

Here, Shields et al. demonstrate that the hematopoietically expressed homeobox gene Hhex is overexpressed in acute myeloid leukemia (AML) and is essential for the initiation and propagation of MLL-ENL-induced AML but dispensable for normal myelopoiesis, indicating a specific requirement for Hhex for leukemic growth. The findings in this study describe for the first time a nonclustered homeobox transcription factor that is essential for AML initiation and maintenance and provide mechanistic insight into these processes.
Keywords: acute myeloid leukemia, transcription factor, self-renewal, homeobox, tumor suppressor
Abstract
Unlike clustered HOX genes, the role of nonclustered homeobox gene family members in hematopoiesis and leukemogenesis has not been extensively studied. Here we found that the hematopoietically expressed homeobox gene Hhex is overexpressed in acute myeloid leukemia (AML) and is essential for the initiation and propagation of MLL-ENL-induced AML but dispensable for normal myelopoiesis, indicating a specific requirement for Hhex for leukemic growth. Loss of Hhex leads to expression of the Cdkn2a-encoded tumor suppressors p16INK4a and p19ARF, which are required for growth arrest and myeloid differentiation following Hhex deletion. Mechanistically, we show that Hhex binds to the Cdkn2a locus and directly interacts with the Polycomb-repressive complex 2 (PRC2) to enable H3K27me3-mediated epigenetic repression. Thus, Hhex is a potential therapeutic target that is specifically required for AML stem cells to repress tumor suppressor pathways and enable continued self-renewal.
In the hematopoietic system, self-renewal capacity is normally restricted to hematopoietic stem cells (HSC) and must be tightly controlled, since aberrant self-renewal is a hallmark of cancer (He et al. 2009). Indeed, a number of leukemia oncoproteins act via inducing self-renewal of either HSCs or more mature progenitors that have normally lost this property. Prototypical examples of this are oncogenic fusion proteins generated by chromosomal translocations at the MLL locus, which occur in ∼5%–6% of acute myeloid leukemia (AML) cases, resulting in reciprocal chromosomal translocations that link MLL to a vast number of fusion partners (Krivtsov and Armstrong 2007). The resultant fusion proteins activate target genes, including those in the HOXA cluster (HOXA5–10) and their essential cofactor, MEIS1, leading to expression of a “HOX code” that has been implicated in leukemia maintenance (Ayton and Cleary 2003; Horton et al. 2005). In addition to this self-renewal network, recent studies have shown that sustained leukaemogenesis by MLL fusion oncogenes requires expression of epigenetic modifiers, including the Polycomb-repressive complex 2 (PRC2) (Neff et al. 2012; Tanaka et al. 2012; Shi et al. 2013). PRC2 is composed of core components, including Suz12 and Eed along with the methyltransferases Ezh1 or Ezh2, which mediate silencing of key target genes via catalysis of the repressive mark H3K27me3 (Margueron and Reinberg 2011).
The above studies have identified important roles for clustered HOX family (class I) homeobox genes in regulation of both normal HSCs and leukemic stem cells (LSCs) (Argiropoulos and Humphries 2007; Alharbi et al. 2013). However, the role of HOX genes in MLL-associated leukemia is complicated by the fact that MLL fusion proteins activate many HOXA cluster genes, and these exhibit partial redundancies in leukemia maintenance (Kumar et al. 2004; So et al. 2004). Recently, evidence has emerged that nonclustered (class II) homeobox genes may also play important roles in MLL-driven leukemia. These include members of the caudal-type homeobox (CDX) subfamily of ParaHox genes that are overexpressed in AML and act upstream of HOX genes to promote their expression (Bansal et al. 2006; Scholl et al. 2007; Rawat et al. 2012). In addition, the H2.0-like homeobox (HLX) gene is overexpressed in most AML cases and promotes leukemogenesis (Kawahara et al. 2012). Together, these studies indicate an important role for both clustered and nonclustered homeobox genes in the initiation and maintenance of AML.
The hematopoietically expressed homeobox gene (Hhex, also known as Prh), encodes a member of the NK-like (NKL) subclass of homeodomain proteins that is thought to function primarily as a transcriptional repressor (Guiral et al. 2001; Swingler et al. 2004; Soufi and Jayaraman 2008). It was first identified via its expression in hematopoietic tissues, being abundantly expressed in HSCs and progenitors before being down-regulated upon differentiation (Crompton et al. 1992; Bedford et al. 1993). In the T-cell lineage, down-regulation of Hhex is crucial, as overexpression causes thymocyte self-renewal and T-cell leukemia in mouse models and is associated with early T-cell precursor ALL (ETP-ALL) in humans (George et al. 2003; McCormack et al. 2010, 2013). Furthermore, a rare chromosomal translocation [t(10;11)(q23;p15)] results in the generation of a NUP98-HHEX fusion oncogene in AML (Jankovic et al. 2008).
Loss of Hhex during embryonic development is lethal due to a failure of liver development, precluding the analysis of hematopoietic development in knockout mice (Keng et al. 2000; Martinez Barbera et al. 2000). However, studies using embryoid body differentiation and blastocyst complementation approaches have defined critical roles for Hhex in the development of definitive HSCs and B cells (Bogue et al. 2003; Guo et al. 2003; Kubo et al. 2005; Paz et al. 2010).
Using Hhex conditional knockout mice (Hunter et al. 2007), we showed recently that Hhex is dispensable for maintenance of adult HSCs and myeloid lineages but essential for the commitment of diverse lymphoid lineages at the stage of the common lymphoid progenitor (CLP) (Jackson et al. 2015). Moreover, Hhex is required for the radioresistance of LSCs in a mouse model of human ETP-ALL (Shields et al. 2015). However, the role of Hhex in myeloid leukemia has not been studied previously. Here we show that Hhex is overexpressed in human AML and is essential for myeloid leukemia driven by the oncogenic fusion protein MLL-ENL as well as its downstream effectors, HoxA9 and Meis1, while being dispensable for normal myelopoiesis. Conditional deletion of Hhex results in loss of PRC2-mediated epigenetic silencing of the Cdkn2a locus, resulting in induction of the Cdkn2a-encoded tumor suppressors p16INK4a and p19ARF. Therefore, targeting Hhex is a potential strategy to induce tumor suppression to specifically inhibit myeloid leukemia.
Results
Hhex is overexpressed in human AML and required for initiation of AML by MLL-ENL
As Hhex regulates the development of definitive HSCs and is highly expressed in human ETP-ALL, we assessed whether its expression is altered in AML patient samples. Using a curated microarray database (HemaExplorer) (Bagger et al. 2013), we found that Hhex is highly expressed in early progenitors before being down-regulated in all lineages except monocytes and B cells (Fig. 1A). Strikingly, expression was twofold to fourfold higher in AML patient samples relative to normal HSCs, irrespective of leukemia subtype, with highest expression found in AML with inv(16)/t(16;16) or t(8;21) translocations (Fig. 1A). Analysis of an independent patient cohort (n = 536) (Verhaak et al. 2009) confirmed high HHEX expression in AMLs with the favorable inv(16)/t(16;16) and t(8;21) karyotypes (Fig. 1B). Consistent with the above observation, HHEX expression was highest in the favorable risk group (Fig. 1C). Patients in this cohort were also assigned into prognostic groups using the European Leukemia Network (ELN) classification (Li et al. 2013). Patients within the intermediate risk groups could be dichotomized into those with better or worse outcomes based on an automatically determined (k-means clustering) (Diffner et al. 2013) HHEX expression threshold (Fig. 1D,E). Five-year survival rates were ∼25% versus ∼50% (Int-1; P = 0.01) and ∼30% versus ∼50% (Int-2; P = 0.05) in high and low HHEX expressors, respectively. These data show that HHEX expression in human AML is context-dependent and adds value to existing prognostic classification systems.
Figure 1.
