°C with rotation (50 µg of chromatin per antibody). Antibodies used were H3 (2.4 µg per ChIP; Active Motif, 39159) and H3K9me3 (2 µg per ChIP; Diagenode, pAB-056-050). After immunoprecipitation, protein A/G beads were washed with RIPA ChIP buffer (10 mM Tris at pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.1% Na-deoxycholate, protease inhibitors). Elution, reverse cross-linking, and proteinase K (Thermo) digestions were performed as a single step as in Lindeman et al. (2009) with modifications. Briefly, 150 µL of elution buffer (20 mM Tris at pH 7.5, 5 mM EDTA, 50 mM NaCl) was added to washed beads and incubated overnight at 68°C. Eluate was removed, and beads were washed in an additional 150 µL of elution buffer for 15 min at 68°C. Eluates were pooled, and DNA was purified by phenol:chloroform:isoamyl alcohol extraction followed by ethanol precipitation. As a final step, DNA was purified using the Zymo ChIP clean and concentration (D5205) kit following the manufacturer's instructions. ChIP results were quantified by ddPCR (Robin et al. 2014) with EvaGreen on a QX100 droplet reader. All concentration values for ChIP DNA samples were normalized to their respective input, and 95% confidence intervals were calculated as per Bio-Rad (pers. comm.). Primer pairs used were as follows: primer 1, 5′-GACGCAGAAGACGGTGATTT-3′ and 5′-TCACCCCTTTCTTTGACTCG-3′; primer 2, 5′-CGCACCACGAGACTATATCC-3′ and 5′-CAGTCTGCCCGTTCTCAGAT-3′; primer 3, 5′-GGCACACTGACACCTCACAC-3′ and 5′-TAACAGACAGGACCCTCAGC-3′; and primer 4, 5′-ACATCTACACCGAAAACCCATC-3′ and 5′-GCGCTCTGCGTTTTAGAGTT-3′.
Zebrafish fertility studies
To assess fertility in zebrafish, wild-type and p53−/− embryos were collected from controlled matings within 2 h of laying. For each genotype, we documented the total number of eggs and the number of unfertilized eggs layed within the first 6 h after laying to obtain the percentage of unfertilized embryos (Supplemetal Table 4). This was performed over three individual trials, each trial containing ≥20 adult zebrafish per genotype. Wild-type and p53−/− adult zebrafish were age-matched.
Mouse genetics and immunohistochemistry
The genetically modified mouse strains used were as follows: p53−/− mice were obtained from Jackson Laboratories, and Alb-c-Myc mice were generously provided by E. Sandgren (University of Wisconsin, Madison, WI). All mouse experiments were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee (IACUC). Immunohistochemistry was performed as previously described (Comerford et al. 2014). Antigen retrieval was performed for 10 min with citrate buffer.
Analysis of IAP and Line1 ORF1 expression in p53 wild-type-driven and p53-null Myc-driven liver tumors was done as follows: Immunohistochemistry was performed on formalin-fixed deparaffinized sections of livers collected from wild-type and tumor-bearing p53+/+:Alb-c-Myc and p53−/−:Alb-c-Myc mice using antibodies specific for mouse IAP gag (1:500) or mouse LINE-1 ORF1p (1:500). The anti-mouse L1 ORF1 antibody was a gift from Alex Bortvin (Carnegie) and Sandy Martin (University of Colorado) (Soper et al. 2008). The anti-mouse IAP gag antibody was a gift from Bryan Cullen (Duke) (Dewannieux et al. 2004). Antibody specificity was verified by performing immunohistochemistry on testis isolated from postnatal day 10 (P10) GASZ+/− and GASZ−/− mice, confirming expression of IAP and LINE-1 ORF1p in GASZ−/− testis (Supplemental Fig. 12; Ma et al. 2009). The GASZ testis sections were a gift from Martin Matzuk (Baylor College of Medicine). Slides were viewed, and images captured and processed using a Leica DMRX microscope and MetaMorph microscopy automation and image analysis software (Molecular Devices).
