

Abstract
Background:
Seizures are recognised in multiple sclerosis (MS), but their true incidence and the mechanism by which they are associated with MS is unclear.
Objective:
The objective of this paper is to determine the lifetime frequency of seizures in the United Kingdom MS Tissue Bank (UKMSTB) population and any pathological features associated with seizures.
Methods:
We evaluated 255 individuals from the UKMSTB. A subset underwent analysis of cortical thickness, grey matter lesion (GML) (type and number) and cortical neuronal numbers (total and GABAergic).
Results:
A total of 37/255 patients had seizures (14.5% lifetime incidence); in 47% they were associated with concurrent infection. In those with seizures, death and wheelchair use occurred earlier and in 59% seizures developed after 15 years of disease. Seizures were associated with Type 1 GMLs and reduced cortical thickness in the middle temporal gyrus. Localised selective GABAergic interneuron loss in layers IV and VI was related to GMLs but was not explained by the presence of inflammation or by mitochondrial dysfunction within Type I GMLs.
Conclusion:
We confirm that seizure frequency rises in MS. Type I GMLs in the temporal lobe underlie a loss of inhibitory interneurons in cortical layers IV and VI and these changes could together with concurrent infection enhance susceptibility to seizures.
Keywords: Multiple sclerosis, seizure, neuropathology, temporal lobe, epilepsy
Introduction
Seizures have been recognised in the context of multiple sclerosis (MS) since the earliest reports of the disease.1 Although the reported prevalence varies, seizures are more frequent than in the general population,2–4 occurring in about 2%–3% of all patients with MS.5–8 The origin, extent and importance of this increase above the baseline incidence is uncertain. Cross-sectional studies show a minimal rise in the incidence of seizures,9 whereas retrospective assessment where MS is present for up to 20 years found an incidence three times above baseline.3,8 Patients with significant disability have a further increase in seizure frequency to 4.25%,10 rising to 7% in those requiring baclofen.11 The picture is further complicated by the fact that some medications prescribed for MS may increase the incidence of seizures (e.g. baclofen), while other therapies (e.g. carbamazepine) may reduce this incidence.
Seizures are a manifestation of excessive and/or hypersynchronous activity of neurons in the brain12 and are seen in the context of a neuronal insult. MS is classically considered an inflammatory demyelinating disease of the white matter (WM). Though the pathological substrate of seizures in MS remains to be determined, the emerging appreciation of cortical pathological processes and neuronal loss in MS13,14 may help to explain the observed increased rate of seizures in MS. Magnetic resonance imaging (MRI) studies have demonstrated new plaques in the juxtacortical and/or cortical regions in patients with generalised convulsions15,16 and also higher cortical-subcortical lesion loads in those with epilepsy,17 suggesting that a relationship existed between the extent of the cortical and subcortical lesions and the presence of seizures. Moreover, it has been demonstrated that relapsing–remitting MS (RRMS) patients with epilepsy have a more severe and rapidly evolving cortical pathology compared with RRMS without epilepsy.18
Unfortunately MRI provides an incomplete assessment of neuronal processes that may be active around the time of onset of epilepsy due to both intrinsic limitations of the technique and because it is performed at a time distant from the seizure event. Neuropathology does allow us to assess neuronal processes in more detail, although as with MRI it is distant from the seizure event. Neuropathological studies have shown changes in GABAergic neurotransmission and interneuron loss that could increase susceptibility to seizures,19,20 and on that basis it has been hypothesised that epileptogenic neuronal foci could result from cortical pathology, which might otherwise be relatively asymptomatic.10,16,17 The United Kingdom (UK) MS Tissue Bank (MSTB) population is unique in that it is recruited throughout the UK, with analysis of the neuropathology ultimately being available to confirm the diagnosis. Using this population we analysed the prevalence of seizures and correlated this with an in-depth study of the possible pathological substrate.
Material and methods
Demographic and clinical data
The UK MS Tissue Bank (UKMSTB) has multi-centre research ethics approval for a national prospective donor scheme (MREC/02/2/39). Once a registered donor has died, a summary of the clinical history is prepared by a clinical neurologist with an interest in MS.21,22 Any history of seizures was noted including the date of seizure onset and the basis on which a diagnosis of seizures was made. Seizures were classified according to the International League Against Epilepsy guidelines.23 Concurrent infection was relevant if a temperature was documented prior to or within 24 hours of admission and/or antibiotics were being taken prior to or on admission and or concurrent infection was documented as a cause of the seizure in the clinical notes. Fifty-one individuals with MS (21 patients with MS and seizures, 30 patients with MS alone without seizures (Table 1)) and eight controls without evidence of neurological disease or neuropathological alterations were chosen sequentially from the UKMSTB based on tissue availability and suitability of tissue preservation.
Table 1.