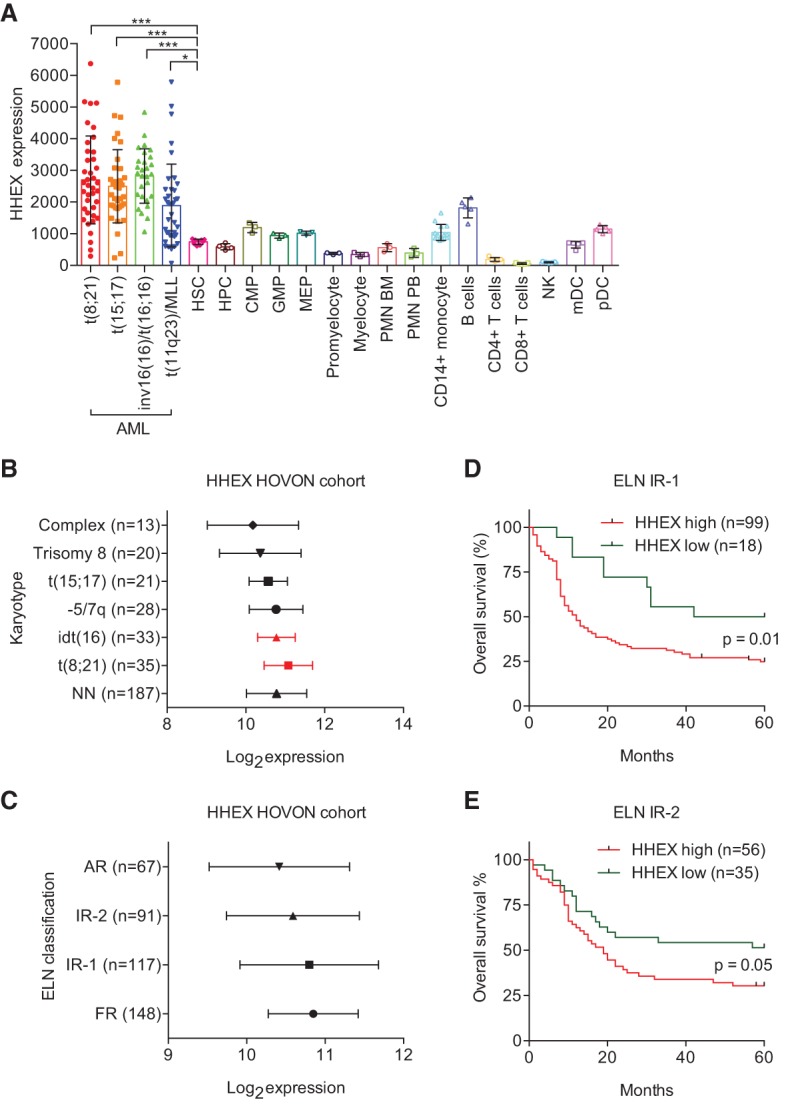
Hhex is overexpressed in human AML and is associated with an adverse outcome in ELN intermediate-1 and intermediate-2 classified AML. (A) Expression of Hhex expression in four AML karyotypes, hematopoietic progenitors, and differentiated cells using the HemaExplorer microarray database. Single points on the graph represent the average HHEX expression of two individual microarray probes for the cell types indicated. (*) P < 0.05; (***) P < 0.001, Student's t-test. (B) HHEX expression levels (Robust Multiarray Average [RMA] normalized; log2) in AML patients from the HOVON (Hemato-Oncologie voor Volwassenen Nederland ) cohort with frequent cytogenetic abnormalities. (NN) Cytogenetically normal AML. (C) HHEX expression levels (RMA normalized; log2) in patients from the HOVON cohort stratified according to ELN classes (Li et al. 2013). (D) Five-year overall survival of patients classified as ELN intermediate-1 with high/low expression (k-means clustering) of HHEX. (E) Five-year overall survival of patients classified as ELN intermediate-2 with high/low expression (k-means clustering) of HHEX. In D and E, P was determined using a log rank (Mantel-Cox) test.
We next sought to determine whether Hhex is required for the development of myeloid leukemia by the MLL-ENL fusion oncogene. As AML induced by MLL fusion oncogenes is initiated from HSCs and myeloid progenitors up to and including granulocyte/monocyte progenitors (GMPs) (Cozzio et al. 2003; Krivtsov et al. 2006), we first tested whether Hhex loss had any effect on the number and function of these cells following Hhex deletion in the bone marrow (BM) of Hhex−/ΔMx mice. Flow cytometric analysis revealed no defects, with a slightly increased frequency of long-term HSCs (LT-HSCs), common myeloid progenitors (CMPs), and myelo-erythroid progenitors (MEPs) in Hhex−/ΔMx mice when compared with Hhex+/fl controls (Supplemental Fig. S1A–C). We next tested the function of myeloid progenitors in Hhex−/ΔMx mice in semisolid agar colony assays. When cultured in a mixture of cytokines (IL-3, stem cell factor [SCF], and EPO), BM from Hhex−/ΔMx mice showed no defect in colony formation, with slightly increased frequencies of granulocyte and macrophage colonies (Supplemental Table S1). Culture in individual cytokines revealed additional differences, with increased numbers of granulocyte colonies in the presence of GM-CSF or IL-3 and a reduction in macrophage colonies in response to GM-CSF or M-CSF (Supplemental Table S1). Overall, these data indicate that Hhex is dispensable for the development and function of myeloid progenitors in vitro and in vivo.
To assess whether Hhex is required for initiation of AML, lineage-depleted BM cells from Hhex−/ΔMx mice were transduced with MSCV-IRES-GFP retroviruses expressing MLL-ENL and injected into irradiated congenic (Ly5.1) recipient mice (Fig. 2A). At the time of injection, Hhex−/ΔMx BM cells showed levels of viral infection (as assessed by GFP fluorescence) comparable with control (Hhex+/fl) BM cells (Supplemental Fig. S2A). Notably, MLL-ENL expression in Hhex+/fl BM led to a significant increase in Hhex mRNA expression compared with transduction with control (MIG) retrovirus (Fig. 2B).
Figure 2.
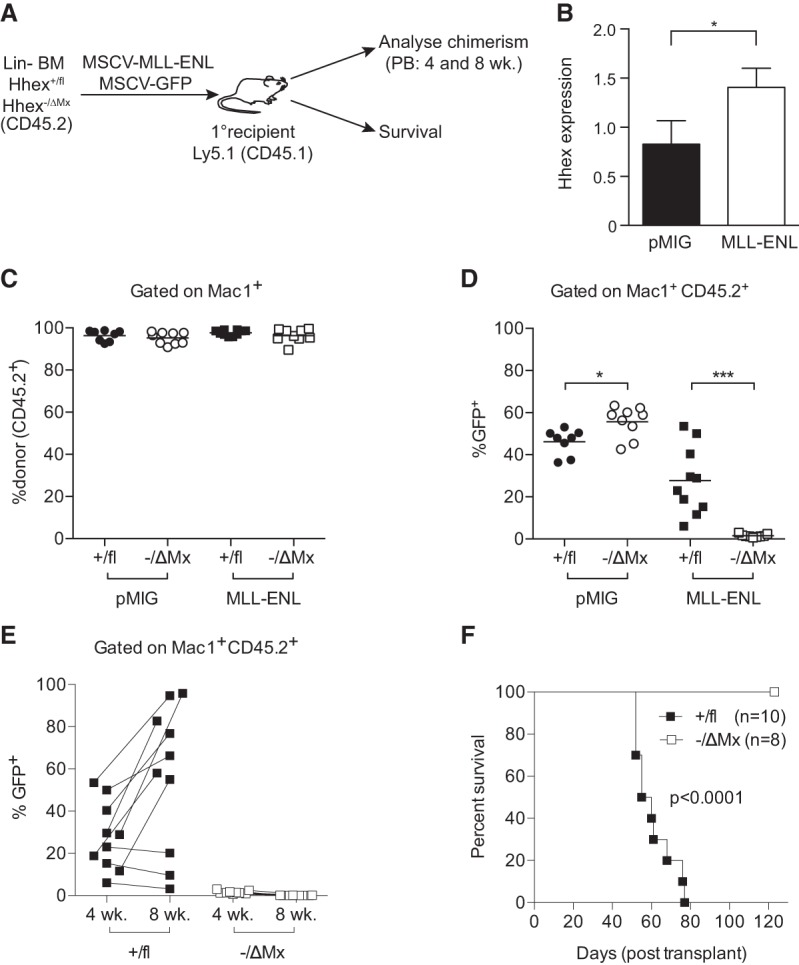
Hhex is required for initiation of AML by MLL-ENL. (A) Schematic diagram of experimental design using Hhex+/fl and Hhex−/ΔMx mice. (B) Hhex expression in virally transduced BM cells at 2 d after transduction as assessed by quantitative PCR. (C–E) Peripheral blood analysis of recipient mice injected with MSCV-IRES-GFP (MIG) and MIG-MLL-ENL transduced lineage-depleted BM from mice of the indicated Hhex genotypes. (C) Percentage of myeloid (Mac1+) cells that are donor-derived (CD45.2+) at 4 wk after transplant. (D) Percentage of donor-derived myeloid cells (Mac1+CD45.2+) that are virally transduced (GFP+) at 4 wk after transplant. Lines show the mean. (E) As in D, showing 4- and 8-wk time points of MLL-ENL transduced BM recipients of the indicated Hhex genotypes. Lines connect sequential samples taken from individual mice. (F) Kaplan-Meier survival curve of recipients of MLL-ENL transduced BM of the indicated Hhex genotypes (analyzed in C–E). Data in C–F are a combination of two separate experiments. (*) P < 0.05; (***) P < 0.001, Student's t-test. In F, P was determined using a log rank (Mantel-Cox) test.
All recipient mice, regardless of the genotype of donor cells, showed complete myeloid repopulation in the peripheral blood 4 wk after transplantation (Fig. 2C). Donor-derived lymphoid cells were reduced in recipients reconstituted with Hhex−/ΔMx BM (data not shown), consistent with our previous report that Hhex is required for lymphoid development downstream from the CLP but is dispensable for normal adult HSC function in transplant assays (Jackson et al. 2015). The levels of transduction with control (MIG) retrovirus was similar in donor myeloid cells derived from either Hhex−/ΔMx or Hhex+/fl BM (Fig. 2D). Transduction with MLL-ENL retrovirus was initially low in both Hhex−/ΔMx and Hhex+/fl BM cells; however, transduced control (Hhex+/fl) BM cells were rapidly expanded both in vitro (Supplemental Fig. S2B) and in vivo, with the peripheral blood of recipient mice containing abundant donor-derived Mac1+GFP+ cells at 4 wk, consistent with the establishment of a preleukemic state (Fig. 2D), which became more severe by 8 wk after transplant (Fig. 2E). In striking contrast, donor-derived Mac1+GFP+ cells were almost completely absent in the peripheral blood of Hhex−/ΔMx MLL-ENL recipient mice at 4 wk after transplant and continued to decline by 8 wk after transplant (Fig. 2D,E). Accordingly, while all Hhex+/fl MLL-ENL recipients succumbed to myeloid leukemia within 11 wk, none of the eight Hhex−/ΔMx MLL-ENL recipients developed leukemia within a 4-mo observation period (Fig. 2F). Thus, while Hhex is dispensable for adult HSC function and myeloid reconstitution, it is essential for the initiation of AML by MLL-ENL.