Relative expression of IAP gag and LINE-1 ORF1p was quantified by assigning a score ranging from 0 (no visible expression) to 5 (maximal expression) based on the intensity of AEC chromagen (red) staining in normal parenchyma (wild-type livers) and in adenomas, hepatocellular tumors (T), and tumor-adjacent livers (TAL) in p53+/+:Alb-c-Myc and p53−/−:Alb-c-Myc mice. Tumor-adjacent liver or intervening stroma that was either negative or low for IAP gag or LINE-1 ORF1p expression served as an internal baseline control (Fig. 7G,H).
Preparation of ovary extracts and Western blot analysis
For each humanized fly strain and p53− control, 20 ovaries were dissected into PBS and then homogenized with a glass pestle in RIPA lysis buffer and protease inhibitor cocktail (Roche). Extract concentration was measured by standard Bradford protein assay. Twenty micrograms of tissue extracts was subjected to 10% SDS-PAGE (NuPAGE, Invitrogen), after which the proteins were transferred to PVDF membrane. The immunoblots were performed overnight at 4°C using the following primary antibodies: Mouse anti-hp53 DO-1 (Santa Cruz Biotechnology) was used at 1:1000 and 1:5000 anti-tubulin (E7, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Bound antibodies were visualized by chemiluminescence ECL Plus kit (Amersham Biosciences/GE Healthcare) using a 1:5000 dilution of anti-mouse IgG (Jackson ImmunoResearch Laboratories, Inc.).
Wilms tumor immunostaining
Archived Wilms tumor samples were collected, processed, and sequenced as previously described (Rakheja et al. 2014). Deparaffinization and immunostaining of p53+ and p53 mutant Wilms tumors were performed in parallel. Antigen retrieval was performed with sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20 at pH 6.0) in a pressure cooker. Tissue sections were then permeabilized for 1 h at room temperature with PBST and then blocked for 2–4 h with blocking solution (5% normal donkey serum, 1% bovine serum albumin in PBST). The mouse anti-human ORF-1 primary antibody (Rodic et al. 2014) was diluted at a concentration of 1:500 in blocking solution, and samples were incubated overnight at 4°C. For fluorescence visualization, Alexa-488 (Invitrogen) secondary antibody was used. After three washes, VectaShield with DAPI (Vector Laboratories) and a coverslip were placed on the slide for microscopy imaging.
RNA-seq analyses
All tumor unaligned colon cancer single-end RNA-seq files generated by the TCGA Research Network (https://tcga-data.nci.nih.gov/tcga; phs000178.v8.p7) for which matched controls were available were downloaded from the Cancer Genomics Hub for inclusion in these studies (The Cancer Genome Atlas Network 2012). A total of 18 patients, eight of which were wild type for p53 and 10 of which were verified mutant for p53, were sequenced. FastQ files were prefiltered to remove adapters, PCR primers, low-quality 3′ and 5′ bases, and low-quality sequences (http://www.bioinformatics.babraham.ac.uk/projects/fastqc; Martin 2011; Schmieder and Edwards 2011). The resulting FastQ files were aligned to the University of California at Santa Cruz (UCSC) hg19 reference genome (http://genome.ucsc.edu) (Karolchik et al. 2014) TopHat (tophat -p 12 --no-coverage-search) (Kim et al. 2013). Duplicate sequences were marked with Picard MarkDuplicates (http://broadinstitute.github.io/picard) and filtered for all subsequent analyses. Repeat element expression was determined using an algorithm based on the methods outlined in Criscione et al. (2014). First, sequencing reads aligning to a single position in the genome (uniquely aligned reads) were identified. Genomic coordinates for these uniquely aligned reads were then compared with the genomic coordinates of all RepeatMasker (http://www.repeatmasker.org) annotated repeat elements present in the genome (http://genome.ucsc.edu, rmsk track). RepeatMasker uses the Smith-Waterman alignment algorithm to scan the genome for repetitive sequences using the RepBase update repeat library (Jurka 2000). Uniquely mapped reads whose alignment coordinates overlap with an annotated repeat were identified and retained. Next, we identified all sequencing reads that aligned to more than one region in the genome (multiply mapped reads) and all sequencing reads that could not be aligned to the reference genome (unmapped reads). These multiply mapped and unmapped