

In this study, Luo et al. find that the AFF family protein AFF3 can specifically bind both gametic differentially DNA-methylated regions (gDMRs) and enhancers within imprinted loci in an allele-specific manner. These results provide the mechanistic details of the control of dosage-critical imprinted gene expression through the regulated binding of the transcription elongation factor AFF3 between a DMR and an enhancer.
Keywords: elongation control, enhancers, transcription
Abstract
Genomic imprinting is a critical developmental process characteristic of parent of origin-specific gene expression. It is well accepted that differentially DNA-methylated regions (DMRs) and enhancers are two major classes of cis-elements determining parent of origin-specific gene expression, with each recruiting different sets of transcription factors. Previously, we identified the AF4/FMR2 (AFF) family protein AFF3 within the transcription elongation complex SEC-L3. Here, we report that AFF3 can specifically bind both gametic DMRs (gDMRs) and enhancers within imprinted loci in an allele-specific manner. We identify the molecular regulators involved in the recruitment of AFF3 to gDMRs and provide mechanistic insight into the requirement of AFF3 at an enhancer for the expression of an ∼200-kb polycistronic transcript within the imprinted Dlk1-Dio3 locus. Our data suggest that the heterochromatic environment at the gDMR reinforces silencing of its related enhancer by controlling the binding and activity of AFF3 in an allele-specific manner. In summary, this study provides molecular details about the regulation of dosage-critical imprinted gene expression through the regulated binding of the transcription elongation factor AFF3 between a DMR and an enhancer.
The vast majority of autosomal genes is expressed from both the maternal and paternal alleles in higher mammals, while a subset of genes regulated by genomic imprinting exhibits parental origin-specific monoallelic expression. Imprinted genes play pivotal roles in regulating growth and cell fate determination during development (Bartolomei and Ferguson-Smith 2011; Barlow and Bartolomei 2014). The abnormal dosage of imprinted gene expression is associated with various human congenital disorders—including Prader-Willi, Angelman, and Beckwith-Wiedemann syndromes—as well as with cancers (Peters 2014). Recent studies have also demonstrated that the epigenetic silencing of maternally expressed genes Meg3, Rian, and Mirg and a cluster of microRNAs (miRNAs) located in the Dlk1-Dio3 locus during cellular reprogramming is accompanied by lowered developmental potential of induced pluripotent stem cells (iPSCs) (Stadtfeld et al. 2010, 2012).
Imprinted genes are generally clustered in large chromatin domains defined as imprinted loci. Imprinted loci commonly bear a CpG-rich DNA sequence, which can be targeted by de novo DNA methyltransferases during spermatogenesis and oogenesis to generate gametic differentially DNA-methylated regions (gDMRs). Some gDMRs experimentally shown to control the imprinted expression of these clustered genes are also known as imprinting control regions (ICRs) (Bartolomei and Ferguson-Smith 2011; Barlow and Bartolomei 2014). The acquired parental allele-specific DNA methylation imprints in oocytes or sperm are maintained after fertilization and erased in primordial germ cells (PGCs) in the gonads (Reik 2007). The differential methylation status of an ICR can lead to the imprinted gene expression pattern through diverse ways. For example, at some imprinted loci, such as Igf2r and Kcnq1, ICRs have methylated imprints on the maternal alleles, whereas the corresponding unmethylated regions on the paternal alleles are the promoters of the imprinted noncoding RNA genes. The transcripts themselves or the process of transcribing these noncoding genes are involved in the allele-specific silencing of genes within the same locus at the chromatin level through either recruiting chromatin-modifying machineries or interfering with RNA polymerase II (Pol II) engagement (Mancini-Dinardo et al. 2006; Nagano et al. 2008; Latos et al. 2012). In contrast, the H19 ICR, with a paternally methylated imprint, controls the imprinting status of H19 and Igf2 through coordinating the function of various cis-regulatory elements, including enhancers, insulators, and promoters (Hark et al. 2000).
Factors that associate with the methylated ICRs to protect the imprint marks from genome-wide epigenetic reprogramming after fertilization and maintain the methylation status during development have been intensively investigated. The Krüppel-associated box (KRAB)-containing zinc finger protein (KRAB-ZFP) ZFP57 specifically binds to the methylated ICRs and interacts with KRAB-associated protein 1 (KAP1), also known as TRIM28. Subsequently, TRIM28 recruits other repressive chromatin-related factors—such as the histone H3 Lys9 methyltransferase SETDB1; the DNA methyltransferases DNMT1, DNMT3A, and DNMT3B; and the heterochromatin-associated protein HP1—to the methylated ICRs to maintain a silent chromatin state (Quenneville et al. 2011; Messerschmidt et al. 2012). However, genomic imprinting is a finely tuned and highly regulated process, and yet-to-be-identified factors might exist to ensure the proper expression of imprinted genes through functionally interacting with cis-elements within the imprinted loci.