Hhex is essential for maintenance of AML induced by MLL-ENL
To determine whether Hhex is required for maintenance of AML by MLL-ENL, we generated ROSA26-CreERT2;Hhex−/fl mice (hereafter termed CreERT2;Hhex−/fl mice), in which deletion of the Hhexfl allele can be induced by tamoxifen-regulated Cre recombinase (hereafter termed HhexΔERT2) (Fig. 3A). BM from these mice was used to generate MLL-ENL-induced myeloid leukemias, as above. Next, 10,000 flow cytometry-sorted leukemia-initiating cells (LICs; GFP+, Mac1+, Kit+) from primary leukemic mice were injected into secondary recipients, and the Hhexfl allele was deleted by two tamoxifen administrations starting at 10 d after transplant (Fig. 3A). Development of fatal leukemia in placebo-treated controls occurred rapidly, within 23 d after transplant (Fig. 3B). Tamoxifen administration led to a small delay in the development of control Hhex+/fl tumors, likely due to Cre-mediated toxicity. In contrast, survival of tamoxifen-treated Hhex−/fl tumor recipients was more than doubled relative to placebo-treated controls (Fig. 3B). Strikingly, genomic PCR analysis revealed that while the Hhexfl allele was completely deleted in relapsed MLL-ENL Hhex+/ΔERT2 leukemias, secondary leukemias developing in recipients of MLL-ENL Hhex−/ΔERT2 LICs were completely nondeleted at the Hhexfl locus (Fig. 3C). Thus, secondary leukemias emanated from a small fraction of Hhexfl (nondeleted) cells that remained after tamoxifen treatment. To verify that Hhexfl deletion is not toxic to normal myeloid cells, mice were reconstituted with BM from CreERT2;Hhex−/fl mice, and, 1 mo later, the Hhexfl allele was deleted by tamoxifen treatment. We saw no selection for nondeleted Hhexfl in myeloid cells in the peripheral blood or BM of these chimeric mice up to 4 mo after tamoxifen treatment (Fig. 3D). Thus, Hhex is dispensable for normal myelopoiesis but critical for establishment and maintenance of MLL-ENL-induced myeloid leukemia.
Figure 3.
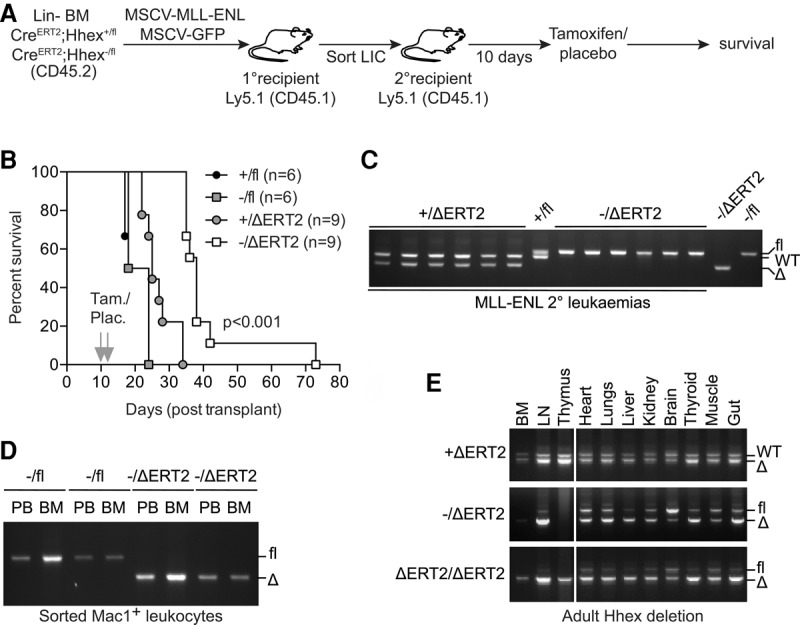
Hhex is required for maintenance of AML but not normal myeloid cells. (A) Schematic diagram of experimental design to test the role of Hhex in leukemia maintenance. (B) Kaplan-Meier survival curve of recipient mice injected with purified LICs (10,000 CD45.2+, Mac1+, GFP+, Kit+ cells) and administered with either tamoxifen (Tam.) or placebo (Plac.). P < 0.001 between Hhex−/fl and Hhex−/ΔERT2 recipient mice using a log rank (Mantel-Cox) test. (C) Hhexfl is selected for in Hhex−/ΔERT2 MLL-ENL secondary leukemias. Genomic DNA was extracted from whole BM from leukemic mice (>95% CD45.2+, Mac1+, GFP+ cells) of the indicated Hhex genotypes and analyzed by PCR. (D) Hhexfl is not selected for in normal myeloid cells. Recipient mice were injected with lineage-depleted CreERT2;Hhex−/fl BM and then administered with either tamoxifen or placebo after 4 wk. Four months later, BM and peripheral blood (PB) were harvested from placebo-treated (−/fl) and tamoxifen-treated (−/ΔERT2) recipients, and CD45.2+, Mac1+ cells were sorted and used for genomic PCR. (E) Hhexfl deletion is tolerated in the whole animal. Eight-week-old mice of the indicated Hhex genotypes were administered tamoxifen, and, 4 wk later, organs were harvested and processed for genomic PCR analysis. (LN) Lymph node.
Next, we assessed the impact of Hhex deletion in the whole animal by administering tamoxifen to CreERT2-Hhex−/fl mice. These mice were closely monitored for 1 mo, during which time no physical signs of illness were evident. Furthermore, no significant pathology was observed in Hhex-null animals (Supplemental Fig. S3), and genomic PCR analysis of various tissues and organs demonstrated recombination of the Hhexfl allele in all tissues (Fig. 3E). Thus, systemic loss of Hhex in adult mice has minimal short-term side effects, further demonstrating the potential for Hhex as a therapeutic target in AML.
To determine the cellular effects of Hhex deletion on MLL-ENL-induced leukaemic cells, we established cell lines from Hhex-deletable, CreERT2-Hhex−/fl primary leukemias. These were then administered with tamoxifen in vitro to induce deletion of the Hhexfl allele. This caused a marked decline in the growth rate of Hhex−/fl leukemia cell lines, leading to the complete elimination of these cells by 2 wk after treatment (Fig. 4A). Seven days after tamoxifen administration, cells lacking Hhex (Hhex−/ΔERT2) had reduced numbers of cycling cells (Fig. 4B) and increased expression of myeloid differentiation markers (Fig. 4C) and showed features of granulocyte/macrophage differentiation (Fig. 4D). Thus, loss of Hhex causes cell cycle arrest of leukemic blasts accompanied by cellular differentiation.
Figure 4.

Loss of Hhex induces cell cycle arrest and differentiation of AML cell lines. (A) Growth of BM-derived MLL-ENL AML cell lines of the indicated Hhex genotypes in the presence or absence of tamoxifen to delete Hhexfl. (B) Cell cycle analysis of MLL-ENL cell lines at day 7 using DAPI staining and flow cytometry. Data are mean + SD of triplicate determinations. (*) P < 0.05; (**) P < 0.01, Student's t-test. (C) Flow cytometric analysis of MLL-ENL cell lines showing granularity (side scatter [SSC]) and differentiation markers at day 7. The blue line indicates untreated cells, and the red line indicates cells treated with tamoxifen. (D) Wright-Giemsa staining of MLL-ENL cell lines at day 7. Bar, 10 µm.
Loss of Hhex causes up-regulation of tumor suppressors in LSCs
To determine the transcriptional alterations caused by loss of Hhex in AML, we performed high-throughput sequencing of RNA derived from MLL-ENL-induced LICs and cell lines following deletion of Hhex. LICs were derived from the BM of MLL-ENL transduced CreERT2;Hhex−/fl recipient mice 1 mo after Hhex deletion. At this point, while the tumor burden had significantly decreased relative to control mice (Supplemental Fig. S4A), we were still able to obtain sufficient numbers of LICs for RNA sequencing. Cell lines were harvested 7 d after Hhex deletion. Analysis of genomic DNA and RNA sequencing data confirmed that both LICs and cell lines were completely deleted at the Hhexfl locus (Supplemental Fig. S4B,C). Differential gene expression analysis demonstrated that 248 genes were differentially expressed (124 up and 124 down) in Hhex-deleted LICs (Supplemental Table S2), while 99 genes were differentially expressed (54 up and 45 down) in Hhex-deleted cell lines (Supplemental Table S3).
We first asked whether Hhex regulates expression of HoxA cluster genes and their cofactors, Pbx3 and Meis1, which are well-characterized drivers of MLL-induced leukemia (Krivtsov et al. 2006). We found that these genes were up-regulated in MLL-ENL LICs and cell lines relative to normal HSC-enriched Lin−Sca+Kit+ (LSK) cells and were not significantly altered by loss of Hhex (Fig. 5A). However, consistent with the loss of self-renewal and enhanced differentiation of MLL-ENL cell lines after Hhex loss, gene set enrichment analysis (GSEA) showed that Hhex-deleted cell lines down-regulate Myc-associated self-renewal programs and up-regulate a myeloid differentiation signature along with p53 target genes (Supplemental Table S4).