Epilepsy in patients with MS.
| ID | Seizure onset (years) | Seizure history, treatment (diagnosed by), secondary to concurrent infection, type of infection | Investigations: EEG (date) | Age of MS onset (years) | Age wheelchair user (years) | Age died (years) |
|---|---|---|---|---|---|---|
| Onset after first symptoms of MS | ||||||
| MS020 | 35 | Blackout (neurologist), no | ‘Moderate bilateral disturbances, paroxysmal with left temporal emphasis’ | 23 | 38 | 60, |
| MS022 | 54 | Tonic clonic (neurologist), no | – | 39 | 49 | 56 |
| MS029 | 56 | Tonic clonic (A+E, neurologist), yes, UTI | – | 29 | 43 | 59 |
| MS037 | 62 | Blackout, incontinent, confused on recovery, anticonvulsants (neurologist), yes, UTI | ‘Diffusely abnormal, more than would be expected in MS’ | 30 | 54 | 76 |
| MS038 | 40 | Tonic clonic, carbamezepine (neurologist), yes, UTIs | – | 24 | 27 | 42 |
| MS040 | 54 | Right-sided focal (neurologist), yes, chest infection | – | 36 | 46 | 58 |
| MS049 | 55 | Tonic clonic, (ITU), no | ‘Right temporal slow waves’ (1965) | 37 | 76 | 76 |
| MS053 | 59 | Tonic clonic (neurologist), no | – | 31 | 53 | 66 |
| MS074* | 63 | Tonic clonic (neurologist), yes, chest infection | – | 28 | 50 | 64 |
| MS076 | 42 | Definite generalised seizures, phenytoin (neurologist), No | ‘Abnormal’ (1983 – at time of diagnosis of MS when had paroxysmal movements – not seizures) | 31 | 45 | 49 |
| MS079* | 49 | Temporal: complex partial, left-sided onset (neurologist), yes, chest infection – cause of death | – | 25 | 35 | 49 |
| MS092 | 26 | Tonic clonic (neurologist), no | – | 21 | 28 | 38 |
| MS104a | 50 | Tonic clonic, carbamezepine (neurologist), yes, UTIs | – | 41 | 46 | 53 |
| MS112a | 57 | Tonic clonic (A+E), yes, UTIs | – | 30 | 35 | 66 |
| MS124 | 28 | Right-sided focal (neurologist), no | – | 25 | 26 | 30 |
| MS127 | 51 | Tonic clonic (neurologist), yes, abscess buttock | – | 28 | 47 | 52 |
| MS131 | 51 | Temporal: partial (neurologist), no | – | 31 | 44 | 56 |
| MS135 | 59 | Temporal: left-sided focal with loss of awareness (neurologist), no | – | 24 | 43 | 61 |
| MS142 | 62 | Right-sided with secondary generalisation (neurologist), yes, UTI | – | 35 | 61 | 67 |
| MS147 | 57 | Right-sided with secondary generalisation (neurologist), no | – | 38 | 52 | 60 |
| MS161 | 62 | Tonic clonic, carbamezepine (neurologist), yes, UTI | – | 42 | - | 69 |
| MS172 | 53 | Tonic clonic (neurologist), no | – | 26 | 46 | 58 |
| MS186 | 47 | Tonic clonic with infections, carbamezepine (neurologist), yes, chest infection | – | 20 | 33 | 57 |
| MS188 | 62 | Generalised, loss of awareness (neurologist), no | – | 35 | 37 | 78 |
| MS197a | 26 | Simple (neurologist), no | – | 24 | 46 | 51 |
| MS222 | 62 | Abdominal spasm, phenytoin and phenobarbitone (neurologist), no | ‘normal’ | 31 | 40 | 70 |
| MS223 | 45 | Tonic clonic (neurologist), Yes, sepsis non-CNS, LP normal | ‘normal’ | 43 | 45 | 45 |
| MS234a | 38 | Tonic clonic (neurologist), yes, UTI | – | 23 | 31 | 39 |
| MS242 | 49 | Tonic clonic × 2, no treatment (neurologist), no | – | 38 | 46 | 57 |
| MS249 | 66 | Tonic clonic × 2, phenytoin (neurologist), yes, UTI | – | 27 | 60 | 69 |
| MS268 | 49 | Tonic clonic, carbamezepine (neurologist), no | – | 46 | 53 | 54 |
| MS272 | 45 | Nocturnal tonic clonic, valproate and lamotrigine (neurologist), no | – | 26 | 44 | 64 |
| MS278 | 23 | Tonic clonic, no treatment (neurologist), no | – | 9 | 23 | 30 |
| MS298 | 71 | Tonic clonic with infection × 1, no treatment (neurologist), yes. sepsis secondary to UTI | – | 29 | 68 | 72 |
| Onset before first symptoms of MS | ||||||
| MS058 | 7 | 3 tonic clonic seizures over 18 months (paediatrician), no | – | 31 | 48 | 52 |
| MS184 | 34 | Blackouts, (neurologist), no | Primary generalised EEG abnormality | 35 | 56 | 74 |
| MS219 | 13 | Generalised, stopped in late teens (paediatrician), no | – | 40 | 48 | 57 |
MS: multiple sclerosis; ID: identification; EEG: electroencephalogram; CNS: central nervous system; ITU: intensive therapy unit; A+E: accident and emergency; UTI: urinary tract infection. aIndicates analysed as part of neuronal density analysis.
Detailed cortical tissue and lesion classification
Paraffin-embedded tissue blocks from precentral, superior frontal and middle temporal gyri were examined. In the patients with MS the degree of inflammation, the extent of demyelination and degree of lesion activity were evaluated as previously described.20,24 Grey matter lesions (GMLs) were characterised into Type I (grey matter (GM)/WM junction) or Type III (subpial), according to the criteria of Peterson et al.25
Immunohistochemistry
From each formaldehyde-fixed, paraffin-embedded tissue block, 5-µm-thick paraffin sections were processed and immunostained as previously described.20 All sections were counterstained with haematoxylin, sealed and viewed with a Nikon E1000M microscope. Images were captured (QImaging) and analysed using Image Pro Plus software (Media Cybernetics).