The AF4/FMR2 (AFF) family members are intrinsically disordered proteins (IDPs) lacking a fixed structure with versatile functions during development and disease pathogenesis (Luo et al. 2012b). AFF proteins can serve as scaffolds for the positive transcription elongation factor (P-TEFb)-containing superelongation complex (SEC) family (Luo et al. 2012a,b). The SEC family is comprised of the AFF4-containing SEC, the AFF2-containing SEC-L2, and the AFF3-containing SEC-L3, with each regulating different sets of genes (Lin et al. 2010; Luo et al. 2012a,b). Of these, SEC is the best characterized, being found occupying highly transcribed genes and regulating rapid gene activation upon exposure to environmental stimuli (Lin et al. 2011; Luo et al. 2012b). To investigate the function of SEC family members in transcriptional regulation, we performed chromatin immunoprecipitation (ChIP) and sequencing (ChIP-seq) studies in mouse embryonic stem cells (mESCs) and found that AFF3 can specifically bind both ICRs and enhancers in an allele-specific manner within the imprinted loci. Our studies demonstrate that AFF3 is recruited to the methylated alleles of ICRs and that the binding of AFF3 to ICRs depends on the sequence-specific DNA-binding factor ZFP57, the corepressor TRIM28, and DNA methyltransferases. We also show that AFF3 is bound to the enhancer upstream of the maternally inherited Meg3/Gtl2 gene within the imprinted Dlk1-Dio3 locus in wild-type ESCs. AFF3 is required for the activity of the enhancer and the expression of an ∼200-kb polycistron encompassing Meg3, Rian, Mirg, and numerous miRNA genes from the active allele. Based on the results, we propose that the methylated ICR-bound factors and/or the heterochromatic environment created could maintain the imprinted status partially through limiting the binding of an active enhancer factor such as AFF3.
Results
AFF3 binds to both gDMRs and enhancers of imprinted genes in mESCs
By performing ChIP-seq in different mESC lines and using two different antibodies, we were able to identify 973 high-confidence AFF3-binding sites. Functional annotation of genes near AFF3 peaks using Genomic Regions Enrichment of Annotations Tool (GREAT) found enrichment for mouse phenotypes related to genomic imprinting (Supplemental Fig. S1A; McLean et al. 2010). For example, AFF3 is enriched at the promoter region of the long noncoding RNA gene Airn, which resides in the gDMR/ICR of Igf2r/Airn locus bearing a maternal-specific methylation imprint. AFF3 binding at the Igf2r/Airn gDMR correlates with the presence of the known gDMR marks of TRIM28, SETDB1, and H3K9me3 (Supplemental Fig. S1B; Quenneville et al. 2011; Latos et al. 2012; Messerschmidt et al. 2012; Zuo et al. 2012). AFF3 is also detected in the intergenic regions within the Dlk1-Dio3 locus, which harbors a paternally methylated gDMR/ICR (Fig. 1A). Two discrete AFF3-binding sites are located 12 kb (distal AFF3 peak) and 10 kb (proximal AFF3 peak) upstream of the promoter region of the maternally expressed gene Meg3 (Fig. 1B). A comparison of our AFF3 ChIP-seq data with ChIP-seq data for the gDMR marks TRIM28 (Rowe et al. 2013), SETDB1 (Bilodeau et al. 2009), and H3K9me3 (Karimi et al. 2011) and the enhancer marks ELL3, p300, and H3K27ac (Lin et al. 2013) demonstrated that the distal AFF3 peak overlaps with the gDMR marks within the Dlk1-Dio3 locus, while the proximal AFF3 peak correlates with a potential enhancer element, referred to here as the “Meg3-distal gDMR” and the “Meg3-proximal enhancer,” respectively (Fig. 1B). Expanding the analysis to other known imprinted loci, we observed that AFF3 is enriched at almost all of the known gDMRs as well as at putative enhancers, including those near the imprinted genes Cdkn1c, Nespas, and Meg3 (Fig. 1C; Supplemental Fig. S1C). We knocked down AFF3 in v6.5 ESCs by lentivirus-mediated shRNA and observed reduced AFF3 signal at these regions, confirming that AFF3 occupies these regulatory regions (Supplemental Fig. S2).
Figure 1.
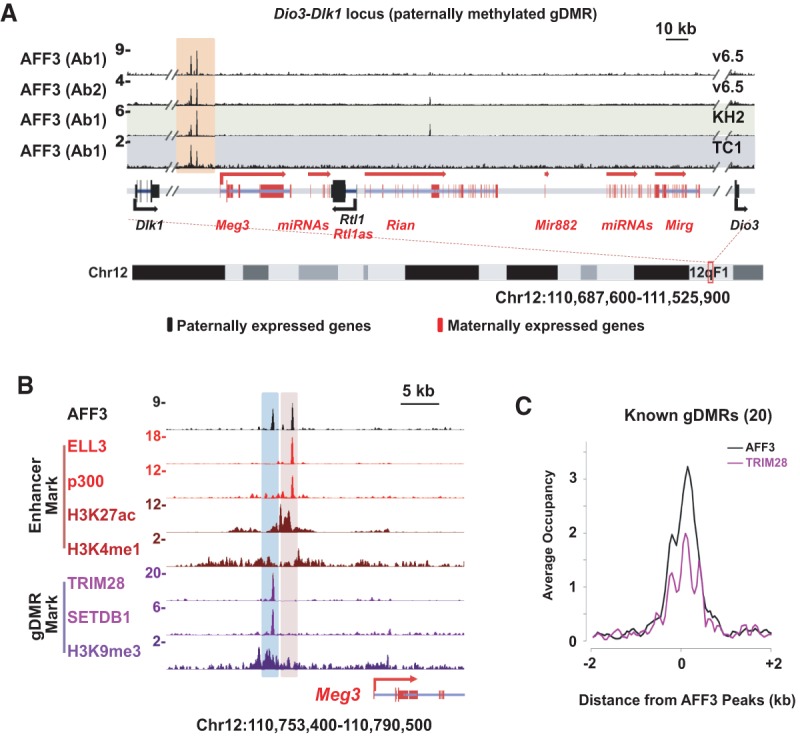