Figure 5.

Up-regulation of p16INK4a and p19ARF tumor suppressor pathways in leukemic cells following Hhex deletion. (A) Maintenance of the MLL-associated self-renewal signature following Hhex deletion. Normalized expression of HoxA and associated genes is shown for wild-type LSK cells and compared with MLL-ENL LICs and cell lines with the indicated Hhex genotype. (B) GSEA showing the association between genes up-regulated following Hhex deletion in LICs and MLL-ENL-induced cell lines. Genes significantly up-regulated in Hhex-deleted LICs were compared in MLL-ENL-induced control (HET) and Hhex-deleted (knockout) cell lines. (Left) In the enrichment plot, skewing to the right indicates that most genes are also up-regulated in Hhex-deleted cell lines. (Right) The heat map shows the top 30 genes most up-regulated in Hhex-deleted cell lines. Cdkn2a and Cdkn2b are indicated by arrowheads. (C) Induction of Cdkn2a expression upon Hhex deletion. Tracks show RNA sequencing coverage of the indicated cell types and Hhex genotypes at the Cdkn2a locus. Units are reads per million mapped reads (RPM). (D,E) Kinetics of p16INK4a and p19ARF expression and cell cycle arrest in Hhex HET and knockout MLL-ENL cell lines following deletion of Hhex. Cells were harvested at the times indicated following tamoxifen (10−9 M) treatment and either lysed in RIPA buffer and processed for immunoblotting with antibodies specific for p16INK4a, p19ARF, and HSP70 (D) or fixed, permeabilized, and stained with DAPI and Ki67-PE-Cy7 for cell cycle analysis by flow cytometry (E). (F) Retroviral transduction of Hhex-null AML cells with Hhex-F suppresses induction of p16INK4a and p19ARF. Hhex HET, Hhex-deleted (knockout), and Hhex-rescued (KO+Hhex-F) cell lines were lysed in RIPA buffer and processed for immunoblotting with antibodies specific for Hhex and as in D.
The above results suggest that Hhex acts independently of HoxA/Meis1 genes to maintain the self-renewal capacity of MLL-ENL-induced leukemias. To test this directly, BM cells from Hhex−/ΔMx mice were transduced with MSCV-HoxA9-Meis1 retroviruses as above and injected into irradiated recipient mice. All control (Hhex+/fl) HoxA9-Meis1 recipient mice succumbed to leukemia within 9 wk; however, leukemia development in Hhex−/ΔMx HoxA9-Meis1 recipients was significantly delayed (Supplemental Fig. S5A). PCR analysis of the BM of Hhex−/ΔMx HoxA9-Meis1 leukemic mice revealed selection of undeleted Hhexfl leukemia cells (Supplemental Fig. S5B). Thus, Hhex is also required for HoxA9/Meis1-driven AML, indicating that it acts independently of HoxA/Meis1 to maintain self-renewal of AML stem cells.
A comparison of differentially expressed genes between Hhex-deleted LICs and cell lines showed that genes up-regulated in Hhex-deleted LICs were generally also up-regulated following Hhex deletion in cell lines (Fig. 5B). In contrast, genes down-regulated in Hhex-deleted LICs and cell lines showed only weak correlation (data not shown). Of the genes up-regulated after Hhex deletion in both LICs and Hhex−/fl cell lines, we noted the cell cycle inhibitors Cdkn2a and Cdkn2b (Fig. 5B,C). As these genes encode potent cell cycle inhibitors, we hypothesized that they may be responsible for the growth arrest following Hhex withdrawal. Furthermore, analysis of LSK cells from Hhex−/ΔMx mice revealed that the levels of Cdkn2a and Cdkn2b expression induced in these cells were extremely small when compared with LICs and cell lines, which may explain why loss of Hhex causes specific loss of LICs while sparing normal HSCs (Fig. 5C).
We next performed immunoblotting experiments to test whether loss of Hhex results in induction of the Cdkn2a-encoded tumor suppressor proteins p16INK4a and p19ARF. This showed abundant expression of both p16INK4a and p19ARF in Hhex−/ΔERT2 MLL-ENL cell lines following tamoxifen treatment (Fig. 5D) that were induced with kinetics similar to the loss of cycling cells (S and G2/M phases) in Hhex-deleted cell lines (Fig. 5E). To determine whether the growth inhibitory effects of Hhex deletion in AML cell lines could be reversed by Hhex re-expression, we transduced pools of Hhex-null AML cells (4 d after tamoxifen treatment) with retroviruses bearing Hhex with a C-terminal Flag epitope tag (termed Hhex-F). Overexpression of Hhex-F in Hhex-null MLL-ENL cells was associated with significant toxicity (Supplemental Fig. S6A). However, stable Hhex-F-rescued cell lines expressing physiological levels of Hhex were selected over time and used in subsequent assays. Hhex-F transduced cells displayed myeloid-specific marker expression profiles similar to Hhex-nondeleted AML cells (Supplemental Fig. S6B) and did not show the up-regulation of p16INK4a and p19ARF observed in Hhex-null AML lines (Fig. 5F). Together, these data suggest that loss of Hhex may restrict growth of AML cells by causing the induction of tumor suppressor pathways.
Deletion of p16INK4a and p19ARF tumor suppressors restores AML growth in the absence of Hhex
To assess the role of tumor suppressor pathways in the growth arrest seen upon Hhex deletion in AML cells, we cloned lentiviral CRISPR vectors (pLentiCRISPR) (Shalem et al. 2014) with guide sequences against Cdkn2a, Cdkn2b, and p53 (Supplemental Table S5). As Cdkn2a encodes both p16INK4a and p19ARF tumor suppressors, we designed guide sequences to target each alternative transcript-coding region independently through their unique first exons as well as together via their shared second exon. A guide sequence against firefly luciferase served as a control. These constructs were used to stably transduce CreERT2;Hhex−/fl cell lines and Hhexfl deleted by tamoxifen treatment. Immunoblotting analysis of Hhex-deleted cells showed a complete absence of p16INK4a and p19ARF upon CRISPR–Cas9-mediated deletion (Fig. 6A). We next tested whether loss of these proteins was sufficient to restore growth of Hhex-deleted MLL-ENL cell lines. CRISPR–Cas9-mediated targeting of p15INK4b, encoded by Cdkn2b, did not rescue the growth of leukemia cells after Hhexfl deletion, with growth arrest occuring after 2 wk (Fig. 6B). While deletion of p16INK4a caused a partial rescue of cell numbers following Hhex deletion, these cells still showed eventual growth arrest (Fig. 6B). In contrast, deletion of p19ARF or combined deletion of both p16INK4a and p19ARF led to an almost complete rescue of cell growth upon Hhex deletion (Fig. 6B). As p19ARF acts in part via the p53 pathway, we also designed guide sequences to target p53. This showed a partial rescue of growth in Hhex-deleted cells, which was less than that seen upon p19ARF deletion, suggesting that p19ARF functions largely independently of p53 in inhibiting AML growth. Furthermore, and consistent with the importance of Hhex-dependent suppression of p16INK4a and p19ARF for the maintenance of AML growth, Hhex−/ΔERT2 cells lacking p16INK4a and p19ARF continued to proliferate and maintained normal levels of myeloid-specific markers (Supplemental Fig. S7A) and blast cell morphology (Supplemental Fig. S7B). Hence, growth arrest of AML cell lines following Hhex deletion is due to induction of tumor suppressors encoded by Cdkn2a.
Figure 6.