reads were then realigned to a custom genome containing only repeat elements with Bowtie (http://genomebiology.com/2009/10/3/R25) such that all possible best alignments for each read were identified and saved. To generate the custom genome used for realignment, we extracted DNA sequences for every Repeat Masker annotated repeat (±15 bases) in the human genome. Sequences for elements in the same subfamily were concatenated into a single “pseudochromosome,” separated by a string of 250 Ns. For example, all sequences for genomic repeats in the AluJb subfamily were represented in a single “AluJb pseudochromosome.” Multiply mapped and unmapped reads where all best realignments were within a single pseudochromosome were assigned to the subfamily of that pseudochromosome. Multiply mapped and unmapped reads where best alignments were found to be more than a single pseudochromosome were assigned to the subfamily of each pseudochromosome to which the read aligned using a fractional approach (1/n, where n = number of psuedochromosomes where the read aligned). This type of analysis is unable to resolve sequencing reads to a single genomic position but does enable calculation of reads aligning to elements in a given subfamily, family, or class. Therefore, we assigned subfamily, family, and class designations to each genomic repetitive element and pseudochromosome from our custom genome based on the classification found in RepBase (Jurka 2000). We added the number of uniquely aligned reads mapping to a single genomic element of a given class, family, or subfamily to the whole or fractional number of multiply mapped and unmapped reads realigning to a pseudochromosome assigned to the same class, family, or subfamily. Read counts were normalized to sequencing depth and presented as reads per million (RPM). All repeat element subfamily, family, and class annotations were based on repeat classifications found in RepBase. The pseudogene class includes RNA, rRNA, snRNA, and tRNA pseudogenes. The simple repeat class includes centromeric and satellite repeats together with a repetitive sequence comprised of an iterated short nucleotide sequence. The mutation status of p53 was determined on the basis of the Protected Mutations data sets available through TCGA (The Cancer Genome Atlas Network 2012) and/or mutations evident at the RNA level.
Microscopy and image processing
FISH confocal images (Fig. 1C) were taken with a Leica TCS SP5 confocal microscope using a 40× objective lens with 3× digital zoom with Leica software. Drosophila ovary (Supplemental Fig. 3D-G′) and Wilms tumor confocal images (Fig. 5A; Supplemental Figs. 5, 6) were taken with a Multiphoton Zeiss LSM780 inverted confocal microscope using a 40× objective lens with 3× digital zoom with Zeiss Zen software. Zebrafish 11-hpf embryo confocal images for ORF1p quantification were taken with a Multiphoton Zeiss LSM780 upright confocal microscope using a 10× objective lens with Zeiss Zen software. Fluorescent images of whole-mount zebrafish were taken on the Zeiss SteREO Discovery V.12 microscope using the 1.50× lens with AxioVision software. Forty-eight-hour-post-fertilization embryos were imaged with 18× zoom, and 11-hpf embryos were imaged with 65× zoom. Antibody staining or FISH signal image comparisons between wild-type and p53− samples were done with the same laser image intensities and master gain settings. Z-stacks were taken at 0.5-µm sections. Z-stacks of images were projected using ImageJ software (National Institutes of Health). Figures were prepared using Adobe Photoshop and Illustrator CS2 (Adobe Systems).
Zebrafish 11-hpf ORF1p image processing
Eleven-hour-post-fertilization embryos were mounted in 1% agarose in PBS. Embryos were imaged on the Multiphoton Zeiss LSM780 upright confocal microscope. To obtain the ORF1p volume using Imaris8 software (Bitplane), the surface of the ORF1p was built using a baseline subtraction of 10,000. To obtain the embryo volume using Imaris8 software (Bitplane), the surface of the embryo autofluorescence was built using a baseline subtraction of 2000. The ORF1p volume was divided by the embryo volume and multiplied by 100 to obtain the ORF1+ volume per embryo in percent (Fig. 3B′). Embryos were imaged for one trial, but similar ORF1p expression patterns between wild-type and p53− embryos were repeated over multiple trials.