AFF3 binds to both gDMRs and enhancers of imprinted genes in mESCs. (A) AFF3 occupies an intergenic region within the Dlk1-Dio3 locus in mESCs. ChIP-seq binding profiles of AFF3 at the Dlk1-Dio3 loci in v6.5, KH2, and TC1 mESCs are shown. Two different antibodies were generated and used to chromatin immunoprecipitate AFF3 in v6.5 cells (indicated by Ab1 and Ab2). The two intergenic AFF3 peaks within the Dlk1-Dio3 locus are highlighted with an orange bar. (B) The distal intergenic peak to the Meg3 transcription start site (TSS) within the Dlk1-Dio3 locus is associated with the gDMR marks TRIM28 (Rowe et al. 2013), SETDB1 (Bilodeau et al. 2009), and H3K9me3 (Karimi et al. 2011), while the proximal peak is associated with the enhancer marks ELL3 (Lin et al. 2013), p300 (official protein symbol EP300) (Creyghton et al. 2010), H3K27ac, and H3K4me1 (Creyghton et al. 2010). Shown are ChIP-seq binding profiles of AFF3, gDMR marks (TRIM28, SETDB1, and H3K9me3), and enhancer marks (ELL3, p300, H3K27ac, and H3K4me1) at the genomic region upstream of the Meg3 TSS. AFF3 ChIP-seq in v6.5 cells using Ab1 is shown. The gDMR region is highlighted with a blue bar, and the enhancer region upstream of the Meg3 TSS is highlighted with a pink bar. Schematic illustrations of the Dlk1-Dio3 locus show the maternally and paternally expressed genes indicated by red and black, respectively. (C) AFF3 co-occupies the gDMRs of imprinted loci with TRIM28. An average occupancy plot of AFF3 and TRIM28 is shown for the known imprinted loci in Supplemental Figure S1C.
AFF3 is recruited to the methylated gDMRs
gDMRs are targeted by parent of origin-dependent DNA methylation (Bartolomei and Ferguson-Smith 2011; Lee and Bartolomei 2013; Barlow and Bartolomei 2014). To understand whether the binding of AFF3 to gDMRs is allele-specific, the methylation status of the AFF3-recruiting allele was assayed by bisulfite sequencing analyses following AFF3 ChIP at the paternally methylated gDMR of the H19/Igf2 locus and the maternally methylated gDMRs for the Peg1 and Snrpn loci. In all cases, regardless of parent of origin, AFF3 was exclusively found at the methylated gDMRs (Fig. 2A). To validate these assays, we performed AFF3 ChIP and methylated DNA immunoprecipitation (MeDIP) quantitative PCR (qPCR) assays in the androgenic (AG) and parthenogenetic (PG) mouse embryonic fibroblast (MEF) cell lines (Hernandez et al. 2003) and confirmed that AFF3 is recruited to the methylated alleles of gDMRs (Fig. 2B,C). ChIP-seq of AFF3 and H3K9me3 in AG and PG MEF cells also demonstrated that AFF3 tends to colocalize with H3K9me3 at gDMRs (Supplemental Fig. S3A). To determine whether DNA methylation is required for the recruitment of AFF3 to gDMRs, we performed AFF3 ChIP in ESCs depleted of the DNA methyltransferases DNMT1, DNMT3A, and DNMT3B, which lack DNA methylation genome-wide, including at gDMRs (Tsumura et al. 2006). AFF3 is enriched at all tested gDMRs in the wild-type cells but not in the triple-knockout cells (DNMT triple knockouts), while the occupancy of AFF3 at the nonmethylated Prkcsh gene promoter is equally detected in both cell lines (Fig. 2D). AFF3 is also lost at gDMRs in ESCs null for TRIM28 or ZFP57, factors known to maintain the heterochromatic environment at gDMRs during early embryonic development (Supplemental Fig. S3B,C). The depletion of DNMTs, TRIM28, or ZFP57 does not reduce the AFF3 RNA or protein level in mESCs (Supplemental Fig. S3D). Furthermore, genome-wide analyses demonstrated that 91 AFF3 peaks, including the majority of the AFF3-occupied known gDMRs, are significantly reduced in ZFP57 knockout ESCs, and ∼90% of them are bound by the ZFP57 cofactor TRIM28 (Fig. 2E; Supplemental Fig. S3E). Some of the 91 sites that do not overlap the known ICRs or other gDMRs listed in the Web resource Web Atlas of Murine Genomic Imprinting and Differential Expression (WAMIDEX) (Schulz et al. 2008) do overlap a subset of novel gDMRs identified from genome-wide DNA methylation mapping, including four experimentally validated maternal gDMRs near Cdh15, Zfp777, Zfp787, and AK008011/Nhlrc1 loci (Proudhon et al. 2012). These same gDMRs were also found to be occupied by ZFP57 (Strogantsev et al. 2015). Therefore, many more ICRs may exist that are regulated by some of the same factors that regulate the known gDMRs. We also performed ChIP-qPCR to analyze the occupancy of TRIM28 and the histone methyltransferase SETDB1 at gDMRs in mESCs after AFF3 depletion. The occupancy of TRIM28 and SETDB1 at the gDMRs was not obviously affected after AFF3 knockdown (Supplemental Fig. S3F,G).
Figure 2.