Quantitative analysis of demyelinated lesions, cortical atrophy and neuronal density
For each tissue block one section was stained for luxol fast blue (LFB)/cresyl fast violet (CFV) and one was immunostained for myelin oligodendrocyte glycoprotein (MOG) and detailed evaluation of WM and GM demyelination and cortical thickness was performed as previously described.20 Cortical thickness was evaluated on paraffin sections from precentral gyrus, superior frontal gyrus and middle temporal gyrus. To obtain a detailed analysis of the neuronal pathology in the GM, quantification of the numerical density of the NeuN+ and GAD67+ cells was performed on serial paraffin sections from the middle temporal and superior frontal gyrus of six MS cases with seizures (a in Table 1, five of whom had seizures associated with concurrent infection) and six MS cases without seizures, compared to the GM of six controls. All cases were randomly chosen and analysed blinded to case identification. To determine mitochondrial respiratory chain complex IV (cytochrome-c oxidase (COX)) and complex II (succinate dehydrogenase (SDH)) activity, COX and SDH histochemistry was performed in serial sections and in single cells as previously described.26
Statistical analysis
Clinical data were analysed using nonparametric tests for the role of concurrent infection. Statistical analysis of the histological data was performed using one-way analysis of variance (ANOVA) and Bonferroni’s multiple comparison tests.
Results
The frequency of seizures is increased in MS in the UKMSTB population
Of 255 individuals with MS from the UKMSTB cohort, 227 (89%) had secondary progressive MS (SPMS), 23 had primary progressive (PPMS) (9%) and five had RRMS (2%). Mean age at MS onset was 32.6±10.5 years (mean±SD, range 9–66 years), 240 (94.1%) required a wheelchair with a mean age of wheelchair use being 48.6±12.8 years (range 18–85 years) and a mean age of death 62.0±12.8 years (range 30–92 years). In the 255 individuals and 15,815 person-years of observation, 37 patients developed epilepsy, giving a 14.5% lifetime incidence (Table 1).
Seizures in MS are commonly transient and associated with concurrent infection
Three patients developed seizures before first symptoms of MS; in those, clinical assessment at diagnosis confirmed retrospectively, supported a clinical diagnosis of primary generalised epilepsy, confirmed in one on electroencephalogram (EEG). EEGs were available for six of 34 remaining patients of which four were abnormal. The diagnosis was made clinically by a neurologist in 94% (32/34 patients). Eight individuals had focal onset whilst the remainder had features supporting secondary generalisation. There was no difference in clinical milestones in those with focal or generalised seizures.
Notably seizures were associated with immediate prior or simultaneous infection in 16/34 (47%) patients who developed seizures after MS onset and in none of those who had seizures prior to MS onset. Concurrent infections were urinary tract infection (UTI) in 10, of which one resulted in sepsis, and chest infections in four, of which one led to death. In addition there was one case of a buttock abscess and one sepsis with an unknown source but a cerebrospinal fluid examination was normal. In those with concurrent infection seizures occurred at a significantly younger age (42.3±16.5 years) compared with those who did not have concurrent infection (54.6±9.5, p < 0.026, Wilcox test); there was no difference otherwise in age of MS onset, age of reaching Expanded Disability Status Scale (EDSS) score of 7.0 and death.
Earlier wheelchair use and death occur in those with seizures and MS though seizures occurred predominantly in the latter phase of MS
The mean age of death was significantly earlier in those with seizures (57.6 vs 62.8, t-test, p < 0.022). Age of death was also earlier in a Kaplan-Meier analysis (Figure 1(a)). The presence of seizures was associated with earlier wheelchair use (44.7 vs 49.3, t-test, p < 0.03), confirmed on Kaplan-Meier analysis (log rank test, p < 0.03). However, there was no difference in the age of MS onset between those with and without seizures. After 10 years of MS, nine developed seizures (24%), and after 15 years 15 developed seizures (41%), the majority developing seizures after 15 years of disease. Plotting the frequency of seizures as related to the proportion of each person’s disease (time from age at MS onset to seizure onset/time from age at MS onset to age died) (Figure 1(b)), confirmed the majority of seizures occur in the latter 20% of the disease course. Plotting the frequency of seizures as it related to the proportion of each patient’s time to require a wheelchair (time from age at MS onset to seizure onset/time from MS onset to age at wheelchair use) demonstrated that the majority of seizures occurred after individuals became wheelchair users (Figure 1(c)).
Figure 1.

Kaplan-Meier survival analysis showed a significantly reduced survival in those with seizures compared to those without ((a), log rank test, *p < 0.01). In (b) seizure onset was quantified for each seizure case as a proportion of when seizures started from disease onset/total disease length (disease onset to death). This demonstrates that the majority of cases occurred in the latter half of their disease. In (c) seizure onset has been quantified from the time from wheelchair use to seizure onset/time from wheelchair use to death. Thus a negative × axis score indicates seizures occurred before wheelchair use started. This illustrates that the majority of seizures occurred after wheelchair use.
Seizures in patients with MS are associated with reduced thickness of the temporal cortex
To examine the neuropathological basis of seizures in MS, three separate cortical gyri: precentral, middle temporal and superior frontal, were studied. We compared individuals with MS and seizures (n = 21) to MS without seizures (n = 30) and controls (n = 8). Cortical thickness was reduced in all three gyri in all MS groups compared to controls but compared to patients with MS alone there was a significant reduction in cortical thickness in patients with MS and seizures in the middle temporal gyrus (Figure 2).