Wilms tumor image processing
Ten fields of view were taken on the Multiphoton Zeiss LSM780 inverted confocal microscope for each tumor and matched normal kidney. Images were deconvoluted with AutoQuant (AutoQuant) software using 10 iterations of three-dimensional deconvolution. The average fluorescence intensity of the ORF1 signal for each field of view was obtained using ImageJ software. The Z-stacks were projected using the sum slices projection type, and the mean gray value was recorded. The average fluorescence intensity of the ORF1 signal was normalized to the nuclei density. To obtain the nuclei volume using Imaris8 software (Bitplane), the surface of the blue channel was built using a baseline subtraction of 15,000. The ORF1 mean gray value was divided by the nuclei volume per field of view. To obtain the normalized fluorescence intensity, all images were then normalized to the average value of all 10 fields of view for the CMC87 sample.
Statistics
For all statistical analysis, data were placed into GraphPad Prism software. For statistics on the ddPCR of TAHRE transcripts on single fly ovaries (Fig. 1B), a one-way ANOVA test was performed on all genotypes, and p53−/− was significantly different from wild type (P-value = 0.0172) and p53Rescue (P-value = 0.0347) at the 95% confidence interval. Quantification of TAHRE FISH (Fig. 1C′) was analyzed using a two-tailed unpaired t-test. At the 99% confidence level, wild type was significantly different from p53−/− (P-value = 0.001). Note that TAHRE FISH on the p53Rescue was performed once, and statistics could not be calculated.
For statistics on the qRT–PCR of retroelement transcripts on bulk ovaries (Fig. 1E,E′), a one-way ANOVA test was performed on all genotypes at the 95% confidence interval. For Idefix, p53−/− was significantly different from wild type (P-value = 0.0278). For TAHRE, p53−/− was significantly different from wild type (P-value = 0.0094) and p53Rescue (P-value = 0.0234). For Burdock, p53−/− was significantly different from wild type (P-value = 0.0028) and p53Rescue (P-value = 0.0031). For HeT-A, p53−/− was significantly different from wild type (P-value = 0.0004). For Gypsy, p53−/− was significantly different from wild type (P-value = 0.0002) and p53Rescue (P-value = 0.0016).
For statistics on the qRT–PCR of retroelement transcripts on wild-type, Spo11+/−;p53−/−, and Spo11−/−;p53−/− ovaries (Fig. 2), a one-way ANOVA test was performed on all genotypes at the 95% confidence interval. For TAHRE, Spo11+/−;p53−/− was significantly different from wild type (P-value = 0.0004) and Spo11−/−;p53−/− (P-value = 0.0035). For HeT-A, Spo11+/−;p53−/− was significantly different from wild type (P-value = 0.0218) at the 95% confidence interval and Spo11−/−;p53−/− (P-value = 0.0803) at the 90% confidence interval. For Idefix, Spo11+/−;p53−/− was significantly different from wild type (P-value = 0.0315) and Spo11−/−;p53−/− (P-value = 0.0164). For Gypsy, Spo11+/−;p53−/− was significantly different from wild type (P-value = 0.0051) and Spo11−/−;p53−/− (P-value = 0.0068). For Burdock, Spo11+/−;p53−/− was significantly different from wild type (P-value = 0.0516) at the 90% confidence interval and Spo11−/−;p53−/− (P-value = 0.006) at the 95% confidence interval.
Quantification of ORF1p expression in wild-type and p53−/− zebrafish injected with the pLRE3-mEGFP reporter (Fig. 3B′) was analyzed using a two-tailed unpaired t-test. At the 99% confidence level, wild type was significantly different from p53−/− (P-value = 0.0025). Quantification of EGFP+ cells in wild-type and p53−/− zebrafish injected with the pLRE3-mEGFP reporter (Fig. 3C′) was analyzed using a two-tailed unpaired t-test. At the 99% confidence level, wild type was significantly different from p53−/− (P-value < 0.0001).