AFF3 is recruited to methylated gDMRs. (A) AFF3 binds to the methylated H19, Snrpn, and Peg1 gDMRs in mESCs. Bisulfite sequencing of whole-cell extract (WCE; input for ChIP) and AFF3 ChIP at the H19, Snrpn, and Peg1 gDMRs. Methylated and unmethylated cytosines are designated by filled and unfilled circles, respectively. Each line indicates a unique DNA clone. In contrast to whole-cell extract, the AFF3 ChIP DNA is predominantly methylated at all tested gDMRs. (B) AFF3 binds to the methylated gDMRs in AG and PG MEF cell lines, shown by ChIP-qPCR. (C) Differential DNA methylation at gDMRs in AG and PG MEF cells is shown by MeDIP-qPCR. The Herc3 gene served as a positive control for the ChIP-qPCR and MeDIP-qPCR experiments. (D) DNA methylation is required for the recruitment of AFF3 to the methylated gDMRs. The association of AFF3 in the DNMT triple knockouts with gDMRs was compared with its association with gDMRs in wild-type ESCs by ChIP-qPCR. The Prkcsh gene served as a positive control for the ChIP-qPCR. The Hba gene served as a negative control for ChIP-qPCR. Error bars represent the standard deviation of two independent measurements of a representative ChIP experiment. (E) ZFP57 is required for the recruitment of AFF3 to gDMRs. MA plot showing changes in AFF3 occupancy in the ZFP57 knockout ESCs. The X-axis shows the log2 geometric average of AFF3 occupancy in the two conditions. The Y-axis shows the log2 fold change between conditions. Each dot represents one of the 973 high-confidence AFF3 peaks. gDMRs are highlighted by black circles. Colored dots indicate a significant change was measured (adjusted P-value < 0.05).
AFF3 regulates the activity of the Meg3 upstream enhancer in an allele-specific manner
Of all of the AFF3 peaks overlapping an annotated gDMR, only one peak exhibits a significant increase in AFF3 occupancy in the ZFP57 knockout cells (Fig. 2E; Supplemental Table 1). This AFF3 peak resides within the intergenic DMR (IG-DMR) of the Dlk1-Dio3 cluster and corresponds to the Meg3-proximal enhancer peak located just 2.4 kb from the Meg3-distal gDMR peak (Figs. 1B, 3A). While bisulfite sequencing of ChIP DNA demonstrated that AFF3 is on the inactive allele at the Meg3-distal gDMR, AFF3 is restricted to the unmethylated allele at the Meg3-proximal enhancer (Fig. 3B,C; Supplemental Fig. S4).
Figure 3.

The activity of the Meg3 upstream enhancer is regulated by allele-specific binding of AFF3. (A) Schematic illustration of the amplified genomic regions of the Meg3-distal gDMR (B) and the Meg3-proximal enhancer (C). AFF3 occupies the Meg3-distal gDMR on the paternal allele (B), while binding to the active Meg3-proximal enhancer on the maternal allele (C). Methylated and unmethylated cytosines are designated by filled and unfilled circles, respectively. Each line indicates a unique DNA clone. (D) AFF3 is required for the activity of the Meg3-proximal enhancer. Shown are ChIP-seq binding profiles of H3K27ac at the genomic regions upstream of the Meg3 gene in mESCs bearing either NonT or AFF3 shRNA. H3K27ac is significantly reduced at the Meg3 upstream enhancer upon AFF3 RNAi. (E) AFF3 is required for the expression of the maternally expressed genes within the Dlk1-Dio3 locus. Shown are RNA sequencing (RNA-seq) track files in the Dlk1-Dio3 locus in mESCs bearing either NonT or AFF3 shRNA. Both AFF3 (F) and H3K27ac (G) levels are increased about twofold in ZFP57-null cells, reflecting the loss of heterochromatin. Shown are ChIP-seq binding profiles of AFF3 (F) and H3K27ac (G) at the genomic region upstream of the Meg3 TSS in both ZFP57 wild-type and knockout ESCs.
Knockdown of AFF3 leads to reduced H3K27ac at the Meg3-proximal enhancer, indicating that AFF3 is required to maintain its active chromatin state (Fig. 3D; Supplemental Table 2). Furthermore, the expression levels of Meg3 and the downstream maternally expressed noncoding genes within the Dlk1-Dio3 locus are also reduced in AFF3 knockdown ESCs (Fig. 3E; Supplemental Fig. S5A), suggesting that an activating function of AFF3 might be exerted from the nearby Meg3-proximal enhancer. In order to confirm that the reduced expression of these maternally expressed genes is only due to reduced expression of the maternal allele, we performed gene expression and single-nucleotide polymorphism (SNP) analyses in TT2 ESCs generated from C57BL/6 × CBA F1 embryos. These studies demonstrate that all expression comes from the maternal allele in control and AFF3 knockdown cells but at reduced levels in AFF3 knockdown cells compared with control cells (Supplemental Fig. S5B,C).
A model for the methylated IG-DMR limiting AFF3 enhancer function
In ZFP57-null ESCs, H3K9me3 (Quenneville et al. 2011) and DNA methylation (Li et al. 2008) are diminished from the IG-DMR on the paternal allele. We found that AFF3 is also lost from the Meg3-distal gDMR in these cells, while AFF3 occupancy doubles at the Meg3-proximal enhancer region (Fig. 3F). AFF3 binding at these regions exhibited similar changes after TRIM28 knockout (Supplemental Figs. S3B, S5D). In ZFP57-null cells, the active enhancer mark H3K27ac is also increased nearly twofold at this region (Fig. 3G). Furthermore, the Meg3, Rian, and Mirg transcripts within this locus are up-regulated approximately twofold relative to wild-type cells (Supplemental Fig. S6A). Gene expression and SNP analyses in TT2 ESCs demonstrate that transcription is originating from both alleles in the absence of ZFP57 (Supplemental Fig. S6B–D).