Figure 2.
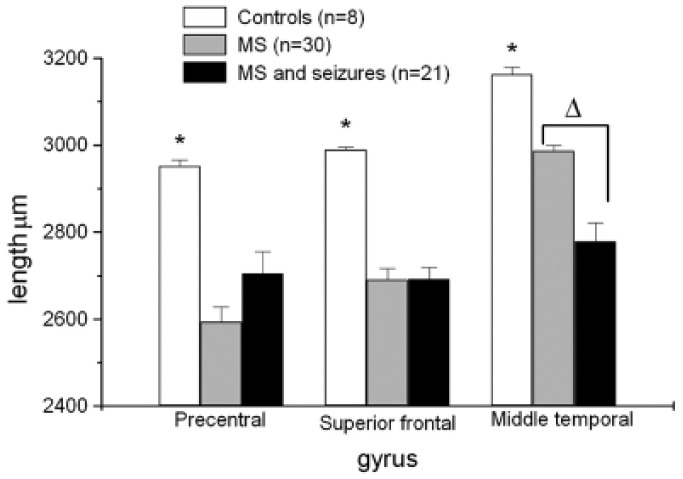
Individuals with MS and seizures (n = 21) were compared to controls (n = 8) and MS alone (n = 30). There were significant reductions in cortical thickness of all gyri in patients with MS compared to controls (mean ± SE).
*p < 0.0015). Compared to patients with MS alone there was a significant reduction in cortical thickness in individuals with MS and seizures in the middle temporal gyrus (Δp < 0.00003).
Seizures in patients with MS are associated with Type 1 GMLs in the temporal cortex
In MS patients with seizures GMLs were always present in the middle temporal gyrus (21/21), significantly above those with MS alone (23/30, p < 0.032). GMLs were not always present in either the precentral or superior frontal gyrus nor were there any differences in the frequency of GMLs between those with and without seizures. Furthermore the increase in GMLs in the middle temporal gyrus was in Type I GMLs (14/21 with MS and seizures, nine of 30 with MS alone, p < 0.01) whereas there was no difference in the frequency of Type III GMLs (Table 2).
Table 2.
GML numbers in the precentral, superiori frontal and middle temporal cortex subdivided into Type I and Type III lesions in MS patients with seizures (n = 21) and with MS alone (n = 30).
| n | Precentral |
Superior frontal |
Middle temporal |
|
|---|---|---|---|---|
| gyrus | gyrus | gyrus | ||
| GMLs of all types present in each patient (%) | ||||
| MS | 30 | 22 (73) | 26 (87) | 23 (77) |
| MS + seizures | 21 | 11 (52) | 16 (76) | 21 (100)a |
| Type I GMLs present in each patient | ||||
| MS | 30 | 11 | 12 | 9 |
| MS + seizures | 21 | 5 | 6 | 14b |
| Type III GMLs present in each patient | ||||
| MS | 30 | 21 | 13 | 13 |
| MS + seizures | 21 | 7 | 9 | 8 |
Lesions were seen in the middle temporal gyrus in 100% of those with MS and seizures, a significant increase above those with MS alone (Fisher’s test, ap < 0.032). A significant increase in Type I GMLs but not Type III GMLs in the middle temporal gyrus was seen in those with seizures compared to those with MS alone (bFisher’s test, p < 0.01) whereas there was no difference in the frequency of Type III GMLs. GML: grey matter lesion; MS: multiple sclerosis.
Selective cortical layer IV and VI interneuron loss occurs in the temporal cortex in those with seizures and MS
A significant reduction in total NeuN+ neuronal numbers was detected in those patients with MS (mean 19%) compared to controls. In particular, there was selective neuronal loss in layers IV (37% vs controls) and VI (48% vs controls) of the middle temporal gyrus in patients with seizures and MS compared to individuals with MS and no seizures. GAD67+ interneuron (Figure 3) cell loss mirrored the neuronal cell loss observed in layers IV and VI in the middle temporal gyrus (Figure 4(b), (d)). No significant differences in neuronal density were found in the superior frontal gyrus (Figure 4(a), (c)).
Figure 3.