For statistics on the ddPCR on TAHRE transcripts in the humanized flies (Fig. 4), a one-way ANOVA test was performed on all genotypes at the 95% confidence interval. R175H was significantly different from Hp53 Rescue1 (P-value = 0.0.0030) and Hp53 Rescue2 (P-value = 0.0059). G245S was significantly different from Hp53 Rescue1 (P-value = 0.0116) and Hp53 Rescue2 (P-value = 0.0333). R248Q was significantly different from Hp53 Rescue1 (P-value = 0.0102) and Hp53 Rescue2 (P-value = 0.0318). R273C was significantly different from Hp53 Rescue1 (P-value = 0.0077) and Hp53 Rescue2 (P-value = 0.0139). R273H was significantly different from Hp53 Rescue1 (P-value = 0.0076) and Hp53 Rescue2 (P-value = 0.0212).
For statistical analysis on normalized fluorescence intensity of Wilms tumors (Fig. 5A′), an ordinary one-way ANOVA was performed on all samples at the 99.9% confidence interval. The wild-type p53 samples (85, 87, and 89) were not significantly different from each other (sample 85 vs. 87, P-value = 0.9789; sample 85 vs. 89, P-value > 0.9999; sample 87 vs. 89, P-value = 0.9659). All wild-type p53 tumors (85, 87, and 89) were significantly different from the p53 mutant tumors (11, 23, and 59) as listed below. Tumor 85 was significantly different from tumors 11 (P-value = 0.0002), 23 (P-value < 0.0001), and 59 (P-value < 0.0001). Tumor 87 was significantly different from tumors 11 (P-value < 0.0001), 23 (P-value < 0.0001), and 59 (P-value < 0.0001). Tumor 89 was significantly different from tumors 11 (P-value = 0.0003), 23 (P-value < 0.0001), and 59 (P-value < 0.0001). For statistical analysis on normalized fluorescence intensity of matched normal tissue (Fig. 4A′), an ordinary one-way ANOVA was performed on all samples at the 99.9% confidence interval. All samples were not significantly different from each other, except sample 89 was significantly different from samples 11 (P-value = 0.0002) and 23 (P-value < 0.0001).
For statistical analysis on normalized fluorescence intensity of the additional Wilms tumors (Fig. 5B), an ordinary one-way ANOVA was performed on all samples at the 99.9% confidence interval. The wild-type p53 samples (3, 5, 7, 25, 29, 83, and 91) were not significantly different from each other. All wild-type p53 tumors (3, 5, 7, 25, 29, 83, and 91) were significantly different from the p53 mutant tumor (23) (P-value < 0.0001).
We performed a multinomial distribution for statistical analysis on the Wilms tumor data (Fig. 5). Four possible outcomes were possible based on ORF1p expression and p53 status, each with a probability of 0.25. The stratification of elevated ORF1p expression and p53− mutations was highly significant (P-value = 0.00000426).
For statistical analysis of mouse IAP gag and L1 ORF1p expression in p53 wild-type-driven and p53-null-Myc-driven liver tumors, a two-tailed unpaired t-test was performed. For IAP gag expression, p53 wild-type tumors were significantly different from p53-null-Myc-driven liver tumors (P-value < 0.0001) at the 95% confidence interval. For L1 ORF1p expression, p53 wild-type tumors were significantly different from p53-null-Myc-driven liver tumors (P-value < 0.0001) at the 95% confidence interval.
Drosophila p53 transcripts between wild-type, p53−, and p53Rescue bulk fly ovaries (Supplemental Fig. 1) were analyzed using an ordinary one-way ANOVA at the 95% confidence interval. p53−/− flies were significantly different from wild type (P-value = 0.0287). p53−/− flies were significantly different from p53Rescue flies (P-value = 0.0167). Wild-type and p53Rescue flies were not significantly different.
Fertility between p53− and p53Rescue flies (Supplemental Table 2) was analyzed using a unpaired t-test with equal SD. p53−/− flies were significantly different from p53Resuce (P-value = 0.0381) at the 95% confidence level.