We next depleted AFF3 in ZFP57-null ESCs, where only enhancer-bound AFF3 is present, and observed reduced expression of the noncoding RNA genes (Supplemental Fig. S7). This finding further confirms that an activating function of AFF3 from the enhancer is regulating the expression of these genes and also suggests that AFF3 is required for full derepression of these genes in the ZFP57-null background. Therefore, AFF3 occupies the enhancer of the active maternal Meg3 allele, regulating its activity and downstream gene expression (Fig. 3A–E), while the methylated IG-DMR, maintained by ZFP57 and its associated heterochromatin machinery, has an inhibitory role in the activity of the nearby Meg3 enhancer, which could be mediated by limiting the binding of AFF3 at the imprinted paternal allele (Fig. 3F,G).
AFF3 regulates the expression of a 210-kb Meg3-to-Mirg transcript within the Dlk1-Dio3 locus
SECs regulate transcription elongation through phosphorylation of the Pol II C-terminal domain (CTD) (Luo et al. 2012a,b). To examine possible elongation functions for AFF3 at the Dlk1-Dio3 locus, we performed Pol II ChIP-seq in AFF3 knockdown cells. Pol II appears to be continuously transcribing from Meg3 to Mirg, and depletion of AFF3 leads to a reduced level of Pol II throughout the locus (Fig. 4A). Furthermore, both total and nascent RNA sequencing (RNA-seq) demonstrate that transcripts are reduced throughout the entire region in AFF3 knockdown cells (Fig. 4B; Supplemental Table 3). Global run-on sequencing (GRO-seq) (Min et al. 2011) and ChIP-seq for markers of transcription elongation also show continuous coverage over this region (Fig. 4C,D). The only apparent transcription start site (TSS), as indicated by enrichment for TBP, NELFA, and H3K4me3, overlaps with the annotated Meg3 gene TSS (Fig. 4E). The continuous coverage of transcription elongation marks and a single start site of transcription for this region can be observed in different tissues and cell types (Supplemental Fig. S8). These findings are in line with previous proposals that these noncoding RNAs might be processed from a single transcript based on these regions having only one predicted promoter element (at the Meg3 locus), that intergenic transcription is detected within the maternally expressed region of this locus, and that deletion of the Meg3 promoter disrupts the expression of the downstream noncoding RNAs (Tierling et al. 2006; Zhou et al. 2010). Together, these data provide strong evidence that these maternally expressed noncoding RNA genes are transcribed as a single, >200-kb, polycistronic transcription unit that requires AFF3 for its expression in mESCs.
Figure 4.

The maternally expressed Meg3 gene is transcribed as a polycistron that is regulated by AFF3 at the transcriptional elongation stage. (A) Pol II is continuously transcribing from Meg3 to Mirg. Depletion of AFF3 leads to the reduction of elongating Pol II in the body of the Meg3 polycistron. (B) AFF3 knockdown leads to reduced nascent RNA at the Meg3 polycistron. GRO-seq (Min et al. 2011) (C) and the ChIP-seq binding profiles (D) of the elongation marks (SPT5 [Rahl et al. 2010], AFF4 [Lin et al. 2011], and H3K36me3 [Marson et al. 2008]) demonstrate continuous transcription of the Meg3 polycistron. (E) The ChIP-seq binding profiles of the TSS marks (TBP [Liu et al. 2011], NELFA [Rahl et al. 2010], and H3K4me3 [Mikkelsen et al. 2007]) indicate that the TSS of the Meg3 polycistron overlaps with the annotated Meg3 TSS. The paternally expressed Rtl1 gene is not transcribed in ESCs. A schematic illustration of the Dlk1-Dio3 locus (with maternally and paternally expressed genes indicated by red and black, respectively) is shown below.
Discussion
Regulation of the maternally expressed genes within the Dlk1-Dio3 locus in pluripotent cells
Our analyses demonstrate that AFF3 binds to the methylated alleles at gDMRs/ICRs, whereas at the Meg3-proximal enhancer, it preferentially binds to the unmethylated allele and promotes target gene transcription. The distinct functional states of AFF3 at gDMRs and at the Meg3 enhancer could be determined by the local chromatin environment. The DNA-binding factor ZFP57 recruits the scaffolding factor TRIM28, the histone H3 Lys9 methyltransferase SETDB1, and the DNA methyltransferases DNMTs, setting up a repressive chromatin environment at gDMRs (Quenneville et al. 2011). The open chromatin signature of the corresponding region on the maternal allele of the IG-DMR could enable an activating function of AFF3 through this enhancer for regulating expression of the Dlk1-Dio3 locus, which is transcribed as a single polycistron. Consistent with these findings, a recent study found that enhancer-like noncoding RNAs emanating from the Dlk1-Dio3 ICR were required for the expression of the maternally expressed genes in the cluster (Kota et al. 2014).