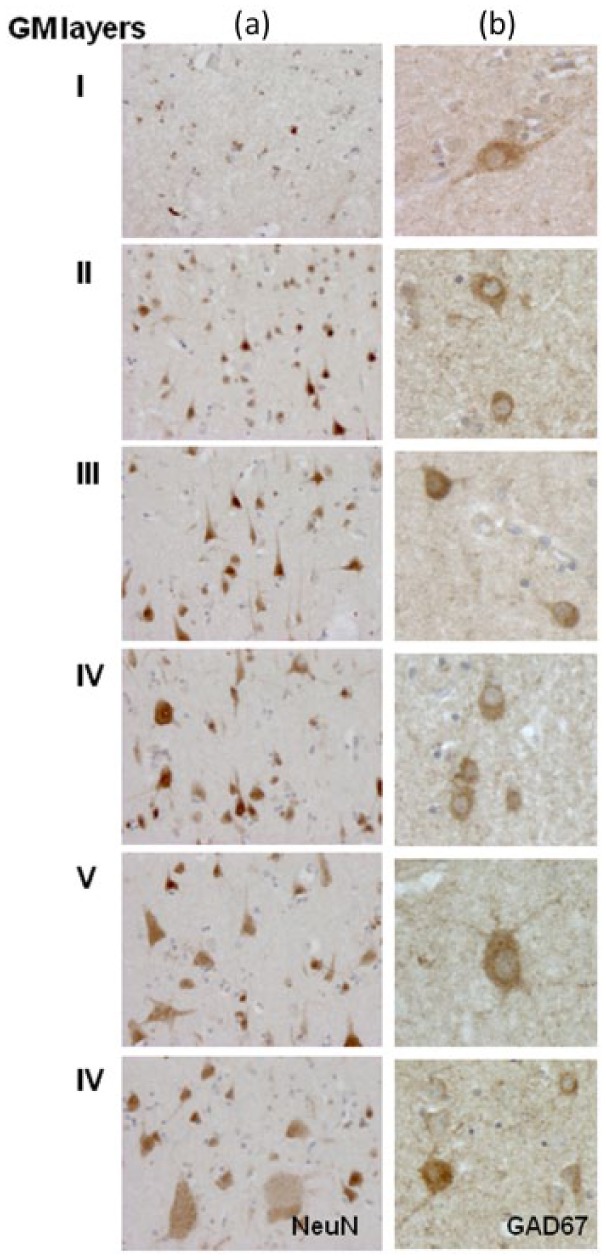
Neuronal distribution in middle temporal cortex. NeuN (column (a)) and GAD67 (column (b)) antibodies were used to localise respectively total neurons and interneurons in the temporal and cortex. Quantification of the numerical density of the NeuN+ nuclei and GAD67+ cells is shown in Figure 4. High magnification of GAD67 immunostaining illustrates the variability in size and shape of the interneuron population in the different cortical layers (column (b)). Original magnification: 100× = column (a), 200× = column (b). GM: grey matter.
Figure 4.

Neuronal cell counts ((a), (c)) and GAD-67+ interneuron cell counts ((b), (d)) were performed in six patients with MS and seizures and six individuals with MS alone in each layer of the precentral ((a), (b)) and middle temporal gyrus ((c), (d)) and compared to six control cases. All cases were randomly chosen and analysed blinded to case identification. The percentage of reduction in counts compared to controls is shown (mean ± SE). There is a significant (*p < 0.001) reduction in the numbers of neurons and interneurons in layers IV and VI of the middle temporal gyrus. MS: multiple sclerosis.
Selective cortical layer IV and VI interneuron loss in the temporal cortex is related to the presence of Type 1 GMLs
Histologically, Type I GMLs in the temporal lobe were significantly more numerous (14/21 vs 9/30, p < 0.01) in MS with seizures than those without (Figure 5(a), (b), Table 2). To determine whether local underlying Type I GMLs within the middle temporal gyrus were associated with the loss of GAD67+ interneurons, neuronal counts were performed in six controls, six patients with MS and seizures and six individuals with MS alone in layers IV (Figure 5(c)) and VI (Figure 5(d)). Counts were performed in the middle temporal gyrus directly overlying a Type I GML and where there was no Type I GML. The interneuron cell loss was significantly increased by 43.4% in layer IV (Figure 5(c)) and 54.3% in layer VI, where there was an underlying lesion compared to where there was no lesion (Figure 5(d)).
Figure 5.
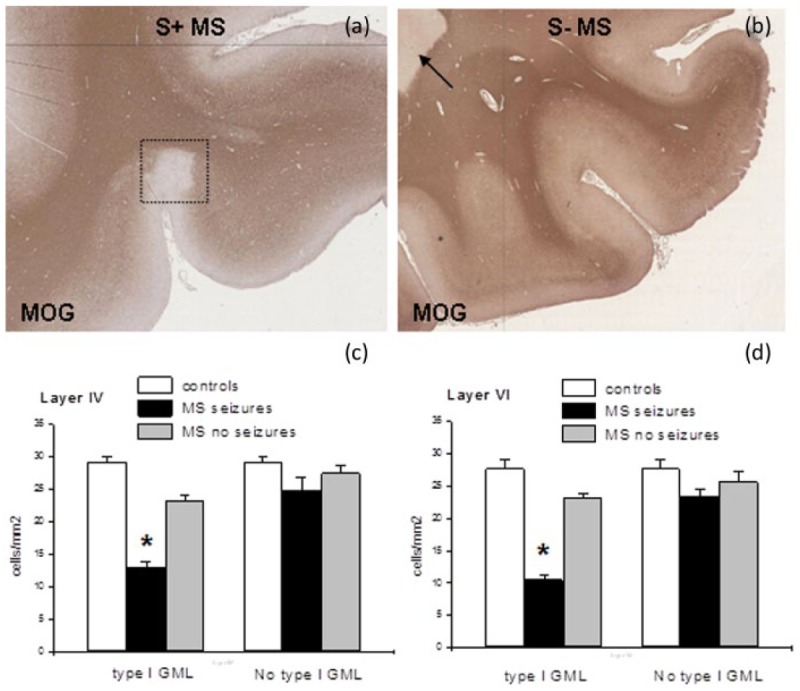
Demyelination in the temporal grey matter (GM) and white matter (WM) of seizure-positive (a) and seizure-negative (b) cases. Immunostaining for the myelin oligodendrocyte protein, MOG, shows a Type I GML (in the rectangle), in a seizure-positive multiple sclerosis (MS) patient (MS234). Conversely, only a circumscribed WM lesion (arrow in (b)) is shown in a seizure-negative patient (MS46). Original magnification: (a), (b) (tiled images) = 100×. Interneuron cell counts were performed in patients with MS with and without seizures as well as in controls in layers IV (c) and VI (d) to assess the effect of a localised Type I GML. Cell counts (mean ± SE) revealed that the interneuron cell loss was significantly (*p < 0.001) reduced in layers IV and VI only local to a Type I GML.