For statistical analysis on egg length (Supplemental Fig. 4A,B), a one-way ANOVA test was performed on all genotypes at the 99.9% confidence interval. aub(HN)/(QC); p53(NS)/(K1) was significantly different from aub(HN)/+ (P-value < 0.0001), aub(HN)(QC) (P-value < 0.0001), and aub(HN)/+; p53(NS)/(K1) (P-value < 0.0001). aub(HN)/+; p53(NS)/(K1) was not significantly different from aub(HN)/+ (P-value > 0.9999) but was significantly different from aub(HN)/(QC) (P-value < 0.0001). aub(HN)/+ was significantly different from aub(HN)/(QC) (P-value < 0.0001). cuff(QQ)/(WM); p53(NS)/(K1) was significantly different from cuff(QQ)/+ (P-value < 0.0001), aub(QQ)/(WM) (P-value < 0.0001), and aub(QQ)/+; p53(NS)/(K1) (P-value < 0.0001). cuff[QQ]/+; p53(NS)/(K1) was not significantly different from cuff(QQ)/+ (P-value = 0.1602) or (QQ)/(WM) (P-value = 0.2810). cuff(QQ)/+ was not significantly different from cuff(QQ)/(WM) (P-value = 0.0085).
Flamenco transcript between wild-type and p53− bulk fly ovaries (Supplemental Fig. 4C) was analyzed using an ordinary one-way ANOVA at the 99% confidence interval. For primer pair 1, p53−/− flies were significantly different from wild type (P-value = 0.0042). For primer pair 2, p53−/− flies were significantly different from wild type (P-value = 0.0037).
Fertility between p53− and wild-type zebrafish (Supplemental Table 4) was analyzed using a unpaired t-test with equal standard deviations. p53−/− zebrafish were significantly different from wild-type zebrafish (P-value = 0.0012) at the 99% confidence level.
Supplementary Material
Acknowledgments
We thank the following for generously providing reagents and materials: Kathleen M. Burns for the human LINE-1 ORF1p antibody, Alex Bortvin and Sandy Martin for the mouse LINE-1 ORF1p antibody, Bryan Cullen for the mouse IAP gag antibody, Mikiko Siomi for α-aubergine and α-armitage antibodies, William Theurkauf for the α-rhino antibody, and Martin Matzuk for the GASZ+/− and GASZ−/− mouse testis sections. We thank the Texas Advanced Computing Center (TACC) at the University of Texas at Austin and the BioHPC at University of Texas Southwestern Medical Center at Dallas for providing high-performance computing resources, and, specifically, Chris Hempel, Nathaniel Mendoza, Doug James, and David Gignac of the TACC, and David Trudgian and Liqiang Wang of the BioHPC for their technical assistance. We thank Daniel Hwang, Corey Timmerman, Chandru Sundarrajan, Sam Glaves, Erin Regan, and Jessica Alatorre for technical assistance, and Ms. Nancy Leff for editorial assistance. We thank the University of Texas Southwestern Live Cell Imaging Core, especially Kate Luby-Phelps and Abhijit Bugde, for assisting in microscopy and image processing. This work was supported by grants to A.W. (T32 GM083831, National Research Service Award 1F31CA189691-01), A.E.J. (T32 5T32DK007307), P.K. (National Research Service Award 1F31GM108472-01), and J.M.A. (National Institutes of Health R01GM072124 and R01GM115682, the Welch Foundation I-1865, Ellison Medical Foundation AG-SS-2743-11, and Cancer Prevention Research Institute of Texas RP110076). Grants from the Children's Medical Center Foundation (Dallas, TX) and a Young Investigator Research Grant from the Society for Pediatric Pathology supported D.R. Grants from the National Institutes of Health (R01CA135731) and the Cancer Prevention and Research Institute of Texas (RP110394) supported J.F.A. K.S.C. is a Damon Runyon-Sohn Pediatric Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-4P-13). J.V.M. is an Investigator of the Howard Hughes Medical Institute. A grant to J.V.M. from the National Institutes of Health (GM060518) supported the generation of the LINE-1 expression constructs used in this study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.266098.115.