Silencing of the maternally expressed genes in the Dlk1-Dio3 locus was linked to “bad” iPSCs that exhibit a compromised ability to generate “all-iPSC” mice (Stadtfeld et al. 2010, 2012). The noncoding transcripts within the locus, including Meg3, Rian, and Mirg, were demonstrated to interact with the key epigenetic regulator Polycomb-repressive complex 2 (PRC2) in mESCs. Decreased occupancy of PRC2 and H3K27me3 was observed over the developmentally regulated genes in MEG3-depleted cells, suggesting the functional importance of the noncoding transcripts in pluripotency (Kaneko et al. 2014). Clearly, the ICR binding of AFF3 is dependent on ZFP57 and/or other factors recruited by ZFP57, while AFF3 binding to the Meg3-proximal enhancer does not require ZFP57. Therefore, it would be interesting to explore the DNA-binding factors that start the path for setting up the open chromatin and recruiting AFF3 to the enhancer region in ESCs. Manipulating the activity of the Meg3 enhancer through its trans-acting factor, AFF3, identified in this study, and the potential pioneer factors bound to this enhancer could facilitate cellular reprogramming for regenerative medicine.
There are only three well-established paternally imprinted loci in mice: the Dlk1-Dio3 locus, the Rasgrf1 locus, and the H19-Igf2 locus. The gDMRs within these paternally imprinted loci are located at intergenic regions, giving them the potential to have enhancer activities when unmethylated. However, the Rasgrf1 and H19-Igf2 loci are either silenced or unstably expressed in mESCs. Only the maternally expressed noncoding RNAs within the Dlk1-Dio3 locus are highly expressed in mESCs, allowing us to investigate the function of AFF3 at the IG-DMR and Meg3-proximal enhancer in these cells.
Sequestration model of AFF3 enhancer function at the IG-DMR
The IG-DMR at the Dlk1-Dio3 locus not only ensures inactivation of the Meg3-proximal enhancer on the paternal allele but also recruits AFF3 to the methylated IG-DMR locus, which could serve to modulate AFF3 recruitment to the active enhancer on the maternal allele, thereby regulating enhancer activity and transcription throughout the Meg3 polycistron (Fig. 5). The evidence that led us to propose that the IG-DMR sequesters AFF3 from the active enhancer is the combination of the following observations. Bisulfite sequencing shows that AFF3 is normally bound to the paternally imprinted ICR and the maternal active Meg3 enhancer located within the IG-DMR. In ZFP57 knockout ESCs, the increased expression of the noncoding RNAs in this region, due to loss of imprinting, is consistent with higher occupancy of AFF3 and H3K27ac at the Meg3-proximal enhancer. However, loss of AFF3 leads to loss of expression of these genes in not only wild-type but also ZFP57 knockout cells, suggesting that AFF3 is a positive regulator of these genes from its only remaining binding site in the region; namely, the Meg3-proximal enhancer. Last, loss of AFF3 in wild-type ESCs leads to loss of H3K27ac at the enhancer, indicating that AFF3 has a function in setting up the open chromatin environment at the enhancer on the active allele, which provides a rationale for why ICR factors could enforce the silent enhancer state in part through sequestering AFF3.
Figure 5.
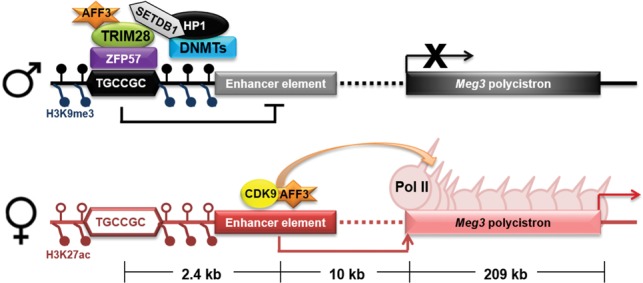
Cartoon model illustrating the locations of IG-DMR-bound AFF3 and Meg3 upstream enhancer-bound AFF3 at the Dlk1-Dio3 locus. Methylation of the IG-DMR inhibits the genesis of an active enhancer on the paternal allele through sequestering AFF3, while AFF3 on the active allele is free to bind the active enhancer on the maternal allele to regulate transcription of the Meg3 polycistron at the elongation stage.
Control of allele-specific expression or silencing of the Meg3 polycistron by differential methylation of identical enhancer elements on different alleles under the same cellular context provides direct evidence for a critical role of enhancer methylation status in gene expression, supporting prior correlations of methylation status at enhancers with altered gene expression profiles, seen not only across various cancer types but also under physiological developmental processes (Aran and Hellman 2013; Hon et al. 2013). Considering the widespread existence of disease- and tissue-specific differential methylation, the mechanism proposed here could be more generally applied to explain the function of DNA methylation and heterochromatin in regulating enhancer activity.
A function of AFF3 in autoimmune diseases
A role for AFF3 in imprinted gene expression in humans is indicated by the existence of SNPs in AFF3 regulatory regions that are associated with human autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes (Stahl et al. 2010; Cen et al. 2012; Wallace et al. 2012). These diseases are themselves linked to changes in imprinted gene expression (Camprubi and Monk 2011), raising the possibility that their etiology could result from misregulation of AFF3 at regulatory regions of imprinted genes. Our proposed mechanism of gDMR-bound silencing factors sequestering AFF3 from its cis enhancer, while, on the other allele, AFF3 occupies the active enhancer to stimulate transcription elongation by an SEC-like complex provides new areas of investigation for understanding the regulation of imprinted gene expression through enhancer activity and transcriptional elongation control in development and disease.