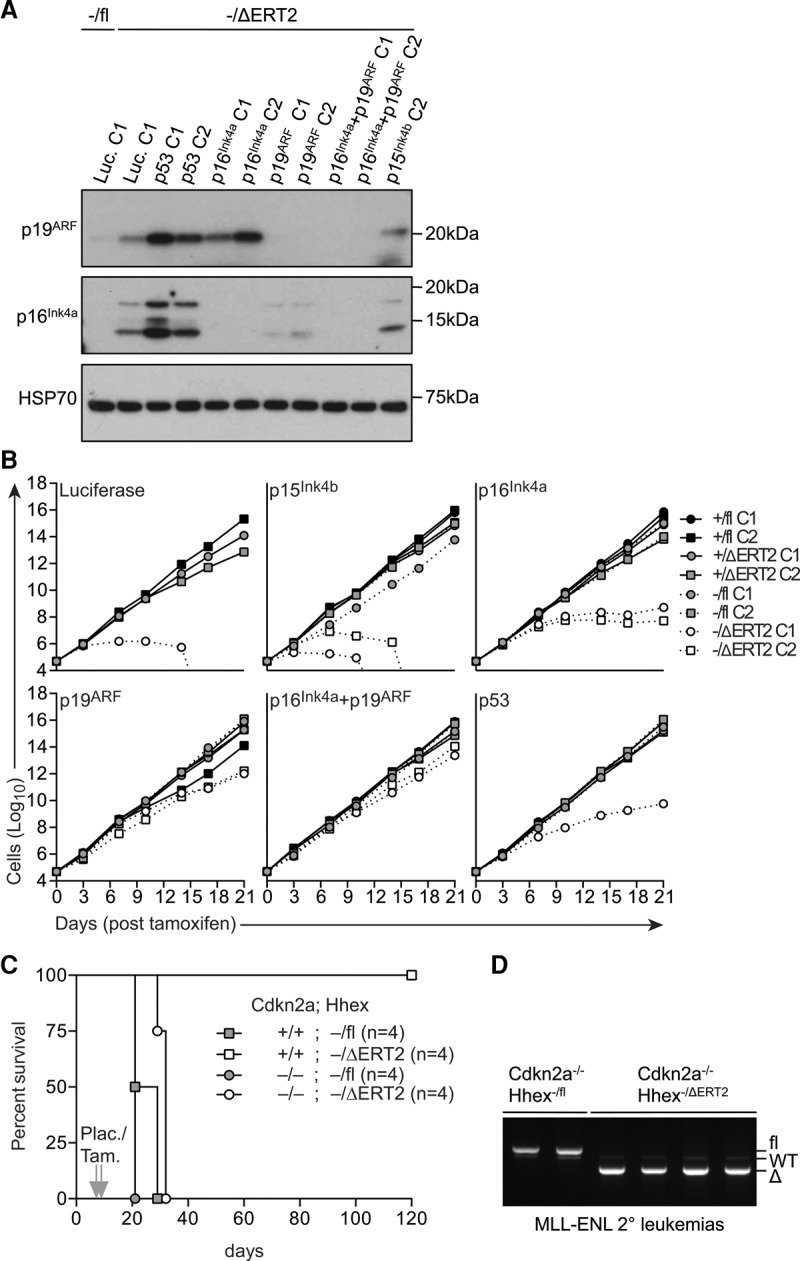
Loss of p16Ink4a and p19ARF rescues leukaemogenesis in the absence of Hhex. (A) Immunoblot of p16Ink4a and p19ARF expression in CreERT2;Hhex−/fl CRISPR/Cas9 cell lines after 7 d in the presence or absence of tamoxifen. (B) Growth of CreERT2;Hhex+/fl and CreERT2;Hhex−/fl CRISPR/Cas9 cell lines in the presence or absence of tamoxifen. In A and B, C1 and C2 refer to individual cell lines generated with unique CRISPR guide sequences for each target gene. (C) Kaplan-Meier survival curve of recipient mice injected with purified LICs (5000 CD45.2+, Mac1+, GFP+, Kit+ cells) of the indicated genotypes and administered with either tamoxifen (Tam.) or placebo (Plac.). P < 0.01 between Cdkn2a+/+;Hhex−/ΔERT2 and Cdkn2a−/−;Hhex−/ΔERT2 recipient mice using a log rank (Mantel-Cox) test. (D) Genomic PCR analysis of BM from leukemic mice in C.
To assess the role of Hhex-dependent suppression of Cdkn2a in the attenuation of MLL-ENL-induced AML in vivo, we generated CreERT2;Hhex−/fl mice on the Cdkn2a knockout background. BM from these mice was used to generate MLL-ENL-induced myeloid leukemias, as above. Next, LICs from leukemic mice were injected into secondary recipients, and Hhexfl was deleted by tamoxifen administration 7 d later. Strikingly, MLL-ENL-induced leukemia developed from Hhex−/ΔERT2 LICs lacking Cdkn2a, in contrast to Cdkn2a wild-type cells that never developed leukemia up to 4 mo after transplant (Fig. 6C). Furthermore, we saw no selection for undeleted Hhexfl in Hhex-deleted, Cdkn2a-null leukemias (Fig. 6D). Thus, loss of Cdkn2a allows maintenance of AML in the absence of Hhex, indicating that the primary effect of Hhex in AML is to repress Cdkn2a.
Hhex represses p16INK4a and p19ARF expression via regulation of PRC2 function
Previous studies have indicated an important role for homeobox transcription factors, including Hoxa9 and Hlx1, in the recruitment of PRC2 to the Cdkn2a/b locus to facilitate transcriptional repression and the maintenenace of self-renewal potential (Martin et al. 2013; Collins et al. 2014). We therefore tested whether Hhex mediates repression of this locus in AML cells via PRC2-mediated epigenetic silencing using antibodies directed against the PRC2-encoded repressive mark H3K27me3 as well as the activating mark H3K4me3 for chromatin immunoprecipitation (ChIP) sequencing. H3K27me3 marks were abundant in Hhex−/fl MLL-ENL cell lines both within the gene body of Cdkn2a (Fig. 7A; Supplemental Fig. S8A) and Cdkn2b (Fig. 7A) and at an enhancer ∼50 kb upstream that has been previously shown to be important for HoxA9-mediated repression of Cdkn2a (Fig. 7A; Collins et al. 2014). Conversely, the H3K27me3 mark was greatly reduced in Hhex-null MLL-ENL cells (Fig. 7A; Supplemental Fig. S8A), including the upstream enhancer region, with re-expression of Hhex-F rescuing the deposition of this mark (Fig. 7A; Supplemental Fig. S8A).
Figure 7.

Hhex is required for the repressive function of PRC2 at the Cdkn2a/b locus. (A) ChIP sequencing analysis showing enrichment of H3K27me3 (red) and H3K4me3 (green) across a broad region of chromosome 4 encompassing Cdkn2a and Cdkn2b in the indicated cell lines. Units are reads per million mapped reads (RPM). (B) The mean difference plot of H3K27me3 (gene body) in MLL-ENL-induced control (−/fl) and Hhex-deleted (−/ΔERT2) cell lines demonstrates that Hhex regulates a discrete set of genes. Blue dots represent genes with significantly decreased H3K27me3, and red dots represent genes with significantly increased H3K27me3 following Hhex deletion. Dotted lines indicate genes that are located at the same loci. (C) Correlation analysis of differentially methylated genes identified in B that also have significant changes (greater than two log fold change) in mRNA expression level between MLL-ENL-induced control (−/fl) and Hhex-deleted (−/ΔERT2) cell lines identified by RNA sequencing. P = 0.0002. (D) Mean difference plot of H3K27me3 (gene body) in Hhex-deleted (−/ΔERT2) cell lines and Hhex-F-rescued −/ΔERT2 cell lines. Differentially methylated genes identified in B are highlighted with green dots. (E) Immunoblotting of whole-cell lysates from control (−/fl) and Hhex-deleted (−/ΔERT2) and Hhex-F-rescued (−/ΔERT2 +Hhex-F) cell lines. (F) Coimmunoprecipitation of Hhex-F and PRC2 component suz12 in −/ΔERT2 + Hhex-F cell lines. (in.) Input (1/50th immunoprecipitation); (IP) M2-Flag immunoprecipitate. (G) Model of Hhex-dependent regulation of Cdkn2a by recruitment of PRC2.
To determine whether Hhex binds to the Cdkn2a locus, we performed ChIP sequencing on Hhex-F transduced Hhex−/ΔERT2 cells (Supplemental Fig. S8A) using an antibody against the Flag epitope. This revealed a peak overlapping a bivalently marked region encompassing Exon1α (Supplemental Fig. S8B) that has been previously shown to be subject to antagonistic regulation by Polycomb and SWI/SNF complexes to modulate the expression of p16INK4a (Wilson et al. 2010). Subsequent ChIP-PCR experiments confirmed that this region is bivalently marked, with loss of Hhex causing a decrease in repressive marks and a reciprocal increase in active marks (Supplemental Fig. S8C), and that Hhex binds directly (Supplemental Fig. S8D).
To establish whether Hhex regulates PRC2-repressed target genes globally, we performed GSEA of our RNA sequencing data from MLL-ENL cell lines using gene sets from two separate studies that have described critical PRC2 targets in MLL-AF9-induced leukemia (Supplemental Table S6; Shi et al. 2013; Xu et al. 2015). We found that, in both cases, loss of Hhex caused significant reactivation of PRC2 targets, including five genes (Cdkn2b, Zmat3, Igf1, Ryk, and Serpine2) that are in common between the two gene sets (Supplemental Fig. S9). Thus, Hhex regulates a subset of PRC2 target genes in addition to Cdkn2a.
Next, to determine whether Hhex affects the PRC2-encoded epigenetic mark H3K27me3 globally, we analyzed this mark in Hhex-null cell lines. Remarkably, we found that loss of Hhex caused a significant change in the deposition of H3K27me3 at only nine separate gene-coding loci containing 13 genes (Fig. 7B). Two genes demonstrated significantly increased H3K27me3 deposition, and 11 genes, including Cdkn2b, showed significantly decreased H3K27me3 deposition (Fig. 7B). All of these genes showed a strong inverse correlation between H3K27me3 marking and RNA expression, confirming them as bona fide Hhex-dependent PRC2 target genes (Fig. 7C). Re-expression of Hhex (Hhex-F) restored the H3K27me3 status of all 13 genes but also resulted in significant changes in the H3K27me3 status of previously unregulated genes, suggesting that Hhex can cause aberrant epigenetic regulation when ectopically expressed (Fig. 7D). However, loss of Hhex did not alter the overall level of H3K27me3, as assessed by Western blotting (Fig. 7E), indicating that Hhex is not required for PRC2 function generally but regulates its function at specific loci that are required for leukemic growth.
To determine whether Hhex interacts with the PRC2 complex, we performed coimmunoprecipitation assays using Flag-tagged Hhex (Hhex-F). This showed that Hhex coprecipitates with the PRC2 core component Suz12 (Fig. 7F). Furthermore, using Suz12 ChIP quantitative PCR analysis, we observed Hhex-dependent enrichment of Suz12 at the Hhex-binding site identified above (Supplemental Fig. S8E). However, the observation that only a small proportion of Suz12 was bound to Hhex may explain why loss of Hhex does not affect PRC2 function globally. Together, these data suggest a model in which Hhex mediates the recruitment of PRC2 to key loci, including Cdkn2a to facilitate epigenetic repression and enable continued cycling of LSCs in AML (Fig. 7G).