References
- Aravin AA, Hannon GJ, Brennecke J. 2007. The Piwi–piRNA pathway provides an adaptive defense in the transposon arms race. Science 318: 761–764. [DOI] [PubMed] [Google Scholar]
- Beauregard A, Curcio MJ, Belfort M. 2008. The take and give between retrotransposable elements and their hosts. Annu Rev Genet 42: 587–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. 2010. LINE-1 retrotransposition activity in human genomes. Cell 141: 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CD, Morris JP, Liu TX, Schulte-Merker S, Kanki JP, et al. 2005. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci 102: 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic O, Fernández-Miñán A, Tena JJ, de la Calle-Mustienes E, Gómez-Skarmeta JL. 2013. The developmental epigenomics toolbox: ChIP-seq and MethylCap-seq profiling of early zebrafish embryos. Methods 62: 207–215. [DOI] [PubMed] [Google Scholar]
- Brouha B, Meischl C, Ostertag E, de Boer M, Zhang Y, Neijens H, Roos D, Kazazian HH Jr. 2002. Evidence consistent with human L1 retrotransposition in maternal meiosis I. Am J Hum Genet 71: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira PE, Richardson SR, Faulkner GJ. 2014. L1 retrotransposons, cancer stem cells and oncogenesis. FEBS J 281: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda J, Genzor P, Bortvin A. 2011. piRNAs, transposon silencing, and germline genome integrity. Mutat Res 714: 95–104. [DOI] [PubMed] [Google Scholar]
- Cole LK, Ross LS. 2001. Apoptosis in the developing zebrafish embryo. Dev Biol 240: 123–142. [DOI] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. 2014. Acetate dependence of tumors. Cell 159: 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH. 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Marchetto MC, Muotri AR, Mu Y, Carson CT, Macia A, Moran JV, Gage FH. 2011. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci 108: 20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N. 2014. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15: 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Preall JB, McGinn J, Hannon GJ. 2013. A transcriptome-wide RNAi screen in the Drosophila ovary reveals factors of the germline piRNA pathway. Mol Cell 50: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Brot A. 2014. “Of apoptosomes and oncogenes: repurposing a death machine and deconstructing the action of p53 mutations.” PhD thesis, Department of Cell Biology, University of Texas Southwestern Medical Center at Dallas, Dallas, TX. [Google Scholar]
- Dewannieux M, Dupressoir A, Harper F, Pierron G, Heidmann T. 2004. Identification of autonomous IAP LTR retrotransposons mobile in mammalian cells. Nat Genet 36: 534–539. [DOI] [PubMed] [Google Scholar]
- Doucet-O'Hare TT, Rodić N, Sharma R, Darbari I, Abril G, Choi JA, Young Ahn J, Cheng Y, Anders RA, Burns KH, et al. 2015. LINE-1 expression and retrotransposition in Barrett's esophagus and esophageal carcinoma. Proc Natl Acad Sci 112: E4894–E4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Gacita A, Wood LD, Ma F, Xing D, Kim M-S, Manda SS, Abril G, Pereira G, Makohon-Moore A, et al. 2015. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res 25: 1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH Jr, Boeke JD. 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87: 905–916. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, Hammer GD, Collins KL, O'Shea KS, Menendez P, et al. 2010. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature 466: 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Dewan A, Zupnick A, Normart R, Gabriel A, Prives C, Levine AJ, Hoh J. 2009. p53 responsive elements in human retrotransposons. Oncogene 28: 3857–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D. 2015. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zheng T, Wang J. 2011. Regulation of fertility by the p53 family members. Genes Cancer 2: 420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illmensee K, Mahowald AP. 1974. Transplantation of posterior polar plasm in Drosophila. Induction of germ cells at the anterior pole of the egg. Proc Natl Acad Sci 71: 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. 2000. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet 16: 418–420. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. 2014. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res 42: D764–D770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana JS, Theurkauf W. 2010. piRNAs, transposon silencing, and Drosophila germline development. J Cell Biol 191: 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Xi H, Li C, Lee S, Xu J, Khurana JS, Zhang F, Schultz N, Koppetsch BS, Nowosielska A, et al. 2009. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell 138: 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R, Ephrussi A. 1994. Germ plasm formation and germ cell determination in Drosophila. Ciba Found Symp 182: 282–296. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Oren M. 2009. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman LC, Vogt-Kielland LT, Aleström P, Collas P. 2009. Fish'n ChIPs: chromatin immunoprecipitation in the zebrafish embryo. Methods Mol Biol 567: 75–86. [DOI] [PubMed] [Google Scholar]