The presence of inflammatory cells or mitochondrial chain activity changes do not explain the interneuron cell loss in layers IV and VI
There was no correlation between the degree of neuronal loss in cortical layers IV and VI and the density of major histocompatibility complex (MHC) class II+-activated microglia and/or CD3+ T lymphocytes present within the GML in the temporal cortex. Mitochondrial respiratory chain complex II and complex IV activity was reduced in a proportion of neurons in Type I GMLs. Both complex II and complex IV activity within NeuN+ cells were significantly reduced in cortical layer VI of Type I GMLs (n = 6) compared with cortical layer VI neurons from controls (n = 6), but there was no difference in those with MS with or without seizures (Table 3).
Table 3.
Complex II and IV activity in Neu+ cells within layer VI of the grey matter within lesions, in normal-appearing grey matter (NAGM) and in controls.
| n | Mitochondrial activity in layer VI (mean ± SD) | |
|---|---|---|
| Complex II | ||
| Type I GML | 28 | 4.16±7.36a |
| NAGM | 29 | 8.35± 4.97 |
| Controls | 20 | 8.26±6.29 |
| Complex IV | ||
| Type I GML | 42 | 1.77±7.77b |
| NAGM | 53 | 5.33±4.01 |
| Controls | 44 | 7.44±5.60 |
There was a significant reduction in complex II (ap = 0.03) and complex IV (bp = 0.001) activity within the Type I GML only. GML: grey matter lesion.
Discussion
This work has shown that seizures are significantly increased in MS over the lifespan of the disease in a population-based cohort. We confirm that seizures increase as the disease progresses, implying seizures are the direct result of the neurodegenerative disease process. Supporting this we show that seizures are associated with earlier use of a wheelchair and an earlier death, but not with early disease onset or early high disease activity.27 The unique advantage of this study is that it has allowed us to show Type I GMLs underlie a local loss of interneurons in cortical layers IV and VI in the temporal lobe in association with seizures. There are some limitations in this study. Firstly we studied only three areas of the brain; this does give us a view of the cortex but is not exhaustive. Secondly we were able to study in detail only six patients with MS and seizures, though five did have seizures associated with concurrent infection.
All population-based cohorts have some bias but can still offer valuable insights.28 The UKMSTB population potentially is biased towards more severe disease as there is a younger mean age of death compared to other populations.29,30 However, the frequency of PPMS is within the range seen in other populations,28 and the high rates of SPMS result from its unique perspective: a diagnosis made at the end of the disease. Here we have confirmed that the risk of seizures in MS is significantly raised,3,8 not only by showing an increased percentage but also through identifying the clinical context of that risk. People with MS who had seizures at any time during their disease took less time from disease onset to require a wheelchair and died earlier, although seizures were predominantly associated with concurrent infection. This confirms that seizure risk increases with more aggressive disease as previously implied.10,11
The fact that seizures occur when cortical damage and neuronal loss is more severe and after a wheelchair is required when the disease is progressive supports a neurodegenerative process underlying seizures. Further support arises from the finding that seizures are not associated with predictors of early inflammatory activity. Given that seizures arise from neuronal dysfunction/damage and are increased in the context of cortical damage in neurodegenerative diseases such as Alzheimer’s,31 this finding makes sense. However, it is not clear whether frequency of seizures or poor seizure control is associated with more severe disease.
Here we have shown for the first time that in those MS cases in which seizures occur there is localised cortical damage within the middle temporal gyrus that could be a result of loss of interneurons in cortical layers IV and VI. This raises two questions: What pathology could underlie this and how could it contribute to seizure generation? Type I GMLs extending from the WM appear to underlie the temporal lobe damage in those with seizures. Although this may produce general effects on cerebral function, we have shown that it is localised to the area above the lesion itself. The findings are consistent with MRI studies in which Type I GMLs were associated with seizures in early MS.10,11 It appears that this anatomical localisation is important as Type III GMLs in the precentral gyrus are associated with a different pattern of cortical layer damage and are not associated with seizures.20 Layer VI cell loss could be as a result of damage from inflammatory cells ‘spilling’ into the GM from their focus in the underlying WM. However, layer IV cell loss was not associated with the presence of any infiltrating inflammatory cells. The pathology in layers IV and VI may arise from damage to the axons of neurons that project through the damaged WM of the Type I lesions to form synapses on interneurons. Loss of GABAergic inhibitory interneuron input from layer IV to excitatory pyramidal neurons in layer V in particular will lead to excessive excitatory output increasing susceptibility to abnormal electrical activity and to seizures.32 We cannot, however, exclude a role for seizures themselves causing the loss of interneurons.
The origin of seizures in MS could have a variety of causes arising from the anatomy of the cell injury and/or the pathology underlying the injury. We did not find any evidence of specific mitochondrial dysfunction in patients with MS and epilepsy despite the known involvement of mitochondria in MS and seizures,19,26 but we did see mitochondrial dysfunction within Type I GMLs in cortical layer VI. Mitochondrial dysfunction is associated with neuronal excitability and glutaminergic activity that could potentially support the generation of seizures.33 Together with the GABAergic interneuron cell loss in layer VI in the middle temporal gyrus, dysfunction of surviving neurons in layer VI due to an energy deficit may play an important role in the pathogenesis of focal-onset seizures in MS patients.