Discussion
As Hhex is a critical regulator of HSC development during embryogenesis that is overexpressed in human AML, we assessed the requirement for this factor in MLL-ENL-induced myeloid leukemia using a conditional knockout model. We found that Hhex is required to initiate MLL-ENL-driven AML and that deletion of Hhex in established leukemia invariably leads to selection for nondeleted clones, indicating that Hhex is essential for development and sustained growth of leukemia. Transcriptome analysis showed that MLL-ENL-induced HoxA overexpression was not affected by Hhex deletion, indicating that Hhex is required independently of the MLL-induced “Hox code” to maintain leukemogenesis. Accordingly, leukemogenesis by collaborating HoxA9 and Meis1 also required Hhex. Together, these results indicate that Hhex acts independently of HoxA/Meis1 genes, which are established drivers of MLL fusion leukemia in this setting (Krivtsov et al. 2006).
By transcriptome analysis, we found that loss of Hhex causes up-regulation of cell cycle inhibitors encoded by Cdkn2a and Cdkn2b in MLL-ENL transformed LICs and cell lines. Remarkably, loss of Cdkn2a, which encodes both p16INK4a and p19ARF, restored the growth of MLL-ENL-induced AML both in vitro and in vivo, with individual targeting indicating that p19ARF is the major mediator of growth arrest following Hhex deletion. As p53 gene disruption only partially rescued growth of AML cells when Hhex was deleted, our data also suggest that the cell cycle inhibitory action of p19ARF is largely independent of its canonical ability to inhibit MDM2 and stabilize p53. Indeed, several p53-independent p19ARF functions have been reported, including sequestration of Myc and E2f1 and attenuation of ribosomal RNA transcription and processing (Sherr 2006; Lessard et al. 2010).
In HSCs and AML stem cells, suppression of Cdkn2a and Cdkn2b is facilitated by Polycomb group (PcG) proteins contained within PRC1 and PRC2 (Lessard and Sauvageau 2003; Hidalgo et al. 2012; Tanaka et al. 2012). PRC2-dependent silencing of Cdkn2a is critical for maintenance of self-renewal capacity, as loss of the core PRC2 component Eed or both of the PRC2 enzymatic components Ezh1 and Ezh2 leads to loss of AML stem cells (Neff et al. 2012; Tanaka et al. 2012; Shi et al. 2013). Furthermore, chemical inhibition of PRC2 function inhibits growth of leukemia driven by MLL fusion proteins (Kim et al. 2013; Xu et al. 2015). However as, PRC2 is also required for normal HSC function, the therapeutic potential of targeting this complex is presently unclear (Xie et al. 2014).
Homeobox transcription factors, including the Hhex-related Hlx1 protein and HOXA9, have recently been implicated in the recruitment of PRCs to the CDKN2A locus in human fibroblast cell lines, which prevents p16INK4a-dependent cell cycle arrest (Martin et al. 2013). Our findings identify Hhex as a crucial homeobox protein that similarly enables PRC2 function at the Cdkn2a locus to maintain the self-renewal capacity of MLL-ENL-induced AML. Hhex is also required for maintaining the H3K27me3 mark across an enhancer region ∼50 kb upstream of Cdkn2b, which was shown to be essential for HoxA9/C/EBPα-dependent suppression of Cdkn2a and Cdkn2b in HoxA9;Meis1-driven AML (Collins et al. 2014). We found that the magnitude of induction of Cdkn2a in AML cell lines and LICs following Hhex loss greatly exceeds that in normal LSK cells. This implies that Hhex-independent mechanisms maintain epigenetic silencing of Cdkn2a in normal HSCs but not LICs, potentially explaining the specific requirement for Hhex in AML stem cells.
In AML patient blasts, Cdkn2a/b is often repressed via H3K27me3 in combination with Cdkn2b promoter methylation (Paul et al. 2010), and low expression of the Cdkn2a-encoded tumor suppressors p16INK4a and p14ARF correlates with poor outcome (Muller-Tidow et al. 2004; de Jonge et al. 2009; Paul et al. 2009). However, as Cdkn2a/b are rarely mutated in AML, targeting epigenetically silenced Cdkn2a is an attractive therapeutic option (LaPak and Burd 2014). This study highlights inhibition of Hhex as a potential strategy to relieve PRC2-mediated epigenetic suppression of Cdkn2a and specifically inhibit self-renewal of AML stem cells.
Material and methods
Mice
All mice used were on a C57BL/6 background. The Hhexfl (Hunter et al. 2007), Hhex− (Bogue et al. 2003), Mx-Cre (Kuhn et al. 1995), ROSA26-CreERT2 (Seibler et al. 2003), and Ink4a/Arf−/− (Serrano et al. 1996) mouse strains have been described. Four-week-old to 6-wk-old CD45.1+ C57BL/6 mice (Ly5.1; Walter and Eliza Hall Institute) were used as recipients in chimeric transplant experiments. To induce expression of the Mx1-Cre allele, polyinosinic–polycytidylic acid sodium salt [poly(I:C); Sigma] dissolved in saline was administered to mice (12 mg per kilogram of mouse body weight) intraperitoneally at 7 wk of age at least 3 wk prior to their use in experiments. To induce expression of the ROSA26-CreERT2 allele, mice were administered 70 µL of tamoxifen (4.2 mg [Sigma] in vehicle; 10% ethanol, 90% peanut oil [Sigma]) by oral gavage on two consecutive days, whereas placebo-treated mice received 70 µL of vehicle control. All experiments were approved by the Walter and Eliza Hall Institute Animal Ethics Committee.
Flow cytometry
Antibodies used in experiments for lineage marker depletion included rat anti-mouse B220 (RA3-6B2), CD3 (KT3-1-1), CD4 (GK1.5-7), CD8 (53.6.7), CD19 (1D3), Gr-1 (RB6-8C5), Mac-1 (M1/70), and TER119 (Ly76), all from the Walter and Eliza Hall Institute Monoclonal Antibody Facility. The exclusion of hematopoietic lineage cells from murine BM was performed using anti-rat antibodies in combination with sheep anti-rat IgG-coated immunomagnetic beads (Invitrogen). Antibodies used for analysis and sorting by flow cytometry included goat anti-rat IgG-Alexa680 (Invitrogen, A21096) and the following rat anti-mouse biotinylated or flurophore-conjugated antibodies purchased from either eBiosciences, Biolegend, BD Pharmingen, or Invitrogen or produced by the Walter and Eliza Hall Institute Monoclonal Antibody Facility: B220 (RA3-6B2), c-Kit (ACK-4), CD4 (GK1.5-7), CD8α (53.6.7), CD16/32-Biotin (24G2), CD19 (1D3), CD34 (RAM34), CD45.1 (A20.1), CD45.2 (S450-015-2), F4/80 (BM8), Flt-3 (A2F10), Gr-1 (RB6-8C5), IL-7Rα (A7R34), Mac-1 (M1/70), Sca-1 (D7), and TER119 (Ly76). Streptavidin-PE-Cy7 and Streptavidin-PerCP-Cy5.5 were purchased from eBiosciences. An FcγR-blocking step was performed prior to staining using 1 mg/mL rat γ-globulin (Jackson ImmunoResearch). For cell cycle analysis, cells were fixed and permeabilized using Cytofix/Cytoperm (BD Pharmingen) and then stained with 10 µg/mL DAPI and, on occasion, anti-Ki67-PE-Cy7 (BD Biosciences, clone 56). Data were acquired on a LSR Fortessa (BD Pharmingen) or LSR II W (BD Pharmingen) flow cytometer and analyzed using FlowJo software (version 9.4.3, Tree Star). Flow cytometric cell sorting was performed using an Aria (BD Pharmingen) device.
Genomic PCR
The efficiency of inducible Hhex deletion was verified by extraction of genomic DNA from fractionated samples lysed in DirectPCR (Viagen Biotech) containing proteinase K (Sigma) overnight at 55°C and subsequent PCR analysis using the 5′-GGTGGGGAGAGGTATTTCTGA-3′, 5′-AGACGCACCACCATCATTTT-3′, and 5′-GAACTAAATTAAGAGGCTGC-3′ oligonucleotides. The presence of floxed, wild-type, and deleted Hhex alleles was evident by the amplification of the 1429-base-pair (bp), 1229-bp, and 929-bp DNA fragments, respectively.
Retroviral transduction and BM transplantation
Lineage-depleted BM was transduced with ecotropic MSCV retroviruses immobilized on 15 µg/mL retronectin (Takara Biosciences) coated nontissue culture-treated plates and cultured in StemPro-34 medium (Invitrogen) supplemented with StemPro nutrient supplement, 10 ng/mL mouse IL-3 (mIL-3), 10 ng/mL mouse IL-6 (mIL-6), 50 ng/mL mouse SCF (mSCF), and 50 ng/mL mouse Flt3 ligand (mFlt3). All cytokines used were produced in-house (Walter and Eliza Hall Institute) with the exception of mIL-3 (Peprotech). Two days after transduction, cells of the equivalent of one-twelfth of the original volume of lineage-depleted BM were injected into lethally irradiated (9.5 Gy) Ly5.1 recipient mice via the tail vein. For leukemia transplantation experiments, BM was harvested from leukemic mice, and LICs (CD45.2+, Mac1+, GFP+, Kit+) were sorted by flow cytometry and injected into sublethally irradiated (6.5 Gy) Ly5.1 recipient mice via the tail vein.