- Link N, Kurtz P, O'Neal M, Garcia-Hughes G, Abrams JM. 2013. A p53 enhancer region regulates target genes through chromatin conformations in cis and in trans. Genes Dev 27: 2433–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WJ, Amatruda JF, Abrams JM. 2009. p53 ancestry: gazing through an evolutionary lens. Nat Rev Cancer 9: 758–762. [DOI] [PubMed] [Google Scholar]
- Lu WJ, Chapo J, Roig I, Abrams JM. 2010. Meiotic recombination provokes functional activation of the p53 regulatory network. Science 328: 1278–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Buchold GM, Greenbaum MP, Roy A, Burns KH, Zhu H, Han DY, Harris RA, Coarfa C, Gunaratne PH, et al. 2009. GASZ is essential for male meiosis and suppression of retrotransposon expression in the male germline. PLoS Genet 5: e1000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Hannon GJ. 2009. Small RNAs as guardians of the genome. Cell 136: 656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J 17: 10–12. [Google Scholar]
- McKim KS, Hayashi-Hagihara A. 1998. mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: evidence that the mechanism for initiating meiotic recombination is conserved. Genes Dev 12: 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida KM, Saito K, Mori T, Kawamura Y, Nagami-Okada T, Inagaki S, Siomi H, Siomi MC. 2007. Gene silencing mechanisms mediated by Aubergine piRNA complexes in Drosophila male gonad. RNA 13: 1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH Jr. 2000. Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res 28: 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JS, Barkett M, McCall K. 2003. Stage-specific regulation of caspase activity in Drosophila oogenesis. Dev Biol 260: 113–123. [DOI] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629. [DOI] [PubMed] [Google Scholar]
- Rakheja D, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. 2014. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. 2015. The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr 3: MDNA3-0061-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, Wright WE. 2014. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev 28: 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodic N, Sharma R, Sharma R, Zampella J, Dai L, Taylor MS, Hruban RH, Iacobuzio-Donahue CA, Maitra A, Torbenson MS, et al. 2014. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol 184: 1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodic N, Steranka JP, Makohon-Moore A, Moyer A, Shen P, Sharma R, Kohutek ZA, Huang CR, Ahn D, Mita P, et al. 2015. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med 21: 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RJ, Weiner MM, Lin H. 2014. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505: 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Ishizu H, Komai M, Kotani H, Kawamura Y, Nishida KM, Siomi H, Siomi MC. 2010. Roles for the Yb body components Armitage and Yb in primary piRNA biogenesis in Drosophila. Genes Dev 24: 2493–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpiz S, Kwon D, Uneva A, Kim M, Klenov M, Rozovsky Y, Georgiev P, Savitsky M, Kalmykova A. 2007. Characterization of Drosophila telomeric retroelement TAHRE: transcription, transpositions, and RNAi-based regulation of expression. Mol Biol Evol 24: 2535–2545. [DOI] [PubMed] [Google Scholar]
- Shpiz S, Olovnikov I, Sergeeva A, Lavrov S, Abramov Y, Savitsky M, Kalmykova A. 2011. Mechanism of the piRNA-mediated silencing of Drosophila telomeric retrotransposons. Nucleic Acids Res 39: 8703–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA. 2011. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12: 246–258. [DOI] [PubMed] [Google Scholar]
- Soper SF, van der Heijden GW, Hardiman TC, Goodheart M, Martin SL, de Boer P, Bortvin A. 2008. Mouse maelstrom, a component of nuage, is essential for spermatogenesis and transposon repression in meiosis. Dev Cell 15: 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T. 2007. p53 alterations in human cancer: more questions than answers. Oncogene 26: 2145–2156. [DOI] [PubMed] [Google Scholar]
- Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3: 1339–1345. [DOI] [PubMed] [Google Scholar]
- Venken KJ, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, et al. 2009. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods 6: 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. 2007. p53 in health and disease. Nat Rev Mol Cell Biol 8: 275–283. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. 2001. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21: 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. 2000. A guide for the laboratory use of zebrafish (Danio rerio). University of Oregon Press, Eugene, OR. [Google Scholar]
- Wylie A, Lu WJ, D'Brot A, Buszczak M, Abrams JM. 2014. p53 activity is selectively licensed in the Drosophila stem cell compartment. Elife 3: e01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.