What is notable here is that 47% of seizures in MS were associated with concurrent infections but not primary central nervous system (CNS) infections. This could be due to infection itself or the associated fever. Peripheral infection drives central inflammation and could potentially facilitate seizures.34 However, febrile seizures are well described in children where they appear, as here, to be based on abnormalities in GABAergic transmission.35 Previous study of MS cortex has found a decrease in presynaptic and postsynaptic components of GABAergic neurotransmission and in the density of inhibitory interneuron processes in MS cortex,19 suggesting that changes in the excitatory/inhibitory balance in the GM could have an important role in seizure induction. Here we have shown these changes in patients with seizures are particularly focused in particular layers in the temporal cortex, and these changes could increase susceptibility to seizures in the context of concurrent infection (Figure 6).
Figure 6.
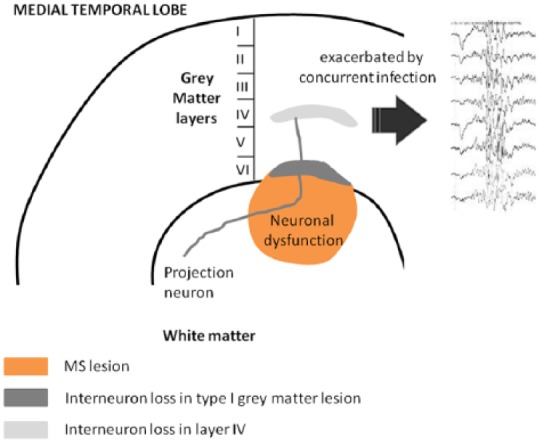
A proposed mechanism of seizure generation in the temporal lobe of patients with MS. A Type I GML together with the associated underlying white matter lesion, evidenced by and in part due to the presence of activated microglia, produce neuronal dysfunction (mitochondrial stress) and cell death (interneuron loss within layer VI). Resulting damage to projecting neurons contributes to the loss of interneurons in layer IV. The resulting loss of inhibition enhances the risk of seizures. Additional stressors including concurrent infection are then more likely to result in the emergence of seizures. MS: multiple sclerosis; GML: grey matter lesion.
Acknowledgments
All tissue samples were supplied by the UKMSTB (www.ukmstissuebank.imperial.ac.uk), funded by the MS Society (registered charity 207495). The authors would like to thank members of the UKMSTB Team (F Roncaroli, S Gentleman, S Fordham and C MacDonald) for assistance in the collection and characterisation of the material used in this study and Dr Alessia Nicotra for help with EEG tracing in Figure 6. RN is grateful for support from the NIHR Biomedical Research Centre funding scheme.
Footnotes
Conflict of interest: None declared.
Funding: This work was supported by a PhD fellowship from S. Gratton (to RM), by the Medical Research Council (G0700356 to RR), the MS Society of Great Britain and Northern Ireland (910/09 to RR, RN), by the Italian MS Foundation grant (FISM 2011/R/23 to RM) and the Italian Ministry of Health grant (GR-2010-2313255 to RM).
Contributor Information
Richard Nicholas, UK Multiple Sclerosis Tissue Bank, Wolfson Neuroscience Laboratories, Imperial College London Faculty of Medicine, Hammersmith Hospital Campus, UK.
Roberta Magliozzi, UK Multiple Sclerosis Tissue Bank, Wolfson Neuroscience Laboratories, Imperial College London Faculty of Medicine, Hammersmith Hospital Campus, UK/Department of Cell Biology and Neuroscience, Istituto Superiore di Sanità, Rome, Italy.
Graham Campbell, Centre for Neuroregeneration, University of Edinburgh, UK.
Don Mahad, Centre for Neuroregeneration, University of Edinburgh, UK/Centre for Clinical Brain Sciences, University of Edinburgh, UK.
Richard Reynolds, UK Multiple Sclerosis Tissue Bank, Wolfson Neuroscience Laboratories, Imperial College London Faculty of Medicine, Hammersmith Hospital Campus, UK.