Quantitative PCR
MLL-ENL and MIG control transduced BM cells were harvested 2 d after transduction, washed, and resuspended in RLT buffer (Qiagen). RNA was purified using the RNeasy minikit (Qiagen) with on-column DNase digestion using the RNase-free DNase set (Qiagen) and then reverse-transcribed using the Transcriptor first strand cDNA synthesis kit (Roche) using 60 μM random hexamer primers. PCR was performed using the SYBR Green PCR master mix (Applied Biosystems) using the following primer pairs for Hhex (5′-CCTCTGCACAAAAGGAAAGG-3′ and 5′-ATTTAGCTCGGCGATTCTGA-3′) and 18S (5′-GTAACCCGTTGAACCCCATT-3′ and 5′-CCATCCAATCGGTAGTAGCG-3′) at 200 μM. Amplification was performed in a LightCycler 480 real-time PCR system (Roche) using standard curves obtained from FDC-P1 cell line cDNA as a reference, and expression values were normalized to 18S.
Cell culture
MLL-ENL Hhex−/fl and MLL-ENL Hhex+/fl leukemia cells were harvested from leukemic recipient mice and cultured in growth medium (IMDM supplemented with 10% FCS with 10 ng/mL mIL-3). Recombination mediated by CreERT2 was induced by adding 10−9 M 4-hydroxy-tamoxifen to the growth medium. After 7 d, cells were harvested for flow cytometric analysis or centrifuged onto a microscope slide and stained with May-Grünwald-Giemsa stain. For viable cell counts, cells were mixed with Accucheck beads (Invitrogen), stained with 1 μg/mL propidium iodide (Sigma), and enumerated by flow cytometry.
HHEX expression in human AML
Expression data and clinical annotations from human AML samples were obtained from Verhaak et al. (2009) and downloaded from Gene Expression Omnibus (GEO; GSE6891). Classification of these samples into prognostic groups following the recommendation of the ELN was obtained from Li et al. (2013). The raw expression files were preprocessed, including background subtractions, quantile normalization, and log2 transformation using Partek Genomics suite (version 6.6). HHEX expression levels were compared between patients of different cytogenetics and with different risk profiles (ELN) using the Mann-Whitney U-test in Graphpad Prism (version 6.05). Patients with high/low HHEX expression levels were identified using an unsupervised k-means clustering approach in Matlab (version R2014b), and their overall survival was compared and visualized using Kaplan-Meier statistics (log rank test) in Graphpad Prism (version 6.05).
CRISPR–CAS9 gene disruption
The first 250 coding nucleotides of mouse p15Ink4b, p16Ink4a, p19ARF, and p53 and firefly luciferase were used for optimized CRISPR design (http://www.crispr.mit.edu) of guide sequences (Supplemental Table S5). Where possible, guide sequences that target the first exon of each gene (shared exon 2 of Cdkn2a was targeted for disruption of both p16Ink4a and p19ARF) were selected with limited probability of off target effects (quality score >50). To generate the BsmBI-adapted ends and the 5′ PAM (NGG) site, the sequence CACCG was added to the 5′ end of the forward oligonucleotide, the sequence ACCC was added to the 5′ end, and a single C was added to the 3′ end of the reverse oligonucleotide. Forward and reverse oligonucleotide pairs for each target were annealed and ligated with BsmBI-restricted lentiCRISPR (Shalem et al. 2014) and confirmed by Sanger sequencing. Ecotropic lentiviruses bearing CRISPR guides were produced in 293T cells and used to transduce MLL-ENL Hhex−/fl and MLL-ENL Hhex+/fl cell lines. Virally transduced cells were selected for by the addition of 1 µg/mL puromycin (Sigma).
Western blotting
Protein samples were prepared from washed cell pellets, lysed in RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS in 10 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM PMSF, 2 mM sodium vanadate, 50 mM sodium fluoride, protease inhibitor cocktail [Roche Applied Bioscience]), and diluted in reducing Laemmli buffer or lysed directly in 4× Laemmli buffer. Proteins were resolved on precast 4%–20% bis-acrylamide gels (Bio-Rad), transferred to PVDF, and immunoblotted with rabbit anti-p16 (M-15; Santa Cruz Biotechnology), rat anti-p19 (p19ARF exon 2; Rockland), rabbit anti-H3K27me3 (Millipore, 07-449), rabbit anti-H3 (Millipore, 05-928), rabbit anti-Hhex, and mouse anti-HSP70 (N6; Walter and Eliza Hall Institute).
ChIP
H3K27me3, H3K4me3, and M2-Flag ChIP was performed using a protocol based on Nelson et al. (2006) with some modifications. Briefly, cells were fixed in 1.42% paraformaldehyde and lysed in immunoprecipitation buffer (1% Triton X-100, 0.5% NP40 in 50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 5 mM EDTA, protease inhibitor cocktail), and the chromatin pellet was harvested by centrifugation. DNA was fragmented in a Covaris Sonicator for 30 min at 4°C, and then protein–DNA complexes were immunoprecipitated from clarified chromatin fractions using 2 µg of H3K27me3 (Millipore, 07-449) or 2 µg of H3K4me3 (Millipore, 07-473) antibodies and 30 µL of protein A-sepharose. For Hhex-Flag ChIP, 30 µL of anti-Flag (M2)-agarose beads (Sigma) was added directly to clarified chromatin fractions. Following reversal of cross-links and RNase I and proteinase K treatments, extracted DNA was purified using the QIAquick purification kit (Qiagen).
RNA sequencing
For RNA sequencing, see the Supplemental Material.
ChIP sequencing
For ChIP sequencing, see the Supplemental Material.
Coimmunoprecipitation
Whole-cell lysates were prepared by resuspending cell pellets in hypotonic buffer (20 mM Tris-HCl at pH 7.5, 10 mM KCl, 3 mM MgCl2, 1 mM PMSF, protease inhibitor cocktail) followed by 20 strokes of a dounce homogenizer. Supernatants were harvested by centrifugation, and nuclear extracts were collected in nuclear extraction buffer (20 mM Tris-HCl at pH 7.5, 300 mM NaCl, 0.5% NP40). Next, 30 µL of anti-Flag (M2)-agarose beads was incubated with pooled fractions for 2 h at 4°C, and beads were washed extensively and resuspended in 2× reducing Laemmli loading buffer. Input (In.) and immunoprecipitation samples were subjected to SDS-PAGE and immunoblotting with the indicated antibodies.
Supplementary Material
Acknowledgments
We thank Ian Majewski for reagents and helpful comments on the manuscript; Jason Corbin and Jasmin McManus for blood analysis; Waruni Abeysekera for aligning ChIP sequencing reads; Sandra Mifsud, Ladina DiRago, Chayanica Nasa, William Stanley, and Stephen Wilcox for technical assistance; Walter and Eliza Hall Institute Bioservices for mouse husbandry; and the Walter and Eliza Hall Institute Flow Cytometry Facility. We also thank Tobias Herold (Department of Internal Medicine III, University Hospital Grosshadern, Ludwig Maximilians-Universität, Munich, Germany) and Peter J.M. Valk (Department of Hematology and Clinical Genetics, Erasmus University Medical Center, Rotterdam, The Netherlands) for providing clinical data. This work was supported by a Program grant (101664 to W.S.A.), Project grants (628386 and 1003391 to M.P.M.), a fellowship (1058344 to W.S.A.), the Independent Research Institutes Infrastructure Support (IRIIS) Scheme from the Australian Government's National Health and Medical Research Council (NHMRC), grants-in-aid from the Cancer Council of Victoria and the Leukemia Foundation of Australia (M.P.M. and S.P.G.), a Future Fellowship from the Australian Research Council (M.P.M.), the Australia Cancer Research Fund (W.S.A.), and a Victorian State Government Operational Infrastructure Support (OIS) Grant.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.268425.115.