Materials and methods
Cell culture
The mESC lines used in this study were grown on irradiated primary MEF feeder layers in 0.1% gelatin-coated tissue culture plates. mESCs, except for DNMT triple-knockout ESCs, were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 15% ES-certified fetal bovine serum (FBS) (Hyclone), recombinant LIF (Millipore), 2 mM L-glutamine, 0.1 mM nonessential amino acids, and 0.1 mM β-mercaptoethanol. For experimental analyses, ESCs were grown for one passage without MEF feeder cells in tissue culture plates for 30 min. The medium used for DNMT triple-knockout ESC culture was Glasgow minimum essential medium (GMEM) with the above supplements. The uniparental MEF cell lines were cultured in DMEM supplemented with 10% FBS (Sigma).
Lentivirus-based RNAi
Mouse AFF3 (RMM4534-NM_010678) shRNA constructs were purchased from Open Biosystems. Mouse ZFP57 (SHCLNG-NM_009559) shRNA and the nontargeting shRNA constructs (SHC002) were purchased from Sigma. Preparation of lentiviral particles and viral transduction were carried out as previously described (Lin et al. 2013). Briefly, 293T cells cultured in DMEM with 10% FBS were cotransfected 24 h after seeding with the shRNA construct or the nontargeting control shRNA, the packaging plasmid PsPAX2, and the envelope plasmid pMD2.G using xTremeGENE-HP (Roche). The medium was changed 16 h after transfection. The supernatants containing lentiviral particles were collected 48 and 72 h after transfection, filtered through a 0.45-µm filter, and concentrated at 18,000 rpm for 2 h at 4°C. ESCs and MEF cells were transduced with concentrated lentiviral particles and polybrene (Sigma). Twenty-four hours after transduction, the cells were subjected to 2 µg/mL puromycin selection for an additional 48 h. Seventy-two hours after transduction, the cells were harvested for the experimental analyses.
Antibodies
The antibodies to 5-methyl-cytosine (5mC) (Eurogentec, BI-MECY-0100), H3K9me3 (Abcam, ab8898), H3K27ac (Abcam, ab4729), ZFP57 (Abcam, ab45341), SETDB1 (Santa Cruz Biotechnology, sc-66884), and RNA Pol II (Santa Cruz Biotechnology, sc-899) are commercially available. Antibodies against AFF3 (Ab#1, antigen, a mixture of two peptides from amino acids 124–237 and amino acids 816–982 of human AFF3) were generated in our laboratory and have been described previously (Luo et al. 2012a). A second antibody against AFF3 (Ab#2, antigen, human AFF3 amino acids 816–982) was also generated in our laboratory. Antigens were expressed as His tag fusion proteins in pET-16b, purified on NTA-agarose according to Qiagen's protocol, and sent to Pocono Rabbit Farm and Laboratory for immunization into rabbits.
qRT–PCR and total RNA-seq library preparation
Total RNA was isolated with RNeasy kit (Qiagen), treated with RNase-free DNase I (New England Biolabs), and repurified with RNeasy. cDNAs were synthesized with the High-Capacity RNA-to-cDNA kit (Applied Biosystems). The expression levels were measured on MyIQ (Bio-Rad) using iQ SYBR Green supermix (Bio-Rad). For total RNA-seq, ribosomal RNA depletion and library preparation were performed with TruSeq sample preparation kit (Illumina).
Chromatin isolation and nascent RNA-seq library preparation
About 1 × 108 fresh ESCs were washed twice in ice-cold PBS. The cells were then resuspended in buffer A (10 mM HEPES at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, proteinase inhibitors [Roche]) and incubated for 15 min on ice. The cell suspension was transferred to a precooled Dounce tissue grinder and homogenized 10 times by using a tight pestle on ice. After centrifugation, the pellet containing enriched nuclei was washed twice in S1 buffer (0.25 M sucrose, 10 mM MgCl2, 10 mM HEPES at pH 7.9, 1 mM DTT, proteinase inhibitors). To obtain the chromatin pellet, the nuclear pellet was then washed in ice-cold NUN buffer (20 mM HEPES at pH 7.9, 7.5 mM MgCl2, 0.2 mM EDTA, 300 mM NaCl, 1 M urea, 1% NP-40, 1 mM DTT, 20 U/mL RNase inhibitor [Ambion], proteinase inhibitors) five times. Chromatin-bound RNA was isolated with RNeasy (Qiagen). The RNA was poly-A-depleted by using oligo(dT) magnetic beads (Invitrogen), treated with RNase-free DNase I, and repurified with RNeasy. Five-hundred nanograms of RNA was used for ribosomal RNA depletion with Ribo-zero Gold (Epicentre), and library preparation was performed with TruSeq Stranded Total RNA-seq sample preparation kit.
ChIP and ChIP-seq library preparation
ChIP assays were performed according to the previously described protocol (Lee et al. 2006). Briefly, 5 × 107 cells were cross-linked by using 1% paraformaldehyde for 10 min at room temperature. Cross-linking was quenched by the addition of glycine. The fixed chromatin was then subjected to sonication and immunoprecipitation with the specified antibody. ChIP-seq libraries were prepared by using ChIP-seq sample preparation kit (Illumina) or KAPA Biosystems HTP library preparation kit.
MeDIP
MeDIP assays were performed according to a previously described protocol (Weber et al. 2007). Briefly, genomic DNA was isolated and purified with DNeasy kit (Qiagen). One microgram of purified genomic DNA was sonicated by using a Diagenode Bioruptor and heat-denatured for 10 min at 95°C. Two-hundred nanograms of sonicated DNA was saved as input. The rest of the fragmented DNA was immunoprecipitated with the antibody against 5mC overnight at 4°C.