References
- 1. Williams GH, Jr, Nosik W, Hunter J. Convulsions as manifestation of multiple sclerosis. JAMA 1952; 150: 990–992. [DOI] [PubMed] [Google Scholar]
- 2. Kelley BJ, Rodriguez M. Seizures in patients with multiple sclerosis: Epidemiology, pathophysiology and management. CNS Drugs 2009; 23: 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olafsson E, Benedikz J, Hauser WA. Risk of epilepsy in patients with multiple sclerosis: A population-based study in Iceland. Epilepsia 1999; 40: 745–747. [DOI] [PubMed] [Google Scholar]
- 4. Poser CM, Brinar VV. Epilepsy and multiple sclerosis. Epilepsy Behav 2003; 4: 6–12. [DOI] [PubMed] [Google Scholar]
- 5. Engelsen BA, Grønning M. Epileptic seizures in patients with multiple sclerosis. Is the prognosis of epilepsy underestimated? Seizure 1997; 6: 377–382. [DOI] [PubMed] [Google Scholar]
- 6. Koch M, Uyttenboogaart M, Polman S, et al. Seizures in multiple sclerosis. Epilepsia 2008; 49: 948–953. [DOI] [PubMed] [Google Scholar]
- 7. Martínez-Juárez IE, López-Meza E, González-Aragón Mdel C, et al. Epilepsy and multiple sclerosis: Increased risk among progressive forms. Epilepsy Res 2009; 84: 250–253. [DOI] [PubMed] [Google Scholar]
- 8. Nicoletti A, Sofia V, Biondi R, et al. Epilepsy and multiple sclerosis in Sicily: A population-based study. Epilepsia 2003; 44: 1445–1448. [DOI] [PubMed] [Google Scholar]
- 9. Nyquist P, Cascino GD, McClelland RL, et al. Incidence of seizures in patients with multiple sclerosis: A population-based study. Mayo Clin Proc 2002; 77: 910–912. [DOI] [PubMed] [Google Scholar]
- 10. Moreau T, Sochurkova D, Lemesle M, et al. Epilepsy in patients with multiple sclerosis: Radiological-clinical correlations. Epilepsia 1998; 39: 893–896. [DOI] [PubMed] [Google Scholar]
- 11. Schuele SU, Kellinghaus C, Shook SJ, et al. Incidence of seizures in patients with multiple sclerosis treated with intrathecal baclofen. Neurology 2005; 64: 1086–1087. [DOI] [PubMed] [Google Scholar]
- 12. ILAE Commission Report. The epidemiology of the epilepsies: Future directions. International League Against Epilepsy. Epilepsia 1997; 38: 614–618. [PubMed] [Google Scholar]
- 13. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005; 128: 2705–2712. [DOI] [PubMed] [Google Scholar]
- 14. Stadelmann C, Albert M, Wegner C, et al. Cortical pathology in multiple sclerosis. Curr Opin Neurol 2008; 21: 229–234. [DOI] [PubMed] [Google Scholar]
- 15. Martínez-Lapiscina EH, Ayuso T, Lacruz F, et al. Cortico-juxtacortical involvement increases risk of epileptic seizures in multiple sclerosis. Acta Neurol Scand 2013; 128: 24–31. [DOI] [PubMed] [Google Scholar]
- 16. Thompson AJ, Kermode AG, Moseley IF, et al. Seizures due to multiple sclerosis: Seven patients with MRI correlations. J Neurol Neurosurg Psychiatry 1993; 56: 1317–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Truyen L, Barkhof F, Frequin ST, et al. Magnetic resonance imaging of epilepsy in multiple sclerosis: A case control study. Implications for treatment trials with 4-aminopyridine. Mult Scler 1996; 1: 213–217. [PubMed] [Google Scholar]
- 18. Calabrese M, Grossi P, Favaretto A, et al. Cortical pathology in multiple sclerosis patients with epilepsy: A 3 year longitudinal study. J Neurol Neurosurg Psychiatry 2012; 83: 49–54. [DOI] [PubMed] [Google Scholar]
- 19. Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 2006; 59: 478–489. [DOI] [PubMed] [Google Scholar]
- 20. Magliozzi R, Howell OW, Roncaroli F, et al. Substantial neuronal loss in the grey matter accompanies the presence of meningeal B-cell follicles in multiple sclerosis. Ann Neurol 2010; 68: 477–493. [DOI] [PubMed] [Google Scholar]
- 21. Allen I, Gentleman S, Graeber M, et al. International Classification of Diseases of the Nervous System – Demyelinating Diseases – Multiple Sclerosis, www.icdns.org/forums (2006, Accessed: 23 December 2014).
- 22. Reynolds R, Nicholas R, Roncaroli F, et al. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol 2011; 17: 1211–1217. [DOI] [PubMed] [Google Scholar]
- 23. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989; 30: 389–399. [DOI] [PubMed] [Google Scholar]
- 24. Choi S, Howell OW, Carassiti D, et al. Does meningeal inflammation play a role in the pathology of primary progressive multiple sclerosis? Brain 2012; 135: 2925–2937. [DOI] [PubMed] [Google Scholar]
- 25. Peterson JW, Bö L, Mörk S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 2001; 50: 389–400. [DOI] [PubMed] [Google Scholar]
- 26. Campbell GR, Ziabreva I, Reeve AK, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 2011; 69: 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: A geographically based study 10: Relapses and long-term disability. Brain 2010; 133: 1914–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hurwitz BJ. Analysis of current multiple sclerosis registries. Neurology 2011; 76 (Suppl 1): S7–S13. [DOI] [PubMed] [Google Scholar]
- 29. Tremlett H, Paty D, Devonshire V. Disability progression in multiple sclerosis is slower than previously reported. Neurology 2006; 66: 172–177. [DOI] [PubMed] [Google Scholar]
- 30. Weinshenker BG, Rice GPA, Noseworthy JH, et al. The natural history of multiple sclerosis: A geographically based study. I. Clinical course and disability. Brain 1989; 112: 133–146. [DOI] [PubMed] [Google Scholar]
- 31. Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med 2010; 12: 71–77. [DOI] [PubMed] [Google Scholar]
- 32. Muzzi P, Camera P, Di Cunto F, et al. Deletion of the citron kinase gene selectively affects the number and distribution of interneurons in barrelfield cortex. J Comp Neurol 2009; 513: 249–264. [DOI] [PubMed] [Google Scholar]
- 33. Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res 2010; 88: 23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 2007; 7: 161–167. [DOI] [PubMed] [Google Scholar]
- 35. Kang JQ, Shen W, Macdonald R. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci 2009; 29: 2845–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]