References
- Alharbi RA, Pettengell R, Pandha HS, Morgan R. 2013. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 27: 1000–1008. [DOI] [PubMed] [Google Scholar]
- Argiropoulos B, Humphries RK. 2007. Hox genes in hematopoiesis and leukemogenesis. Oncogene 26: 6766–6776. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. 2003. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17: 2298–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagger FO, Rapin N, Theilgaard-Monch K, Kaczkowski B, Thoren LA, Jendholm J, Winther O, Porse BT. 2013. HemaExplorer: a database of mRNA expression profiles in normal and malignant haematopoiesis. Nucleic Acids Res 41: D1034–D1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal D, Scholl C, Frohling S, McDowell E, Lee BH, Dohner K, Ernst P, Davidson AJ, Daley GQ, Zon LI, et al. 2006. Cdx4 dysregulates Hox gene expression and generates acute myeloid leukemia alone and in cooperation with Meis1a in a murine model. Proc Natl Acad Sci 103: 16924–16929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford FK, Ashworth A, Enver T, Wiedemann LM. 1993. HEX: a novel homeobox gene expressed during haematopoiesis and conserved between mouse and human. Nucleic Acids Res 21: 1245–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogue CW, Zhang PX, McGrath J, Jacobs HC, Fuleihan RL. 2003. Impaired B cell development and function in mice with a targeted disruption of the homeobox gene Hex. Proc Natl Acad Sci 100: 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T, Figueroa M, Muntean AG, Hess JL. 2014. C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci 111: 9899–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. 2003. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 17: 3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton MR, Bartlett TJ, MacGregor AD, Manfioletti G, Buratti E, Giancotti V, Goodwin GH. 1992. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res 20: 5661–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge HJ, de Bont ES, Valk PJ, Schuringa JJ, Kies M, Woolthuis CM, Delwel R, Veeger NJ, Vellenga E, Lowenberg B, et al. 2009. AML at older age: age-related gene expression profiles reveal a paradoxical down-regulation of p16INK4A mRNA with prognostic significance. Blood 114: 2869–2877. [DOI] [PubMed] [Google Scholar]
- Diffner E, Beck D, Gudgin E, Thoms JA, Knezevic K, Pridans C, Foster S, Goode D, Lim WK, Boelen L, et al. 2013. Activity of a heptad of transcription factors is associated with stem cell programs and clinical outcome in acute myeloid leukemia. Blood 121: 2289–2300. [DOI] [PubMed] [Google Scholar]
- George A, Morse HC III, Justice MJ. 2003. The homeobox gene Hex induces T-cell-derived lymphomas when overexpressed in hematopoietic precursor cells. Oncogene 22: 6764–6773. [DOI] [PubMed] [Google Scholar]
- Guiral M, Bess K, Goodwin G, Jayaraman PS. 2001. PRH represses transcription in hematopoietic cells by at least two independent mechanisms. J Biol Chem 276: 2961–2970. [DOI] [PubMed] [Google Scholar]
- Guo Y, Chan R, Ramsey H, Li W, Xie X, Shelley WC, Martinez-Barbera JP, Bort B, Zaret K, Yoder M, et al. 2003. The homeoprotein Hex is required for hemangioblast differentiation. Blood 102: 2428–2435. [DOI] [PubMed] [Google Scholar]
- He S, Nakada D, Morrison SJ. 2009. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol 25: 377–406. [DOI] [PubMed] [Google Scholar]
- Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nunez J, Martinez F, Dominguez O, Torres M, Gonzalez S. 2012. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 11: 649–662. [DOI] [PubMed] [Google Scholar]
- Horton SJ, Grier DG, McGonigle GJ, Thompson A, Morrow M, De Silva I, Moulding DA, Kioussis D, Lappin TR, Brady HJ, et al. 2005. Continuous MLL-ENL expression is necessary to establish a ‘Hox Code’ and maintain immortalization of hematopoietic progenitor cells. Cancer Res 65: 9245–9252. [DOI] [PubMed] [Google Scholar]
- Hunter MP, Wilson CM, Jiang X, Cong R, Vasavada H, Kaestner KH, Bogue CW. 2007. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Dev Biol 308: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JT, Nasa C, Shi W, Huntington ND, Bogue CW, Alexander WS, McCormack MP. 2015. A crucial role for the homeodomain transcription factor Hhex in lymphopoiesis. Blood 125: 803–814. [DOI] [PubMed] [Google Scholar]
- Jankovic D, Gorello P, Liu T, Ehret S, La Starza R, Desjobert C, Baty F, Brutsche M, Jayaraman PS, Santoro A, et al. 2008. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood 111: 5672–5682. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Pandolfi A, Bartholdy B, Barreyro L, Will B, Roth M, Okoye-Okafor UC, Todorova TI, Figueroa ME, Melnick A, et al. 2012. H2.0-like homeobox regulates early hematopoiesis and promotes acute myeloid leukemia. Cancer Cell 22: 194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng VW, Yagi H, Ikawa M, Nagano T, Myint Z, Yamada K, Tanaka T, Sato A, Muramatsu I, Okabe M, et al. 2000. Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem Biophys Res Commun 276: 1155–1161. [DOI] [PubMed] [Google Scholar]
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. 2013. Targeted disruption of the EZH2–EED complex inhibits EZH2-dependent cancer. Nat Chem Biol 9: 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. 2007. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7: 823–833. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. 2006. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442: 818–822. [DOI] [PubMed] [Google Scholar]
- Kubo A, Chen V, Kennedy M, Zahradka E, Daley GQ, Keller G. 2005. The homeobox gene HEX regulates proliferation and differentiation of hemangioblasts and endothelial cells during ES cell differentiation. Blood 105: 4590–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. 1995. Inducible gene targeting in mice. Science 269: 1427–1429. [DOI] [PubMed] [Google Scholar]
- Kumar AR, Hudson WA, Chen W, Nishiuchi R, Yao Q, Kersey JH. 2004. Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood 103: 1823–1828. [DOI] [PubMed] [Google Scholar]
- LaPak KM, Burd CE. 2014. The molecular balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res 12: 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. 2003. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255–260. [DOI] [PubMed] [Google Scholar]
- Lessard F, Morin F, Ivanchuk S, Langlois F, Stefanovsky V, Rutka J, Moss T. 2010. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol Cell 38: 539–550. [DOI] [PubMed] [Google Scholar]
- Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, Mansmann U, Radmacher MD, Maharry KS, Sun M, et al. 2013. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol 31: 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N, Popov N, Aguilo F, O'Loghlen A, Raguz S, Snijders AP, Dharmalingam G, Li S, Thymiakou E, Carroll T, et al. 2013. Interplay between Homeobox proteins and Polycomb repressive complexes in p16INK(4)a regulation. EMBO J 32: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Barbera JP, Clements M, Thomas P, Rodriguez T, Meloy D, Kioussis D, Beddington RS. 2000. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127: 2433–2445. [DOI] [PubMed] [Google Scholar]
- McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, Jane SM, Curtis DJ. 2010. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 327: 879–883. [DOI] [PubMed] [Google Scholar]
- McCormack MP, Shields BJ, Jackson JT, Nasa C, Shi W, Slater NJ, Tremblay CS, Rabbitts TH, Curtis DJ. 2013. Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood 122: 2093–2103. [DOI] [PubMed] [Google Scholar]
- Muller-Tidow C, Metzelder SK, Buerger H, Packeisen J, Ganser A, Heil G, Kugler K, Adiguzel G, Schwable J, Steffen B, et al. 2004. Expression of the p14ARF tumor suppressor predicts survival in acute myeloid leukemia. Leukemia 18: 720–726. [DOI] [PubMed] [Google Scholar]
- Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, Xie H, Orkin SH, Armstrong SA. 2012. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci 109: 5028–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185. [DOI] [PubMed] [Google Scholar]
- Paul E, Paul E, Uggla B, Deneberg S, Bengtzen S, Hermansson M, Dahlman I, Rosenquist R, Wiman KG, Nahi H. 2009. Low p14ARF expression in de novo acute myeloid leukemia with normal karyotype is associated with poor survival. Leuk Lymphoma 50: 1512–1518. [DOI] [PubMed] [Google Scholar]
- Paul TA, Bies J, Small D, Wolff L. 2010. Signatures of polycomb repression and reduced H3K4 trimethylation are associated with p15INK4b DNA methylation in AML. Blood 115: 3098–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz H, Lynch MR, Bogue CW, Gasson JC. 2010. The homeobox gene Hhex regulates the earliest stages of definitive hematopoiesis. Blood 116: 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat VP, Humphries RK, Buske C. 2012. Beyond Hox: the role of ParaHox genes in normal and malignant hematopoiesis. Blood 120: 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl C, Bansal D, Dohner K, Eiwen K, Huntly BJ, Lee BH, Rucker FG, Schlenk RF, Bullinger L, Dohner H, et al. 2007. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J Clin Invest 117: 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibler J, Zevnik B, Kuter-Luks B, Andreas S, Kern H, Hennek T, Rode A, Heimann C, Faust N, Kauselmann G, et al. 2003. Rapid generation of inducible mouse mutants. Nucleic Acids Res 31: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. 1996. Role of the INK4a locus in tumor suppression and cell mortality. Cell 85: 27–37. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. 2006. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 6: 663–673. [DOI] [PubMed] [Google Scholar]
- Shi J, Wang E, Zuber J, Rappaport A, Taylor M, Johns C, Lowe SW, Vakoc CR. 2013. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras(G12D) acute myeloid leukemia. Oncogene 32: 930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields BJ, Alserihi R, Nasa C, Bogue C, Alexander WS, McCormack MP. 2015. Hhex regulates Kit to promote radioresistance of self-renewing thymocytes in Lmo2-transgenic mice. Leukemia 29: 927–938. [DOI] [PubMed] [Google Scholar]
- So CW, Karsunky H, Wong P, Weissman IL, Cleary ML. 2004. Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood 103: 3192–3199. [DOI] [PubMed] [Google Scholar]
- Soufi A, Jayaraman PS. 2008. PRH/Hex: an oligomeric transcription factor and multifunctional regulator of cell fate. Biochem J 412: 399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingler TE, Bess KL, Yao J, Stifani S, Jayaraman PS. 2004. The proline-rich homeodomain protein recruits members of the Groucho/Transducin-like enhancer of split protein family to co-repress transcription in hematopoietic cells. J Biol Chem 279: 34938–34947. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, Suzuki Y, Sugano S, Nakaseko C, Yokote K, et al. 2012. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 120: 1107–1117. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Lowenberg B, Delwel R, Valk PJ. 2009. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 94: 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. 2010. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18: 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. 2014. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, et al. 2015. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 125: 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.