Supplementary Material
Acknowledgments
We are grateful to Professor Didier Trono, Professor Stuart Orkin, Professor Colin Stewart, Professor Xiajun Li, and Professor Matthew Lorincz for providing cell lines for this study. We also thank the Molecular Biology core facility at the Stowers Institute for creating and sequencing libraries for next-generation sequencing, and the Stowers Tissue Culture facility for cell culture. We are also thankful to Laura Shilatifard for editorial assistance. Z.L. was supported as a Fellow of the Leukemia and Lymphoma Society. The study was supported in part by grant R01GM069905 from the National Institute of Health to A.S.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.270413.115.
References
- Aran D, Hellman A. 2013. DNA methylation of transcriptional enhancers and cancer predisposition. Cell 154: 11–13. [DOI] [PubMed] [Google Scholar]
- Barlow DP, Bartolomei MS. 2014. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol 6: a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei MS, Ferguson-Smith AC. 2011. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol 3: a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA. 2009. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev 23: 2484–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camprubi C, Monk D. 2011. Does genomic imprinting play a role in autoimmunity? Adv Exp Med Biol 711: 103–116. [DOI] [PubMed] [Google Scholar]
- Cen H, Leng RX, Wang W, Zhou M, Feng CC, Chen GM, Li R, Pan HF, Li XP, Ye DQ. 2012. Association of AFF1 rs340630 and AFF3 rs10865035 polymorphisms with systemic lupus erythematosus in a Chinese population. Immunogenetics 64: 935–938. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405: 486–489. [DOI] [PubMed] [Google Scholar]
- Hernandez L, Kozlov S, Piras G, Stewart CL. 2003. Paternal and maternal genomes confer opposite effects on proliferation, cell-cycle length, senescence, and tumor formation. Proc Natl Acad Sci 100: 13344–13349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. 2013. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet 45: 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldaña-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. 2014. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53: 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. 2011. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8: 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota SK, Lleres D, Bouschet T, Hirasawa R, Marchand A, Begon-Pescia C, Sanli I, Arnaud P, Journot L, Girardot M, et al. 2014. ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1-Dio3 domain. Dev Cell 31: 19–33. [DOI] [PubMed] [Google Scholar]
- Latos PA, Pauler FM, Koerner MV, Şenergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, et al. 2012. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338: 1469–1472. [DOI] [PubMed] [Google Scholar]
- Lee JT, Bartolomei MS. 2013. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152: 1308–1323. [DOI] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA. 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc 1: 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. 2008. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. 2010. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37: 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, Shilatifard A. 2011. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev 25: 1486–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Garruss AS, Luo Z, Guo F, Shilatifard A. 2013. The RNA Pol II elongation factor Ell3 marks enhancers in ES cells and primes future gene activation. Cell 152: 144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Scannell DR, Eisen MB, Tjian R. 2011. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell 146: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. 2012a. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol 32: 2608–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, Shilatifard A. 2012b. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol 13: 543–547. [DOI] [PubMed] [Google Scholar]
- Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. 2006. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 20: 1268–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. 2008. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134: 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. 2012. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335: 1499–1502. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. 2011. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev 25: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. 2008. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322: 1717–1720. [DOI] [PubMed] [Google Scholar]
- Peters J. 2014. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet 15: 517–530. [DOI] [PubMed] [Google Scholar]
- Proudhon C, Duffie R, Ajjan S, Cowley M, Iranzo J, Carbajosa G, Saadeh H, Holland ML, Oakey RJ, Rakyan VK, et al. 2012. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol Cell 47: 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. 2011. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell 44: 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. 2010. c-Myc regulates transcriptional pause release. Cell 141: 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W. 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447: 425–432. [DOI] [PubMed] [Google Scholar]
- Rowe HM, Kapopoulou A, Corsinotti A, Fasching L, Macfarlan TS, Tarabay Y, Viville S, Jakobsson J, Pfaff SL, Trono D. 2013. TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res 23: 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R, Woodfine K, Menheniott TR, Bourc'his D, Bestor T, Oakey RJ. 2008. WAMIDEX: a Web atlas of murine genomic imprinting and differential expression. Epigenetics 3: 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. 2010. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465: 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Ferrari F, Choi J, Walsh RM, Chen T, Ooi SS, Kim SY, Bestor TH, Shioda T, et al. 2012. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat Genet 44: 398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, et al. 2010. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 42: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strogantsev R, Krueger F, Yamazawa K, Shi H, Gould P, Goldman-Roberts M, McEwen K, Sun B, Pedersen R, Ferguson-Smith AC. 2015. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol 16: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierling S, Dalbert S, Schoppenhorst S, Tsai CE, Oliger S, Ferguson-Smith AC, Paulsen M, Walter J. 2006. High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics 87: 225–235. [DOI] [PubMed] [Google Scholar]
- Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, et al. 2006. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11: 805–814. [DOI] [PubMed] [Google Scholar]
- Wallace C, Rotival M, Cooper JD, Rice CM, Yang JH, McNeill M, Smyth DJ, Niblett D, Cambien F, Tiret L, et al. 2012. Statistical colocalization of monocyte gene expression and genetic risk variants for type 1 diabetes. Hum Mol Genet 21: 2815–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schübeler D. 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Cheunsuchon P, Nakayama Y, Lawlor MW, Zhong Y, Rice KA, Zhang L, Zhang X, Gordon FE, Lidov HG, et al. 2010. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 137: 2643–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, et al. 2012. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem 287: 2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.